

Abstract
Over the course of the human lifespan, somatic DNA mutations accumulate in healthy tissues. This process has been most clearly described in blood and bone marrow, esophagus, colon and skin, but cumulative DNA damage likely affects all tissues of the body. While most acquired genetic variants have no discernable functional consequences, some randomly occurring somatic mutations confer a relative fitness advantage on a single stem cell and its progeny compared to surrounding cells, which may lead to progressive expansion of a clone (i.e., a genetically identical group of cells). When these mutations occur in a cell with the capacity to self-renew and expand, the mutations persist, and such clonal expansion is a risk factor for further mutation acquisition and clonal evolution.
Hematopoietic stem cells are a special case of clonal expansion, because both the stem cells and their blood cell progeny circulate in large numbers, and these cells are not subject to some of the anatomical restrictions that characterize other tissues in which somatic mutations conferring a fitness advantage also occur. Therefore, clonally restricted hematopoiesis can have biological and clinical consequences that are distinct from clonal expansions in other tissues. Such consequences include not just clonal progression to overt myeloid neoplasia (or, less commonly, to lymphoid neoplasia) driven by acquisition of secondary mutations in the cells of the expanded clone, but also cardiovascular events, as described further below – and most likely other diseases that are influenced by aberrant function of mutant blood cells.
If we can understand in more detail how clonal hematopoiesis arises and how clonal selection and expansion occur, as well as develop strategies to avert the clinical consequences associated with clonal hematopoiesis, this may both improve public health and yield more general insights into the biology of aging.
Keywords: clonal hematopoiesis, clonal evolution, myelodysplastic syndromes, somatic mosaicism, biology of aging
Graphical Abstract
Clonal hematopoiesis: an overview
One of the most unexpected and interesting developments in the biology of human aging in recent years has been the discovery of the ubiquity of somatic mosaicism in various tissues of older persons.1–4 Clonal expansion in aging tissues is often driven by some of the same acquired mutations conferring relative fitness that have been previously associated with overt malignancy in those same tissues, yet most people with these mutations do not have malignancy and will never develop a neoplasm.4,5
Due to the availability of large numbers of cells for serial assessments, blood is especially amenable to study when it comes to investigating somatic mosaicism, with next-generation sequencing platforms providing a method for rapid and sensitive detection of somatic mutations, either in a targeted fashion or via whole exome or whole genome approaches. As a result, understanding of the prevalence, pathobiology and clinical associations of clonal expansion of hematopoietic cells is advancing rapidly, and this has served as a model for parallel processes in other tissues. 6–8
While terminology related to this quickly-evolving field is in flux, as discussed further below, the term “clonal hematopoiesis” has been the most widely applied shorthand for the state in which one hematopoietic stem cell or progenitor has acquired a somatic mutation that confer an advantage relative to neighboring cells, and then contributes an outsized proportion of blood cell production in comparison to representation among surviving hematopoietic stem cells.
This biological state (not a disease per se) has emerged as a risk factor for both hematological neoplasia and for other health problems, including cardiovascular events. 9 In many cases of acute leukemia, in contrast, expansion of clonal cells also occurs, but these cells fail to differentiate and merely inhibit hematopoiesis, and residual blood cell production largely depends on remaining healthy non-clonal cells.10
One of two specific genes – DNMT3A and TET2, encoding proteins both of which have roles in DNA methylation – is somatically mutated in most instances of clonal hematopoiesis (Figure 1).6–8 These mutations can be detected at a DNA variant allele frequency (VAF, i.e. proportion of abnormal DNA sequencing reads relative to total reads) of at least 2% in the blood of more than 10% of people by age 70 years. Since these mutations are heterozygous and loss of heterozygosity is uncommon with these particular alleles, a 2% VAF means that 4% of blood cells are derived from a single clone – a dramatic expansion, given that adult humans have 50,000–200,000 hematopoietic stem cells, so that a neutral somatic variant would be observed at a VAF on the order of 1 × 10−5 (0.00001%). In some cases of clonal hematopoiesis, VAFs as high as 40–50% are observed, which means that in such cases a single somatically altered stem cell is giving rise to the majority of the affected person’s circulating blood cells.11
Figure 1:

The most common mutations in clonal hematopoiesis of indeterminate potential (CHIP) include DNMT3A and TET2, followed by ASXL1, JAK2, and TP53. IDH1/2 mutations are less common. Dozens of other mutations occur, but these are relatively rare.
Clonal hematopoiesis predisposes both to overt cancer diagnosable using conventional World Health Organization (WHO) clinicopathological criteria12, similar to somatic expansions in other tissues, but is also associated with increased all-cause mortality and a variety of clinical phenotypes.13 Another gene, TP53, is less commonly mutated than DNMT3A or TET2, but is a strong predictor of clonal evolution, especially in the presence of cytotoxic chemotherapy or radiotherapy that result in suppression of normal hematopoiesis at the expense of clonal selection of a chemo- and radio-resistant TP53 mutant clone.14–16
This review summarizes some of the recent developments in clonal hematopoiesis as an exemplar of pre-malignant changes during human aging. Clonal hematopoiesis must be understood not as an isolated phenomenon but within the context of somatic mutation acquisition in all tissues throughout the human lifespan. The extent to which clonal states are also a risk factor for various non-neoplastic disorders, or altered patterns of response to specific therapies for hematological and other diseases, is an area of active research.
A brief history and evolution of terminology related to clonal hematopoiesis
The term “clone” was first used in an agricultural context in 1903 by botanists Herbert Webber and Orator Fuller Cook to describe asexual propagation of plants by transplant or cuttings.17 In 1912, plant geneticist George Harrison Shull proposed expanding the term to animals; tadpoles were the first non-plant organism cloned in 1952. By the 1970s, use of the term “clone” had expanded to describe any group of genetically identical cells or nucleic acid strands, not just whole organisms.18
From the 1990s onward, study of mutation serial mutation acquisition in blood cells paralleled analysis of clonality.19 Analysis of non-random X chromosome inactivation in women with informative markers had been used by Philip Fialkow and others since the 1960s to demonstrate clonality in myeloid neoplasms (e.g., that myeloproliferative neoplasms are clonal), and skewed X inactivation had been found to be associated with aging in apparently healthy women.20–22 In 2012, Lambert Busque and his colleagues detected mutations in TET2, a gene that had just been linked to myeloid neoplasia in 2009, in 5% of older women with skewed X chromosome inactivation, but did not find mutations in any younger women or older women with balanced X-chromosome inactivation.23 In the 1990s, apparently healthy individuals had been noted to have very low levels of BCL translocations that are commonly associated with lymphoma detectable in the blood in the absence of a lymphoid neoplasm, or BCR-ABL fusions associated with chronic myeloid leukemia but without leukemia – albeit only transiently, and at very low levels (<1% VAF), unlike the more stable and higher allelic burden (>5% VAF) TET2 mutations.24,25
Also in 2012, two series reported a high prevalence of large structural variations of chromosomes (>1% in the general population older than age 50), and such somatic mosaicism was associated both with all-cause mortality and also the development of hematologic malignancies.26,27 In 2014, three groups reported data from a series of whole exome sequencing studies that had been performed for other purposes, such as to look for predisposing germline alleles to schizophrenia, diabetes and other prevalent conditions.6–8 Because blood-derived DNA had been used for these experiments, the analyses were repurposed to study clonal hematopoiesis, collectively in more than 30,000 individuals. This led to the finding that clonal hematopoiesis is common with aging and also that clonal hematopoiesis results from a relatively restricted set of genes, including genes commonly associated with hematologic malignancies.28
In these 2014 series, most mutations observed were in single genes and with a median VAF of almost 10%.7 The three most common genes mutated were DMMT3A, TET2, and ASXL1; JAK2 and TP53 were the next most frequently mutated, and there was a long list of rarer mutations present in <1% of individuals with clonal hematopoiesis.29 Since whole exome sequencing is relatively insensitive to small clones (Figure 2), subsequent studies were performed using error corrected targeted sequencing with a much greater depth of coverage, and these studies demonstrated that clonal hematopoiesis with clones at <1% VAF is almost universal with aging.30,31 However it is unclear whether these tiny clones have clinical importance, as the larger clones detected in the whole exome sequencing analyses do.
Figure 2:

The prevalence of clonal hematopoiesis depends on the analytical sensitivity of the technique used to detect it. With very sensitive techniques, such as error-corrected methods that can detect populations with a variant allele frequency (VAF) of <0.01%, clonal hematopoiesis is almost universally observed by middle age. Whole-exome and whole-genome sequencing techniques typically have less depth of coverage and sensitivity, and may not detect clonal hematopoiesis below 7–10% VAF. Targeted sequencing platforms with >100x depth of coverage may be able to detect clonal hematopoiesis at the 1–2% VAF level and in this setting the prevalence of clonal hematopoiesis exceeds 10% by age 65–70 years Adapted from reference 1, with permission.
Clonal hematopoiesis with aging was also described without any known leukemia-associated driver mutation such as the ones listed above, either because the current known catalog of driver mutations is incomplete, because of mutations in the non-coding genome, or, more speculatively, because epigenetic changes without accompanying DNA mutation may also lead to clonal drift or expansion.6,32 In addition, initiating clonal changes were detected in large datasets of AML patients years before the AML emerged.31,33,34
Recognizing a need for a term to describe this biological phenomenon, in 2015 we proposed the “clonal hematopoiesis of indeterminate potential” (CHIP) both to highlight the potential for this state to progress to a hematological neoplasm (~0.5% risk per year), and to distinguish this clonal state from WHO-criteria diagnosable disorders such as myelodysplastic syndromes (MDS), which remain defined by the presence of clinically meaningful cytopenias (by definition, absent in CHIP), dysplastic blood and marrow cell morphology, excess myeloblasts, or somatic chromosomal variants exclusive of several non-specific alterations like loss of the Y chromosome or trisomy 8.35 Our proposed CHIP definition required individuals to have a VAF of at least 2%, which reflected both technical limitations of next-generation sequencing platforms commonly used in clinical laboratories, but also the unclear prognostic significance of smaller clones detected only with error-corrected or other very sensitive sequencing techniques.
Aging-related clonal hematopoiesis (ARCH) is another term that has been used to describe an overlapping set of states and that does not require a specific VAF or the presence of a putative leukemia driver mutation.36 “Clonal hematopoiesis” in turn is a shorthand that is commonly used to describe all these states, although this usage is somewhat problematic both because overt hematologic malignancies are also defined by a clonally restricted hematopoietic process, while healthy hematopoiesis in turn is a consequence of the cumulative contribution of tens of thousands of different clones.37
Clonal expansion in other tissues
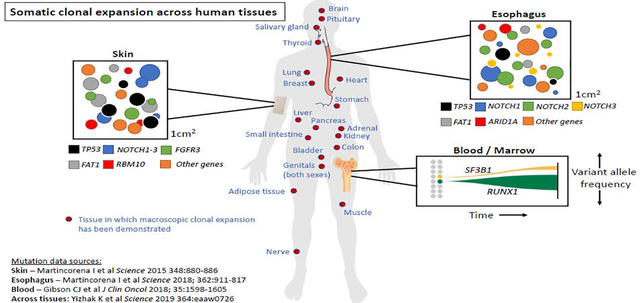
Somatic mosaicism in hematopoietic tissues is just one example of clonal expansion associated with acquisition of somatic mutations, which most likely occurs in every tissue.5 While hematopoietic stem cells acquire about 1 exonic mutation every 10 years10, the rate of acquisition of mutations in other tissues is less clear (but likely a similar order of magnitude, given that the common epithelial cancers are all most frequent beyond age 50).
For example, when esophageal tissue was sampled from 9 normal donors and subject to deep sequencing, age-associated acquisition of somatic mutations was observed in genes linked to esophageal malignancy, including NOTCH1, NOTCH2, NOTCH3 ARID1A, and even TP53.38 Similarly, an RNA sequencing analysis of 29 normal tissues collected from 500 people as part of the Genotype-Tissue Expression (GTEx) project showed clonal expansion in all tissues examined; skin, esophagus, and lung had the largest number of somatic mutations.5
A recent detailed study of clonal architecture in the liver that included whole genome sequencing of 482 microdissections of 100–500 hepatocytes from 5 normal and 9 cirrhotic livers indicated a high incidence of clonal outgrowth in association with fibrosis and cirrhosis, suggesting that the tissue microenvironment may play an important role in initiation or expansion of emergent clones.39 Experiments supporting this hypothesis with respect to clonal hematopoiesis are described further below.
A particularly interesting case is the brain. A dual-platform analysis of 102 genes in 173 adult human brain samples with >5000-fold depth of sequencing demonstrated somatic mutations and macroscopic islands of mutated neurons in 50% of brains tested, and the most common mutations observed were in the same two genes that are most frequent in clonal hematopoiesis, DNMT3A and TET2.40 While detection of these specific mutations may be due to contamination of sequenced tissue by blood, the somatic mutations might also be in the microglia. Microglia are not derived from hematopoietic progenitors, but share some features with hematopoiesis-derived cells, including descent from the primitive yolk sac and a tissue scavenging role that resembles that of tissue macrophages.
Thus, it is clear that random accumulation of mutations over time that, in some cases, influences fitness of a certain cell and its progeny and contributes to clonal expansion, is not at all unique to hematological cells. However, clonal expansion of hematopoietic tissue has distinct consequences because of the way blood cells circulate in large numbers and interact with other tissues, as described below.1
We do not yet know the rate of acquisition of persistent somatic mutations or the prevalence of clonal hematopoiesis, or clonal expansion in other tissues, in any species besides humans. Clonal hematopoiesis might be expected to occur in non-human primates, due to similar hematopoiesis compared to humans and a long lifespan, but is thus far poorly studied.41 Clonality testing in veterinary clinical practice is only in its infancy and further developments are awaited.42
Risk factors for development of clonal hematopoiesis: germline and environment
The mechanisms by which clonal hematopoiesis arises may give insight into why some individuals have clonal expansion earlier or to a greater degree than others. Among the mechanisms of somatic mutation acquisition, the most prevalent by far appears to be single base pair change as a result of the ubiquitous spontaneous deamination of methylated cytosine bases at CpG dinucleotides, resulting in a thymine substitution which may avoid DNA repair, rather than the more easily recognized uracil that is created by spontaneous deamination of unmethylated cytosine.43 The consequence of this type of DNA mis-repair is a stable thymine:adenine base pair in a daughter cell, rather than the parental cytosine:guanine paring. As a result, C to T transitions are by far the most common variant detected both in clonal hematopoiesis and in clonal expansion in other tissues. Replication errors by DNA polymerase, errors introduced as a result of mis-repaired double-strand DNA breaks leading to small insertion-deletions, and large chromosomal structural rearrangements also occur, but are less common than single base pair changes.
While clonal emergence with aging may be driven to some extent by chance/stochastic processes, several germline variants have been associated with an increased likelihood of clonal hematopoiesis. For instance, germline loss of the DNA glycosylase Methyl-CpG Binding Domain 4 (MBD4), an enzyme that contributes to repair of DNA damage resulting from 5 methyl-cytosine deamination described above, not only increases the risk of clonal hematopoiesis and of MDS/AML, but also results in a more than 30-fold higher mutation burden (enriched for C to T transitions) in AML cells in affected individuals compared to AML generally.44 MBD4 germline loss is rare, however, representing only 9 cases of among 10,683 TCGA database patients, and it may be that other more common but less highly penetrant germline DNA repair polymorphisms contribute to the risk clonal hematopoiesis.
An analysis of mosaic chromosomal alterations in 151,202-participant UK Biobank demonstrated 3 loci at which germline variants were associated with cis chromosomal deletions or with loss of heterozygosity: MPL (encoding the thrombopoietin receptor) and 2 genes about which much less is known, FRA10B and TM2D3-TARSL2.45 Another germline variant predisposing to aging-associated clonal hematopoiesis emerged from an analysis of 11,262 participants in the Iceland deCODE project, in which the presence of more than 20 mosaic somatic variants was used to classify patients as whole genome sequencing “outliers” and therefore having clonal hematopoiesis, even in the absence of a mutation in a putative leukemia driver gene.32 In the deCODE population, a germline intron 3 deletion of the gene encoding telomerase reverse transcriptase (TERT), important for telomere length maintenance, was found to predispose to clonal hematopoiesis (p=7.4 × 10−12, with an odds ratio of 1.37) -- yet perhaps surprisingly this variant was not associated with shorter telomeres in affected individuals. Finally, in another UK Biobank analysis, somatic loss of the Y chromosome in leukocytes was associated with more than 150 genetic variants (and with an increased likelihood of developing non-myeloid cancers including prostate adenocarcinoma, germ cell tumors and renal cell carcinoma), and this finding was validated in an additional population of 757,114 men of Japanese or European ancestry. 46
Further insight into germline predisposition to clonal hematopoiesis emerged from a German study of 500 allogeneic hematopoietic cell transplant related donors aged 55 years or above.47 Clonal hematopoiesis was present in 16% of donors, with a median VAF of 5.9%, and was associated with more chronic graft-versus-host disease and also a trend towards higher non-relapse mortality – but lower neoplastic disease relapse, such that the net overall survival was unaffected by the clonal hematopoiesis state of the donor. Intriguingly, clonal hematopoiesis was present in 19.2% of related donors for recipients with myeloid malignancies compared to 6.3% for siblings with lymphoid neoplasms. Given the myeloid bias of clonal evolution in CHIP, this suggests a mutual germline predisposition to clonal states in both donor and recipient, with a possible contribution from a shared environment.
Clonal hematopoiesis is slightly less common in people of Hispanic ethnicity, and is seen with increased frequency in males and in cigarette smokers.6,7 The environment may also have an influence on the likelihood of acquiring clonal hematopoiesis. The effect of the environment on clonal outgrowth is also supported by a murine model in which Tet2 null mice (which at baseline have increased gut permeability to bacteria compared to wild-type mice) underwent either intestinal barrier disruption to increase bacterial translocation or administration of a toll-like receptor 2 agonist, mimicking systemic bacterial infection.48 The mice treated in this way developed myeloproliferation with a severity that correlated with serum interleukin-6 levels. In contrast, Tet2 null mice that underwent intestinal detoxification or were raised in a germ-free environment mostly remained healthy. There are no data yet about the effect of the microbiome on clonal hematopoiesis in humans.
Risk factors for clonal expansion may differ from those contributing to clone origination. For instance, chemotherapy or radiotherapy suppress normal hematopoiesis to a greater extent than they do hematopoiesis from cells with clonal mutations of TP53 or PPM1D, and patients with TP53 mutant clonal hematopoiesis who undergo cytotoxic therapy are at greatly increased risk for therapy related myeloid neoplasms.49 The fact that in many cases clonal hematopoiesis is stable for years indicates that the expansion is being held in check, either because of a limitation on the micro-environmental niche that can be occupied by these cells until they acquire a stronger leukemic driver mutation, or else because of an endogenous immune response.
Clinical consequences of CHIP: neoplasia
CHIP predisposes to neoplasia, mostly (but not exclusively) myeloid neoplasms.13 This is thought to usually result from acquisition of a stronger driver mutation on a background CHIP clone. For instance, an illustrative case was described in 2018 in which 45-year-old man had CHIP with three different truncating TET2 mutations and a frame shifting ASXL1 mutation all at >20% VAF, then acquired a RHOA mutation and developed angioimmunoblastic T-cell lymphoma.11 After successful treatment of the lymphoma, the following year the same patient acquired an NPM1 mutation on the same CHIP background and developed AML.
There is overlap between clonal hematopoiesis and the concept of minimal residual disease. In a European cooperative group study of patients treated with intensive therapy for acute myeloid leukemia, for instance, treated patients in whom the only detectable mutation after achieving clinical-pathological complete remission was a “CHIP mutation” (DNMT3A, TET2 or ASXL1) had a low incidence of recurrent disease after 4 years, whereas most of those who still had detectable stronger leukemia drivers (eg FLT3, IDH1/2, NPM1, RUNX1) soon relapsed.50
In the context of acquired aplastic anemia, the pathophysiology of which is usually T-cell directed immune attack against hematopoietic stem cells, clonal mutations frequently emerge, again likely due to relative fitness of cells bearing those mutations compared to neighboring cells, albeit in a markedly abnormal marrow environment.51 Mutations in PIGA, BCORL1, or BCOR are associated with a response to anti-T-cell immunotherapy and a better prognosis, while other clonal mutations such as DNMT3A or Ras pathway mutations are associated with non-response to immunotherapy and with a greater likelihood of eventual clonal progression to MDS or another state.
After stem cell transplantation, donor CHIP (i.e., a mutant clone derived from the allogeneic donor that engrafts in the recipient) contributes to cytopenias, and may also result in donor-derived leukemia.52 After autologous hematopoietic cell transplant, too, clonal hematopoiesis is associated with adverse outcomes.53,54
Clonal hematopoiesis can cause diagnostic classification confusion in patients with cytopenias and a non-diagnostic bone marrow morphology. The high frequency of CHIP in the general older population means that some patients may have cytopenias and mild morphologic changes due to reactive non-clonal cause together with an unrelated clonal process, which may cause uncertainty about the relative contribution of the clonal process to the cytopenias.55 Given the large number of factors other than clonal hematopoiesis that can contribute to cytopenias, it may be difficult for clinicians to thoroughly rule out a concomitant non-clonal process in a patient who has both unexplained cytopenias and clonal hematopoiesis.
Individuals with unexplained cytopenias who have clonal mutations in genes associated with CHIP and MDS, however, are at increased risk for subsequent progression, with mutations in spliceosome genes and those with multiple mutations at high VAF (>20%) at greatest risk. Among patients with unexplained cytopenias, one multi-center analysis indicated that a coexistent clonal mutation resulted in a hazard ratio of 13 for progression to WHO-diagnosable MDS or another myeloid malignancy, compared to patients with idiopathic cytopenias and no mutation.56
Clinical consequences of CHIP: other disorders
Individuals with CHIP also have an increased risk of a cardiovascular event.57–59 This risk (hazard ratio ~1.8 compared to controls without clonal hematopoiesis) is of a similar order of magnitude to well-established modifiable cardiovascular risk factors including cigarette smoking, hypertension, and hyperlipidemia. The pathophysiology of this risk has been extensively reviewed elsewhere57,58,60, but briefly, murine models indicate that clonally-derived monocytes and macrophages infiltrate atherosclerotic plaques and set up a pro-inflammatory reaction that recruits more pro-inflammatory macrophages, injures the endothelium, and accelerates atherogenesis. This process can be blocked either in experimental models with NLRP3 inflammasome inhibition, or clinically with anti-cytokine therapy.59,61 In a randomized, multi-center placebo-controlled clinical trial of more than 10,000 patients who had experienced myocardial infarction and still had an elevated C-reactive protein, treatment with canakinumab, an antibody against interleukin 1-β, reduced recurrent cardiovascular events compared to placebo, and the greatest relative risk reduction with canakinumab therapy was seen in those patients who had CHIP, especially TET2 mutant clonal hematopoiesis.62,63
Cardiovascular outcomes beyond atherosclerosis also appear to be influenced by CHIP. For instance, in one analysis of CHIP-associated JAK2 mutations, the risk of venous thrombosis was markedly increased compared to those without JAK2 clonal hematopoiesis.64 Congestive heart failure outcomes are also worse among patients with CHIP, probably due to locally altered myocardial remodeling driven by pro-inflammatory clonal macrophages.65
In one study of 83 geriatric patients, CHIP was associated with increased frailty and increased levels of interleukin-6, tumor necrosis factor-α and interleukin-8 – though whether this association represents cause-and-effect or merely co-existence of different markers of accelerated aging is unclear. 66 One exceptional responder to chimeric antigen receptor T-cell therapy resulted from disruption of a copy of TET2 in one of the engineered T cells, accompanied by cell expansion.67 At present, it is not clear whether clonal hematopoiesis is associated with other clinical conditions besides neoplasia and cardiovascular events. 68
The high frequency with which patients are found to have CHIP both during genetic analysis of non-myeloid neoplasms14,29, including germline testing of hereditary breast and other cancer syndromes, has prompted creation of clinics dedicated to patients with clonal hematopoiesis and other hematologic malignancy precursor conditions.69 Our institute started a dedicated hematological neoplasm precursor condition clinic in 2019 in order to counsel such patients, who are understandably often quite anxious when they learn they have a mutation that can predispose to leukemia but is more likely to contribute to a myocardial infarction or stroke. In the future, it is essential to design interventions both to try to eliminate emergent clones and to try to reduce the cardiovascular and other consequences of clonally derived cells.
Key unanswered questions
Despite the rapid progress in this field, a number of important questions remain about clonal hematopoiesis, and by extension about somatic variation in other tissues. In addition to the range of disease states associated clonal hematopoiesis, the reason for long-term stability of clones in some patients and the drivers of both clonal emergence and clonal evolution are incompletely understood. The relative risk of different CHIP mutations both for cardiovascular disease and clonal progression is also not well understood, and it is unclear whether clonal expansion can result purely from epigenetic changes without somatic DNA changes at either the chromosome or single nucleotide levels. In addition, clinical consequences of clonal expansion in other tissues beyond predisposing to neoplasia risk is almost entirely unknown.
Conclusion
Clonal hematopoiesis is just one example of clonal expansion due to somatic mutation acquisition in aging tissue – but it is a clonal expansion with special consequences including pro-inflammatory interactions with non-hematopoietic cells, since blood cells circulate and are not subject to the same anatomical restrictions that characterize clonal expansion in other tissues, which in contrast would be expected to have only more local effects. Given the high frequency with which clonal hematopoiesis is observed and the magnitude of cardiovascular risk associated with clonal hematopoiesis, the possibility of using anti-inflammatory approaches to prevent primary or secondary cardiovascular events and improve patient outcomes is attractive from a public health standpoint. Many investigative groups are actively searching for additional clinical associations.
Highlights.
Imbalanced hematopoiesis in which one clone or a small group of clones bearing somatic mutations contribute an outsized proportion of blood cell production, commonly termed “clonal hematopoiesis”, is a frequent finding in human aging
Clonal hematopoiesis is a risk factor for hematological neoplasia, and the high prevalence of clonal hematopoiesis in older persons may cause diagnostic confusion in patients with unexplained blood cytopenias and lack of definitive diagnostic findings
Pre-malignant clonal expansion also occurs widely in other tissues besides blood and marrow, but clonal hematopoiesis has unique biological properties and clinical implications compared to clonal expansion in other tissues because blood cells circulate; these properties include inflammatory pro-atherogenic interactions between circulating clonal monocytes/macrophages bearing somatic mutations and the vascular endothelium
Certain germline alleles predispose to clonal hematopoiesis, and likely environmental factors influence the probability of clonal emergence as well, but this is incompletely understood at present
Acknowledgements
Funding: DPS and BLE are supported by the Edward P. Evans Foundation and by NIH SPORE P50CA206963. BLE is a Howard Hughes investigator and is supported by the Deerfield Foundation and by NIH P01CA066996 and NIH R01HL082945. DPS is also supported by the James & Lois Champy Fund.
Disclosures/Acknowledgments: DPS: data safety monitoring committee or consulting related to clinical trials: Celgene, H3 Biosciences, Janssen, Onconova, Otsuka, Acceleron. BLE has received research funding from Celgene and Deerfield. He has received consulting fees from GRAIL, and he serves on the scientific advisory boards for and holds equity in Skyhawk Therapeutics and Exo Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science 2019;366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoang ML, Kinde I, Tomasetti C, et al. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc Natl Acad Sci U S A 2016;113:9846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blokzijl F, de Ligt J, Jager M, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016;538:260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martincorena I Somatic mutation and clonal expansions in human tissues. Genome Med 2019;11:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yizhak K, Aguet F, Kim J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019;364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genovese G, Kähler AK, Rose SA, et al. Clonal hematopoiesis and cancer risk in blood derived DNA sequence. N Engl J Med 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luis TC, Wilkinson AC, Beerman I, Jaiswal S, Shlush LI. Biological implications of clonal hematopoiesis. Exp Hematol 2019;77:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012;150:264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiacci E, Venanzi A, Ascani S, et al. High-Risk Clonal Hematopoiesis as the Origin of AITL and NPM1-Mutated AML. N Engl J Med 2018;379:981–4. [DOI] [PubMed] [Google Scholar]
- 12.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405. [DOI] [PubMed] [Google Scholar]
- 13.Bowman RL, Busque L, Levine RL. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 2018;22:157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coombs CC, Zehir A, Devlin SM, et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017;21:374–82 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi K, Wang F, Kantarjian H, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol 2017;18:100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillis NK, Ball M, Zhang Q, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol 2017;18:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webber HJ. New Horticultural and Agricultural Terms. Science 1903;18:501–3. [DOI] [PubMed] [Google Scholar]
- 18.Steensma DP. The origin and evolution of the term “clone”. Leuk Res 2017;57:97–101. [DOI] [PubMed] [Google Scholar]
- 19.Busque L, Mio R, Mattioli J, et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood 1996;88:59–65. [PubMed] [Google Scholar]
- 20.Fey MF, Liechti-Gallati S, von Rohr A, et al. Clonality and X-inactivation patterns in hematopoietic cell populations detected by the highly informative M27 beta DNA probe. Blood 1994;83:931–8. [PubMed] [Google Scholar]
- 21.Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med 1976;295:913–6. [DOI] [PubMed] [Google Scholar]
- 22.Barr RD, Fialkow PJ. Clonal origin of chronic myelocytic leukemia. N Engl J Med 1973;289:307–9. [DOI] [PubMed] [Google Scholar]
- 23.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012;44:1179–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Hernandez AM, Shibata D, Cortopassi GA. BCL2 translocation frequency rises with age in humans. Proc Natl Acad Sci U S A 1994;91:8910–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood 1995;86:3118–22. [PubMed] [Google Scholar]
- 26.Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet 2012;44:642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet 2012;44:651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 2016;374:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y, Ulrich BC, Supplee J, et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res 2018;24:4437–43. [DOI] [PubMed] [Google Scholar]
- 30.McKerrell T, Park N, Moreno T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis Cell Reports 2015:Available online 26 February 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zink F, Stacey SN, Norddahl GL, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017;130:742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abelson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018;559:400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014;506:328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shlush LI. Age-related clonal hematopoiesis. Blood 2018;131:496–504. [DOI] [PubMed] [Google Scholar]
- 37.Gibson CJ, Steensma DP. New Insights from Studies of Clonal Hematopoiesis. Clin Cancer Res 2018. [DOI] [PubMed] [Google Scholar]
- 38.Martincorena I, Fowler JC, Wabik A, et al. Somatic mutant clones colonize the human esophagus with age. Science 2018;362:911–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunner SF, Roberts ND, Wylie LA, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019;574:538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keogh MJ, Wei W, Aryaman J, et al. High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat Commun 2018;9:4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu KR, Espinoza DA, Wu C, et al. The impact of aging on primate hematopoiesis as interrogated by clonal tracking. Blood 2018;131:1195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keller SM, Vernau W, Moore PF. Clonality Testing in Veterinary Medicine: A Review With Diagnostic Guidelines. Vet Pathol 2016;53:711–25. [DOI] [PubMed] [Google Scholar]
- 43.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanders MA, Chew E, Flensburg C, et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood 2018;132:1526–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loh PR, Genovese G, Handsaker RE, et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 2018;559:350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson DJ, Genovese G, Halvardson J, et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frick M, Chan W, Arends CM, et al. Role of Donor Clonal Hematopoiesis in Allogeneic Hematopoietic Stem-Cell Transplantation. J Clin Oncol 2019;37:375–85. [DOI] [PubMed] [Google Scholar]
- 48.Meisel M, Hinterleitner R, Pacis A, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018;557:580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu JI, Dayaram T, Tovy A, et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018;23:700–13 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N Engl J Med 2018;378:1189–99. [DOI] [PubMed] [Google Scholar]
- 51.Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med 2015;373:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibson CJ, Kennedy JA, Nikiforow S, et al. Donor-engrafted CHIP is common among stem cell transplant recipients with unexplained cytopenias. Blood 2017;130:91–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gibson CJ, Lindsley RC, Tchekmedyian V, et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J Clin Oncol 2017;35:1598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mouhieddine TH, Park J, Redd R, et al. Abstract 2954: Immunomodulator maintenance post autologous stem cell transplant predicts better outcome in multiple myeloma patients with clonal hematopoiesis of indeterminate potential. Cancer Res 2018;78:2954-. [Google Scholar]
- 55.Kwok B, Hall JM, Witte JS, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015;126:2355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malcovati L, Galli A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017;129:3371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khetarpal SA, Qamar A, Bick AG, et al. Clonal Hematopoiesis of Indeterminate Potential Reshapes Age-Related CVD: JACC Review Topic of the Week. J Am Coll Cardiol 2019;74:578–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Libby P, Sidlow R, Lin AE, et al. Clonal Hematopoiesis: Crossroads of Aging, Cardiovascular Disease, and Cancer: JACC Review Topic of the Week. J Am Coll Cardiol 2019;74:567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 2017;377:111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017;355:842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017;377:1119–31. [DOI] [PubMed] [Google Scholar]
- 63.Svensson EC, Madar A, Campbell CD, et al. Abstract 15111: TET2-Driven Clonal Hematopoiesis Predicts Enhanced Response to Canakinumab in the CANTOS Trial: An Exploratory Analysis. Circulation 2018;138:A15111–A. [Google Scholar]
- 64.Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dorsheimer L, Assmus B, Rasper T, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol 2019;4:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cook EK, Izukawa T, Young S, et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv 2019;3:2482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fraietta JA, Nobles CL, Sammons MA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018;558:307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arends CM, Weiss M, Christen F, et al. Clonal hematopoiesis in patients with ANCA-associated vasculitis. Haematologica 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolton KL, Gillis NK, Coombs CC, et al. Managing Clonal Hematopoiesis in Patients With Solid Tumors. J Clin Oncol 2019;37:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]