

Abstract
Trichloroethylene (TCE) is an industrial solvent and a common environmental contaminant detected in thousands of hazardous waste sites. Risk of exposure is a concern for workers in occupations that use TCE as well as for residents who live near industries that use TCE or who live near TCE-contaminated sites. Although renal, hepatic and carcinogenic effects of TCE have been documented, less is known about TCE impacts on reproductive functions despite epidemiology reports associating maternal TCE exposure with adverse pregnancy outcomes. Toxicological evidence suggests that the placenta mediates at least some of the adverse pregnancy outcomes associated with TCE exposure. Toxicology studies show that the TCE metabolite, S-(1,2-dichlorovinyl)-L-cysteine (DCVC) generates toxic effects such as mitochondrial dysfunction, apoptosis, oxidative stress, and release of prostaglandins and pro-inflammatory cytokines in placental cell lines. Each of these mechanisms of toxicity have significant implications for placental functions and, thus, ultimately the health of mother and developing child. Despite these findings there remain significant gaps in our knowledge about effects of TCE on the placenta, including effects on specific placental cell types and functions as well as sex differences in response to TCE exposure. Due to the critical role that the placenta plays in pregnancy, future research addressing some of these knowledge gaps could lead to significant gains in public health.
Keywords: Trichloroethylene, trichloroethene, placenta, toxicity, adverse pregnancy outcomes
Graphical Abstract
Environmental significance.
Because of its continued industrial use and improper disposal, trichloroethylene (TCE) is a common environmental contaminant detected in thousands of hazardous waste sites. Due to widespread and persistent environmental contamination, TCE poses a threat for human exposure through drinking contaminated water and inhaling volatilized compound. Extensive scientific research has demonstrated potent organ-specific TCE toxicity. Recent epidemiological studies report associations between maternal TCE exposure and adverse pregnancy outcomes. TCE toxicity to the placenta may contribute to adverse pregnancy outcomes due to the placenta’s critical role during gestation. Better understanding of TCE impacts on the placenta may provide critical information for assessing and ultimately controlling risks to pregnant women from TCE exposure. This perspective details important research being conducted into the effects of TCE and its reactive metabolites on placenta and provides recommendations for future research.
Introduction
Pregnant women are exposed to a large number of environmental contaminants.1 For example, perfluorooctanyl sulfonate (PFOS), several polychlorinated biphenyls (PCBs), and several polybrominated diphenyl ethers (OBDEs) were detected in serum of 99–100% of pregnant women included in the U.S. Centers for Disease Control and Prevention (CDC) National Biomonitoring Program.1 During pregnancy, the placenta provides the essential interface for exchange of oxygen, nutrients, and metabolic wastes between mother and fetus.2 In addition, the placenta becomes the major source of hormones necessary for pregnancy maintenance, parturition, and maternal and fetal health.3–5 Because the placenta is critically important for pregnancy success, disruption of placental development and function is an important health concern.5 Indeed, the summary report from a recent workshop sponsored by the U.S. Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, concludes that, “the majority of adverse pregnancy outcomes can trace their origin to the placenta.”6 Although various environmental contaminants have been detected in placental tissue,7 our understanding of toxicant effects on placenta is limited. This article explores placental toxicity from exposure to trichloroethylene (TCE), a common and persistent environmental pollutant,8 in the context of implications for risks to pregnancy. Challenges specifically related to study of the placental as a toxicant target and of trichloroethylene in particular are discussed.
Adverse pregnancy outcomes: a global public health crisis
Adverse pregnancy outcomes are a significant public health concern, especially preterm birth (<37 weeks gestation) and low birth weight (<2500 g).9, 10 Globally, complications from preterm birth are the number one leading cause of death in children under 5 years of age,11 with approximately 15 million or 11% of infants born preterm each year.9 The U.S. has the highest infant mortality rate compared with other developed countries,11, 12 and disorders related to preterm birth and low birth weight contribute to the majority of infant deaths.13 Preterm births continued to rise in the U.S., after a brief respite, to 10.02% in 2018.14 Likewise, the U.S. incidence of low birth weight increased to 8.28% by 2017.10 Babies born preterm or with low birth weight are more likely to require intensive and prolonged hospitalization, and are at increased risk for chronic health complications.10, 15, 16 The increased incidence of these adverse pregnancy outcomes and their potential significance for long-term health consequences have resulted in widespread recognition that the high rates of preterm birth and low birth weight constitute a national and global public health crisis.11, 17–19
Contribution of environmental contaminants to adverse pregnancy outcomes
Despite considerable research efforts and identification of risk factors associated with adverse pregnancy outcomes, such as genetic factors, infections, contraindicated maternal behaviors and chronic maternal conditions, the specific pathophysiology underlying most adverse pregnancy complications outcomes remains poorly understood.18 Exposure to environmental contaminants are increasingly thought to contribute to adverse pregnancy outcomes. Indeed, epidemiological studies have detected associations between maternal exposures to environmental contaminants and increased risk for adverse pregnancy outcomes.20, 21 For example, studies have associated exposure to fine air particulate matter (PM 2.5) with increased risk of pre-eclampsia and preterm birth.22, 23 Other studies reported that exposure to relatively low concentrations of lead and cadmium were associated with increased preterm birth and small head circumference.24, 25 Similarly, maternal exposure to polyfluoroalkyl substances (PFAS) was positively associated with preterm birth.26 Although some studies have established links between environmental contaminants and adverse pregnancy outcomes, many studies have reported conflicting results on the same contaminants.20 For example, despite dozens of studies on the subject, an association between maternal environmental tobacco smoke exposure and preterm birth remains inconclusive.27 Despite reasonable evidence that maternal exposure to environmental contaminants contribute to elevated risks of adverse pregnancy outcomes, further investigation into the biological plausibility and specific toxicological mechanism of different environmental contaminants is warranted.
Importance of placenta as a toxicant target organ
Adverse pregnancy outcomes are strongly linked to disorders of the placenta.6 A leading cause of fetal death in 2014 in the U.S. was identified as “complications of placenta, cord and membranes,”28 and complications of the placenta are included in the most prevalent identifiable cause of fetal death in the U.S.12, 28 Furthermore, recent reviews identify the placenta as a key factor in preterm birth29 and fetal growth restriction.30 In addition, adaptation of the placenta to changes in its environment is a proposed to play an important role in developmental origins of adult health and disease.2, 5, 6
Because the placenta is perfused with maternal blood at a rate up to 700 ml/min by term,31 any toxicants that are circulating in the mother’s blood are efficiently delivered to the placenta, increasing opportunity for toxicant effects in this organ. In addition, toxicants can move across the placenta between maternal and fetal blood supplies by mechanisms that include simple passive diffusion and transporter-mediated processes, thereby contributing to increased opportunity for placental toxicant exposure and effects.32 Moreover, some toxicants can preferentially bioaccumulate in the placenta, as shown for bisphenol A,33, 34 some PCBs,35 some co-planar dioxins,36 and cadmium and other metals.35
Because of its significant endocrine functions,3, 5 the placenta is vulnerable to endocrine disruption.37, 38 Likewise, because the placenta synthesizes and releases cytokines and prostaglandins, molecules that regulate placental function as well as the onset of labor,3, 4 disruption of the balance of placental cytokines and prostaglandins could lead to preterm birth or other adverse pregnancy outcomes.5, 6, 39, 40 Moreover, it has been suggested that early placental formation and development, particularly remodeling of the uterine spiral arteries, may be especially vulnerable to environmental insult.5, 38, 41, 42 Recent reviews provide evidence that placental insufficiency, which is thought to be a symptom of failed spiral artery remodeling, is known or strongly suspected to contribute to intrauterine growth restriction,30, 43–45 preterm birth,29, 46 and pre-eclampsia.45, 47, 48
Because proper placental development and function are vital for pregnancy health and success, improved understanding of chemical impacts on the placenta would provide critical insight into environmental risks for pregnancy. Recent reviews highlight the incomplete and uneven knowledge of placental toxicology, with a tendency to focus on endocrine disrupting chemicals.38, 49
Trichloroethylene is a pervasive and persistent environmental contaminant
Trichloroethylene (TCE) is an anthropogenic volatile organic chemical with a legacy of environmental contamination.50 Historically, TCE was used for degreasing metal parts and as a solvent in the manufacture of textiles, lubricants, paints, paint strippers, pesticides and more.50 Today, over 80% of TCE production is used as a chemical intermediate in closed-system refrigerant manufacturing processes, specifically HFC-134a, a refrigerant used in car air conditioning systems.50, 51 The other current primary use of TCE is in vapor metal degreasing, although the U.S. Environmental Protection Agency (EPA) proposed banning this application.52 Within the U.S., the state of Indiana reported the highest release of TCE into the environment from industrial sources, accounting for 35% of total reported releases in the U.S. in 2017 and four times more than the next highest state, Pennsylvania.50, 53 Due to its low vapor pressure, TCE readily volatilizes into air. In addition, TCE is denser than water, allowing it to form subsurface plumes as a dense non-aqueous phase liquid (DNAPL). As a result of its chemical properties, long-standing industrial use, and past disposal practices, TCE is a common and persistent environmental pollutant found in soil, air and water.8 It contaminates 1055 of 1750 current and former U.S. EPA-designated National Priorities List (Superfund) sites,50, 53 and is ranked number 16 in the National Priorities List of the U.S. Agency for Toxic Substances and Disease Registry (ATSDR).50, 53
Exposure to trichloroethylene
Exposure in air
Monitoring data from the U.S. EPA indicates that TCE is commonly detected in ambient air, albeit at low concentrations (typically < 1 ppb).50 Still, high ambient air concentrations have been reported, such as 49 ppb at a site in Oregon in 2011, suggesting that point sources of emissions may elevate air concentrations locally.50 In addition, indoor air TCE concentrations can become elevated due to TCE release from building materials and household products, volatilization from tap water use (e.g., showering), and migration from surrounding contamination into buildings (vapor intrusion). For example, a recent study detected blood concentrations of TCE that were 50 times higher in people exposed through residential vapor intrusion (>1.6 μg M−3 indoor air concentration) compared with residents of households with no detectable TCE vapor intrusion.54 Numerous studies report higher TCE concentrations in indoor air compared to outdoor air, even for structures located near point source emitters and when no TCE was detected in outdoor air, as summarized.50 In addition, inhalation exposure is a concern for occupations that involve TCE, particularly those that use TCE in vapor degreasing operations. The U.S. Occupational Safety and Health Administration (OSHA) limits TCE inhalation exposure in the workplace: the permissible exposure limit (PEL) for TCE in air averaged over an 8-hour workday is 100 ppm with an acceptable maximum level of 300 ppm TCE for no more than 5 minutes of any 2-hour period.55
Exposure in drinking water
TCE exposure can also occur through ingestion of contaminated drinking water. The U.S. EPA has set a maximum contaminant level of 5 ppb in drinking water,56 whereas the World Health Organization recommends 20 ppb as a drinking water guideline value.57 In 2005, the last year of the most recent review of public water systems by the U.S. EPA, TCE was detected in 14.8% of public water supplies that used a surface water source compared to 4.9% that relied on ground water as the source.50 In the latter review, median levels were 1.6 ppb and 1.1 ppb for public water from surface and ground water sources, respectively, with 159 ppb reported as the highest TCE concentration for a public water supply. A notable contamination of drinking water occurred at the U.S. military base Camp Lejeune in North Carolina in the 1970’s and 1980’s.58 TCE contaminated drinking water that was provided to family housing on the base, with a peak concentration of 1,400 ppb detected in 1982.59 Health effects continue to be studied in the TCE-exposed Camp Lejeune population.58
Exposure of pregnant women
Because of the pervasive presence of TCE as an air, water and soil contaminant, and its continued use in industrial processes, pregnant women are likely exposed to TCE. However, estimates of prevalence of exposure of pregnant women are not available.
Trichloroethylene metabolism
The low-molecular weight, lipophilic, and nonpolar properties of TCE allow it to be quickly absorbed into the blood stream from the lungs, gastrointestinal tract, and skin.60 Once in the blood stream, the chemical distributes rapidly to highly perfused organs,60 including liver, kidney and lungs where it is biotransformed. Biotransformation is important for converting TCE to reactive metabolites that can damage cellular macromolecules.61 There are two major metabolic pathways for TCE in mammals, each pathway involving multiple steps of biotransformation (Figure 1).
Figure 1. Simplified depiction of two TCE biotransformation pathways.
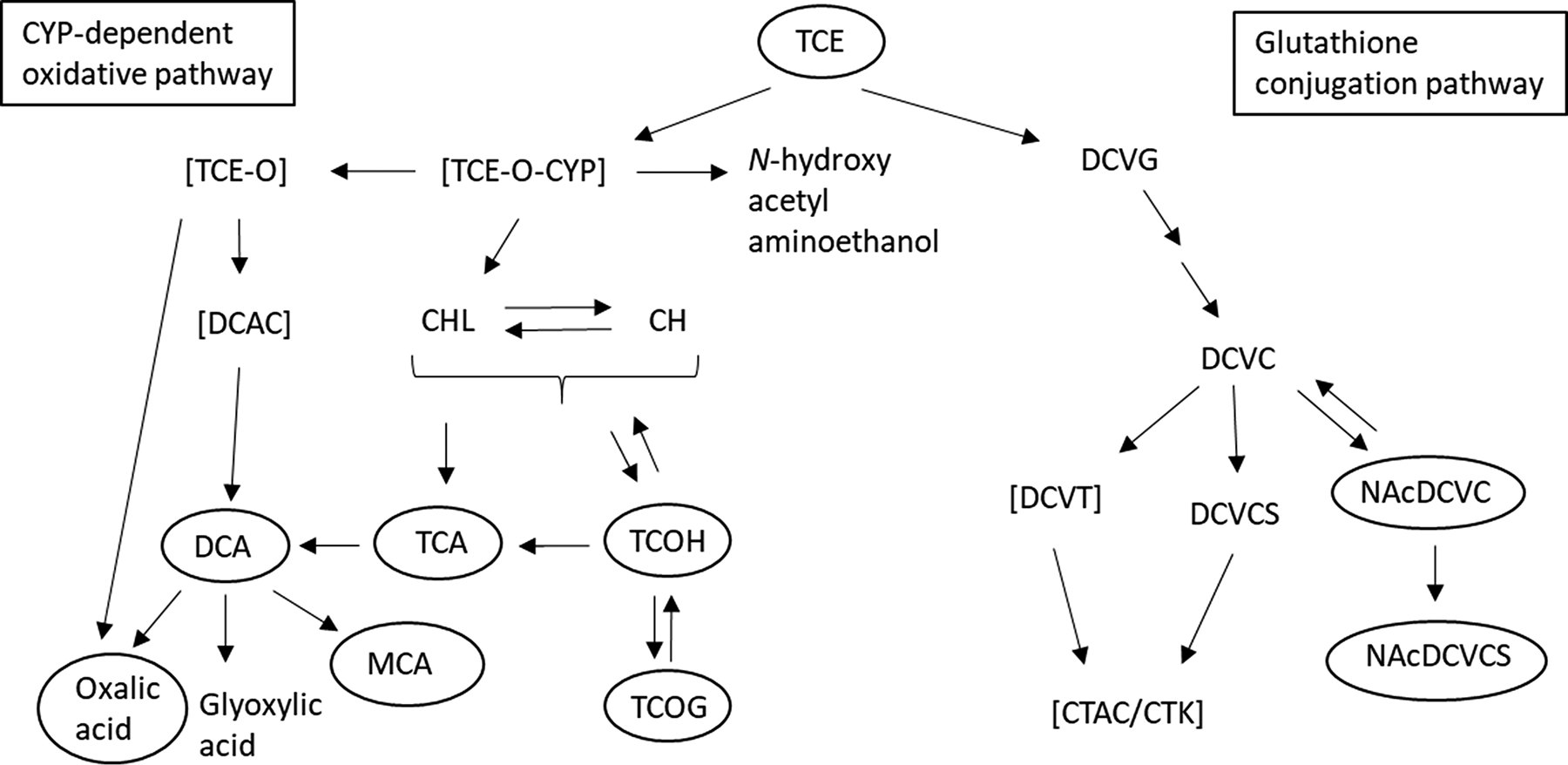
TCE is metabolized through the CYP oxidation pathway or the glutathione conjugation pathway. Metabolites that are unstable are depicted in brackets whereas metabolites that can be found in urine are circled. With the exception of all unstable metabolites and DCVCS, all other metabolites are systemically available.61 Abbreviations: CYP, cytochrome P450; TCE-O, TCE-epoxide; TCE-O-CYP, CYP-bound TCE-epoxide; DCAC, dichloroacetyl chloride; CHL, chloral; CH, chloral hydrate; DCA, dichloroacetic acid; TCA, trichloroacetic acid; TCOH, trichloroethanol; TCOG, trichloroethanol glucuronide; MCA, monochloroacetic acid; DCVG, S-(1,2-dichlorovinyl)glutathione; DCVC, S-(1,2-dichlorovinyl)-L-cysteine; DCVCS, DCVC sulfoxide; NAcDCVC, N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine; NAcDCVCS, NAcDCVC sulfoxide; CTAC, chlorothionoacetyl chloride; CTK, chlorothioketene. This figure was modified from Lash et al.61
Occurring primarily in the liver, an important metabolic pathway of TCE is oxidation by several CYP family of enzymes (notably CYP1A1, CYP1A2, CYP2E1 and CYP3A4) to generate a TCE-epoxide-CYP (TCE-O-CYP) intermediate. The TCE-O-CYP intermediate can generate N-hydroxy-acetyl-aminoethanol, chloral (CHL)/chloral hydrate (CH) or TCE-epoxide (TCE-O), which can convert to oxalic acid (OA) or dichloroacetyl chloride (DCAC). CHL/CH is subsequently biotransformed in multiple steps to the several key oxidative metabolites: dichloroacetate (DCA), trichloroacetate (TCA), monochloroacetate (MCA), trichloroethanol (TCOH), and the glucuronide of TCOH, trichloroethanol glucuronide (TCOG).60, 62 Whereas unstable metabolites from the CYP oxidative pathway include TCE-O-CYP, TCE-O, and DCAC, CH and CHL have been shown to be mutagenic and genotoxic.63
In the other important metabolic pathway, TCE is conjugated to glutathione (GSH) as S-(1,2-dichlorovinyl) glutathione (DCVG). The DCVG is further metabolized to S-(1,2-dichlorovinyl)-L-cysteine (DCVC), which can be metabolized to S-(1,2-dichlorovinyl)thiol (DCVT), N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine (NAcDCVC), or DCVC sulfoxide (DCVCS).60, 62 NAcDCVC can be converted to NAcDCVC sulfoxide (NAcDCVCS) by CYP3A enzymes, and DCVT or DCVCS can spontaneously rearrange to chlorothionoacetyl chloride (CTAC) or chlorothioketene (CTK), two unstable compounds that are readily interconverted.63 In addition to CTAC and CTK, other known unstable metabolites of the glutathione conjugation metabolic pathway of TCE include DCVT.61 Several downstream metabolites of DCVC – DCVT, CTAC, CTK, and NAcDCVCS – are toxic and mutagenic.61, 64
With the exceptions of TCE-O, DCAC, DCVT, DCVCS, CTK, and CTAC, all other TCE metabolites are systemically available.63 TCE itself has a relatively short plasma half-life of 3–4 days in humans, depending on exposure duration and concentration. Although some TCE is exhaled in its parent form, most TCE is metabolized with elimination from the body in bile and urine.60, 65 TCE, OA, DCA, TCA, MCA, TCOH, TCOG, NAcDCVC, and NAcDCVCS are known to be recovered in urine, whereas hepatic DCVG can be readily excreted into plasma or bile.63 Therefore, based on the numerous stable TCE metabolites formed by the CYP oxidative pathway that are recovered in urine, the CYP pathway is known as the quantitatively dominant TCE metabolic pathway. This contrasts with the glutathione conjugation metabolic pathway, which forms fewer metabolites recovered in urine but more unstable and reactive metabolites.61
Considerable differences exist among species, tissue, strain within a species, and sex for TCE metabolism, influencing susceptibility to TCE toxicity. Mice are known to have a higher maximal rate of CYP oxidative metabolism than rats, and rats have a higher maximal rate of CYP oxidative metabolism compared to humans.61 Similarly, mouse tissues exhibit higher glutathione conjugation rates compared to equivalent rat tissues,66 and humans have lower activity of γ-glutamyltransferase, which metabolizes DCVG,60–62 compared to mice and rats.67 In different organs of mice, liver cells more effectively metabolize CH/CHL via UDP-glucuronosyltransferase compared to lung cells.68 In rats, liver cells have higher GSH conjugation rates compared to kidney cells.66 Among 15 different mouse strains, exposure to the same dose of TCE produces variable serum levels of DCVG.69 Finally, sex-specific difference includes higher abundance of TCE metabolites from the CYP pathway in liver of male compared to female rats, as well as evidence suggesting a faster breakdown of DCVG in males compared to females rats.70 Because CYPs can be differentially expressed between males and females,71 inter-individual differences among humans in TCE metabolism can be quite large. TCE metabolism can also be modified by lifestyle factors. For example, ethanol is a known modulator of CYP2E1 expression.72
Placental transfer and metabolism of trichloroethylene
Any TCE and TCE metabolites circulating in the mother’s blood are efficiently delivered to the placenta. Several studies have demonstrated that TCE and its metabolites readily cross the placenta. In women, early studies that were conducted when TCE was used as an anesthetic gas in obstetric surgery detected TCE in the umbilical vein and umbilical artery, indicating placental transfer.73, 74 Ghantous et al. demonstrated that unmetabolized radiolabeled TCE as well as radiolabeled TCA derived from TCE metabolism, were detected in the placenta, amniotic fluid and fetal tissues of TCE inhalation-exposed pregnant mice at all stages of gestation.75 Notably, the glutathione-pathway metabolite DCVC has not been measured directly in placental tissue, which may be attributed to its reactivity and short half-life.76
Toxicants that are lipophilic, such as TCE, can transfer across the placenta by simple passive diffusion.77 On the other hand, various transporters could potentially facilitate placental transfer of some non-lipophilic TCE metabolites.77, 78 For example, the TCE metabolites DCA and TCA, which circulate in maternal blood,61 could possibly be transported by placental organic anionic transporters. Additionally, DCVC, which is available systemically61 and resembles a small peptide in structure, could be transported by placental peptide transferases. Some transporters involved in TCE tissue distribution have been characterized in kidney.61 However, no research to date has specifically investigated the role of transporters in placental transfer of TCE or its metabolites.
In addition to providing evidence of placental transfer, the findings of Ghantous et al. support occurrence of TCE metabolism in the placenta or fetus independent of maternal metabolism.75 Consistent with the latter findings, the placenta contains a variety of enzymes capable of metabolizing TCE and its metabolites. In regard to the CYP-dependent oxidative pathway, a range of CYPs are expressed in human placenta.79 These include CYP1A1 and low levels of CYP2E1,79 two CYPs that produce the TCE-O-CYP intermediate from TCE,61 the first step of CYP-dependent TCE metabolism. Low activity of aldehyde dehydrogenase, the enzyme that converts CH/CHL into TCA as part of the CYP-dependent pathway,61 has also been detected in rat placenta.80 In regard to the GSH conjugation pathway of TCE metabolism, the human placenta expresses various glutathione S-transferases (GSTs),81, 82 the enzymes known to metabolize TCE into DCVG.61 The human placenta also contains active γ-glutamyltransferase,83 one of the two enzymes that converts DCVG into DCVC.61 Cysteinyl-glycine dipeptidase, the other enzyme necessary for DCVG conversion to DCVC,61 is thought to be relevant to reproduction for its role in GSH metabolism84 but has not been detected in placenta to date.
Non-reproductive adverse health effects associated with trichloroethylene exposure
TCE has been extensively studied as a renal and liver toxicant because these organs are the primary sites of TCE metabolism via glutathione conjugation and cytochrome P450, respectively, in the body.8, 60, 70, 85 TCE was recently re-classified by the U.S. EPA,86 U.S. National Toxicology Program (NTP),51, 87 and International Agency for Research on Cancer (IARC)88 as a known human carcinogen. This re-classification was based, in part, on epidemiology studies associating TCE exposure with elevated risk of renal cancer8, 89 and reports that the glutathione conjugation TCE metabolite, S-(1,2-dichlorovinyl)-L-cysteine (DCVC), causes nephrotoxicity.60, 90 In addition to liver and kidney toxicity, TCE-induced health effects have been reported in epidemiology and animal studies that indicate neurotoxicity, respiratory distress, and gastrointestinal irritation.8, 62, 91–94
Association of maternal trichloroethylene exposure with adverse pregnancy outcomes
Human studies
Although many studies have established TCE as a potent renal and liver toxicant,8 TCE effects on pregnancy outcomes remain understudied and inconclusive. Epidemiology studies of associations between TCE exposure and adverse pregnancy outcomes report inconsistent findings. Exposure to drinking water contaminated with TCE and other volatile organic chemicals was associated with low birth weight in Woburn, Massachusetts, when exposure in the year of birth as well as the year prior to birth were included in the analysis, but not when the analysis was restricted to the year pregnancy ended.95 A review of birth records and state databases in northern New Jersey found evidence of an association between TCE in drinking water and low birth weight at term, but not for small for gestational age (weight below the 10th percentile for the gestational age) and very low birth weight (≤1500 g).96 A study of women exposed to TCE via drinking water in Tucson, Arizona, found a positive association with very low birth weight but no significant association with low birth weight (≤2500 g).97 In a more recent vapor intrusion study of pregnant women exposed to TCE via indoor air, positive associations were observed for TCE exposure with low birth weight, small for gestational age, and low birth weight at term.98, 99 Moreover, a study of pregnant women residing at Camp Lejeune, North Carolina, found that TCE exposure via drinking water was associated with measures indicative of impaired fetal growth, including low birth weight at term, reduced mean birth weight, and small for gestational age.99
At least some of the inconsistency in findings of published epidemiology reports may be related to chemical co-exposures that are common with TCE. As noted by the U.S. ATSDR, studies of associations between human health effects and TCE exposure have involved exposure to multiple chemicals but failed to assess possible confounding by chemical co-exposures.50 Future research of TCE-exposed human populations will likely benefit from advances in methods to study co-exposures, which has become a priory of the National Institute of Environmental Health Sciences.100, 101
Animal studies
Exposure of Wistar rats to 100 ppm in air from gestation day 8–21 for 4 hours per day significantly reduced fetal weight at term by approximately 9%.102 The dose of the prior study is similar to the U.S. OSHA PEL standard of 100 ppm TCE in air averaged over an 8-hour workday. Likewise, in a study of Wistar rats exposed to 480 mg TCE/kg body weight/day during mid-pregnancy (gestation day 6–16) via a food treat, fetal weights were reduced approximately 10% on gestation day 16 without significant reduction of maternal body, liver, or kidney weights.103 However, a study with Sprague-Dawley rats exposed by oral gavage to 500 mg TCE/kg body weight/day on gestation days 6–15 found no significant reduction in fetal weight on gestation day 21.104 The doses used in the prior studies with oral exposure are equivalent to an exposure within an order of magnitude of the U.S. OSHA PEL but are several orders of magnitude higher than expected from drinking water exposures. Differences in findings among the rodent studies could be related to differences in strain of rat, exposure regimen, and gestational age at fetal assessment. Regardless, there is sufficient evidence to warrant further evaluation of TCE effects on pregnancy outcomes in toxicology and epidemiology studies.
Placental toxicity of trichloroethylene
Several lines of evidence support the placenta as a target of TCE toxicity, as discussed in this section below and illustrated in Figure 2. This evidence includes toxicokinetics, biotransformation, activation of biochemical and cellular responses, and disruption of placental functions that can lead to adverse pregnancy outcomes, supported by epidemiology studies.
Figure 2.
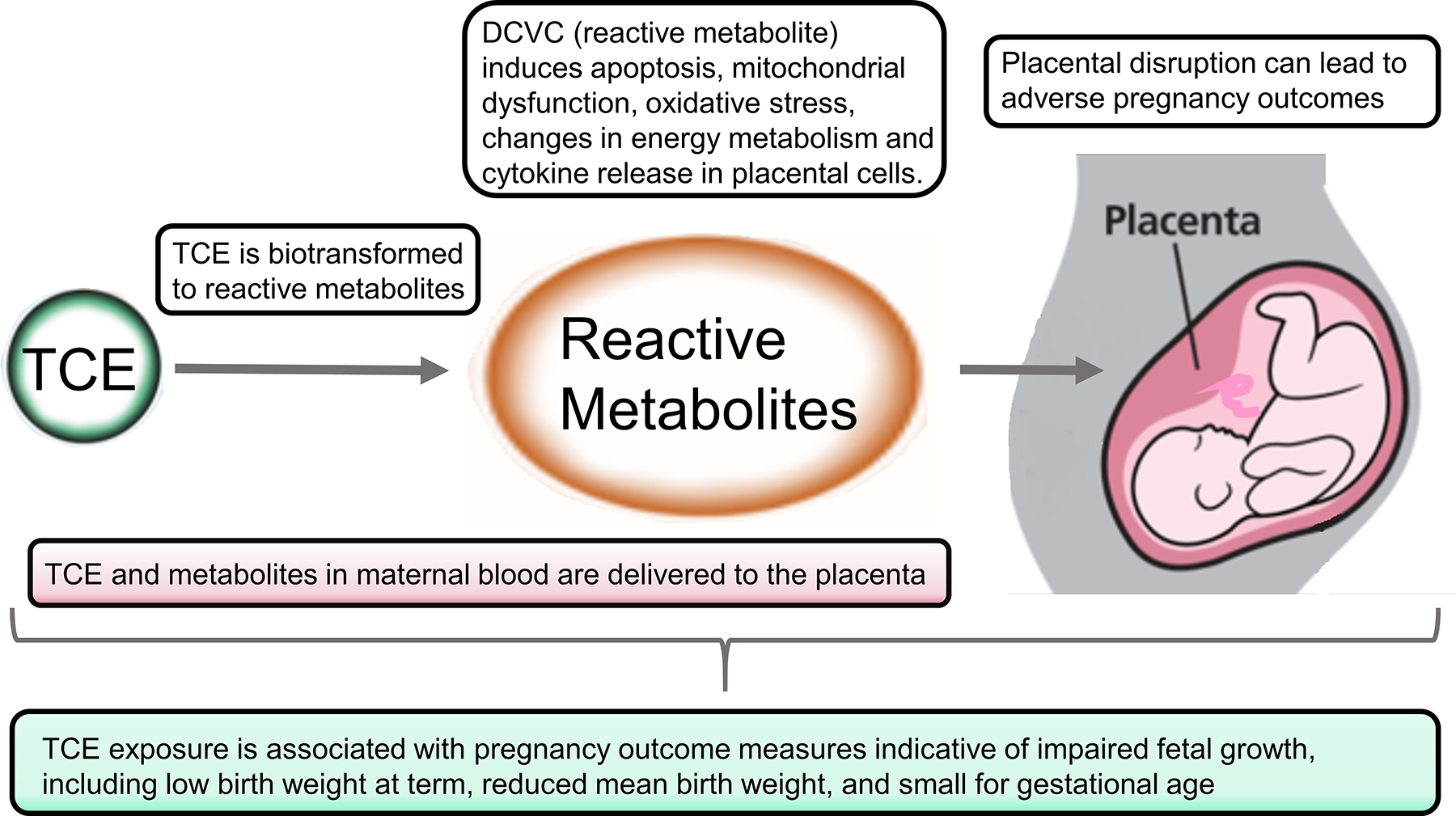
Key mechanistic evidence suggesting the placenta is a target of TCE toxicity.
Placental toxicity of the trichloroethylene metabolite DCVC
Experiments in our laboratory indicate that the glutathione pathway metabolite DCVC is particularly toxic to placental cells at low concentrations.105–107 The DCVC concentrations of 5–20 μM used in the placental cell studies encompass the average maximum concentration of 13.4 μM DCVG measured in the blood of female volunteers exposed to 100 ppm TCE in air for 4 hours.108 The TCE concentration used in the latter human exposure study is the U.S. PEL concentration limit for occupational TCE exposure averaged over an 8-hour workday.55 Because DCVG is the precursor of DCVC and precursor conversion to DCVC appears to be in a 1:1 stoichiometric ratio,60 the range of DCVC concentrations used in placental cell experiments is relevant for human occupational exposures. DCVC disrupts placental cells through multiple molecular and cellular pathways, as described below. In contrast, we observed only nominal cytotoxicity in placental cells exposed to the oxidative metabolites TCA and DCA, even at much higher concentrations than DCVC toxic concentrations (unpublished).
DCVC stimulates pro-inflammatory response in placental cells
DCVC appears to disturb the balance between pro- and anti-inflammatory pathways in placental cells. In a human extravillous trophoblast model, the HTR-8/SVneo cell line, 10 and 20 μM DCVC stimulated generation of reactive oxygen species and the synthesis and release of the pro-inflammatory cytokine IL-6 after 10 and 24 h of exposure, respectively.107 The DCVC-stimulated IL-6 release from HTR-8/SVneo cells was blocked by treatment with aminooxyacetic acid (AOAA), a renal cysteine conjugate β-lyase (β-lyase) inhibitor,109, 110 suggesting that the DCVC-stimulated release of IL-6 was due to a reactive metabolite of DCVC generated via β-lyase activity, consistent with reports of DCVC toxicity in other cell types.61, 109, 110 In addition, DCVC-stimulated release of IL-6 and IL6 mRNA expression was strongly inhibited by antioxidant treatments,107 implicating a role for reactive oxygen species in the IL-6 response, also consistent with findings in other cell types.111, 112 These results suggest that TCE and its metabolites can activate pro-inflammatory pathways in the placenta, potentially via the generation of reactive oxygen species. These findings have significant implications for pregnancy, as an overall imbalance in pro- versus anti-inflammatory signaling in gestational tissues and maternal circulation is thought to be an important contributor to several adverse pregnancy outcomes such as preeclampsia, preterm birth and spontaneous abortion.113, 114
DCVC disrupts mitochondrial function and membrane potential
In addition to the findings on reactive oxygen species and the pro-inflammatory response, the prior study with HTR-8/SVneo cells determined that DCVC disrupted the mitochondrial membrane potential with exposures to 5, 10 and 20 μM for 4, 10 and 24 h.107 Moreover, the mitochondrial membrane transition pore inhibitor bongkrekic acid significantly attenuated but did not completely inhibit IL-6 release.107 Because mitochondrial dysfunction and depolarization of the mitochondrial membrane are potential initiators as well as responses of increased cellular generation of reactive oxygen species, subsequent studies focused on DCVC effects on mitochondria, cellular energy production and lipid peroxidation.
In follow-up studies, our laboratory recently showed that 20 μM DCVC exposure for 12 h causes adaptive changes to macronutrient and energy metabolism pathway utilization in order to maintain adequate ATP levels.115 Consistent with energy metabolism disruptions, mitochondrial dysfunction (e.g., proton leak, energy coupling deficiency, and dissipated mitochondrial membrane potential) was also measured in the same cell line, as early as six hours after initiating treatment.106 In addition, mitochondrial DNA content (a proxy for mitochondrial copy number) was reduced after 12 hours of 20 μM DCVC exposure.106 Lastly, we reported that DCVC elevated levels of the lipid peroxidation byproduct malondialdehyde.105 Because optimal mitochondrial functioning is critical for placental extravillous trophoblast cells due to their sizable energy requirements for biological processes such as tissue remodeling,116–118 mitochondrial disruption could have significant implications for placental health. Indeed, the most widely-reported aspects of mitochondrial dysfunction associated with abnormal placentae are mitochondria-generated reactive oxygen species, lipid peroxidation, and mitochondrial DNA content, as reviewed.119, 120 Given the critical role that mitochondria play during pregnancy, the effects of DCVC on these processes warrant further study.
DCVC stimulates intrinsic and extrinsic apoptosis pathways
Our laboratory has also shown that DCVC concentrations as low as 20 μM for 12 h induces apoptosis via both the receptor-mediated extrinsic and mitochondrial-mediated intrinsic apoptotic signaling pathways in the HTR-8/SVneo extravillous trophoblast model.105 Apoptotic cell death can severely impair tissue development and function if it occurs excessively or prematurely. Apoptosis is a programmed cell death that is specifically characterized by signal-induced, energy-dependent breakdown and condensation of nuclei, and fragmented cellular components packaged into apoptotic vesicles in preparation for phagocytosis by immune system cells.121 During pregnancy, apoptosis plays a vital role in normal implantation, placentation and remodeling of decidual tissue and maternal spiral arteries in early pregnancy.121, 122 However, excessive apoptosis can disrupt placental development, causing shallow extravillous trophoblast invasion into the uterine wall and insufficient spiral artery remodeling.48, 123–127 If severe, such changes can lead to pregnancy disorders such as pre-eclampsia and intrauterine growth restriction122, 128–130 Additionally, increased expression of apoptosis-associated genes has been reported in pregnancies with recurrent miscarriages and failure of blastocyst implantation.131, 132 Because ample evidence links elevated apoptosis to abnormal placental development, induction of apoptosis in placental extravillous trophoblasts could be a mechanism by which TCE and its metabolites (e.g., DCVC) contribute to poor placentation and early pregnancy disorders if this occurs in vivo.
Table 1 summarizes various mechanisms of TCE (and associated metabolite) induced placental toxicity that have been identified thus far. No other published studies were identified that investigated the toxicity of TCE or its metabolites in placenta. It should be noted that which mechanisms predominate likely depend on a number of factors such as level and duration of TCE exposure.
Table 1.
Mechanisms of TCE toxicity observed in placental models
| Observed mechanism of TCE (or TCE metabolite) toxicity | Model utilized | Potential impact on placenta | Relevant Pregnancy outcome | References |
|---|---|---|---|---|
| Changes in energy metabolism pathways and macronutrient utilization by cells | HTR-8 cell line | Impaired trophoblast function during tissue remodeling; macronutrients diverted away from fetus to maintain placental sufficiency. | Preeclampsia; intrauterine growth restriction | 115 |
| Mitochondrial dysfunction; impaired mitochondrial adaptability | HTR-8 cell line | Insufficient metabolic energy capacity during tissue remodeling; impaired mitochondrial adaptability | Preeclampsia; intrauterine growth restriction | 106, 107 |
| Apoptosis; lipid peroxidation | HTR-8 cell line | Insufficient extravillous trophoblast invasion, inadequate remodeling of spiral arteries | Preterm birth; preeclampsia | 105 |
| Immunological responses; cytotoxicity | HTR-8 cell line | Increase in pro-inflammatory cytokines (e.g. IL-6), overall shift toward pro-inflammatory state in gestational compartment | Preterm birth; preeclampsia | 107 |
| Oxidative stress | HTR-8 cell line; pregnant Wistar rats | Increased ROS in placental cells leading to activation of inflammatory pathways | Preterm birth, preeclampsia | 103, 107 |
| Reduced fetal weight | Pregnant Wistar rats | Changes in macronutrient delivery to fetus | Intrauterine growth restriction; small-for-gestational-age; low birth weight | 103 |
Challenges for advancing understanding of TCE toxicity to placenta
Limitations of experimental models for studying TCE toxicity
One of the most significant challenges for studying placental toxicity is the lack of an adequately relevant animal model.133 There is considerable variation among placental mammals in uterine and placental structure.134–136 For example, the uterus of mice and rats has two uterine horns, whereas the human uterus has a single cavity. Moreover, placental differences can include variations in morphology (e.g., discoid in humans and rodents, but cotyledonary in sheep), the degree of invasion into the uterine wall (e.g., deeply invasive in humans and rodents, but superficial in rabbits and sheep), and the histology of cells and cell layers that separate maternal and fetal blood (e.g., humans and rabbits have different histology compared to rodents and sheep).134–136
In addition, the endocrine functions of the placenta are critical for pregnancy,137 yet are widely different across species.138 As one example, the human placenta acquires the major role of sex steroid hormone production in the first trimester, whereas critical sex hormone production in rats and mice remains in the ovary throughout pregnancy. Moreover, although prostaglandins have a central role in the initiation of labor in mammals,139 prostaglandin F2α initiates parturition in mice by stimulating cell death of hormone-synthesizing cells in the ovary, a parturition initiation mechanism that is absent in women.3, 140 Despite the lack of an animal species that accurately models human pregnancy, our understanding of human pregnancy has been advanced by utilization of animal models, in particular mouse, rat, and sheep, as long as limitations due to inter-species differences are recognized.135, 141 Moreover, because of the absence of an adequate animal model, placental toxicity investigations must rely on a combination of in vitro approaches in addition to animal experimentation, each with its own compromises.142
The ideal in vitro model for placental toxicity experimentation would involve normal human placental cells and tissue. However, as an internal organ the human placenta is not ethically accessible for study until delivery. Deliveries do not occur on a normal work schedule, making acquisition of fresh placental tissue difficult for in vitro models that require live tissue or cells. In addition, because most available placental tissue is from normal term deliveries, experimental findings may be limited and may not reflect responses of the placenta of early and mid-gestation non-labor pregnancy. Placental tissue from first trimester elective pregnancy terminations have contributed valuable insights into early placentation processes like extravillous trophoblast invasion.143 However, research using tissue from elective pregnancy terminations may be limited by restrictions of research on human fetal tissue. In vitro models span the use of intact placental cotyledon diffusion, villous explant cultures, primary cell cultures, and cell lines. In addition, new models are in development to create a “placenta on a chip” that use microfluidic technologies.144, 145
Despite their usefulness, in vitro models of human placental cells and tissues have shortcomings.146 Most notably, once removed from the body the cells and tissues are no longer part of integrated physiologic systems, and re-creation of the normal milieu in vitro is approximated, at best. Furthermore, tissue disruption to isolate cells for primary monoculture – as well as culture conditions – removes cells from their 3-dimenstional tissue structure and isolates them from other cells with which there is normally contact. Whereas cell lines free the researcher from constraints of tissue availability, derivation from cancerous tissue or induction of immortalization extensively alters genetic and epigenetic profiles.147–149 Common human placental cell lines such as HTR-8/SVneo, BeWo, JEG-3, and JAR typically retain some but not all unique functional characteristics and molecular markers of trophoblasts and other placental cells in vivo.150 For example, HTR-8/SVneo cells express several markers of extravillous trophoblasts151 but only express the major histocompatibility protein HLA-G, a marker of extravillous trophoblast cells, when grown on Matrigel.152 Because in vitro models often fail to retain complex cell-to-cell and tissue-tissue interactions present in vivo, toxicological assessments with the models must be interpreted with caution.
Combined maternal-fetal origins of placenta
The developmental origin and structural complexity of the placenta create further challenges for understanding toxicant impacts. Foremost, the placenta is an organ that arises from a combination of fetal and maternal cells. As a consequence, toxicants may behave differently in maternal tissues compared to placenta due to genetic differences contributed by the father through the fetus. With respect to TCE metabolism, including bioactivation to harmful reactive metabolites and detoxication to stable excretable metabolites, genetic differences play a key role in determining individual susceptibility.61 In particular, genetic differences in genes coding for metabolic enzymes could lead to variations in metabolism within the placenta, depending on maternal or fetal cell origin.
In addition to different genotypes, the combined fetal and maternal origin means that a placenta is not necessarily a female tissue, but rather has a fetal sex which can be male or female. Sex differences are observable not only at the phenotypic level of man versus woman, but resonate at the cellular, molecular, and genetic levels, as well. Likewise, sexual dimorphism is evident in placenta.153 Thus, toxicants such as TCE, which demonstrates sex-specific toxicity,61 may exert different effects in placental cells of male fetal origin versus female fetal origin.
Heterogeneity of cell types in placenta
Another challenge for identifying TCE toxicity in the placenta lies in the heterogeneity of cell types that make up this multifunctional organ. The placenta contains a variety of cell types with diverse functions.5 For example, cytotrophoblasts are proliferative cells that provide structural support to the core of placental villi and can differentiate into specialized trophoblasts that invade and remodel uterine interstitial tissue, spiral arteries, and glands. In addition, cytotrophoblasts fuse to form syncytiotrophoblasts that provide important hormone synthesis and transport functions.154 In addition, Hofbauer cells are resident placental macrophages of fetal origin that have roles in placental differentiation, placental blood vessel formation, immunoregulation, and more.155 With respect to TCE, most studies to date have investigated effects of TCE or its metabolites on extravillous trophoblasts,105–107, 115 placental tissue,103 or extra placental membranes,156 despite the potential for significant impacts on pregnancy from disruptions in other cell types. Further research investigating the impacts of TCE on specific placental cell types and on cell-cell interactions using in vitro models of human tissue would provide important insight into the mechanism of TCE placental toxicity.
Exposure assessment challenges
Regarding human studies, perhaps the greatest challenge remains in exposure assessment. Identification and quantification of exposure of pregnant women to TCE is challenging, in part, due to lack of validated biomarker(s) specific for TCE exposure. In particular, measurement of the reactive, short-lived, non-excretable intermediate metabolites of TCE glutathione pathway remains problematic, yet this pathway appears to be most relevant for placenta. Identification and measurement of exact protein, nucleic acid, and lipid adducts resulting from formation of the reactive metabolites also remain a challenge. The ability to readily detect such adducts would be crucial to understanding molecular initiating events of TCE metabolites, inform additional mechanisms of action, and help TCE exposure assessment. Consequently, these challenges hamper environmental epidemiology studies with the need to often use imprecise methods for TCE exposure assessment, including estimation, surrogate/proxy measures, and geographic location. More definitive, specific, and easily applicable exposure tools that allow individual exposure estimation would improve human studies of TCE impacts on adverse pregnancy outcomes.
Finally, other structurally similar volatile organic compounds (VOCs) such as perchloroethylene are also associated with adverse pregnancy outcomes99 and have not been adequately evaluated for placental toxicity. Given their structural similarity and biotransformation to some of the same metabolites, it is plausible that perchloroethylene and possibly other VOCs could act through similar mechanisms as TCE in the placenta.
Summary and Conclusions
Due to its widespread use and presence in contaminated waste sites, TCE exposure is a public health concern with exposures occurring both occupationally and through the broader environment. Although certain toxic effects related to TCE exposure are documented, less is known about reproductive and developmental endpoints. Current evidence suggests that some TCE metabolites have toxic effects on the placenta, with significant health implications for exposed mothers and fetuses. However, there remain significant gaps in our understanding that limit our ability to assess the hazard that TCE presents to placental health. Future research could clarify effects on specific placental cell types and functions, the effect of fetal sex on response to TCE exposure, the true extent of genetic variation in TCE metabolism and kinetics, as well as the potential for synergistic or additive toxicity based on co-exposures with other chemicals. In addition, improved methods of assessing exposures would provide better epidemiological assessments of the risks posed by TCE. Similar gaps in our knowledge exist for structurally similar VOC compounds found in the environment (e.g., perchloroethylene), and future studies should assess these compounds for impacts on placental health. Due to the widespread nature of these chemicals as environmental contaminants and the critical role that the placenta plays in lifelong fetal and maternal health, a more thorough understanding of potential placental toxicity of TCE and other VOCs could lead to significant gains in public health.
Acknowledgments
We gratefully acknowledge laboratory members Iman Hassan, Erica Boldenow, and Kelly A. Hogan, and colleagues Dave Bridges, Justin A. Colacino, and Kelly M. Bakulski for assistance and discussions that invariably contributed to this work. We thank Faith Bjork and Monica Smolinski for laboratory support.
Funding Information
This work was supported by the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH), with a research project to RL-C, (P42ES017198), training grant fellowship support to ERE, SMH, and ALS (T32ES007062), and supplementary project support from the Michigan Center for Lifestage Environmental Exposure and Disease (P30ES017885). This work was also supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), NIH with training grant support to ALS (T32HD07934). Additional fellowship support for SMH was from the Michigan Institute for Clinical & Health Research funded by the National Center for Advancing Translational Sciences (NCATS), NIH (UL1 TR002240). Support from a University of Michigan Rackham Graduate Student Research Grant is also gratefully acknowledged. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIEHS, NICHD, NIH, or the University of Michigan.
Footnotes
Conflict of Interest
The authors declare that there are no conflicts of interest.
References
- 1.Woodruff TJ, Zota AR and Schwartz JM, Environmental chemicals in pregnant women in the United States: NHANES 2003–2004, Environ Health Perspect, 2011, 119, 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tarrade A, Panchenko P, Junien C and Gabory A, Placental contribution to nutritional programming of health and diseases: epigenetics and sexual dimorphism, J Exp Biol, 2015, 218, 50–58. [DOI] [PubMed] [Google Scholar]
- 3.Vannuccini S, Bocchi C, Severi FM, Challis JR and Petraglia F, Endocrinology of human parturition, Annales d’endocrinologie, 2016, 77, 105–113. [DOI] [PubMed] [Google Scholar]
- 4.Smith R, Parturition N Engl J Med, 2007, 356, 271–283. [DOI] [PubMed] [Google Scholar]
- 5.Burton GJ, Fowden AL and Thornburg KL, Placental Origins of Chronic Disease, Physiol Rev, 2016, 96, 1509–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ilekis JV, Tsilou E, Fisher S, Abrahams VM, Soares MJ, Cross JC, Zamudio S, Illsley NP, Myatt L, Colvis C, Costantine MM, Haas DM, Sadovsky Y, Weiner C, Rytting E and Bidwell G, Placental origins of adverse pregnancy outcomes: potential molecular targets: an Executive Workshop Summary of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, Am J Obstet Gynecol, 2016, 215, S1–S46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitro SD, Johnson T and Zota AR, Cumulative Chemical Exposures During Pregnancy and Early Development, Curr Environ Health Rep, 2015, 2, 367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiu WA, Jinot J, Scott CS, Makris SL, Cooper GS, Dzubow RC, Bale AS, Evans MV, Guyton KZ, Keshava N, Lipscomb JC, Barone S, Fox JF, Gwinn MR, Schaum J and Caldwell JC, Human health effects of trichloroethylene: key findings and scientific issues, Environmental health perspectives, 2013, 121, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blencowe H, Cousens S, Oestergaard MZ, Chou D, Moller AB, Narwal R, Adler A, Vera Garcia C, Rohde S, Say L and Lawn JE, National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications, Lancet, 2012, 379, 2162–2172. [DOI] [PubMed] [Google Scholar]
- 10.Martin JA, Hamilton BE, Osterman MJK, Driscoll AK and Drake P, Births: Final data for 2017, National Vital Statistics Reports; Hyattsville, MD: National Center for Health Statistics in Medicine, 2018, 67. [PubMed] [Google Scholar]
- 11.Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, Lawn JE, Cousens S, Mathers C and Black RE, Global, regional, and national causes of under-5 mortality in 2000–15: an updated systematic analysis with implications for the Sustainable Development Goals, Lancet, 2016, 388, 3027–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen A, Oster E and Williams H, Why Is Infant Mortality Higher in the United States Than in Europe?, American economic journal. Economic policy, 2016, 8, 89–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heron M, Deaths: Leading Causes for 2016, Natl Vital Stat Rep, 2018, 67, 1–77. [PubMed] [Google Scholar]
- 14.Martin JA, Hamilton BE and Osterman MJK, Births in the United States, 2018. NCHS Data Brief, no 346.Journal, 2019. [PubMed] [Google Scholar]
- 15.Martin JA, Kirmeyer S, Osterman M and Shepherd RA, Born a bit too early: Recent trends in late preterm births, National Vital Statistics Reports; Hyattsville, MD: National Center for Health Statistics in Medicine, 2009, 57. [PubMed] [Google Scholar]
- 16.Institute of Medicine of the National Academies Committee on Understanding Premature Birth and Assuring Healthy Outcomes, in Preterm Birth: Causes, Consequences, and Prevention (Prepublication Copy), eds. Behrman RE and Butler A. Stith, The National Academies Press, Washington, DC, 2006. [Google Scholar]
- 17.March of Dimes, March of Dimes 2019 Report Card Shines Spotlight on The U.S. Maternal And Infant Health Crisis, https://www.marchofdimes.org/news/march-of-dimes-2019-report-card-shines-spotlight-on-the-u-s-maternal-and-infant-health-crisis.aspx, (accessed Nov 4, 2019).
- 18.March of Dimes, The Partnership for Maternal Newborn and Child Health (PMNCH), Save the Children and World Health Organization (WHO), Born Too Soon: The Global Action Report on Preterm Birth, World Health Organization, Geneva, 2012. [Google Scholar]
- 19.DPHP (Office of Disease Prevention and Health Promotion), Healthy People 2020: Maternal, Infant, and Child Health, https://www.healthypeople.gov/2020/topics-objectives/topic/maternal-infant-and-child-health, (accessed Nov 3, 2019).
- 20.Ferguson KK, O’Neill MS and Meeker JD, Environmental contaminant exposures and preterm birth: a comprehensive review, J Toxicol Environ Health B Crit Rev, 2013, 16, 69–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porpora MG, Piacenti I, Scaramuzzino S, Masciullo L, Rech F and Panici PB, Environmental Contaminants Exposure and Preterm Birth: A Systematic Review, Toxics, 2019, 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jedrychowski WA, Perera FP, Maugeri U, Spengler J, Mroz E, Flak E, Stigter L, Majewska R, Kaim I, Sowa A and Jacek R, Prohypertensive effect of gestational personal exposure to fine particulate matter. Prospective cohort study in non-smoking and non-obese pregnant women, Cardiovasc Toxicol, 2012, 12, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C, Sun J, Liu Y, Liang H, Wang M, Wang C and Shi T, Different exposure levels of fine particulate matter and preterm birth: a meta-analysis based on cohort studies, Environ Sci Pollut Res Int, 2017, 24, 17976–17984. [DOI] [PubMed] [Google Scholar]
- 24.Taylor CM, Golding J and Emond AM, Adverse effects of maternal lead levels on birth outcomes in the ALSPAC study: a prospective birth cohort study, BJOG, 2015, 122, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor CM, Golding J and Emond AM, Moderate Prenatal Cadmium Exposure and Adverse Birth Outcomes: a Role for Sex-Specific Differences?, Paediatr Perinat Epidemiol, 2016, 30, 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sagiv SK, Rifas-Shiman SL, Fleisch AF, Webster TF, Calafat AM, Ye X, Gillman MW and Oken E, Early-Pregnancy Plasma Concentrations of Perfluoroalkyl Substances and Birth Outcomes in Project Viva: Confounded by Pregnancy Hemodynamics?, Am J Epidemiol, 2018, 187, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elkin ER and O’Neill MS, Trends in Environmental Tobacco Smoke (ETS) Exposure and Preterm Birth: Use of Smoking Bans and Direct ETS Exposure Assessments in Study Designs, Chem Res Toxicol, 2017, 30, 1376–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoyert DL and Gregory EC, Cause of Fetal Death: Data From the Fetal Death Report, 2014, Natl Vital Stat Rep, 2016, 65, 1–25. [PubMed] [Google Scholar]
- 29.Morgan TK, Role of the Placenta in Preterm Birth: A Review, Am J Perinatol, 2016, 33, 258–266. [DOI] [PubMed] [Google Scholar]
- 30.Burton GJ and Jauniaux E, Pathophysiology of placental-derived fetal growth restriction, Am J Obstet Gynecol, 2018, 218, S745–s761. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y and Zhao S, in Vascular Biology of the Placenta, Morgan & Claypool Life Sciences, San Rafael, CA, 2010. [PubMed] [Google Scholar]
- 32.Myren M, Mose T, Mathiesen L and Knudsen LE, The human placenta--an alternative for studying foetal exposure, Toxicol In Vitro, 2007, 21, 1332–1340. [DOI] [PubMed] [Google Scholar]
- 33.Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M and Chahoud I, Parent bisphenol A accumulation in the human maternal-fetal-placental unit, Environ Health Perspect, 2002, 110, A703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeda Y, Liu X, Sumiyoshi M, Matsushima A, Shimohigashi M and Shimohigashi Y, Placenta expressing the greatest quantity of bisphenol A receptor ERR{gamma} among the human reproductive tissues: Predominant expression of type-1 ERRgamma isoform, J Biochem, 2009, 146, 113–122. [DOI] [PubMed] [Google Scholar]
- 35.Needham LL, Grandjean P, Heinzow B, Jorgensen PJ, Nielsen F, Patterson DG Jr., Sjodin A, Turner WE and Weihe P, Partition of environmental chemicals between maternal and fetal blood and tissues, Environ Sci Technol, 2011, 45, 1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki G, Nakano M and Nakano S, Distribution of PCDDs/PCDFs and Co-PCBs in human maternal blood, cord blood, placenta, milk, and adipose tissue: dioxins showing high toxic equivalency factor accumulate in the placenta, Biosci Biotechnol Biochem, 2005, 69, 1836–1847. [DOI] [PubMed] [Google Scholar]
- 37.Robins JC, Marsit CJ, Padbury JF and Sharma SS, Endocrine disruptors, environmental oxygen, epigenetics and pregnancy, Front Biosci (Elite Ed), 2011, 3, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang C, Song G and Lim W, A mechanism for the effect of endocrine disrupting chemicals on placentation, Chemosphere, 2019, 231, 326–336. [DOI] [PubMed] [Google Scholar]
- 39.Peiris HN, Vaswani K, Almughlliq F, Koh YQ and Mitchell MD, Review: Eicosanoids in preterm labor and delivery: Potential roles of exosomes in eicosanoid functions, Placenta, 2017, 54, 95–103. [DOI] [PubMed] [Google Scholar]
- 40.National Children’s Study Placenta Consortium, Nanes JA, Xia Y, Dassanayake R, Jones RM, Li A, Stodgell CJ, Walker C, Szabo S, Leuthner S, Durkin MS, Moye J and Miller RK, Selected persistent organic pollutants in human placental tissue from the United States, Chemosphere, 2014, 106, 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lala PK and Chakraborty C, Factors regulating trophoblast migration and invasiveness: possible derangements contributing to pre-eclampsia and fetal injury, Placenta, 2003, 24, 575–587. [DOI] [PubMed] [Google Scholar]
- 42.Mikelson CK, Troisi J, LaLonde A, Symes SJK, Thurston SW, DiRe LM, David Adair C, Miller RK and Richards SM, Placental concentrations of essential, toxic, and understudied metals and relationships with birth outcomes in Chattanooga, TN, Environ Res, 2019, 168, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baschat AA, Fetal responses to placental insufficiency: an update, BJOG, 2004, 111, 1031–1041. [DOI] [PubMed] [Google Scholar]
- 44.Gagnon R, Placental insufficiency and its consequences, Eur J Obstet Gynecol Reprod Biol, 2003, 110 Suppl 1, S99–107. [DOI] [PubMed] [Google Scholar]
- 45.Roberts DJ and Post MD, The placenta in pre-eclampsia and intrauterine growth restriction, J Clin Pathol, 2008, 61, 1254–1260. [DOI] [PubMed] [Google Scholar]
- 46.Morgan T, Placental Insufficiency Is a Leading Cause of Preterm Labor, NewReviews, 2014, 15, 5618–e5525. [Google Scholar]
- 47.Steegers EA, von Dadelszen P, Duvekot JJ and Pijnenborg R, Pre-eclampsia, Lancet, 2010, 376, 631–644. [DOI] [PubMed] [Google Scholar]
- 48.Brosens IA, Robertson WB and Dixon HG, The role of the spiral arteries in the pathogenesis of preeclampsia, Obstet Gynecol Annu, 1972, 1, 177–191. [PubMed] [Google Scholar]
- 49.Strakovsky RS and Schantz SL, Using Experimental Models to Assess Effects of Bisphenol A (BPA) and Phthalates on the Placenta: Challenges and Perspectives, Toxicol Sci, 2018, 166, 250–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.ATSDR (Agency for Toxic Substances and Disease Registry), Toxicological Profile for Trichloroethylene (TCE), U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA, 2019. [PubMed] [Google Scholar]
- 51.NTP (National Toxicology Program), Monograph on Trichloroethylene https://ntp.niehs.nih.gov/ntp/roc/monographs/finaltce_508.pdf, (accessed 11/8/2019).
- 52.U.S. EPA (Environmental Protection Agency), Trichloroethylene (TCE); Regulation of Use in Vapor Degreasing Under TSCA Section 6(a), Federal Register, 2017, 82, 7432–7461. [Google Scholar]
- 53.U.S. EPA (Environmental Protection Agency), Superfund: National Priorities List (NPL), https://www.epa.gov/superfund/superfund-national-priorities-list-npl, (accessed Nov 5, 2019).
- 54.Archer NP, Bradford CM, Villanacci JF, Crain NE, Corsi RL, Chambers DM, Burk T and Blount BC, Relationship between vapor intrusion and human exposure to trichloroethylene, J Environ Sci Health A Tox Hazard Subst Environ Eng, 2015, 50, 1360–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.OSHA (Occupational Safety and Health Administration), Annotated OSHA Z-2 Table, https://www.osha.gov/dsg/annotated-pels/tablez-2.html, (accessed June 26, 2019, 2019).
- 56.U.S. EPA (Environmental Protection Agency), National Primary Drinking Water Regulations https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations, (accessed January 15, 2020).
- 57.World Health Organization (WHO), Guidelines for drinking-water quality, fourth edition, World Health Organization, Geneva, Switzerland, fourth edn., 2011. [Google Scholar]
- 58.National Research Council (US) Committee on Contaminated Drinking Water at Camp Lejeune, Contaminated Water Supplies at Camp Lejeune: Assessing Potential Health Effects, National Academies Press, Washington DC, 2009. [PubMed] [Google Scholar]
- 59.Maslia ML, Aral MM, Ruckart PZ and Bove FJ, Reconstructing Historical VOC Concentrations in Drinking Water for Epidemiological Studies at a U.S. Military Base: Summary of Results, Water (Basel), 2016, 8, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lash LH, Fisher JW, Lipscomb JC and Parker JC, Metabolism of trichloroethylene, Environ Health Perspect, 2000, 108 Suppl 2, 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lash LH, Chiu WA, Guyton KZ and Rusyn I, Trichloroethylene biotransformation and its role in mutagenicity, carcinogenicity and target organ toxicity, Mutat Res Rev Mutat Res, 2014, 762, 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu D, Trichloroethylene Toxicity, http://www.atsdr.cdc.gov/csem/tce/docs/tce.pdf, 2016).
- 63.Rusyn I, Chiu WA, Lash LH, Kromhout H, Hansen J and Guyton KZ, Trichloroethylene: Mechanistic, epidemiologic and other supporting evidence of carcinogenic hazard, Pharmacol Ther, 2014, 141, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Werner M, Birner G and Dekant W, Sulfoxidation of mercapturic acids derived from tri- and tetrachloroethene by cytochromes P450 3A: a bioactivation reaction in addition to deacetylation and cysteine conjugate beta-lyase mediated cleavage, Chem Res Toxicol, 1996, 9, 41–49. [DOI] [PubMed] [Google Scholar]
- 65.Forkert PG, Lash L, Tardif R, Tanphaichitr N, Vandevoort C and Moussa M, Identification of trichloroethylene and its metabolites in human seminal fluid of workers exposed to trichloroethylene, Drug metabolism and disposition: the biological fate of chemicals, 2003, 31, 306–311. [DOI] [PubMed] [Google Scholar]
- 66.Lash LH, Xu Y, Elfarra AA, Duescher RJ and Parker JC, Glutathione-dependent metabolism of trichloroethylene in isolated liver and kidney cells of rats and its role in mitochondrial and cellular toxicity, Drug Metab Dispos, 1995, 23, 846–853. [PubMed] [Google Scholar]
- 67.Hinchman CA and Ballatori N, Glutathione-degrading capacities of liver and kidney in different species, Biochem Pharmacol, 1990, 40, 1131–1135. [DOI] [PubMed] [Google Scholar]
- 68.Odum J, Foster JR and Green T, A mechanism for the development of Clara cell lesions in the mouse lung after exposure to trichloroethylene, Chem Biol Interact, 1992, 83, 135–153. [DOI] [PubMed] [Google Scholar]
- 69.Bradford BU, Lock EF, Kosyk O, Kim S, Uehara T, Harbourt D, DeSimone M, Threadgill DW, Tryndyak V, Pogribny IP, Bleyle L, Koop DR and Rusyn I, Interstrain differences in the liver effects of trichloroethylene in a multistrain panel of inbred mice, Toxicol Sci, 2011, 120, 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lash LH, Putt DA and Parker JC, Metabolism and tissue distribution of orally administered trichloroethylene in male and female rats: identification of glutathione- and cytochrome P-450-derived metabolites in liver, kidney, blood, and urine, J Toxicol Environ Health A, 2006, 69, 1285–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Waxman DJ and Holloway MG, Sex differences in the expression of hepatic drug metabolizing enzymes, Molecular pharmacology, 2009, 76, 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bradford BU, Kono H, Isayama F, Kosyk O, Wheeler MD, Akiyama TE, Bleye L, Krausz KW, Gonzalez FJ, Koop DR and Rusyn I, Cytochrome P450 CYP2E1, but not nicotinamide adenine dinucleotide phosphate oxidase, is required for ethanol-induced oxidative DNA damage in rodent liver, Hepatology, 2005, 41, 336–344. [DOI] [PubMed] [Google Scholar]
- 73.Beppu K, Transmission of the anesthetic agents through the placenta in painless delivery and their effects on newborn infants, Keio J Med, 1968, 17, 81–107. [DOI] [PubMed] [Google Scholar]
- 74.Laham S, Studies on placental transfer. Trichlorethylene, IMS Ind Med Surg, 1970, 39, 46–49. [PubMed] [Google Scholar]
- 75.Ghantous H, Danielsson BR, Dencker L, Gorczak J and Vesterberg O, Trichloroacetic acid accumulates in murine amniotic fluid after tri- and tetrachloroethylene inhalation, Acta Pharmacol Toxicol (Copenh), 1986, 58, 105–114. [DOI] [PubMed] [Google Scholar]
- 76.Elfarra AA, in Drug Metabolism and Transport: Molecular Methods and Mechanisms, ed. Lash L, Humana Press Inc., Totowa, New Jersey, 2005, ch. 1. [Google Scholar]
- 77.Griffiths SK and Campbell JP, Placental structure, function and drug transfer, Continuing Education in Anaesthesia Critical Care & Pain, 2015, 15, 84–89. [Google Scholar]
- 78.Leazer TM and Klaassen CD, The presence of xenobiotic transporters in rat placenta, Drug Metab Dispos, 2003, 31, 153–167. [DOI] [PubMed] [Google Scholar]
- 79.Hakkola J, Pelkonen O, Pasanen M and Raunio H, Xenobiotic-metabolizing cytochrome P450 enzymes in the human feto-placental unit: role in intrauterine toxicity, Critical reviews in toxicology, 1998, 28, 35–72. [DOI] [PubMed] [Google Scholar]
- 80.Sanchis R and Guerri C, Alcohol-metabolizing enzymes in placenta and fetal liver: effect of chronic ethanol intake, Alcohol Clin Exp Res, 1986, 10, 39–44. [DOI] [PubMed] [Google Scholar]
- 81.Obolenskaya MY, Teplyuk NM, Divi RL, Poirier MC, Filimonova NB, Zadrozna M and Pasanen MJ, Human placental glutathione S-transferase activity and polycyclic aromatic hydrocarbon DNA adducts as biomarkers for environmental oxidative stress in placentas from pregnant women living in radioactivity- and chemically-polluted regions, Toxicol Lett, 2010, 196, 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prade L, Huber R, Manoharan TH, Fahl WE and Reuter W, Structures of class pi glutathione S-transferase from human placenta in complex with substrate, transition-state analogue and inhibitor, Structure, 1997, 5, 1287–1295. [DOI] [PubMed] [Google Scholar]
- 83.Walker FB, Hoblit DL, Cunningham FG and Combes B, Gamma glutamyl transpeptidase in normal pregnancy, Obstet Gynecol, 1974, 43, 745–749. [PubMed] [Google Scholar]
- 84.Knapen MF, Zusterzeel PL, Peters WH and Steegers EA, Glutathione and glutathione-related enzymes in reproduction. A review, Eur J Obstet Gynecol Reprod Biol, 1999, 82, 171–184. [DOI] [PubMed] [Google Scholar]
- 85.Waters EM, Gerstner HB and Huff JE, Trichloroethylene. I. An overview, J Toxicol Environ Health, 1977, 2, 671–707. [DOI] [PubMed] [Google Scholar]
- 86.U.S. EPA (Environmental Protection Agency), Toxicological Review of Trichloroethylene (CAS No. 79-01-6), National Center for Environmental Assessment, Washtingon D.C., 2011. [Google Scholar]
- 87.NTP (National Toxicology Program), Report on Carcinogens, Fourteenth Edition, U.S. Department of Health and Human Services, Public Health Service, Research Triangle Park, NC, 2016. [Google Scholar]
- 88.IARC (International Agency for Research on Cancer), Carcinogenicity of trichloroethylene tetrachloroethylene, and other chlorinated agents, International Agency for Research on Cancer, Lyon, France, 2014. [Google Scholar]
- 89.Scott CS and Jinot J, Trichloroethylene and cancer: systematic and quantitative review of epidemiologic evidence for identifying hazards, Int J Environ Res Public Health, 2011, 8, 4238–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jaffe DR, Gandolfi AJ and Nagle RB, Chronic toxicity of S-(trans-1,2-dichlorovinyl)-L-cysteine in mice, J Appl Toxicol, 1984, 4, 315–319. [DOI] [PubMed] [Google Scholar]
- 91.Reif JS, Burch JB, Nuckols JR, Metzger L, Ellington D and Anger WK, Neurobehavioral effects of exposure to trichloroethylene through a municipal water supply, Environ Res, 2003, 93, 248–258. [DOI] [PubMed] [Google Scholar]
- 92.Lock EA and Reed CJ, Trichloroethylene: mechanisms of renal toxicity and renal cancer and relevance to risk assessment, Toxicol Sci, 2006, 91, 313–331. [DOI] [PubMed] [Google Scholar]
- 93.Rufer ES, Hacker TA, Flentke GR, Drake VJ, Brody MJ, Lough J and Smith SM, Altered cardiac function and ventricular septal defect in avian embryos exposed to low-dose trichloroethylene, Toxicol Sci, 2010, 113, 444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ramdhan DH, Kamijima M, Yamada N, Ito Y, Yanagiba Y, Nakamura D, Okamura A, Ichihara G, Aoyama T, Gonzalez FJ and Nakajima T, Molecular mechanism of trichloroethylene-induced hepatotoxicity mediated by CYP2E1, Toxicol Appl Pharmacol, 2008, 231, 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lagakos S, Wessen B and Zelen M, An analysis of contaminated well water and health effects in Woburn, Massachusetts Journal of the American Statistical Association, 1986, 81, 583–596. [Google Scholar]
- 96.Bove FJ, Fulcomer MC, Klotz JB, Esmart J, Dufficy EM and Savrin JE, Public drinking water contamination and birth outcomes, Am J Epidemiol, 1995, 141, 850–862. [DOI] [PubMed] [Google Scholar]
- 97.Rodenbeck SE, Sanderson LM and Rene A, Maternal exposure to trichloroethylene in drinking water and birth-weight outcomes, Arch Environ Health, 2000, 55, 188–194. [DOI] [PubMed] [Google Scholar]
- 98.Forand SP, Lewis-Michl EL and Gomez MI, Adverse birth outcomes and maternal exposure to trichloroethylene and tetrachloroethylene through soil vapor intrusion in New York State, Environ Health Perspect, 2012, 120, 616–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruckart PZ, Bove FJ and Maslia M, Evaluation of contaminated drinking water and preterm birth, small for gestational age, and birth weight at Marine Corps Base Camp Lejeune, North Carolina: a cross-sectional study, Environ Health, 2014, 13, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Taylor KW, Joubert BR, Braun JM, Dilworth C, Gennings C, Hauser R, Heindel JJ, Rider CV, Webster TF and Carlin DJ, Statistical Approaches for Assessing Health Effects of Environmental Chemical Mixtures in Epidemiology: Lessons from an Innovative Workshop, Environmental Health Perspectives, 2016, 124, A227–A229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.National Institute of Environmental Health Sciences (NIEHS), 2018–2023 Strategic Plan. Advancing Environmental Health Sciences. Improving Health. (NIH Publication No. 18-ES-7935), 2018.
- 102.Healy TE, Poole TR and Hopper A, Rat fetal development and maternal exposure to trichloroethylene 100 p.p.m, Br J Anaesth, 1982, 54, 337–341. [DOI] [PubMed] [Google Scholar]
- 103.Loch-Caruso R, Hassan I, Harris SM, Kumar A, Bjork F and Lash LH, Trichloroethylene exposure in mid-pregnancy decreased fetal weight and increased placental markers of oxidative stress in rats, Reprod Toxicol, 2019, 83, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fisher JW, Channel SR, Eggers JS, Johnson PD, MacMahon KL, Goodyear CD, Sudberry GL, Warren DA, Latendresse JR and Graeter LJ, Trichloroethylene, trichloroacetic acid, and dichloroacetic acid: do they affect fetal rat heart development?, Int J Toxicol, 2001, 20, 257–267. [DOI] [PubMed] [Google Scholar]
- 105.Elkin ER, Harris SM and Loch-Caruso R, Trichloroethylene metabolite S-(1,2-dichlorovinyl)-l-cysteine induces lipid peroxidation-associated apoptosis via the intrinsic and extrinsic apoptosis pathways in a first-trimester placental cell line, Toxicol Appl Pharmacol, 2018, 338, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Elkin ER, Bridges D and Loch-Caruso R, The trichloroethylene metabolite S-(1,2-dichlorovinyl)-L-cysteine induces progressive mitochondrial dysfunction in HTR-8/SVneo trophoblasts, Toxicology, 2019, DOI: 10.1016/j.tox.2019.152283, 152283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hassan I, Kumar AM, Park HR, Lash LH and Loch-Caruso R, Reactive Oxygen Stimulation of Interleukin-6 Release in the Human Trophoblast Cell Line HTR-8/SVneo by the Trichlorethylene Metabolite S-(1,2-Dichloro)-l-Cysteine, Biology of reproduction, 2016, 95, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lash LH, Putt DA, Brashear WT, Abbas R, Parker JC and Fisher JW, Identification of S-(1,2-dichlorovinyl)glutathione in the blood of human volunteers exposed to trichloroethylene, J Toxicol Environ Health A, 1999, 56, 1–21. [DOI] [PubMed] [Google Scholar]
- 109.Walsh Clang CM and Aleo MD, Mechanistic analysis of S-(1,2-dichlorovinyl)-L-cysteine-induced cataractogenesis in vitro, Toxicol Appl Pharmacol, 1997, 146, 144–155. [DOI] [PubMed] [Google Scholar]
- 110.McGoldrick TA, Lock EA, Rodilla V and Hawksworth GM, Renal cysteine conjugate C-S lyase mediated toxicity of halogenated alkenes in primary cultures of human and rat proximal tubular cells, Arch Toxicol, 2003, 77, 365–370. [DOI] [PubMed] [Google Scholar]
- 111.van de Water B, Zoeteweij JP, de Bont HJ, Mulder GJ and Nagelkerke JF, Role of mitochondrial Ca2+ in the oxidative stress-induced dissipation of the mitochondrial membrane potential. Studies in isolated proximal tubular cells using the nephrotoxin 1,2-dichlorovinyl-L-cysteine, J Biol Chem, 1994, 269, 14546–14552. [PubMed] [Google Scholar]
- 112.Chen JC, Stevens JL, Trifillis AL and Jones TW, Renal cysteine conjugate beta-lyase-mediated toxicity studied with primary cultures of human proximal tubular cells, Toxicol Appl Pharmacol, 1990, 103, 463–473. [DOI] [PubMed] [Google Scholar]
- 113.Raghupathy R and Kalinka J, Cytokine imbalance in pregnancy complications and its modulation, Front Biosci, 2008, 13, 985–994. [DOI] [PubMed] [Google Scholar]
- 114.Vrachnis N, Vitoratos N, Iliodromiti Z, Sifakis S, Deligeoroglou E and Creatsas G, Intrauterine inflammation and preterm delivery, Ann N Y Acad Sci, 2010, 1205, 118–122. [DOI] [PubMed] [Google Scholar]
- 115.Elkin ER, Bridges D, Harris SM and Loch-Caruso RK, Exposure to the trichloroethylene metabolite S-(1,2-dichlorovinyl)-L-cysteine causes compensatory changes to macronutrient utilization and energy metabolism in placental HTR-8/SVneo cells, Chem Res Toxicol, 2020, DOI: 10.1021/acs.chemrestox.9b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Murray AJ, Oxygen delivery and fetal-placental growth: beyond a question of supply and demand?, Placenta, 2012, 33 Suppl 2, e16–22. [DOI] [PubMed] [Google Scholar]
- 117.Vaughan OR and Fowden AL, Placental metabolism: substrate requirements and the response to stress, Reprod Domest Anim, 2016, 51 Suppl 2, 25–35. [DOI] [PubMed] [Google Scholar]
- 118.Mando C, De Palma C, Stampalija T, Anelli GM, Figus M, Novielli C, Parisi F, Clementi E, Ferrazzi E and Cetin I, Placental mitochondrial content and function in intrauterine growth restriction and preeclampsia, American journal of physiology. Endocrinology and metabolism, 2014, 306, E404–413. [DOI] [PubMed] [Google Scholar]
- 119.Holland O, Dekker Nitert M, Gallo LA, Vejzovic M, Fisher JJ and Perkins AV, Review: Placental mitochondrial function and structure in gestational disorders, Placenta, 2017, 54, 2–9. [DOI] [PubMed] [Google Scholar]
- 120.Gupta S, Agarwal A and Sharma RK, The role of placental oxidative stress and lipid peroxidation in preeclampsia, Obstet Gynecol Surv, 2005, 60, 807–816. [DOI] [PubMed] [Google Scholar]
- 121.Sharp AN, Heazell AE, Crocker IP and Mor G, Placental apoptosis in health and disease, Am J Reprod Immunol, 2010, 64, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smith SC, Baker PN and Symonds EM, Increased placental apoptosis in intrauterine growth restriction, Am J Obstet Gynecol, 1997, 177, 1395–1401. [DOI] [PubMed] [Google Scholar]
- 123.Caniggia I, Grisaru-Gravnosky S, Kuliszewsky M, Post M and Lye SJ, Inhibition of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic pregnancies, J Clin Invest, 1999, 103, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pennington KA, Schlitt JM, Jackson DL, Schulz LC and Schust DJ, Preeclampsia: multiple approaches for a multifactorial disease, Dis Model Mech, 2012, 5, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Aardema MW, Oosterhof H, Timmer A, van Rooy I and Aarnoudse JG, Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses, Placenta, 2001, 22, 405–411. [DOI] [PubMed] [Google Scholar]
- 126.Meekins JW, Pijnenborg R, Hanssens M, McFadyen IR and van Asshe A, A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies, Br J Obstet Gynaecol, 1994, 101, 669–674. [DOI] [PubMed] [Google Scholar]
- 127.Kadyrov M, Kingdom JC and Huppertz B, Divergent trophoblast invasion and apoptosis in placental bed spiral arteries from pregnancies complicated by maternal anemia and early-onset preeclampsia/intrauterine growth restriction, Am J Obstet Gynecol, 2006, 194, 557–563. [DOI] [PubMed] [Google Scholar]
- 128.DiFederico E, Genbacev O and Fisher SJ, Preeclampsia is associated with widespread apoptosis of placental cytotrophoblasts within the uterine wall, Am J Pathol, 1999, 155, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Genbacev O, DiFederico E, McMaster M and Fisher SJ, Invasive cytotrophoblast apoptosis in pre-eclampsia, Hum Reprod, 1999, 14 Suppl 2, 59–66. [DOI] [PubMed] [Google Scholar]
- 130.Reister F, Frank HG, Kingdom JC, Heyl W, Kaufmann P, Rath W and Huppertz B, Macrophage-induced apoptosis limits endovascular trophoblast invasion in the uterine wall of preeclamptic women, Lab Invest, 2001, 81, 1143–1152. [DOI] [PubMed] [Google Scholar]
- 131.Rull K, Tomberg K, Koks S, Mannik J, Mols M, Sirotkina M, Varv S and Laan M, Increased placental expression and maternal serum levels of apoptosis-inducing TRAIL in recurrent miscarriage, Placenta, 2013, 34, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jurisicova A, Antenos M, Varmuza S, Tilly JL and Casper RF, Expression of apoptosis-related genes during human preimplantation embryo development: potential roles for the Harakiri gene product and Caspase-3 in blastomere fragmentation, Mol Hum Reprod, 2003, 9, 133–141. [DOI] [PubMed] [Google Scholar]
- 133.Schmidt A, Morales-Prieto DM, Pastuschek J, Fröhlich K and Markert UR, Only humans have human placentas: molecular differences between mice and humans, Journal of Reproductive Immunology, 2015, 108, 65–71. [DOI] [PubMed] [Google Scholar]
- 134.Chavatte-Palmer P and Tarrade A, Placentation in different mammalian species, Annales d’endocrinologie, 2016, 77, 67–74. [DOI] [PubMed] [Google Scholar]
- 135.Grigsby PL, Animal Models to Study Placental Development and Function throughout Normal and Dysfunctional Human Pregnancy, Semin Reprod Med, 2016, 34, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wooding P and Burton G, Springer-Verlag; Berlin, Heidelberg: 2008, ch. Chapter 8. Haemochorial Placentation: Mouse, Rabbit, Man, Apes, Monkeys. [Google Scholar]
- 137.Tal R, Taylor HS, Burney RO, Mooney SB and Giudice LC, in Endotext, eds. Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A and Wilson DP, MDText.com, Inc., South Dartmouth (MA), 2015. [Google Scholar]
- 138.Malassiné A, Frendo JL and Evain-Brion D, A comparison of placental development and endocrine functions between the human and mouse model, Human reproduction update, 2003, 9, 531–539. [DOI] [PubMed] [Google Scholar]
- 139.Olson DM and Ammann C, Role of the prostaglandins in labour and prostaglandin receptor inhibitors in the prevention of preterm labour, Front Biosci, 2007, 12, 1329–1343. [DOI] [PubMed] [Google Scholar]
- 140.Challis JR, Lye SJ and Gibb W, Prostaglandins and parturition, Ann N Y Acad Sci, 1997, 828, 254–267. [DOI] [PubMed] [Google Scholar]
- 141.Napso T, Yong HEJ, Lopez-Tello J and Sferruzzi-Perri AN, The Role of Placental Hormones in Mediating Maternal Adaptations to Support Pregnancy and Lactation, Frontiers in Physiology, 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fry RC, Bangma J, Szilagyi J and Rager JE, Developing novel in vitro methods for the risk assessment of developmental and placental toxicants in the environment, Toxicol Appl Pharmacol, 2019, 378, 114635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miller RK, Genbacev O, Turner MA, Aplin JD, Caniggia I and Huppertz B, Human placental explants in culture: approaches and assessments, Placenta, 2005, 26, 439–448. [DOI] [PubMed] [Google Scholar]
- 144.Lee JS, Romero R, Han YM, Kim HC, Kim CJ, Hong J-S and Huh D, Placenta-on-a-chip: a novel platform to study the biology of the human placenta, The Journal of Maternal-Fetal & Neonatal Medicine, 2016, 29, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Blundell C, Yi YS, Ma L, Tess ER, Farrell MJ, Georgescu A, Aleksunes LM and Huh D, Placental Drug Transport-on-a-Chip: A Microengineered In Vitro Model of Transporter-Mediated Drug Efflux in the Human Placental Barrier, Advanced healthcare materials, 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Myllynen P and Vähäkangas K, Placental transfer and metabolism: An overview of the experimental models utilizing human placental tissue, Toxicology in Vitro, 2013, 27, 507–512. [DOI] [PubMed] [Google Scholar]
- 147.Nestor CE, Ottaviano R, Reinhardt D, Cruickshanks HA, Mjoseng HK, McPherson RC, Lentini A, Thomson JP, Dunican DS, Pennings S, Anderton SM, Benson M and Meehan RR, Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems, Genome Biol, 2015, 16, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bilban M, Tauber S, Haslinger P, Pollheimer J, Saleh L, Pehamberger H, Wagner O and Knofler M, Trophoblast invasion: assessment of cellular models using gene expression signatures, Placenta, 2010, 31, 989–996. [DOI] [PubMed] [Google Scholar]
- 149.Novakovic B, Gordon L, Wong NC, Moffett A, Manuelpillai U, Craig JM, Sharkey A and Saffery R, Wide-ranging DNA methylation differences of primary trophoblast cell populations and derived cell lines: implications and opportunities for understanding trophoblast function, Molecular human reproduction, 2011, 17, 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huckle WR, in Progress in Molecular Biology and Translational Science, ed. Huckle WR, Academic Press, 2017, vol. 145, pp. 29–37. [DOI] [PubMed] [Google Scholar]
- 151.Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N and Lala PK, Establishment and characterization of first trimester human trophoblast cells with extended lifespan, Experimental cell research, 1993, 206, 204–211. [DOI] [PubMed] [Google Scholar]
- 152.Kilburn BA, Wang J, Duniec-Dmuchowski ZM, Leach RE, Romero R and Armant DR, Extracellular matrix composition and hypoxia regulate the expression of HLA-G and integrins in a human trophoblast cell line, Biology of reproduction, 2000, 62, 739–747. [DOI] [PubMed] [Google Scholar]
- 153.Kalisch-Smith JI, Simmons DG, Dickinson H and Moritz KM, Review: Sexual dimorphism in the formation, function and adaptation of the placenta, Placenta, 2017, 54, 10–16. [DOI] [PubMed] [Google Scholar]
- 154.Costa MA, The endocrine function of human placenta: an overview, Reprod Biomed Online, 2016, 32, 14–43. [DOI] [PubMed] [Google Scholar]
- 155.Reyes L, Wolffe B and Golos T, in Macrophages Results and Problems in Cell Differentiation, ed. Kloc M, Springer International Publishing, New York, NY, 2017, vol. 62, pp. 45–60. [DOI] [PubMed] [Google Scholar]
- 156.Boldenow E, Hassan I, Chames MC, Xi C and Loch-Caruso R, The trichloroethylene metabolite S-(1,2-dichlorovinyl)-l-cysteine but not trichloroacetate inhibits pathogen-stimulated TNF-alpha in human extraplacental membranes in vitro, Reprod Toxicol, 2015, 52, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]