

Abstract
Background & Aims:
Trehalose is a disaccharide that might be used in treatment of cardiometabolic diseases. However, trehalose consumption promotes expansion of Clostridioides difficile ribotypes that metabolize trehalose via trehalose-6-phosphate hydrolase (treA). Furthermore, brush border and renal trehalases can reduce the efficacy of trehalose by cleaving it into monosaccharides. We investigated whether a trehalase-resistant analogue of trehalose (lactotrehalose) has the same metabolic effects of trehalose without expanding C difficile.
Methods:
We performed studies with HEK293 and Caco2 cells, primary hepatocytes from mice, and human intestinal organoids. Glucose transporters were overexpressed in HEK293 cells and glucose transport was quantified. Primary hepatocytes were cultured with or without trehalose or lactotrehalose and gene expression patterns were analyzed. C57B6/J mice were given oral antibiotics and trehalose or lactotrehalose in drinking water, or only water (control), followed by gavage with the virulent C difficile ribotype 027 (CD027); fecal samples were analyzed for ToxA or ToxB by ELISA. Other mice were given trehalose or lactotrehalose in drinking water for 2 days before placement on a chow or 60% fructose diet for 10 days. Liver tissues were collected and analyzed by histologic, serum biochemical, and RNAseq, autophagic flux, and thermogenesis analyses. We quantified portal trehalose and lactotrehalose bioavailability by gas chromatography mass spectrometry. Fecal microbiomes were analyzed by 16S rRNA sequencing and principal component analyses.
Results:
Lactotrehalose and trehalose each blocked glucose transport in HEK293 cells, and induced a gene expression pattern associated with fasting in primary hepatocytes. Compared with mice on the chow diet, mice on the high-fructose diet had increased circulating cholesterol, higher ratios of liver weight:body weight, hepatic lipid accumulation (steatosis), and liver gene expression patterns of carbohydrate-responsive de novo lipogenesis. Mice given lactotrehalose while on the high-fructose diet did not develop any of these features and had increased whole-body caloric expenditure compared with mice given trehalose or water and fed a high-fructose diet. Livers from mice given lactotrehalose had increased transcription of genes that regulate mitochondrial energy metabolism compared with liver from mice given trehalose or controls. Lactotrehalose was bioavailable in venous and portal circulation and fecal samples. Lactotrehalose reduced fecal markers of microbial branched chain amino acid biosynthesis and increased expression of microbial genes that regulate insulin signaling. In mice given antibiotics followed by CD027, neither lactotrehalose nor trehalose increased levels of the bacteria or its toxin in stool—in fact, trehalose reduced the abundance of CD027 in stool. Lactotrehalose and trehalose reduced markers of inflammation in rectal tissue following CD027 infection.
Conclusions:
Lactotrehalose is a trehalase-resistant analogue that increases metabolic parameters, compared with trehalose, without increasing the abundance or virulence of C difficile strain CD027. Trehalase-resistant trehalose analogues might be developed as next-generation fasting-mimetics for treatment of diabetes and nonalcoholic fatty liver disease.
Keywords: FGF21, mouse model, NASH, NAFLD
Lay Summary:
We identified a molecule with a structure similar to that of sugar that speeds metabolism and reduces fatty liver but does not promote expansion of pathogenic bacteria in the intestine.
Graphical Abstract
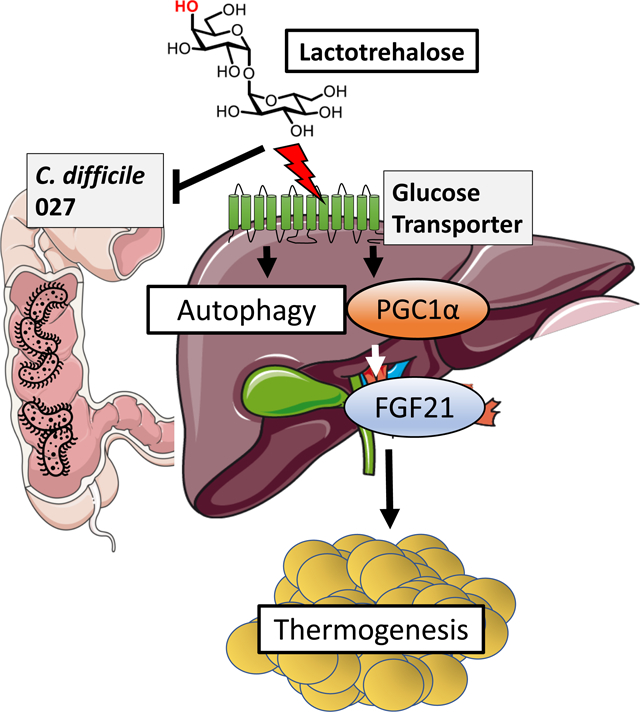
INTRODUCTION
Trehalose is a naturally-occurring glucose-glucose disaccharide that is present in plants and many invertebrates1. In addition, trehalose is used extensively as a drug excipient and food additive. Beyond its broad utility, trehalose is recognized as a promising autophagy-inducing therapeutic agent in humans and rodent models against cardiometabolic2–5 and neurodegenerative disorders6–8. Trehalose is thus in a class of fasting-mimetics1, which recapitulate the well-described effects of fasting and caloric restriction on metabolic diseases9, 10. Specifically, trehalose activates canonical hepatic fasting-like responses, including hepatocyte autophagic flux, transcription factor EB and PPARγ coactivator-1α activation, and hepatic fibroblast growth factor 21 release1, 4, 11, 12. These responses improve mitochondrial function and total energy expenditure, reduce diet-induced hepatic steatosis, and enhance insulin sensitivity11, 13, 14. Targeting the hepatocyte is an attractive therapeutic strategy, in light of its primary function to coordinate gluconeogenesis and ketogenesis and upregulate fatty acid beta oxidation during fasting by mobilizing peripheral fatty acids15.
Clostridioides difficile (C difficile) causes a common nosocomial infection that is characterized by pseudomembranous colorectal inflammation16–21. Increased detection of C difficile infections with ribotype CD027 recently sparked questions regarding the selective origins of this strain22, 23. CD027 expresses a virulence factor, trehalose-6-phosphate hydrolase (treA), which enables CD027 to utilize the disaccharide, trehalose, as a carbon source for growth in low-glucose environments22, 23. TreA facilitates trehalose utilization by hydrolyzing trehalose into two disaccharides: glucose, and glucose-6-phosphate24. Recent work demonstrated that trehalose promotes treA-expressing C difficile ribotypes in vitro, including CD02722, 23. This is important, because trehalose gained United States FDA ‘generally safe’ status in 2000, and is now a common food additive and nutraceutical supplement. The temporal association between this FDA designation and increased CD027 detection in nosocomial outbreaks worldwide prompted the hypothesis that dietary trehalose played a major role in the increased incidence of CD027 infection22, 23. This hypothesis persists despite a marked decrease in CD027 incidence between 2010 and 201425, and despite the fact that normal human stool glucose content is sufficiently high to preclude selective pressure for additional carbon utilization26. In addition, enterally administered trehalose undergoes significant first-pass enterohepatic clearance3, 4, which may attenuate its therapeutic effects on target organs outside the enterohepatic circulation (e.g. the central nervous system). Overall, these concerns generate a need for fasting-mimetic trehalose analogues that retain or extend trehalose therapeutic efficacy, and yet resist brush border and microbial degradation.
The trehalose analogue lactotrehalose (α-D-galactopyranosyl-(1,1)-α-D-glucopyranoside), is a trehalase-resistant trehalose analogue24, 27, 28, which differs from trehalose only in its hydroxyl group orientation at carbon 424, 29. Here, we demonstrate that lactotrehalose conveys enhanced fasting-mimetic properties when compared to native trehalose. Indeed, lactotrehalose exhibited enhanced extrahepatic bioavailability, and induced hepatocyte fasting-like responses, including FGF21 release, PGC1a expression and autophagic flux. Lactotrehalose also reduced fructose-induced hepatic triglyceride accumulation, enhanced whole-body basal thermo-genesis, and increased hepatic mitochondrial oxidative phosphorylation more robustly than native trehalose. Surprisingly, in an in vivo CD027 infection model, neither trehalose nor lactotrehalose enhanced CD027 proliferation, toxin A/B accumulation, or treA virulence factor expression in vivo. Moreover, both trehalose and lactotrehalose suppressed CD027-induced rectal inflammatory marker gene expression. The data suggest that lactotrehalose is a potent fasting-mimetic trehalose analogue that extends the metabolic effects of trehalose without augmenting C difficile virulence.
METHODS
Primary hepatocyte isolation and in vitro assays
Primary hepatocyte isolation and assays were performed precisely as reported previously30. Briefly, wild-type C57 B6/J mice were sacrificed and collagenase was perfused through the portal vein. Dissociated hepatocytes were separated from non-hepatocytes by differential sedimentation and plated on collagen-coated plates 16h prior to treatment and subsequent assay.
Animals
Studies were performed on male 6–8wk-old C57BL/6J mice obtained from Jackson Laboratories (Bar Harbor, ME) unless otherwise indicated. All procedures were performed in accordance with the approved guidelines by the Animal Studies Committee at Washington University School of Medicine.
Portal Vein Disaccharide Measurements
Wild-type mice were placed nil per os 1 hr prior to oral gavage in unanesthetized with vehicle or 200–400 mg/kg trehalose or LT. Mice were anesthetized by inhaled isofluorane immediately prior to terminal portal venous blood sampling via open laparotomy. The time from anesthetic administration to complete blood draw was approximately 60 seconds. The diaphragm and chest cavity (and thus appropriate intrathoracic negative pressure) remained intact during the blood draw.
CD027 Infection
Briefly 6wk-old C57B6/J mice were treated with oral antibiotics (all from Sigma Aldrich, St. Louis, MO): 0.4mg/mL kanamycin, 0.215mg/mL metronidazole, 0.045mg/mL vancomycin, 0.035mg/mL gentamicin, and 850U/mL colistin for 5 days in the drinking water, ad libitum. On the 5th day intraperitoneal clindamycin (30mg/kg) was administered in addition to the oral antibiotic regimen. Following antibiotic treatment, mice were fed sterile water, 3% (w/v) trehalose or 3% (w/v) lactotrehalose ad libitum for 48hrs prior to CD027 (strain 4118) gavage at 105 CFU in 100uL phosphate-buffered saline per mouse. Mice were monitored 4 days prior to sacrifice and terminal analysis. ToxA and ToxB ELISA (Cat. # ABIN1098189) was obtained from Antibodies-online.com. The assay was performed on cecal content precisely per manufacturer specification.
In Vivo Diet-Induced Steatosis
6–8wk-old C57B6/J male mice were treated with or without 3% lactotrehalose in drinking water, ad libitum, 2 days prior to initiating chow or 60% fructose diet (Teklad #TD89247) for 10 days. Histological analysis by oil red-O staining in liver frozen sections, serum biochemical analyses, and hepatic gene expression was performed precisely as we have reported previously4, 13, 14, 30, 31.
Metabolic Cages
Indirect calorimetry experiments were performed in an 8-module indirect calorimeter (TSE Systems) as described previously11, 13, 14, 30–32.
Fluorescence Resonance Energy Transfer
We performed FRET assays to measure glucose uptake in subconfluent HEK293 cells was performed precisely as described recently33.
Immunoblotting
Immunoblotting was performed as described. We obtained p62 antibody from Abcam; LC3B antibody from Novus Biologicals (NB-100–2200) and GAPDH antibody from Cell Signaling Technologies (#5174).
qRT-PCR
Real-time, quantitative reverse transcriptase PCR was performed as described4. Primers used are in Supplemental Figure 9.
GC-MS
GC-MS was performed as previously reported with minor modifications4. Derivatized samples were analyzed on an Agilent 7890A gas chromatograph interfaced to an Agilent 5975C mass spectrometer. The GC column used for the study was a HP-5MS (30 m, 0.25mm internal diameter, 0.25um film coating P.J. Cobert St. Louis, MO).
Quantitative respirometry
Oroboros respirometry was performed as reported with minor modifications32. Mitochondria were isolated from saponin-permeabilized resected liver tissue prior to mitochondrial isolation and respirometry. Data were normalized to input liver tissue weight.
RNAseq
RNAseq was performed by the Washington University Genome Technology Access Center (GTAC) as described13, 14.
Microbiome analysis
Microbial analysis was performed precisely as in prior reports34.
Statistics
Data were analyzed using GraphPad Prism version 7.0. P < 0.05 was defined as statistically significant. Data presented as box plots whiskers show the minimum and maximum, and boxes extend from the first to the third quartiles with midlines at the medians. Each biological replicate represents one animal. Statistical significance is represented as follows: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns, not significant.
RESULTS
We determined the effect of lactotrehalose on glucose uptake in 293 cells expressing either class I GLUTs35 (GLUTs 1–4), or GLUT8 (a key GLUT that modulates hepatocyte fasting signal transduction30–32) on a genetic background in which we knocked down endogenous GLUT133, 36. We quantified glucose transport using a soluble intracellular glucose sensor, which activates fluorescence resonance energy transmission (FRET) upon cytoplasmic glucose entry33, 36. Glucose induced FRET activity, as defined by increased cyan fluorescence:yellow fluorescence emission ratio33 (Supp. Fig. 1). By comparison, the GLUT inhibitor, cytochalasin B (200 μM), completely inhibited glucose-induced FRET activation (Supp. Fig. 1). Consistent with prior radio-labeled glucose analogue uptake data using trehalose as a GLUT inhibitor4, 37, lactotrehalose and trehalose (200mM) each partly inhibited glucose-induced FRET activation (Supp. Fig. 1). However, trehalose was generally a more potent inhibitor than lactotrehalose for each of the class I GLUTs, but not for GLUT8 (Supp. Fig. 1).
We next asked if lactotrehalose induces a fasting-like, glucose-deprived state in hepatocytes. We treated isolated primary murine hepatocytes with or without trehalose or lactotrehalose (100mM each), and assayed fasting gene expression. These genes include the peroxisome proliferator antigen receptor γ (PPARγ) coactivator-1α, and its downstream transcriptional target, fibroblast growth factor 21 (FGF21)38–41, Arg214, and ALOXE313. Lactotrehalose increased PGC1α, FGF21, Arg2 and ALOXE3, as well as activating transcription factor 4 (ATF4) and glucose-regulated protein 78 (GRP78)42 expression to a greater extent than trehalose (Fig. 1B and 1C). Energetic rescue by pyruvate partly reversed lactotrehalose-induced ALOXE3 and GRP78 expression (Figs. 1B and 1C).
Figure 1. Enhanced hepatocyte fasting-like signaling and autophagic responses to lactotrehalose in vitro and in vivo.

A. Chair conformations of trehalose and lactotrehalose demonstrating differences only in the hydroxyl group orientation at the 4-carbon position (in red). B. and C. qRT-PCR analysis of fasting- or stress-induced factors in murine hepatocytes after treatment with or without trehalose or lactotrehalose. D. Enhanced FGF21 and LC3B-II accumulation in murine hepatocytes treated with trehalose or lactotrehalose. E. In vitro triglyceride and cholesterol quantification in serum- and glucose-starved primary murine hepatocytes treated with or without 10mM fructose 24h, with or without trehalose or lactotrehalose. F. qRT-PCR analysis showing expression of fasting-induced factors upon fructose treatment with or without 10mM trehalose 24h with or without trehalose or lactotrehalose. G. Left, Liver weight:body weight ratio in mice treated with chow or high-fructose diet (HFrD, 60% fructose, 10 days). Middle, densitometric quantification of lipid staining by oil red-O from a minimum 5 random high-powered fields per mouse liver, 3–5 mice per group. Right, H&E staining of paraffin-embedded sections and oil red-O staining of frozen sections from mice fed chow or HFrD with or without 3% lactotrehalose in water (ad libitum). Scale bar: 100 μm. H. Left, canonical de novo lipogenesis pathway in liver. Right, qRT-PCR quantification of canonical de novo lipogenesis pathway intermediaries. ns, not significantly different. *, ****, P < 0.05, or 0.0001 by two-tailed t-test with Bonferroni-Dunn post hoc correction for multiple comparisons.
During fasting, hepatocytes secrete the antidiabetic hepatokine, FGF2139–41, 43, and increase autophagic flux4, 44 to enhance hepatic and extrahepatic energy metabolism44, 45. Trehalose and lactotrehalose induced LC3B-II accumulation to similar extents versus untreated hepatocytes. This suggested similar LC3B-I lipidation to the LC3B-II form on nascent autophagosomal membranes46. In contrast, lactotrehalose induced FGF21 protein expression to a greater extent than trehalose in isolated primary hepatocytes (Fig. 1D). In contrast, we also quantified lactotrehalose transcriptional responses in kidney (HEK293) and colonic (Caco2) cell lines, and in small bowel enteroids obtained from the healthy margin of small bowel resections. Neither trehalose nor lactotrehalose (24h treatment) affected expression of any fasting-induced intermediaries: PGC1α, FGF21 or Arg2, and no dose-dependent suppression of the post-prandial enterokine, FGF19, at physiologically relevant trehalose and lactotrehalose concentrations in HEK293 cells (< 10mM in peripheral circulation4, Supp. Fig 2). In Caco2 cultures, trehalose and lactotrehalose induced FGF21 gene expression, but did not affect FGF19 expression (Supp. Fig. 2). In enteroids, trehalose and lactotrehalose suppressed FGF1943.
We next tested whether lactotrehalose attenuates hepatocyte triglyceride (TG) and cholesterol accumulation in response to fructose overload (Fig. 1E). Fructose significantly increased hepatocyte TG and cholesterol accumulation, whereas trehalose and lactotrehalose each abrogated fructose-induced lipid accumulation (Fig. 1E). Additionally, 10 mM fructose in vitro suppresses expression of hepatocyte FGF21, PGC1α, and Arg2 gene expression, relative to fructose-free cultures (Fig. 1F). However, both trehalose and lactotrehalose reversed fructose-suppressed expression of these genes (Fig. 1F).
We then fed 6wk-old male mice 3% lactotrehalose in water ad libitum 2 days prior to 60% fructose diet (HFrD) or isocaloric standard chow (10 days). Lactotrehalose-treated mice do not differ from water-fed controls with regard to total water intake or stool volume, frequency or consistency (not shown). HFrD increases circulating cholesterol (Supp. Fig. 3), liver weight:body-weight ratio (Fig. 1G) and hepatic neutral lipid accumulation, as ascertained by oil red-O staining in frozen liver sections (Fig. 1G). Moreover, HFrD induced hepatocyte carbohydrate-responsive de novo lipogenic gene expression47 (Fig. 1H). In contrast, lactotrehalose treatment reduces circulating cholesterol (Supp. Fig. 3), liver weight:body weight ratio, and hepatic lipid accumulation and transcriptional upregulation of de novo lipogenesis (Fig. 1G, 1H, Supp. Fig. 3).
We next explored the broader effects of lactotrehalose and trehalose on liver transcriptomics. We treated mice with sterile water, 3% trehalose or lactotrehalose in drinking water (ad libitum, 5 days). Again, mice treated with trehalose or lactotrehalose do not differ from sterile water-fed control mice with regard to water intake or stool character (not shown). Liver transcriptomics in trehalose- and lactotrehalose-treated mice reveals distinctive global hepatic transcriptional responses (Fig. 2A, 2B, 2C), which resolve in three-dimensional principal component analysis (Fig. 2B). The pathways that are most significantly upregulated (significance threshold: P < 0.05) when comparing lactotrehalose versus trehalose treatment are enriched for mitochondrial gene expression, oxidative phosphorylation, mitochondrial respiratory chain complex assembly, electron transport chain, mitochondrial translation, and ATP synthesis (Fig. 2D). Lactotrehalose also upregulates translation and peptide biosynthetic processes to a greater extent than trehalose (Fig. 2D). Together, lactotrehalose induces enhanced transcriptional responses consistent with enhanced mitochondrial energy metabolism versus control and native trehalose-treated livers.
Figure 2. Distinct transcriptomic responses to trehalose and lactotrehalose.

A. Heat map based on RNA sequencing data demonstrating significant (P < 0.05, trehalose vs. lactotrehalose) gene-level differences in livers from mice treated with trehalose and lactotrehalose grouped via unsupervised clustering. B. Venn diagram (above) and 3-dimensional principal component analysis (below) demonstrating distinct transcriptomic changes in livers from trehalose-treated versus lactotrehalose-treated and untreated mice. C. Volcano plot comparing gene expression changes at the P < 0.05, 1.5 log(fold-change) threshold in livers from control vs. trehalose, control vs. lactotrehalose, and trehalose vs. lactotrehalose-treated mice. D. Gene Ontology and Kyo-to Expression of Genes and Genomes pathway analysis of pathways significantly upregulated in lactotrehalose vs. trehalose-treated mouse liver (P < 0.05, lactotrehalose vs. trehalose-treated livers).
We next performed functional metabolic assays in vivo to confirm or refute these hepatic transcriptomic data. We compared mitochondrial function by Oroboros respirometry in livers from mice treated with vehicle, trehalose, or lactotrehalose (3% in water, ad libitum, 5 days). Complex I + II mitochondrial respiration and maximal mitochondrial respiration are activated in livers of lactotrehalose-treated mice when compared with livers from both untreated and trehalose-treated mice (Fig. 3A). Indirect calorimetry reveals concomitantly increased whole-animal O2-CO2 exchange and heat generation (Fig. 3B). Trehalose increases whole-body O2-CO2 exchange and basal thermogenesis versus untreated mice, and lactotrehalose induces heat generation to a greater extent than both vehicle and native trehalose throughout light and dark cycles (Fig. 3B and 3C). This occurs despite lower locomotion in lactotrehalose-treated mice (Fig. 3C). Trehalose and lactotrehalose each increased circulating FGF21 (Fig. 3D). In addition, lactotrehalose reduces serum low-density lipoprotein cholesterol (LDL-C, Supp. Fig. 4A) and a trend toward reduced total cholesterol in 5-day lactotrehalose-treated mice when compared with both control- and trehalose-fed mice. There was no difference in water consumption in any group, as determined by indirect calorimeter water consumption rates (Supp. Fig. 4B).
Figure 3. Enhanced liver and whole-body oxidative metabolism in lactotrehalose-treated mice.

A. Quantitative (Oroboros) respirometry in isolated mitochondria derived from livers of mice treated with oral trehalose or lactotrehalose (3% in water, ad libitum, 48 h). B. Mean heat versus time tracing in mice treated with or with- out trehalose or lactotrehalose (3% in water, ad libitum, 5 days). White and grey background denote light and dark cycle periods. C. Oxygen-carbon dioxide exchange, movement, respiratory exchange ratio (RER) and heat area-under-curve quantifications in mice treated in (B). D. Enzyme-linked immunoassay quantification of FGF21 peptide in plasma from mice treated with or without trehalose (3% in water ad libitum, 2 days). In box-whisker plots: middle bar represents the dataset median; boxes represent 25%ile and 75%ile lines; whiskers represent maximum and minimum values in the dataset. *, **, ***, ****, P < 0.05, 0.01, 0.001, or 0.0001 versus comparison group by two-tailed t-test with Bonferroni-Dunn post hoc correction for multiple comparisons.
We next examined lactotrehalose bioavailability. We used gas chromatography and mass spectrometry to define the peak portal vein concentration of trehalose and lactotrehalose 15’ after oral trehalose or lactotrehalose gavage (200 or 400 mg/kg). Both trehalose dosages were previously safely administered to humans, and correlate with the reduction in cardiometabolic risk factors in patients48, 49. Peak portal trehalose after low dose gavage is 237mM, (± SEM, ± 127mM, p = 0.09 vs. control); high-dose gavage is 667mM ± 281mM (p < 0.05 vs. control). Strikingly, mean peak portal lactotrehalose after low-dose gavage is 341mM ± 233mM (p < 0.05 vs. control), whereas the mean lactotrehalose after high-dose gavage is 1.37M ± 0.45M (Fig. 4A, (p < 0.001 vs. control)). Therefore, lactotrehalose had increased peak portal circulation bioavailability, and greater bioavailability at a lower gavage dosing.
Figure 4. Rapid oral bioavailability of trehalose and lactotrehalose in the hepatic portal circulation.

A. Gas Chromatography and Mass Spectrometry (GC-MS) quantification of acute portal venous [trehalose] (left) or [lactotrehalose] (right) 15’ after oral gavage of either sterile water or 200–400mg/kg trehalose or lactotrehalose. B. GC-MS quantification of steady-state peripheral [trehalose] or [lactotrehalose] after 2- and 5-day exposure to trehalose (red dots) or lactotrehalose (blue dots) (3% in water ad libitum). C. GC-MS quantification of steady-state fecal [trehalose] or [lactotrehalose] after 5-day exposure to trehalose (red dots) or lactotrehalose (blue dots) (3% in water ad libitum, 5 days). *, ***, P < 0.05, 0.001 versus comparison group by two-tailed t-test with Bonferroni-Dunn post hoc correction for multiple comparisons.
We then quantified steady-state peripheral venous and fecal trehalose and lactotrehalose from control, trehalose-fed and lactotrehalose-fed mice (Fig. 4B and 4C). Trehalose and lactotrehalose in peripheral circulation are detected minimally above background, 2- and 5-days post-treatment (3% in drinking water ad libitum, Fig. 4B). Basal trehalose is detectable in the venous circulation of untreated mice (Fig. 4B) because trehalose is a component of plant-based rodent chow (Fig. 4B), whereas lactotrehalose is not. In stool, steady-state trehalose in trehalose-fed mouse feces is four-fold above basal levels (Fig. 4C, 1.7 pmol/mg feces vs. 0.4 pmol/mg). Fecal lactotrehalose was 90 pmol/mg feces vs. 0 pmol/mg in lactotrehalose-unexposed mice). Therefore, lactotrehalose is bioavailable in the hepatic portal and enteric microenvironments after enteral administration.
Fasting by itself modulates the bacterial microbiome, which may modify mammalian metabolism50. We thus examined trehalose and lactotrehalose effects on fecal bacterial microbial communities in mice. We observed microbial shifts as determined by principal component analyses in mice treated with oral 3% trehalose or lactotrehalose in drinking water (ad libitum) compared to sterile water-fed controls (Fig. 5A). Lactotrehalose produced a more robust Bray Curtis OTU distance shift versus control and trehalose-fed mice (Fig. 5A). Also, trehalose and lactotrehalose significantly increased genus-level Shannon and Simpson diversity indices (Supp. Fig. 5). Notably, lactotrehalose induces a shift toward more Dehalobacterium (Fig. 5B), which mirrors con-served structural changes in the microbiota of mice and other species subjected to prolonged fasting51. We did not detect resident C difficile in uninfected, antibiotic-naïve mice, and neither trehalose nor lactotrehalose altered colonic C difficile populations in the gut, even after up to 3 wk feeding (3% disaccharide in water, ad libitum, Supp. Fig. 6).
Figure 5. Differential microbial community shifts in mice treated with trehalose and lactotrehalose.

A. Bray Curtis genera and OTU principal component analyses. B. Cladogram illustrating bacterial taxa differences in control versus lactotrehalose-treated mice. Colored nodes from the center to the periphery represent phylum (p), class (c), order (o), family (f), and genus (g) level differences detected between control (red) and lactotrehalose-treated mice (green). C. Metabolic pathway heat map showing bacterial gene expression pathways significantly (threshold P < 0.05, trehalose vs. lactotrehalose) altered in mice treated with or without trehalose or lactotrehalose (3% in water ad libitum, 5 days). Each column represents data from stool obtained from a distinct biological replicate.
Trehalose and lactotrehalose also differentially induced microbial metabolic pathway gene expression, aligned by unsupervised clustering (Fig. 5C). For example, lactotrehalose suppressed microbial gene expression associated with branched chain amino acid biosynthesis52–54 and limonene degradation55, 56 (Fig. 5C). Conversely, lactotrehalose induces more pronounced increases microbial community gene expression in insulin signaling, and in xylene57, 58, dioxin,59 and bisphenol59 degradation (Fig. 5C). These microbial community and pathway shifts represent potentially favorable metabolic associations with regard to diabetic and NAFLD risk.
Trehalose promotes the growth of epidemic CD027 in vitro22, 23, 60, 61. If true in humans, the beneficial effects of trehalose would need to be weighed against the theoretical risks of trehalose consumption. We hypothesized that lactotrehalose does not promote CD027-induced colorectal inflammation. We first treated mice with broad-spectrum antibiotics (intraperitoneal clindamycin, oral colistin, gentamicin, kanamycin, vancomycin and metro-nidazole). Then, we gavaged saline or 105 CFU CD027 (Strain 4118), and analyzed fecal C difficile toxins A and B, and both fecal and rectal tissue inflammatory marker genes. We quantified CD027 burden and TreA expression four days after gavage (Fig. 6A). All CD027-infected groups lost body weight relative to uninfected groups, but no difference was observed in body weight among treated and untreated infected mice (Supp. Fig. 7A). In addition, neither stool mass nor total rectal and cecal content mass (quantified at sacrifice 72h post-infection in a distinct cohort) differed in mice preceding or following CD027 infection, irrespective of trehalose or LT exposure (Supp. Fig. 7B). Neither trehalose nor lactotrehalose treatment increased CD027 burden in stool, as quantified by 16S rDNA PCR. In fact, trehalose significantly reduced CD027 abundance (Fig. 6B). Trehalose and lactotrehalose both produced trends toward lower ToxA and ToxB expression, as determined by enzyme immunoassay (EIA). Indeed, lactotrehalose significantly suppressed ToxA (Fig. 6C). Trehalose and lactotrehalose treatment appeared to suppress TreA virulence factor expression, although the degree of suppression is not statistically significant (Fig. 6D). No difference in mortality was observed in any group (data not shown), and histological rectal inflammation was minimal in all groups (Supp. Fig. 7C). We observed higher inflammatory response markers Kc and interleukin (IL)-6 in stool of trehalose-treated mice versus untreated CD-infected controls, whereas lactotrehalose significantly inhibited CD-induced fecal Kc and IL6 expression (Fig 6E). Both lactotrehalose and trehalose inhibited rectal inflammatory markers following CD027 infection. This includes marker genes IL-1β, S100A8 and S100A9, which were trending or significantly lower in rectal tissue derived from both trehalose and lactotrehalose-fed mice (Fig. 6E) when compared with untreated, CD027-infected mice (Fig. 6E).
Figure 6. lactotrehalose reduces toxin and rectal inflammatory gene expression following CD027 infection in vivo.

A. Experimental design for CD027 infection and analysis. B. Quantification of CD027:total 16S ribosomal DNA ratio in stool from mice 96 h after CD027 infection following pre-treatment with or without trehalose or lactotrehalose (3% in water, ad libitum administered 2d prior to infection). C. ELISA-based C difficile Toxin A and B quantification in cecal contents from mice 72 h after CD027 infection following pre-treatment with or without trehalose or lactotrehalose (3% in water, ad libitum administered 2d prior to infection). D. and E. qRT-PCR quantification of treA and stool inflammatory marker gene expression in mice 96 h after CD027 infection following pre-treatment with or without trehalose or lactotrehalose. Marker gene expression was normalized against total 16S expression in each sample. F. Rectal tissue inflammatory marker gene mRNA expression by qRT-PCR analysis from mice infected with CD027 treated with or without trehalose or lactotrehalose. *, **, ***, ****, P < 0.05, 0.01, 0.001, or 0.0001 by two-tailed t-test with Bonferroni-Dunn post hoc correction for multiple comparisons.
DISCUSSION
Trehalose is well characterized with regard to its autophagy-inducing and fasting-mimetic effects. Each of these properties of trehalose contribute to varying degrees to its therapeutic effects in neurodegenerative, retinal and metabolic diseases5–8. Recent data suggesting that trehalases limit oral trehalose bioavailability3, 4 and safety23 raised concerns over its translational viability. Yet, the continued rise in prevalence of cardiometabolic disease prompts demand for novel metabolic therapies. Trehalose is a fasting-mimetic compound, but its full mechanistic actions are unclear, and the contexts in which it has clinical utility are not fully defined. Although it seems premature to dismiss translatability of trehalose to human clinical use, degradation-resistant trehalose analogues offer the opportunity to minimize safety and bioavailability concerns, and yet retain or extend the therapeutic potential of trehalose.
Here, we demonstrate three key findings that span interests in metabolism, gastroenterology, and microbiology. First, lactotrehalose meets or exceeds the cellular, molecular, and physiological fasting-mimetic effects of native trehalose. Second, we demonstrate divergent microbial and hepatic transcriptomic effects, which suggests that these compounds exert overlapping and divergent effects. Third, lactotrehalose does not promote CD027 expansion in an in vivo CD027 infection model. Lactotrehalose therefore represents a novel therapy against metabolic disease, and other conditions that native trehalose impacts.
Fecal bacterial microbiome analyses suggest that both trehalose and lactotrehalose induce broad, community and metabolic shifts that are probably favorable to mammalian metabolism51, 62. Indeed, lactotrehalose suppressed gene pathways related to biosynthesis of branched chain amino acids, which have increasingly clear roles in diabetes pathogenesis52–54. Lactotrehalose also suppressed gene pathways related to degradation of limonene, which are proposed to have therapeutic effects on hyperglycemia and dyslipidemia55, 56. Conversely, lactotrehalose induced more pronounced upregulations versus trehalose in gene pathways regarding insulin signaling, and in degradation of xylene57, 58, dioxin59 and bisphenols59, each of which is involved in NAFLD/NASH as well as diabetes pathogenesis. Although these associations do not yet permit us to conclude that such microbial shifts are mechanistically involved in the therapeutic effects of trehalose and lactotrehalose, subsequent work may further elucidate disaccharide effects on intestinal microbiota.
Two observations here are relatively unexpected. First, lactotrehalose was not a more potent GLUT inhibitor than trehalose. And yet, trehalase resistance makes lactotrehalose more bioavailable. This may partly explain the enhanced fasting-mimetic effects of lactotrehalose in vitro and in vivo. Furthermore, lactotrehalose and trehalose exhibited comparable GLUT8 inhibitory efficacy. Thus, it is also plausible that altering the trehalose 4-carbon hydroxyl group in trehalose by itself confers properties of the ligand that activate fasting signaling, in-dependent of altering glucose transport. This so-called ‘transceptor’ hypothesis was previously proposed for GLUT235, 63 and GLUT812 in hepatocytes. The other unexpected finding is that trehalose inhibits CD027 prolif-eration by 16S rDNA quantification, and that it did not increase fecal TreA expression. We interpret our in vivo data to suggest that the proposed virulence-promoting effects of native trehalose on C difficile virulence may not be as consequential as previously proposed. We make this interpretation in light of some key experimental differences versus prior C difficile models23. First, we delivered 10-fold more CFU than in prior work23. Secondly, we quantified CD027 toxin, colonization by CD027-specific DNA, mucosal histology, CD027 toxin quantification, and inflammatory markers of colorectal inflammation as our key outcome measures. Nevertheless, unless and until more substantial data in support of the adverse interaction between trehalose and C difficile emerge, the promise of trehalose and its analogues in treating or preventing the metabolic, vascular and neurologic consequences of metabolic disorders should not be dismissed.
The hepatocyte-centered mechanism for trehalose and its analogues is based on prior observations that ablating canonical hepatic fasting signaling intermediaries abrogates the therapeutic effects of trehalose1, 4, 11. The proximal mechanisms regarding trehalose action at the plasma membrane, however, remain an opportunity for further exploration. Prior to this work, key data supporting a GLUT-mediated mechanism for trehalose at the hepatocyte membrane included: (i.) trehalose blocks glucose and fructose transport in vitro4; (ii.) GLUT8 over-expression reverses trehalose-induced hepatocyte autophagy4; (iii.) feeding pyruvate distal to the GLUT reverses trehalose-induced fasting-like signaling4, 14, and (iv.) unbiased in silico modeling demonstrated that trehalose occludes the glucose binding domain in the transporter’s open-inward conformation as its most energetically favorable GLUT8 interaction12. Two additional evidences in vivo support a GLUT blockade mechanism of action. First, we demonstrate therapeutic redundancy between trehalose and liver-specific GLUT8 deletion with regard to hepatic lipid accumulation (Supp. Fig. 8). Second, portal trehalose and lactotrehalose concentrations exceed efficacious concentrations in vitro1. We do acknowledge that these portal trehalose and LT concentrations may provide an overestimate, due to factors including animal stress and glucocorticoid status, anesthesia, splanchnic and portal blood flow, fed fasting status, and timing of sampling64. Therefore, we do not rule out additional mechanisms of trehalose action in the hepatocyte based on these data. Certainly, a GLUT-based mechanism might only partly explain the full effects of these disaccharides, and GLUT inhibition may be a mechanism unique to trehalose effects on the hepatocyte1, 65. Accordingly, we did not observe side-effects that should be considered when targeting GLUTs. In contrast with other bona fide GLUT inhibitors66, or with GLUT mutation syndromes such as Fanconi-Bickel Syndrome67, we did not observe important off-target effects for trehalose or lactotrehalose, including glucose intolerance or glycogen storage defects12. The lack of such off-target effects likely highlights the tissue and transporter selectivity of trehalose and lactotrehalose, which is ultimately owed to first-pass enterohepatic clearance. Furthermore, we view these data as an impetus to also screen for high-affinity, isoform-selective small-molecule GLUT inhibitors that also undergo high first-pass enterohepatic metabolism. The benefit of this approach could be potentially greater specificity and perhaps tolerability68 in comparison to disaccharide analogues.
In conclusion, our findings indicate that concerns regarding the infection-promoting risks of native trehalose warrant further, careful consideration. Moreover, we define lactotrehalose as a next-generation analogue that inhibits CD027-induced rectal inflammation, and yet extends the favorable metabolic effects of native trehalose.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT:
Trehalose is a dietary disaccharide that might be used to treat cardiometabolic or neurodegenerative diseases. However, there is evidence that it promotes expansion of hypervirulent C difficile (ribotype 027).
NEW FINDINGS:
We identified a trehalose analogue, lactotrehalose, that induces hepatic gene expression patterns associated with fasting, increases liver and whole-body oxidative capacity vs trehalose, and blocks hepatic fat accumulation without promoting C difficile pathogenicity.
LIMITATIONS:
These studies were performed in mice; further studies of lacotrehalose are needed in humans.
IMPACT:
Trehalase-resistant trehalose analogues might be developed as fasting-mimetics for treatment of diabetes and NAFLD.
Grant Support:
BJD: NIH R56 DK115764 (BJD), AGA-Gilead Sciences Research Scholar Award in Liver Disease, the AGA-Allergan Research Award in Non-Alcoholic Fatty Liver Disease, the Washington University Digestive Disease Research Core Center P30DK52574, Peer Reviewed Medical Research Program under Award W81XWH-17-1-0133, Longer Life Foundation, NIH/National Center for Advancing Translational Sciences (NCATS) grant #UL1TR002345, the Children’s Discovery Institute (MI-FR-2014-426), the Washington University Diabetes Research Center P30DK020579, and the Robert Wood Johnson Foundation. BMS: NIH R15 AI117670, Camille and Henry Dreyfus Foundation (TH-17-034). PIT: NIH P30DK052574 (Biobank Core). KS, UDW and SVC: United States Department of Agriculture (USDA) Agricultural Research Service CRIS 6026-51000-010-05S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: TH is employed by Hayashibara, Co., which manufactures trehalose.
Writing Assistance: None
REFERENCES
- 1.Zhang Y, DeBosch BJ. Using trehalose to prevent and treat metabolic function: effectiveness and mechanisms. Current Opinion in Clinical Nutrition and Metabolic Care 2019;22:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim Y-M, Lim H, Hur KY, et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nature Communications 2014;5:4934. [DOI] [PubMed] [Google Scholar]
- 3.Sergin I, Evans TD, Zhang X, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun 2017;8:15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBosch BJ, Heitmeier MR, Mayer AL, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. 2016;9:ra21–ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mardones P, Rubinsztein DC, Hetz C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Science Signaling 2016;9:fs2. [DOI] [PubMed] [Google Scholar]
- 6.Rusmini P, Cortese K, Crippa V, et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2018:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menzies FM, Fleming A, Caricasole A, et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017;93:1015–1034. [DOI] [PubMed] [Google Scholar]
- 8.Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007;282:5641–52. [DOI] [PubMed] [Google Scholar]
- 9.Wei M, Brandhorst S, Shelehchi M, et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Science Translational Medicine 2017;9:eaai8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Longo VD, Panda S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metabolism 2016;23:1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Higgins CB, Mayer AL, et al. TFEB-dependent induction of thermogenesis by the hepatocyte SLC2A inhibitor trehalose AU - Zhang, Yiming. Autophagy 2018;14:1959–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayer AL, Higgins CB, Heitmeier MR, et al. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Scientific Reports 2016;6:38586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higgins CB, Zhang Y, Mayer AL, et al. Hepatocyte ALOXE3 is induced during adaptive fasting and enhances insulin sensitivity by activating hepatic PPARγ. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang YHC, Fortune HM, Chen P, Stothard AI, Mayer AL, Swarts BM and DeBosch BJ. Hepatocyte arginase 2 is sufficient to convey the therapeutic metabolic effects of fasting. Nature Communications 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patterson RE, Laughlin GA, LaCroix AZ, et al. Intermittent Fasting and Human Metabolic Health. J Acad Nutr Diet 2015;115:1203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Endres BT, Begum K, Sun H, et al. Epidemic Clostridioides difficile Ribotype 027 Lineages: Comparisons of Texas Versus Worldwide Strains. Open Forum Infectious Diseases 2019;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mogle JA, Santhosh K, Rao K, et al. Clostridium difficile Ribotype 027: Relationship to Age, Detectability of Toxins A or B in Stool With Rapid Testing, Severe Infection, and Mortality. Clinical Infectious Diseases 2015;61:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walk ST, Micic D, Jain R, et al. Clostridium difficile ribotype does not predict severe infection. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 2012;55:1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Feghaly RE, Stauber JL, Deych E, et al. Markers of Intestinal Inflammation, Not Bacterial Burden, Correlate With Clinical Outcomes in Clostridium difficile Infection. 2013;56:1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stang AS, Trudeau M, Vanderkooi OG, et al. Diagnostic Interpretation Guidance for Pediatric Enteric Pathogens: A Modified Delphi Consensus Process. Canadian Journal of Infectious Diseases and Medical Microbiology 2018;2018:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson JI, Weir WH, Crowley JR, et al. Metabolomic networks connect host-microbiome processes to human Clostridioides difficile infections. The Journal of Clinical Investigation 2019;129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins J, Danhof H, Britton RA. The role of trehalose in the global spread of epidemic Clostridium difficile. Gut Microbes 2018:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins J, Robinson C, Danhof H, et al. Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature 2018;553:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danielson ND, Collins J, Stothard AI, et al. Degradation-resistant trehalose analogues block utilization of trehalose by hypervirulent Clostridioides difficile. Chemical Communications 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snydman DR, McDermott LA, Jenkins SG, et al. Epidemiologic Trends in Clostridium difficile Isolate Ribotypes in United States from 2010 to 2014. Open Forum Infectious Diseases 2017;4:S391–S391. [Google Scholar]
- 26.Rivero-Marcotegui A, Olivera-Olmedo JE, Valverde-Visus FS, et al. Water, fat, nitrogen, and sugar content in feces: reference intervals in children. Clinical Chemistry 1998;44:1540. [PubMed] [Google Scholar]
- 27.Kim H-M, Chang Y-K, Ryu S-I, et al. Enzymatic synthesis of a galactose-containing trehalose analogue disaccharide by Pyrococcus horikoshii trehalose-synthesizing glycosyltransferase: Inhibitory effects on several disaccharidase activities. Journal of Molecular Catalysis B: Enzymatic 2007;49:98–103. [Google Scholar]
- 28.Ryu S-I, Kim J-E, Huong NT, et al. Molecular cloning and characterization of trehalose synthase from Thermotoga maritima DSM3109: Syntheses of trehalose disaccharide analogues and NDP-glucoses. Enzyme and Microbial Technology 2010;47:249–256. [Google Scholar]
- 29.O’Neill MK, Piligian BF, Olson CD, et al. Tailoring Trehalose for Biomedical and Biotechnological Applications. Pure Appl Chem 2017;89:1223–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Debosch BJ, Chen Z, Saben JL, et al. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J Biol Chem 2014;289:10989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeBosch BJ, Chen Z, Finck BN, et al. Glucose transporter-8 (GLUT8) mediates glucose intolerance and dyslipidemia in high-fructose diet-fed male mice. Mol Endocrinol 2013;27:1887–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayer AL, Higgins CB, Feng EH, et al. Enhanced Hepatic PPARα Activity Links GLUT8 Deficiency to Augmented Peripheral Fasting Responses in Male Mice. Endocrinology 2018;159:2110–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraft TE, Heitmeier MR, Putanko M, et al. A Novel Fluorescence Resonance Energy Transfer-Based Screen in High-Throughput Format To Identify Inhibitors of Malarial and Human Glucose Transporters. 2016;60:7407–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wankhade UD, Zhong Y, Kang P, et al. Maternal High-Fat Diet Programs Offspring Liver Steatosis in a Sexually Dimorphic Manner in Association with Changes in Gut Microbial Ecology in Mice. Scientific Reports 2018;8:16502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorens B, Mueckler M. Glucose transporters in the 21st Century. 2010;298:E141–E145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hou BH, Takanaga H, Grossmann G, et al. Optical sensors for monitoring dynamic changes of intracellular metabolite levels in mammalian cells. Nat Protoc 2011;6:1818–33. [DOI] [PubMed] [Google Scholar]
- 37.Xu C, Chen X, Sheng W, Yang P Trehalose restores functional autophagy suppressed by high glucose. Reproductive Toxicology 2019;epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Austin S, St-Pierre J. PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 2012;125:4963–71. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Xie Y, Berglund ED, et al. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. Elife 2012;1:e00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Potthoff MJ, Inagaki T, Satapati S, et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci U S A 2009;106:10853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Potthoff MJ. FGF21 and metabolic disease in 2016: A new frontier in FGF21 biology. Nat Rev Endocrinol 2017;13:74–76. [DOI] [PubMed] [Google Scholar]
- 42.Ogborn DI, McKay BR, Crane JD, et al. The unfolded protein response is triggered following a single, unaccustomed resistance-exercise bout. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2014;307:R664–R669. [DOI] [PubMed] [Google Scholar]
- 43.Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev 2012;26:312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madrigal-Matute J, Cuervo AM. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016;150:328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ke PY. Diverse Functions of Autophagy in Liver Physiology and Liver Diseases. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy; 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. The Journal of clinical investigation 2008;118:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshizane C, Mizote A, Yamada M, et al. Glycemic, insulinemic and incretin responses after oral trehalose ingestion in healthy subjects. Nutr J 2017;16:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mizote A, Yamada M, Yoshizane C, et al. Daily Intake of Trehalose Is Effective in the Prevention of Lifestyle-Related Diseases in Individuals with Risk Factors for Metabolic Syndrome. Journal of Nutritional Science and Vitaminology 2016;62:380–387. [DOI] [PubMed] [Google Scholar]
- 50.Haas JT, Staels B. Fasting the Microbiota to Improve Metabolism? Cell Metabolism 2017;26:584–585. [DOI] [PubMed] [Google Scholar]
- 51.Kohl KD, Dearing MD, Passement CA, et al. Unique and shared responses of the gut microbiota to prolonged fasting: a comparative study across five classes of vertebrate hosts. FEMS Microbiology Ecology 2014;90:883–894. [DOI] [PubMed] [Google Scholar]
- 52.Rebholz CM, Yu B, Zheng Z, et al. Serum metabolomic profile of incident diabetes. Diabetologia 2018;61:1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bloomgarden Z Diabetes and branched-chain amino acids: What is the link? 2018;10:350–352. [DOI] [PubMed] [Google Scholar]
- 54.Holeček MJN, Metabolism. Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. 2018;15:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernández del Río R, O’Hara ME, Holt A, et al. Volatile Biomarkers in Breath Associated With Liver Cirrhosis — Comparisons of Pre- and Post-liver Transplant Breath Samples. EBioMedicine 2015;2:1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jing L, Zhang Y, Fan S, et al. Preventive and ameliorating effects of citrus d-limonene on dyslipidemia and hyperglycemia in mice with high-fat diet-induced obesity. European Journal of Pharmacology 2013;715:46–55. [DOI] [PubMed] [Google Scholar]
- 57.Lee J, Ngo J, Blake D, et al. Improved predictive models for plasma glucose estimation from multi-linear regression analysis of exhaled volatile organic compounds. Journal of applied physiology (Bethesda, Md.: 1985) 2009;107:155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cotrim HP, De Freitas LAR, Freitas C, et al. Clinical and histopathological features of NASH in workers exposed to chemicals with or without associated metabolic conditions. Liver International 2004;24:131–135. [DOI] [PubMed] [Google Scholar]
- 59.Gore AC, Fenton SE, Chappell VA, et al. Executive Summary to EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocrine Reviews 2015;36:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abbasi J Did a Sugar Called Trehalose Contribute to the Clostridium difficile Epidemic?Did Trehalose Contribute to the C. difficile Epidemic?Did Trehalose Contribute to the C. difficile Epidemic? JAMA 2018;319:1425–1426. [DOI] [PubMed] [Google Scholar]
- 61.Du Toit A Clostridium difficile is sweet on trehalose. Nature Reviews Microbiology 2018;16:64. [DOI] [PubMed] [Google Scholar]
- 62.Beli E, Yan Y, Moldovan L, et al. Restructuring of the Gut Microbiome by Intermittent Fasting Prevents Retinopathy and Prolongs Survival indb/dbMice. Diabetes 2018;67:1867–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guillemain G, Loizeau M, Pincon-Raymond M, et al. The large intracytoplasmic loop of the glucose transporter GLUT2 is involved in glucose signaling in hepatic cells. 2000;113:841–847. [DOI] [PubMed] [Google Scholar]
- 64.Kimura RE, LaPine TR, Manford Gooch W. Portal Venous and Aortic Glucose and Lactate Changes in a Chronically Catheterized Rat. Pediatric Research 1988;23:235–240. [DOI] [PubMed] [Google Scholar]
- 65.Lee HJ, Yoon YS, Lee SJ. Mechanism of neuroprotection by trehalose: controversy surrounding autophagy induction. Cell Death Dis 2018;9:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hruz PW. HIV protease inhibitors and insulin resistance: lessons from in-vitro, rodent and healthy human volunteer models. Current opinion in HIV and AIDS 2008;3:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thorens B GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015;58:221–232. [DOI] [PubMed] [Google Scholar]
- 68.Bergoz R Trehalose Malabsorption Causing Intolerance to Mushrooms: Report of a probable case. Gastroenterology 1971;60:909–912. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.