

Abstract
Background & Aims
Early-onset colorectal cancer (CRC, in persons younger than 50 years old) is increasing in incidence; yet, in the absence of a family history of CRC, this population lacks harmonized recommendations for prevention. We aimed to determine whether a polygenic risk score (PRS) developed from 95 CRC-associated common genetic risk variants was associated with risk for early-onset CRC.
Methods
We studied risk for CRC associated with a weighted PRS in 12,197 participants younger than 50 years old vs 95,865 participants 50 years or older. PRS was calculated based on single-nucleotide polymorphisms associated with CRC in a large-scale genome-wide association study as of January 2019. Participants were pooled from 3 large consortia that provided clinical and genotyping data: the Colon Cancer Family Registry, the Colorectal Transdisciplinary study, and the Genetics and Epidemiology of Colorectal Cancer Consortium and were all of genetically defined European descent. Findings were replicated in an independent cohort of 72,573 participants.
Results
Overall associations with CRC per standard deviation of PRS were significant for early-onset cancer, and were stronger compared with late-onset cancer (P for interaction=.01); when we compared the highest PRS quartile with the lowest, risk increased 3.7-fold for early-onset CRC (95% CI, 3.28–4.24) vs 2.9-fold for late-onset CRC (95% CI, 2.80–3.04). This association was strongest for participants without a first-degree family history of CRC (P for interaction=5.61×10−5). When we compared the highest with the lowest quartiles in this group, risk increased 4.3-fold for early-onset CRC (95% CI, 3.61–5.01) vs 2.9-fold for late-onset CRC (95% CI, 2.70–3.00). Sensitivity analyses were consistent with these findings.
Conclusions
In an analysis of associations with CRC per standard deviation of PRS, we found the cumulative burden of CRC-associated common genetic variants to associate with early-onset cancer, and to be more strongly associated with early-onset than late-onset cancer—particularly in the absence of CRC family history. Analyses of PRS, along with environmental and lifestyle risk factors, might identify younger individuals who would benefit from preventative measures.
Keywords: colon cancer, SNP, penetrance, EOCRC
Lay Summary
Genetic variants associated with risk of colorectal cancer are more strongly associated with development of early-onset compared with late-onset colorectal cancer.
Graphical Abstract
Introduction
Colorectal cancer (CRC) incidence and mortality have been declining in the U.S. over the last several decades.1 These reductions are largely attributed to successes in CRC early detection, surveillance, and treatment for this disease.2, 3 In contrast to these overall trends, the incidence of CRC in individuals less than 50 years of age (early-onset disease) has been increasing in the U.S. and elsewhere:4 early-onset CRC incidence in the U.S. has increased by an average of 1.8% annually from 1992–2012, and is projected to account for 10% to 25% of newly-diagnosed CRC by 2030.1, 5–10 Furthermore, early-onset CRC tends to present with higher pathologic grade, distant disease, and a greater incidence of recurrence and metastatic disease.5 In response to this newly recognized disease burden, the US Preventative Services Task Force,11 the American Cancer Society,12 the U.S. Multi-Society Task Force on Colorectal Cancer13 and other professional bodies14 have initiated discussions on the merits of revising recent consensus CRC prevention guidelines to include early detection of average-risk individuals younger than 50 years of age. While the American Cancer Society recommends lowering the screening age to 45 years for individuals at average risk,12 others recommend targeting only high-risk groups for early detection.13, 15
Weighing against the potential benefits of CRC early detection and prevention programs targeted to those aged younger than 50 years are concerns about adverse side effects and associated costs.14, 16 New approaches to disease prevention in younger adults are warranted, and assessing germline genetic variants, along with other known risk factors, could facilitate tailored early detection of high risk individuals due to their genetic makeup and lifestyle. To date, genetic research on factors associated with early-onset CRC has been limited largely to the rare monogenic, high-penetrance genetic syndromes associated with this disease in high-risk families, while the frequently occurring low-penetrance polymorphisms have been understudied.
Here, we report on CRC risks for early (<50 years of age) and late-onset disease (≥50 years of age) associated with a polygenic risk score (PRS) developed from 95 common genetic risk variants identified in previous CRC genome-wide association studies (GWAS). Our research provides the first substantive evidence that early-onset CRC exhibits differential genetic risks, compared with late-onset disease, due to low-penetrance, common genetic polymorphisms. The findings of our research may contribute to the identification of individuals susceptible to early-onset CRC for tailored early detection or other preventive interventions.
Methods
Study Participants
We studied 108,062 participants in the discovery dataset, including 50,023 CRC cases and 58,039 controls. Participants for this study were pooled from three large consortia that provided clinical and genotyping data: the Colon Cancer Family Registry (CCFR), the Colorectal Transdisciplinary (CORECT) Study, and the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO) (Table 1 and Table S1) (for additional study information, see earlier publications17–20). All analyses were restricted to participants of genetically defined European descent. Family history of CRC was ascertained through self-report or interviewer-administered questionnaire, and defined as having one or more first-degree relatives with CRC. Participant recruitment across all studies occurred between the 1990’s and the early 2010’s. All study participants provided written informed consent and studies were approved by their respective Institutional Review Boards (see Supplementary Information).
Table 1:
Baseline study characteristics of the discovery and replication datasets
| Discovery dataset | Replication dataset | |||||||
|---|---|---|---|---|---|---|---|---|
| Cases (N=50,023) | Controls (N=58,039) | All participants | CRC Cases | |||||
| <50 Years-Old | ≥50 Years-Old | <50 Years-Old | ≥50 Years-Old | Eligible cohort | CRC cases | <50 Years-Old | ≥50 Years-Old | |
| N | 5479 | 44544 | 6718 | 51321 | 72573 | 1093 | 25 | 1068 |
| Age, Mean (SD) | 43.1 (5.6) | 66.5 (8.7) | 41.3 (7.2) | 65.3 (8.3) | 71.5 (13.1) | 73.1 (10.8) | 45.2 (3.3) | 73.7 (10.1) |
| Sex, N (%) | ||||||||
| Male | 2767 (50.5) | 24145 (54.2) | 3272 (48.7) | 26886 (52.4) | 30160 (41.6) | 526 (48.1) | 9 (36.0) | 517 (48.4) |
| Female | 2706 (49.4) | 20336 (45.7) | 3446 (51.3) | 24435 (47.6) | 42413 (58.4) | 567 (51.9) | 16 (64.0) | 551 (51.6) |
| Missing | 6 (0.1) | 63 (0.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Family History of CRC, N (%) | ||||||||
| Yes | 944 (17.2) | 5558 (12.5) | 578 (8.6) | 5330 (10.4) | 6956 (9.6) | 204 (18.7) | 7 (28.0) | 197 (18.4) |
| No | 3159 (57.7) | 24028 (53.9) | 4130 (61.5) | 28317 (55.2) | 65617 (90.4) | 889 (81.3) | 18 (72.0) | 871 (81.6) |
| Missing | 1376 (25.1) | 14958 (33.6) | 2010 (29.9) | 17674 (34.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Tumor Site, N (%) | ||||||||
| Proximal Colon | 1231 (22.5) | 12978 (29.1) | -- | -- | -- | -- | -- | -- |
| Distal Colon | 1442 (26.3) | 12036 (27.0) | -- | -- | -- | -- | -- | -- |
| Rectum | 1920 (35.0) | 12918 (29.0) | -- | -- | -- | -- | -- | -- |
| Missing | 886 (16.2) | 6612 (14.8) | -- | -- | -- | -- | -- | -- |
| PRS, N (%) | ||||||||
| Quartile 1 | 693 (12.6) | 6227 (14.0) | 1659 (24.7) | 12863 (25.1) | 18175 (25.0) | 163 (14.9) | 2 (8.0) | 161 (15.1) |
| Quartile 2 | 1048 (19.1) | 8824 (19.8) | 1666 (24.8) | 12848 (25.0) | 18150 (25.0) | 232 (21.2) | 4 (16.0) | 228 (21.3) |
| Quartile 3 | 1396 (25.5) | 11877 (26.7) | 1674 (24.9) | 12824 (25.0) | 18132 (25.0) | 287 (26.3) | 7 (28.0) | 280 (26.2) |
| Quartile 4 | 2342 (42.7) | 17616 (39.5) | 1719 (25.6) | 12786 (24.9) | 18116 (25.0) | 411 (37.6) | 12 (48.0) | 399 (37.4) |
Genotyping and SNP Selection
We included 95 CRC-risk-associated SNPs that reached genome-wide significance (p ≤ 5×10−8), in large-scale GWAS, as of January, 2019. No new discovery of CRC-related SNPs was carried out here. Individual participant and genotype data for the 95 SNPs were extracted from GWAS and imputed to the Haplotype Reference Consortium panel, which provides high-quality, accurate imputation for variants with a minor allele frequency as low as 0.1%.21 For details, see Huyghe et al.17 Additional information on SNPs can be located in Table S2.
Statistical Analysis
For cases and controls, we compared baseline participant characteristics between individuals who had a reference age of <50 years to those with a reference age of ≥50 years of age. For cases, reference age was defined as the age of diagnosis of first primary CRC. For controls, reference age was defined as the age at selection.
Genotyped SNPs were coded as 0, 1, or 2 copies of the risk allele. Imputed SNPs were coded for the expected number of copies of the risk allele, as imputed dosages. Potential population substructure within the GECCO, CCFR, and CORECT studies was accounted for through adjustment by principal components of genetic ancestry. To develop the weighted PRS, we used log-odds ratios derived from the literature for 55 of the SNPs, and for the remaining 40 SNPs that were first identified within this discovery dataset, we computed log-odds ratios from a regression model fit with CRC as the outcome (1 vs. 0) and the following independent variables: 95 SNPs, age (in years), sex, principal components, and genotype platform. For the 40 SNPs identified within this discovery dataset, we then implemented a conservative winner’s curse adjustment of the log-odds ratios from the risk model, using Zhong and Prentice’s approach.22 We then weighted the PRS for individuals, by multiplying the number of risk alleles for each SNP by their adjusted log-odds ratios, summing and recoding as a percentile based on the distribution in the controls. The final PRS was modelled as a continuous variable per 1 standard deviation (SD), transformed to the standard normal distribution. Odds ratios and 95% confidence intervals were also estimated comparing quartiles of PRS.
We used unconditional logistic regression to assess the association between the PRS and CRC for those with a reference age <50 years and for those with a reference age ≥50 years. All models additionally included sex, reference age in years, principal components, and genotype platform. Further adjustment by study was not warranted as extensive genome-wide analyses with and without adjusting for study have been conducted, with the results being consistent.17 To test for differences in associations across age, an interaction term was included for age category (<50, ≥50) and PRS (continuous). Models were also examined separately by first-degree family history of CRC. We evaluated the discriminatory accuracy of the risk prediction models by calculating the area under the receiver operating characteristic curve (AUC) for 5-year diagnostic age groups, adjusting for sex, PCs, and genotype platform, using the adjusted.ROC function from the R Package ROCt.
For the larger group with no first-degree family history of CRC, additional sub-group analyses were performed including estimation of CRC risk within specific reference-age groups (15–39, 40–49, 50–59, 60–69, and 70–79 years) and by disease site (proximal colon, distal colon, and rectum). The interaction term used to assess differences in associations across age categories consisted of age as a continuous variable and PRS (continuous). Multinomial logistic regression was used to assess risk differentials by disease site within age strata. Analyses were completed using the R statistical software program version 3.5.1.
Sensitivity Analyses
Replication accounting for cases with Lynch syndrome
Screening of colorectal cancer cases for the presence of Lynch syndrome was systematically carried out for CRC cases recruited through the Ohio State University Medical Center (OSUMC) (Table S1: HNPCC, OCCPI, and OSUMC) as described in detail elsewhere23–25. All cases were screened for MMR deficiency using immunohistochemical analysis. Cases with probable characteristics of Lynch syndrome were subjected to additional genetic testing for conclusively determining a diagnosis of Lynch syndrome based on the presence of one or more germline high penetrance mutations in DNA mismatch repair genes (MLH1, MSH2, MSH6, PMS2) or the EPCAM gene.
Using unconditional logistic regression in these studies, we evaluated the association between the PRS and CRC for those aged <50 years and for those ≥50 years of age, with consideration of Lynch syndrome status among cases. All models additionally included sex, reference age in years, and principal components. To test for differences in associations across age, an interaction term was included for age category (<50, ≥50) and PRS (continuous).
Replication in an independent cohort
To independently replicate the association of this PRS with younger and older-onset CRC, we studied all 72,573 participants of European ancestry who were genotyped in the Research Program on Genes, Environment and Health (RPGEH), a cohort comprised of Kaiser Permanente Northern California (KPNC) health plan members.26, 27 This cohort was not included in the discovery of any of the 95 CRC genetic risk variants. Cancer history was determined from initiation of health plan membership by linkage to the KPNC Cancer Registry, which adheres to the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program standards.
Family history of CRC, defined as having one or more first-degree relatives with CRC, was ascertained through a baseline study questionnaire, electronic family history data in the medical records, and International Classification of Disease codes Z80.0 (Family history of malignant neoplasm of digestive organs) and V16.0 (Cancer family history, gastrointestinal tract). Analyses were restricted to participants of genetically defined European descent. All study participants provided written informed consent, and the study was approved by the Kaiser Permanente Northern California Institutional Review Board.
RPGEH biospecimens were genotyped using the Affymetrix Axiom platform. Details on the calling and quality control can be found elsewhere.28 Consistent with genetic data in the discovery set, we imputed the genotyped data to the Haplotype Reference Consortium. To develop the PRS for this replication, we used 94 SNPs from the discovery dataset, as described above, and, for 1 unmatched SNP (rs755229494), we included the best available surrogate (rs112334046, R2=0.40, MAF=0.0026).
For the longitudinal replication cohort, we employed Cox proportional hazards models to assess the association of PRS with CRC, which was not feasible for the discovery dataset since it included case-control data. The coefficients from the model fit with 95 SNPs in the discovery dataset were used to fit the PRS in the replication analysis, thereby reducing potential for overfitting. The observed time was defined from the age of initial KPNC enrollment to the earliest of age at CRC diagnosis, death or end of follow-up (the RPGEH cohort was followed until December 31, 2016). The replication models also included sex and principal components to account for potential population substructure. Estimates of absolute risk are inferred using Kaplan-Meier plots produced using RPGEH data.
Results
Early-onset CRC cases (N=5,479) had a mean age at diagnosis of 43.1 years, while the older-onset cases (N=44,544) had a mean age at diagnosis of 66.5 years (Table 1). Men and women were approximately equally represented across cases and controls. A first-degree family history of CRC, among those ascertained for family history, was reported for 17.2% of early-onset and 12.5% for late-onset CRC cases, and, respectively, for 8.6% of younger and 10.4% for older controls. Family history information was missing for >25% of participants; all of whom were from 9 studies that did not query participants on family history and therefore were not included in our family history-specific analyses. Younger onset cases tended to have fewer proximal colon tumors and a greater preponderance of tumors in the rectum. Both early-onset and late-onset CRC cases showed marked skewing toward higher PRS values compared with controls, when represented as quartiles (Table 1) and as a continuous score (Figure S1).
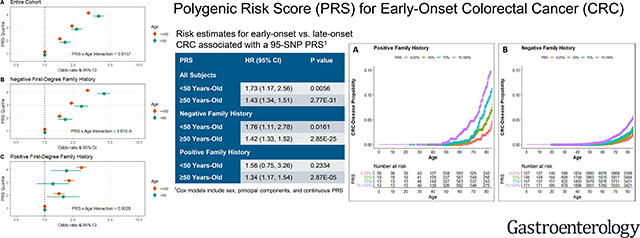
We found that associations with risk for CRC per SD of PRS were significant among participants <50 years of age, and were stronger compared with participants aged ≥50 years (P for interaction = 0.01). Contrasting the highest PRS quartile with the lowest, risks were 3.7-fold higher (OR: 3.73; 95% CI: 3.28, 4.24) for early-onset CRC and 2.9-fold higher (OR: 2.92; 95% CI: 2.80, 3.04) for late-onset disease (Table 2 and Figure 1A). For the larger group of participants who reported a negative first-degree family history of CRC, PRS-associated risks for CRC among participants aged <50 years were also stronger than those for individuals aged ≥50 years (P for interaction = 5.61×10−5); risks comparing the highest with the lowest quartile of PRS were 4.3-fold (OR: 4.26; 95% CI: 3.61, 5.01) for early-onset CRC and 2.9-fold (OR: 2.85; 95% CI: 2.70, 3.00) for late-onset disease (Table 2 and Figure 1B). In contrast, for the smaller group of participants who reported a positive first-degree family history of CRC, risks per SD of PRS tended to be greater for older individuals (P for interaction = 0.003); risks in the highest quartile for PRS were 1.7-fold (OR: 1.70; 95% CI: 1.17, 2.47) for early-onset CRC, and 2.5-fold (OR: 2.47; 95% CI: 2.18, 2.79) for late-onset disease (Table 2 and Figure 1C). The discriminatory capabilities for prediction (i.e., AUC) of these models across the entire age spectrum tended to be highest for early-onset individuals without a family history of CRC, ranging from 0.64 to 0.65 (Table S3).
Table 2:
Risk estimates for early-onset versus late-onset CRC associated with a 95-SNP PRS in the discovery dataseta
| PRS | N (cases) | N (controls) | OR (95% CI) | P value | P value for interactionb |
|---|---|---|---|---|---|
| All Subjects | 0.0137 | ||||
| <50 Years-Old per 1 SD | 5479 | 6718 | 1.64 (1.57, 1.72) | 6.00E-107 | |
| Quartile 1 (ref) | 693 | 1659 | 1.00 | ||
| Quartile 2 | 1048 | 1666 | 1.64 (1.43, 1.89) | 2.07E-12 | |
| Quartile 3 | 1396 | 1674 | 2.19 (1.91, 2.50) | 2.17E-30 | |
| Quartile 4 | 2342 | 1719 | 3.73 (3.28, 4.24) | 1.13E-89 | |
| ≥50 Years-Old per 1 SD | 44544 | 51321 | 1.52 (1.50, 1.54) | < 2.23E-308 | |
| Quartile 1 (ref) | 6227 | 12863 | 1.00 | ||
| Quartile 2 | 8824 | 12848 | 1.45 (1.39, 1.51) | 8.55E-62 | |
| Quartile 3 | 11877 | 12824 | 1.95 (1.87, 2.03) | 1.37E-208 | |
| Quartile 4 | 17616 | 12786 | 2.92 (2.80, 3.04) | < 2.23E-308 | |
| Negative Family History | 5.61E-05 | ||||
| <50 Years-Old per 1 SD | 3159 | 4130 | 1.74 (1.65, 1.84) | 1.33E-81 | |
| Quartile 1 (ref) | 388 | 1085 | 1.00 | ||
| Quartile 2 | 601 | 1025 | 1.66 (1.39, 1.98) | 1.58E-08 | |
| Quartile 3 | 820 | 1001 | 2.46 (2.07, 2.92) | 3.37E-25 | |
| Quartile 4 | 1350 | 1019 | 4.26 (3.61, 5.01) | 3.65E-67 | |
| ≥50 Years-Old per 1 SD | 24028 | 28317 | 1.50 (1.47, 1.53) | < 2.23E-308 | |
| Quartile 1 (ref) | 3529 | 7341 | 1.00 | ||
| Quartile 2 | 4869 | 7083 | 1.44 (1.36, 1.53) | 1.85E-36 | |
| Quartile 3 | 6494 | 7058 | 1.92 (1.82, 2.03) | 6.17E-119 | |
| Quartile 4 | 9136 | 6835 | 2.85 (2.70, 3.00) | < 2.23E-308 | |
| Positive Family History | 0.0028 | ||||
| <50 Years-Old per 1 SD | 944 | 578 | 1.19 (1.05, 1.35) | 0.0063 | |
| Quartile 1 (ref) | 133 | 105 | 1.00 | ||
| Quartile 2 | 203 | 133 | 1.58 (1.05, 2.36) | 0.0265 | |
| Quartile 3 | 208 | 152 | 1.22 (0.82, 1.83) | 0.3277 | |
| Quartile 4 | 400 | 188 | 1.70 (1.17, 2.47) | 0.0052 | |
| ≥50 Years-Old per 1 SD | 5558 | 5330 | 1.42 (1.36, 1.48) | 7.02E-57 | |
| Quartile 1 (ref) | 690 | 1134 | 1.00 | ||
| Quartile 2 | 1037 | 1264 | 1.42 (1.24, 1.63) | 5.85E-07 | |
| Quartile 3 | 1478 | 1343 | 1.81 (1.59, 2.07) | 8.44E-19 | |
| Quartile 4 | 2353 | 1589 | 2.47 (2.18, 2.79) | 2.70E-45 | |
The logistic regression models include age, sex, principal components, genotype platform, and polygenic risk score.
P value produced from interaction term with continuous PRS (per SD) and age (<50 versus ≥50 years).
Figure 1:

Risk estimates for early-onset versus late-onset CRC associated with a 95-SNP PRS in the discovery dataset. (A) Model includes all study participants regardless of first-degree family history of CRC. (B) Model includes study participants without a first-degree family history of CRC. (C) Model includes study participants with a first-degree family history of CRC. Models were adjusted for age, sex, principal components, genotype platform, and polygenic risk score. The interaction p-value reported was produced from a model including an interaction term with a continuous PRS (per SD) and age (<50 years versus ≥50 years).
As the PRS displayed the strongest association for early-onset CRC without a first-degree family history, we investigated whether certain subgroups could account for these strong effects. When stratified further by age at diagnosis, CRC risks were 1.7-fold (OR per SD of PRS: 1.74; 95% CI: 1.55, 1.96) for those diagnosed aged 15–39 years and 1.8-fold (OR per SD of PRS: 1.75; 95% CI: 1.64, 1.87) for those diagnosed aged 40–49 years of age. For participants diagnosed at ≥50 years of age, the related CRC risks were 1.6-fold (OR per SD of PRS: 1.60; 95% CI: 1.54, 1.67) for participants aged 50–59 years, 1.5-fold (OR per SD of PRS: 1.52; 95% CI: 1.48, 1.57) for individuals 60–69 years old, and 1.4-fold (OR per SD of PRS: 1.44; 95% CI: 1.39, 1.49) for those diagnosed between 70–79 years, with age and PRS exhibiting statistical interaction across the entire study age range (Table S4, P for interaction = 3.44×10−10). Furthermore, as found for all cancer sites (Table 2 and Figure 1), the PRS was also more strongly associated with risks for early-onset, compared with late-onset, cancers of the proximal colon, distal colon and rectum (Table S5 and Figure S2), with the greatest risk differentials observed for cancers of the distal colon and rectum (Table S6).
Sensitivity Analyses
Replication accounting for cases with Lynch syndrome
A total of 37 Lynch cases <50 years of age (6.4%, among 574 cases) and 54 Lynch cases ≥50 years of age (2.1%, among 2525 cases) were identified in the Ohio-based studies. Removing Lynch cases from the analysis demonstrated that the relatively small number of these cases did not substantially impact the relationship of PRS with CRC (Table 3). After exclusion of Lynch cases, risks for early-onset CRC per SD of PRS remained similarly increased in participants <50 years of age (OR per SD of PRS: 1.82; 95% CI: 1.61, 2.06) and were greater compared with participants aged ≥50 years (OR per SD of PRS: 1.49; 95% CI: 1.39, 1.60; P for interaction = 0.01). These trends held particularly for participants who reported a negative first-degree family history of CRC (aged <50 years, OR per SD of PRS: 1.83; 95% CI: 1.60, 2.09; aged ≥50 years, OR per SD of PRS: 1.46; 95% CI: 1.35,1.57; P for interaction = 0.01).
Table 3:
Risk estimates for early-onset versus late-onset CRC associated with a 95-SNP PRS among participants with and without Lynch Syndrome, in the Ohio cohorta
| PRS per 1 SD | N (cases) | N (controls) | OR (95% CI) | P value | P value for interactionb |
|---|---|---|---|---|---|
| Including Lynch and Non-Lynch Cases | |||||
| All Subjects | 0.0369 | ||||
| <50 Years-Old | 574 | 979 | 1.73 (1.54, 1.95) | 1.39E-19 | |
| ≥50 Years-Old | 2525 | 1463 | 1.47 (1.37, 1.58) | 1.77E-28 | |
| Negative Family History | 0.0106 | ||||
| <50 Years-Old | 449 | 931 | 1.81 (1.59, 2.07) | 9.64E-19 | |
| ≥50 Years-Old | 1885 | 1271 | 1.45 (1.34, 1.56) | 1.16E-21 | |
| Positive Family History | 0.1517 | ||||
| <50 Years-Old | 106 | 48 | 1.28 (0.84, 1.97) | 0.2530 | |
| ≥50 Years-Old | 565 | 192 | 1.55 (1.30, 1.84) | 1.12E-06 | |
| Excluding Lynch Cases | |||||
| All Subjects | 0.0149 | ||||
| <50 Years-Old | 537 | 979 | 1.82 (1.61, 2.06) | 2.63E-21 | |
| ≥50 Years-Old | 2471 | 1463 | 1.49 (1.39, 1.60) | 1.11E-29 | |
| Negative Family History | 0.0107 | ||||
| <50 Years-Old | 438 | 931 | 1.83 (1.60, 2.09) | 7.50E-19 | |
| ≥50 Years-Old | 1856 | 1271 | 1.46 (1.35, 1.57) | 4.30E-22 | |
| Positive Family History | 0.5627 | ||||
| <50 Years-Old | 80 | 48 | 1.53 (0.98, 2.41) | 0.0635 | |
| ≥50 Years-Old | 540 | 192 | 1.61 (1.34, 1.92) | 2.34E-07 | |
The logistic regression models include age, sex, principal components, and polygenic risk score.
P value produced from interaction term with continuous PRS (per SD) and age (<50 versus ≥50 years).
Replication in an independent cohort
In RPGEH, early-onset CRC cases (N=25) had a mean age of 45.2 years, while the older-onset cases (N=1,068) had a mean age of 73.7 years (Table 1). More women participated than men. A first-degree family history of CRC was reported for 28.0% of early-onset and 18.4% of late-onset CRC cases, compared to 9.6% for the cohort overall. Consistent with the discovery dataset, the distributions of PRS for both early and late-onset CRC cases were skewed towards higher PRS quartiles compared with controls. Right-censoring was due to either death (15%, N=11,165) or lost to follow-up (1%, N=735).
Hazard ratio estimates for PRS and CRC in the independent replication (Table 4) were consistent with findings from the discovery dataset (Table 2), overall (aged <50 years, HR per SD of PRS: 1.73; 95% CI: 1.17, 2.56; aged ≥50 years, HR per SD of PRS: 1.43; 95% CI: 1.34, 1.51) and for individuals who reported a negative first-degree family history of CRC (aged <50 years, HR per SD of PRS: 1.76; 95% CI: 1.11, 2.78; aged ≥50 years, HR per SD of PRS: 1.42; 95% CI: 1.33, 1.52). Although the effects seen for younger and older individuals were consistent with our primary analysis, the specific evaluation of whether these effects differ by age (<50 vs. age ≥50 years) was underpowered in RPGEH, due to the limited number of early-onset CRC cases in this cohort. Numbers of early-onset CRC among individuals with a first-degree family history of CRC in the replication dataset were too few for a meaningful interpretation of the analysis. Kaplan-Meier survival plots, stratified by family history, are displayed in Figure 2, consistent with the hypothesized PRS-related probability gradients across the full age range.
Table 4:
Risk estimates for early-onset versus late-onset CRC associated with a 95-SNP PRS in the RPGEH replication cohorta
| PRS | N in eligible cohort | N (cases) | HR (95% CI) | P value | P value for interactionb |
|---|---|---|---|---|---|
| All Subjects | 0.3291 | ||||
| <50 Years-Old per 1 SD | 26983 | 25 | 1.73 (1.17, 2.56) | 0.0056 | |
| ≥50 Years-Old per 1 SD | 67792 | 1068 | 1.43 (1.34, 1.51) | 2.77E-31 | |
| Negative Family History | 0.3681 | ||||
| <50 Years-Old per 1 SD | 24472 | 18 | 1.76 (1.11, 2.78) | 0.0161 | |
| ≥50 Years-Old per 1 SD | 61129 | 871 | 1.42 (1.33, 1.52) | 2.85E-25 | |
| Positive Family History | 0.6920 | ||||
| <50 Years-Old per 1 SD | 2511 | 7 | 1.56 (0.75, 3.26) | 0.2334 | |
| ≥50 Years-Old per 1 SD | 6668 | 202 | 1.34 (1.17, 1.54) | 2.87E-05 | |
The Cox models include sex, principal components, and polygenic risk score.
P value produced from interaction term with continuous PRS (per SD) and age (<50 versus ≥50 years).
Figure 2:

Absolute risk estimates of being diagnosed with CRC across the age stratum by PRS percentile among individuals in the RPGEH cohort. (A) Among individuals with a first-degree relative with CRC. (B) Among individuals without a family history of CRC.
Discussion
Our study, including more than 50,000 CRC cases and 50,000 controls, demonstrated that a PRS, derived from common genetic variants, successfully identifies participants at increased risk for early-onset CRC, particularly among individuals without a family history of CRC; additionally, the PRS was more strongly associated with early-onset cancer compared with late-onset CRC. The PRS-associated risks were found for early-onset cancer of the proximal and distal colon, and the rectum, with a modest increased propensity for the non-proximal cancers. We confirmed the overall findings for early-onset CRC in a sub-study from Ohio, where Lynch syndrome cases were excluded from the analysis. The results from these case-control studies were also supported by a smaller, prospective study that showed increased PRS-associated risks for early-onset CRC, particularly in those negative for CRC family history. Our findings may have important clinical relevance, as they could contribute, along with other lifestyle and environmental risk factors, to tailored screening in people aged <50 years who are currently not targeted for early detection and for whom CRC rates have increased over the last decades.
The development of a PRS to evaluate the overall predictive power of common risk loci for CRC has previously been carried out;29–31 however, few studies evaluated specifically for association of common polymorphisms with early-onset CRC.32–36 These smaller studies, involving 10 to 33 SNPs, pointed to some individual loci differentially associated with early-onset CRC; however, our much larger study, which included 95 loci identified from GWAS (Table S2), showed that risks related to an individual’s cumulative genetic risk profile for at-risk alleles, as reflected in the PRS, were much greater than the contributions of individual SNPs. A caveat to using these 95 variants in a PRS intended for discriminating early-onset CRC risk is that they are produced from GWAS analyses not specific to early-onset disease; adequately powered GWAS analyses specific for early-onset CRC have yet to be performed. Therefore, although our PRS positively identifies those at heightened risk for early-onset CRC, there is still room for improving its discriminatory accuracy. Furthermore, combining a genetic PRS with lifestyle and environmental risk factors could potentially contribute to even greater precision in identification of individuals who may benefit from earlier onset CRC screening.37
Given that early-onset CRC is increasing in incidence and is commonly diagnosed at later stages, which carries a poorer prognosis, recommendations have been made to lower the screening age to 45 for individuals at average-risk.12 Consideration of early detection for early-onset cancer is dependent, however, on a number of factors, including differentials in CRC risk in absolute terms, projected benefits, potential harms such as colonic perforation, and costs; therefore, potentially tempering some enthusiasm for lowering the CRC screening age and calling for identification of high-risk groups for more targeted early detection.16, 38, 39 Our study highlights the potential utility of a PRS in CRC risk stratification for people <50 years of age, which might inform precision cancer screening in this population that currently lacks consistent early detection recommendations, particularly for those without a family history of CRC.
This study is unique in the large size of the study population, particularly for those <50 years of age, allowing for evaluation of PRS-related risks overall, and by family history, refined age groups, and tumor site. Major results for association of the PRS with early-onset cancer were also replicated in an independent community-based cohort, although the number of early-onset cases in that cohort was limited. Limitations of our study include the lack of CRC family history information on a substantial subset of study participants; however, missingness was defined by study and therefore unlikely to introduce bias. Also, our PRS was generated and validated in individuals of European ancestry, currently limiting its applicability for different ancestral groups, until a PRS is developed and validated in diverse populations. Another limitation is that we did not systematically take into account the genetic mutations related to Lynch and other rarer hereditary cancer syndromes;23, 34, 40–42 however, our sensitivity analysis, in the Ohio investigations where this information was systematically assessed, indicated that risks associated with PRS remained very similar after the removal of Lynch cases from the analysis.
Nevertheless, further research is needed on the combined utility for risk prediction of rare and common variants in those with or without a family history of CRC as it can be expected that accounting for both PRS and high penetrance genes will further improve risk stratification.43, 44 There remains more to be discovered about the genetics of CRC, particularly for early-onset disease, as substantial heritability for CRC remains unexplained and genetic effects are typically stronger for early-onset diesase.45, 46 As more risk loci will be discovered, the predictive power of the PRS is expected to further improve, and to be tested in clinical trials.
In conclusion, we demonstrated that a PRS, derived from common genetic variants, successfully stratifies individuals for early onset CRC based on genetic risk, particularly among individuals who report a negative first-degree family history of CRC. Furthermore, the associations between the PRS and CRC are greater for young-onset than for older-onset disease. The PRS may contribute, along with lifestyle and environmental risk profiling, toward prioritizing individuals at increased susceptibility to early-onset CRC for personalized screening regimens or other intervention strategies. Early-onset CRC is increasing in the US and elsewhere; by selecting high-risk individuals <50 years of age, we can reduce the burden on early detection programs and potentially provide more individualized prevention approaches.
Supplementary Material
What you need to know.
BACKGROUND AND CONTEXT
Although early-onset colorectal cancer (CRC, in persons younger than 50 years old) is increasing in incidence; yet, in the absence of a family history of CRC, this population lacks harmonized recommendations for prevention.
NEW FINDINGS
In an analysis of associations with CRC per standard deviation of polygenic risk score (PRS), we found the cumulative burden of CRC-associated common genetic variants to associate with early-onset cancer, and to be more strongly associated with early-onset than late-onset cancer—particularly in the absence of CRC family history.
LIMITATIONS
Further studies are needed to identify genetic factors associated with early-onset CRC.
IMPACT
Analyses of PRS, along with environmental and lifestyle risk factors, might identify younger individuals who would benefit from preventative measures.
Acknowledgements
ASTERISK: We are very grateful to Dr. Bruno Buecher without whom this project would not have existed. We also thank all those who agreed to participate in this study, including the patients and the healthy control persons, as well as all the physicians, technicians and students.
COLON and NQplus: the authors would like to thank the COLON and NQplus investigators at Wageningen University & Research and the involved clinicians in the participating hospitals.
CORSA: We thank all those who agreed to participate in the CORSA study, including the patients and the control persons, as well as all the physicians and students.
CPS-II: The authors thank the CPS-II participants and Study Management Group for their invaluable contributions to this research. The authors would also like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program.
Czech Republic CCS: We are thankful to all clinicians in major hospitals in the Czech Republic, without whom the study would not be practicable. We are also sincerely grateful to all patients participating in this study.
DACHS: We thank all participants and cooperating clinicians, and Ute Handte-Daub, Utz Benscheid, Muhabbet Celik and Ursula Eilber for excellent technical assistance.
EDRN: We acknowledge all the following contributors to the development of the resource: University of Pittsburgh School of Medicine, Department of Gastroenterology, Hepatology and Nutrition: Lynda Dzubinski; University of Pittsburgh School of Medicine, Department of Pathology: Michelle Bisceglia; and University of Pittsburgh School of Medicine, Department of Biomedical Informatics.
EPICOLON: We are sincerely grateful to all patients participating in this study who were recruited as part of the EPICOLON project. We acknowledge the Spanish National DNA Bank, Biobank of Hospital Clínic-IDIBAPS and Biobanco Vasco for the availability of the samples. The work was carried out (in part) at the Esther Koplowitz Centre, Barcelona.
Harvard cohorts: The study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. We would like to thank the participants and staff of the HPFS, NHS, and PHS for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
LCCS: We acknowledge the contributions of Jennifer Barrett, Robin Waxman, Gillian Smith and Emma Northwood in conducting this study.
NCCCS I & II: We would like to thank the study participants, and the NC Colorectal Cancer Study staff.
NSHDS: We thank all participants in the NSHDS cohorts, the staff at the Department of Biobank Research, Umeå University, the staff at Biobanken norr, Västerbotten County Council, and the scientists managing the Northern Sweden Diet Database. We also thank Prof. Richard Palmqvist and Dr. Björn Gylling at the Department of Medical Biosciences, and Dr. Robin Myte, formerly at the Department of Radiation Sciences, all at Umeå University, Sweden, for their valuable contributions in defining the nested case-control study.
PLCO: The authors thank the PLCO Cancer Screening Trial screening center investigators and the staff from Information Management Services Inc and Westat Inc. Most importantly, we thank the study participants for their contributions that made this study possible.
PMH: The authors would like to thank the study participants and staff of the Hormones and Colon Cancer study.
SEARCH: SEARCH team.
UK Biobank: This research has been conducted using the UK Biobank Resource under Application Number 8614.
WHI: The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf.
Funding:
ASTERISK: a Hospital Clinical Research Program (PHRC-BRD09/C) from the University Hospital Center of Nantes (CHU de Nantes) and supported by the Regional Council of Pays de la Loire, the Groupement des Entreprises Françaises dans la Lutte contre le Cancer (GEFLUC), the Association Anne de Bretagne Génétique and the Ligue Régionale Contre le Cancer (LRCC).
The ATBC Study was supported by the US Public Health Service contracts (N01-CN-45165, N01-RC-45035, N01-RC-37004, and HHSN261201000006C) from the National Cancer Institute.
COLO2&3: National Institutes of Health (R01 CA60987).
ColoCare: This work was supported by the National Institutes of Health (grant numbers R01 CA189184 (Li/Ulrich), U01 CA206110 (Ulrich/Li/Siegel/Figueireido/Colditz, 2P30CA015704-40 (Gilliland), R01 CA207371 (Ulrich/Li)), NIH P30 CA42014 (Ulrich), the Matthias Lackas-Foundation, the German Consortium for Translational Cancer Research, and by TRANSCAN (JTC2012-MetaboCCC, JTC2013-FOCUS).
The Colon Cancer Family Registry (CFR) Illumina GWAS was supported by funding from the National Cancer Institute, National Institutes of Health (grant numbers U01 CA122839, R01 CA143247). The Colon CFR/CORECT Affymetrix Axiom GWAS and OncoArray GWAS were supported by funding from National Cancer Institute, National Institutes of Health (grant number U19 CA148107 to S Gruber). The Colon CFR participant recruitment and collection of data and biospecimens used in this study were supported by the National Cancer Institute, National Institutes of Health (grant number U01 CA167551) and through cooperative agreements with the following Colon CFR centers: Australasian Colorectal Cancer Family Registry (NCI/NIH grant numbers U01 CA074778 and U01/U24 CA097735), USC Consortium Colorectal Cancer Family Registry (NCI/NIH grant numbers U01/U24 CA074799), Mayo Clinic Cooperative Family Registry for Colon Cancer Studies (NCI/NIH grant number U01/U24 CA074800), Ontario Familial Colorectal Cancer Registry (NCI/NIH grant number U01/U24 CA074783), Seattle Colorectal Cancer Family Registry (NCI/NIH grant number U01/U24 CA074794), and University of Hawaii Colorectal Cancer Family Registry (NCI/NIH grant number U01/U24 CA074806), Additional support for case ascertainment was provided from the Surveillance, Epidemiology and End Results (SEER) Program of the National Cancer Institute to Fred Hutchinson Cancer Research Center (Control Nos. N01-CN-67009 and N01-PC-35142, and Contract No. HHSN2612013000121), the Hawai’i Department of Health (Control Nos. N01-PC-67001 and N01-PC-35137, and Contract No. HHSN26120100037C, and the California Department of Public Health (contracts HHSN261201000035C awarded to the University of Southern California, and the following state cancer registries: AZ, CO, MN, NC, NH, and by the Victoria Cancer Registry and Ontario Cancer Registry.
COLON: The COLON study is sponsored by Wereld Kanker Onderzoek Fonds, including funds from grant 2014/1179 as part of the World Cancer Research Fund International Regular Grant Programme, by Alpe d’Huzes and the Dutch Cancer Society (UM 2012–5653, UW 2013-5927, UW2015-7946), and by TRANSCAN (JTC2012-MetaboCCC, JTC2013-FOCUS). The NQplus study is sponsored by a ZonMW investment grant (98-10030); by PREVIEW, the project PREVention of diabetes through lifestyle intervention and population studies in Europe and around the World (PREVIEW) project which received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant no. 312057; by funds from TI Food and Nutrition (cardiovascular health theme), a public–private partnership on precompetitive research in food and nutrition; and by FOODBALL, the Food Biomarker Alliance, a project from JPI Healthy Diet for a Healthy Life.
Colorectal Cancer Transdisciplinary (CORECT) Study: The CORECT Study was supported by the National Cancer Institute, National Institutes of Health (NCI/NIH), U.S. Department of Health and Human Services (grant numbers U19 CA148107, R01 CA81488, P30 CA014089, R01 CA197350,; P01 CA196569; R01 CA201407) and National Institutes of Environmental Health Sciences, National Institutes of Health (grant number T32 ES013678).
CORSA: This study was funded by FFG BRIDGE (grant 829675, to Andrea Gsur), the “Herzfelder’sche Familienstiftung” (grant to Andrea Gsur) and was supported by COST Actions BM1206 and CA17118.
CPS-II: The American Cancer Society funds the creation, maintenance, and updating of the Cancer Prevention Study-II (CPS-II) cohort. This study was conducted with Institutional Review Board approval.
CRCGEN: Colorectal Cancer Genetics & Genomics, Spanish study was supported by Instituto de Salud Carlos III, co-funded by FEDER funds –a way to build Europe– (grants PI14-613 and PI09-1286), Agency for Management of University and Research Grants (AGAUR) of the Catalan Government (grant 2017SGR723), and Junta de Castilla y León (grant LE22A10-2). Sample collection of this work was supported by the Xarxa de Bancs de Tumors de Catalunya sponsored by Pla Director d’Oncología de Catalunya (XBTC), Plataforma Biobancos PT13/0010/0013 and ICOBIOBANC, sponsored by the Catalan Institute of Oncology.
Czech Republic CCS: This work was supported by the Grant Agency of the Czech Republic (grants CZ GA CR: GAP304/10/1286 and 1585) and by the Grant Agency of the Ministry of Health of the Czech Republic (grants AZV 15-27580A and AZV 17-30920A).
DACHS: This work was supported by the German Research Council (BR 1704/6-1, BR 1704/6-3, BR 1704/6-4, CH 117/1-1, HO 5117/2-1, HE 5998/2-1, KL 2354/3-1, RO 2270/8-1 and BR 1704/17-1), the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany, and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A and 01ER1505B).
DALS: National Institutes of Health (R01 CA48998 to M. L. Slattery).
EDRN: This work is funded and supported by the NCI, EDRN Grant (U01 CA 84968-06).
EPIC: The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF), Deutsche Krebshilfe, Deutsches Krebsforschungszentrum and Federal Ministry of Education and Research (Germany); the Hellenic Health Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); ERC-2009-AdG 232997 and Nordforsk, Nordic Centre of Excellence programme on Food, Nutrition and Health (Norway); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom).
EPICOLON: This work was supported by grants from Fondo de Investigación Sanitaria/FEDER (PI08/0024, PI08/1276, PS09/02368, P111/00219, PI11/00681, PI14/00173, PI14/00230, PI17/00509, 17/00878, Acción Transversal de Cáncer), Xunta de Galicia (PGIDIT07PXIB9101209PR), Ministerio de Economia y Competitividad (SAF07-64873, SAF 2010-19273, SAF2014-54453R), Fundación Científica de la Asociación Española contra el Cáncer (GCB13131592CAST), Beca Grupo de Trabajo “Oncología” AEG (Asociación Española de Gastroenterología), Fundación Privada Olga Torres, FP7 CHIBCHA Consortium, Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR, Generalitat de Catalunya, 2014SGR135, 2014SGR255, 2017SGR21, 2017SGR653), Catalan Tumour Bank Network (Pla Director d’Oncologia, Generalitat de Catalunya), PERIS (SLT002/16/00398, Generalitat de Catalunya), CERCA Programme (Generalitat de Catalunya) and COST Action BM1206. CIBERehd is funded by the Instituto de Salud Carlos III.
ESTHER/VERDI. This work was supported by grants from the Baden-Württemberg Ministry of Science, Research and Arts and the German Cancer Aid.
Harvard cohorts (HPFS, NHS, PHS): HPFS is supported by the National Institutes of Health (P01 CA055075, UM1 CA167552, U01 CA167552, R01 CA137178, R01 CA151993, R35 CA197735, K07 CA190673, and P50 CA127003), NHS by the National Institutes of Health (R01 CA137178, P01 CA087969, UM1 CA186107, R01 CA151993, R35 CA197735, K07 CA190673, and P50 CA127003) and PHS by the National Institutes of Health (R01 CA042182).
Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO): National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (U01 CA164930, R01 CA201407). This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA015704.
Kentucky: This work was supported by the following grant support: 1) Clinical Investigator Award from Damon Runyon Cancer Research Foundation (CI-8) and 2) NCI R01CA136726; and, we would like to acknowledge the staff at the Kentucky Cancer Registry
Kiel: This work was supported by institutional funds from the Medical Faculties of the Christian-Albrechts University Kiel and the Technical University Dresden.
LCCS: The Leeds Colorectal Cancer Study was funded by the Food Standards Agency and Cancer Research UK Programme Award (C588/A19167).
MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 509348, 209057, 251553 and 504711 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database.
MEC: National Institutes of Health (R37 CA54281, P01 CA033619, and R01 CA063464).
MECC: This work was supported by the National Institutes of Health, U.S. Department of Health and Human Services (R01 CA81488 to SBG and GR).
MSKCC: The work at Sloan Kettering in New York was supported by the Robert and Kate Niehaus Center for Inherited Cancer Genomics and the Romeo Milio Foundation. Moffitt: This work was supported by funding from the National Institutes of Health (grant numbers R01 CA189184, P30 CA076292), Florida Department of Health Bankhead-Coley Grant 09BN-13, and the University of South Florida Oehler Foundation. Moffitt contributions were supported in part by the Total Cancer Care Initiative, Collaborative Data Services Core, and Tissue Core at the H. Lee Moffitt Cancer Center & Research Institute, a National Cancer Institute-designated Comprehensive Cancer Center (grant number P30 CA076292).
NCCCS I & II: We acknowledge funding support for this project from the National Institutes of Health, R01 CA66635 and P30 DK034987.
NFCCR: This work was supported by an Interdisciplinary Health Research Team award from the Canadian Institutes of Health Research (CRT 43821); the National Institutes of Health, U.S. Department of Health and Human Serivces (U01 CA74783); and National Cancer Institute of Canada grants (18223 and 18226). The authors wish to acknowledge the contribution of Alexandre Belisle and the genotyping team of the McGill University and Génome Québec Innovation Centre, Montréal, Canada, for genotyping the Sequenom panel in the NFCCR samples. Funding was provided to Michael O. Woods by the Canadian Cancer Society Research Institute.
NSHDS: Swedish Research Council; Swedish Cancer Society; Cutting-Edge Research Grant and other grants from the County Council of Västerbotten, Sweden; Wallenberg Centre for Molecular Medicine at Umeå University; Lion’s Cancer Research Foundation at Umeå University; the Cancer Research Foundation in Northern Sweden; and the Faculty of Medicine, Umeå University, Umeå, Sweden.
OFCCR: National Institutes of Health, through funding allocated to the Ontario Registry for Studies of Familial Colorectal Cancer (U01 CA074783); see CCFR section above. Additional funding toward genetic analyses of OFCCR includes the Ontario Research Fund, the Canadian Institutes of Health Research, and the Ontario Institute for Cancer Research, through generous support from the Ontario Ministry of Research and Innovation.
OSUMC: Funding was provided by Pelotonia and the NCI (CA16058 and CA67941).
PLCO: Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. Funding was provided by National Institutes of Health (NIH), Genes, Environment and Health Initiative (GEI) Z01 CP 010200, NIH U01 HG004446, and NIH GEI U01 HG 004438.
PMH: National Institutes of Health (R01 CA076366 to P.A. Newcomb).
RPGEH: Data used in this study were provided by the Kaiser Permanente Research Bank (KPRB) from the KPRB collection, which includes the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH) and the Genetic Epidemiology Research on Adult Health and Aging (GERA) data, funded by the National Institutes of Health [RC2 AG036607 (Schaefer and Risch)], The Ellison Medical Foundation, and the Kaiser Permanente Community Benefits Program. Access to data used in this study may be obtained by application to the KPRB via ResearchBankAccess@kp.org.
SEARCH: The University of Cambridge has received salary support in respect of PDPP from the NHS in the East of England through the Clinical Academic Reserve. Cancer Research UK (C490/A16561); the UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge.
The Swedish Low-risk Colorectal Cancer Study: The study was supported by grants from the Swedish research council; K2015-55X-22674-01-4, K2008-55X-20157-03-3, K2006-72X-20157-01-2 and the Stockholm County Council (ALF project).
Swedish Mammography Cohort and Cohort of Swedish Men: This work is supported by the Swedish Research Council /Infrastructure grant, the Swedish Cancer Foundation, and the Karolinska Institutés Distinguished Professor Award to Alicja Wolk.
UK Biobank: This research has been conducted using the UK Biobank Resource under Application Number 8614
VITAL: National Institutes of Health (K05 CA154337).
WHI: The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C.
Grant Support: National Cancer Institute, National Institutes of Health (Grants: R03-CA215775-01A1, R01-CA206279-03).
Footnotes
Study descriptions:
Sample numbers of our discovery dataset study participants contributing to the whole-genome sequencing are provided in Table S1. The discovery dataset analyses comprised existing genotyping and clinical data from 42 studies that have been described in detail previously13–16. Newly generated genotype data from the Research Program on Genes, Environment and Health (RPGEH) cohort formed our replication dataset.
Disclosures: No potential conflicts (financial, professional, or personal) relevant to the manuscript.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Phillips KA, Liang SY, Ladabaum U, et al. Trends in colonoscopy for colorectal cancer screening. Med Care 2007;45:160–7. [DOI] [PubMed] [Google Scholar]
- 3.Cress RD, Morris C, Ellison GL, et al. Secular changes in colorectal cancer incidence by subsite, stage at diagnosis, and race/ethnicity, 1992–2001. Cancer 2006;107:1142–52. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Torre LA, Soerjomataram I, et al. Global patterns and trends in colorectal cancer incidence in young adults. Gut 2019:gutjnl-2019–319511. [DOI] [PubMed] [Google Scholar]
- 5.Yeo H, Betel D, Abelson JS, et al. Early-onset colorectal cancer is distinct from traditional colorectal cancer. Clin Colorectal Cancer 2017;16:293–299.e6. [DOI] [PubMed] [Google Scholar]
- 6.Bailey CE, Hu CY, You YN, et al. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975–2010. JAMA Surg 2015;150:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy CC, Singal AG, Baron JA, et al. Decrease in incidence of young-onset colorectal cancer before recent increase. Gastroenterology 2018;155:1716–1719.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feletto E, Yu XQ, Lew J-B, et al. Trends in colon and rectal cancer incidence in australia from 1982 to 2014: analysis of data on over 375,000 cases. Cancer Epidemiology Biomarkers & Prevention 2018. [DOI] [PubMed] [Google Scholar]
- 9.Brenner DR, Ruan Y, Shaw E, et al. Increasing colorectal cancer incidence trends among younger adults in Canada. Prev Med 2017;105:345–349. [DOI] [PubMed] [Google Scholar]
- 10.Siegel RL, Jemal A, Ward EM. Increase in incidence of colorectal cancer among young men and women in the United States. Cancer Epidemiol Biomarkers Prev 2009;18:1695–8. [DOI] [PubMed] [Google Scholar]
- 11.Knudsen AB, Zauber AG, Rutter CM, et al. Estimation of benefits, burden, and harms of colorectal cancer screening strategies: modeling study for the US Preventive Services Task Force. Jama 2016;315:2595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf AMD, Fontham ETH, Church TR, et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA: A Cancer Journal for Clinicians 2018;68:250–281. [DOI] [PubMed] [Google Scholar]
- 13.Rex DK, Boland CR, Dominitz JA, et al. Colorectal cancer screening: recommendations for physicians and patients from the U.S. Multi-Society Task Force on colorectal cancer. Gastroenterology 2017;153:307–323. [DOI] [PubMed] [Google Scholar]
- 14.Corley DA, Peek RM Jr. When should guidelines change? A clarion call for evidence regarding the benefits and risks of screening for colorectal cancer at earlier ages. Gastroenterology 2018;155:947–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. JAMA 2016;315:2564–2575. [DOI] [PubMed] [Google Scholar]
- 16.Liang PS, Allison J, Ladabaum U, et al. Potential intended and unintended consequences of recommending initiation of colorectal cancer screening at age 45 years. Gastroenterology 2018;155:950–954. [DOI] [PubMed] [Google Scholar]
- 17.Huyghe JR, Bien SA, Harrison TA, et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nature Genetics 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schumacher FR, Schmit SL, Jiao S, et al. Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat Commun 2015;6:7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peters U, Jiao S, Schumacher FR, et al. Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 2013;144:799–807.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmit SL, Edlund CK, Schumacher FR, et al. Novel common genetic susceptibility loci for colorectal cancer. J Natl Cancer Inst 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016;48:1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong H, Prentice RL. Bias-reduced estimators and confidence intervals for odds ratios in genome-wide association studies. Biostatistics 2008;9:621–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncology 2017;3:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851–60. [DOI] [PubMed] [Google Scholar]
- 25.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008;26:5783–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banda Y, Kvale MN, Hoffmann TJ, et al. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics 2015;200:1285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kvale MN, Hesselson S, Hoffmann TJ, et al. Genotyping informatics and quality control for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics 2015;200:1051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffmann TJ, Kvale MN, Hesselson SE, et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics 2011;98:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunlop MG, Tenesa A, Farrington SM, et al. Cumulative impact of common genetic variants and other risk factors on colorectal cancer risk in 42,103 individuals. Gut 2013;62:871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenkins MA, Makalic E, Dowty JG, et al. Quantifying the utility of single nucleotide polymorphisms to guide colorectal cancer screening. Future Oncol 2016;12:503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu L, Jeon J, Brenner H, et al. A model to determine colorectal cancer risk using common genetic susceptibility loci. Gastroenterology 2015;148:1330–9.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He J, Wilkens LR, Stram DO, et al. Generalizability and epidemiologic characterization of eleven colorectal cancer GWAS hits in multiple populations. Cancer Epidemiol Biomarkers Prev 2011;20:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Holst S, Picelli S, Edler D, et al. Association studies on 11 published colorectal cancer risk loci. Br J Cancer 2010;103:575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giráldez MD, López-Dóriga A, Bujanda L, et al. Susceptibility genetic variants associated with early-onset colorectal cancer. Carcinogenesis 2012;33:613–619. [DOI] [PubMed] [Google Scholar]
- 35.Song N, Shin A, Park JW, et al. Common risk variants for colorectal cancer: an evaluation of associations with age at cancer onset. Sci Rep 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Middeldorp A, Jagmohan-Changur S, van Eijk R, et al. Enrichment of low penetrance susceptibility loci in a Dutch familial colorectal cancer cohort. Cancer Epidemiol Biomarkers Prev 2009;18:3062–7. [DOI] [PubMed] [Google Scholar]
- 37.Jeon J, Du M, Schoen RE, et al. Determining risk of colorectal cancer and starting age of screening based on lifestyle, environmental, and genetic factors. Gastroenterology 2018;154:2152–2164.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy CC, Sanoff HK, Stitzenberg KB, et al. RE: Colorectal cancer incidence patterns in the United States, 1974–2013. JNCI: Journal of the National Cancer Institute 2017;109:djx104–djx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warren JL, Klabunde CN, Mariotto AB, et al. Adverse events after outpatient colonoscopy in the Medicare population. Ann Intern Med 2009;150:849–57, w152. [DOI] [PubMed] [Google Scholar]
- 40.Jasperson KW, Tuohy TM, Neklason DW, et al. Hereditary and familial colon cancer. Gastroenterology 2010;138:2044–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pinto C, Veiga I, Pinheiro M, et al. MSH6 germline mutations in early-onset colorectal cancer patients without family history of the disease. Br J Cancer 2006;95:752–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Voer RM, Hahn MM, Mensenkamp AR, et al. Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci Rep 2015;5:14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whiffin N, Dobbins SE, Hosking FJ, et al. Deciphering the genetic architecture of low-penetrance susceptibility to colorectal cancer. Hum Mol Genet 2013;22:5075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wray NR, Purcell SM, Visscher PM. Synthetic associations created by rare variants do not explain most GWAS results. PLoS Biol 2011;9:e1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiao S, Peters U, Berndt S, et al. Estimating the heritability of colorectal cancer. Hum Mol Genet 2014;23:3898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaitlen N, Kraft P. Heritability in the genome-wide association era. Hum Genet 2012;131:1655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.