

Abstract
Objective:
Restoring calcium sensor protein (S100A1) activity in failing hearts poses a promising therapeutic strategy. We hypothesize that cardiac overexpression of the S100A1 gene mediated by a double-stranded adeno-associated virus (scAAV) results in better functional and molecular improvements compared to the single-stranded virus (ssAAV).
Methods:
Heart failure was induced by coronary artery ligation. Then, intramyocardial injections of saline, AAV9 empty caspid, scAAV9.S100A1, and ssAAV9.S100A1 were performed. Ten weeks post-infarction, all rats received cardiac evaluation; serum and tissue were collected for genetic analysis, cytokine profiling, and assessments of mitochondrial function and structure.
Results:
Overexpression of AAV9.S100A1 improved systolic and diastolic function. Compared to control, ejection fraction was greater in treated groups (54.8% versus 32.3%, p<0.05). Similarly, end-diastolic volume index was significantly less in the treated group than in control (1.14 versus 1.59ml/cm2), while fractional shortening was greater in treated groups than control (26% versus 38%, p<0.05). Interestingly, cardiac mechanics were significantly better when treated with double stranded virus compared to single stranded. Quantitative polymerase chain reaction demonstrated robust transfection of myocardium with the S100A1 gene, with more infection in the self-complimentary group compared to the single-stranded group (5.68±0.44 versus 4.09±0.25 log10 genome copies per 100 ng DNA; p<0.0001). Concentrations of the inflammatory cytokines were elevated in the ssAAV9/S100A1 group compared to the scAAV9/S100A1. Assessment of mitochondrial respiration and morphology demonstrated that injection of self-complementary vector saved both mitochondrial structure and function.
Conclusion:
Gene therapy of S100A1 can prevent pathological post-myocardial infarction remodeling and decrease inflammatory response in ischemic heart failure.
Keywords: gene therapy, adeno-associated virus, post-myocardial remodeling, inflammation, mitochondria, cytokines
Central Picture:
Self-complimentary AAV transfects the nucleus faster and prevents post-MI remodeling.
Central Message:
Gene delivery with calcium cycling protein mediated by double-stranded AAV vector increases myocardial transfection and prevents pathological post-myocardial infarction remodeling in heart failure.
Perspective statement:
In cardiac gene therapy clinical trials, it was demonstrated that single-stranded DNA gene delivery vectors did not effectively transduce into target cells. Gene overexpression of calcium binding protein, S100A1 gene, mediated by double-stranded virus can increase myocardial transfection, improve cardiac function and reduce inflammation in a rodent model of ischemic heart failure.
Introduction
Adverse ventricular remodeling after myocardial infarction (MI) is a complex process regulated by neurohormonal mechanisms, cytokines, oxidative stress, and genetic factors. This process, accompanied by the loss of viable myocardium, promotes a chain of molecular, cellular and physiological consequences that eventually lead to heart failure (HF). For gene-based therapy of HF to be successful, it should target specific cardiomyocyte processes involved in key molecular mechanisms that can reduce myocardial deterioration and alter HF outcomes. The main molecular targets of failing myocardium include excitation-contraction coupling with calcium cycling pathways, β-adrenergic receptor signaling, apoptosis with extracellular matrix alterations, regeneration capacity and myofilament structure1.
In this paper, we focused on dysregulation of calcium cycling transport, which occurs mostly via the activities of the sarcoplasmic reticulum Ca2+-ATPase (Serca2a)/phospholamban complex. In cardiomyocytes the EF-hand Ca2+ sensor protein S100A1 regulates Serca2a/phospholamban function via diminishing diastolic calcium leak and reinforcing the function of ryanodine receptors during systole. Thus, restoring S100A1 activity in failing hearts poses a promising DNA-based strategy2, 3. The goal of this study was to test the hypothesis that gene-transfected cardiac overexpression of S100A1 prevents adverse postinfarction remodeling and that gene construct mediated by double-stranded (sc) adeno-associated virus (AAV) vector causes better functional and molecular improvement compared to single-stranded (ss) AAV vector genome.
Methods
Animal studies and myocardial infarction model
All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (NIH, No.85–23). Following MI creation by ligation of the proximal left anterior descending artery (LAD), rats were randomized into 5 groups and received intramyocardial injections in the antero-lateral left ventricular (LV) wall with 300 μl of (1) saline control, (Saline, n=10); (2) AAV9/null (empty vector) (Null, n=10); (3) 1.2×1011 genome copies (gc) of ssAAV9.S100A1 (SS; n=10); or (4) 1.2×1011 gc of scAAV9.S100A1 (SC; n=10). In the fifth group (5) rats received a thoracotomy with no MI creation (Sham; n=10). Ten weeks after infarction, all rats were humanely euthanized, and tissue was collected for analysis. Details are available in Appendix 1.
Adeno-associated virus production
Vector production, harvest, purification, and testing were done from cell lysates by the Penn Vector Core (http://www.med.upenn.edu/gtp/vectorcore/). Details are available in Appendix 1.
Cardiac function assessment
Evaluation of heart function was performed by transthoracic echocardiography (TTE). All animals underwent TTE at baseline and 10 weeks after MI. All data were analyzed blindly by a PhD post-doc specialist in echocardiography. Details are available in Appendix 1.
Real-time quantitative PCR detection of S100A1 mRNA and calculation of genome copies per cell & histopathology, tissue collection and calculation of MI size and percentage of fibrosis.
Sections from infarct, border and remote zones along with liver were processed for histological, molecular and genetic analysis. Details are available in Appendix 1.
Cytokine analyte profiling and measurement of reactive oxygen species markers: malondialdehyde and 4-hydroxynonenal
Details are available in Appendix 1.
Tissue collection and calculation of MI size and percentage of fibrosis
Randomized sections from the infarct zone, border zone and remote zone were processed for further histological and molecular analysis to quantify the extent of the MI scarring and to assess the percentage of fibrosis. Details are available in Appendix 1.
Mitochondrial isolation and measurement of mitochondrial respiration
Mitochondria were isolated from different areas of myocardium immediately after euthanasia. Cardiac mitochondrial function was assessed by measuring state 3 and state 4 mitochondrial respiration. Details are available in Appendix 1.
Electron microscopy and assessment of mitochondrial density and fractional volume
Immediately after removal, cardiac samples were placed in Karnovsky’s fixative and left for 24 h at 4°C. Details are available in Appendix 1.
Statistical analysis
Continuous variables were tested for normality using skewness and kurtosis, applied to each data set, prior to further statistical testing. Outcomes are presented as mean ± SD or median (IQR) based on normality. Details are available in Appendix 1.
Results
Characterization of heart function
Global cardiac function demonstrated no major differences between all groups at baseline level. Ten weeks after surgery, ischemic injury in the two control groups (Saline and Null) provoked significant decrease of systolic contractile function as evidenced by impaired EF, stroke volume (SV) index, fractional shortening (FS) and marked LV systolic and diastolic enlargement. In contrast, in both treatment groups (SS and SC), S100A1 improved systolic and diastolic function: EF (44.6% and 54.8%); FS (31% and 38%) when compared to control groups (p<0.05). Analysis of LV dimensions demonstrated decreased LV end-systolic and end-diastolic volumes after S100A1 gene therapy with double stranded vector. It should be noted that improvement in many parameters of cardiac mechanics and LV dimensions were significantly better in the group treated with the scAAV compared to the ssAAV group (p<0.05) (Table 1). Overall, 14 rats were lost (28% mortality), 8 of which died within 24 hours, and 6 of which died after developing heart failure. Based on initial experimental results, we began using antiarrhythmic agents (lidocaine IV, 2 mg/kg) and achieved better survival. In addition, no animals died from heart failure in the treatment groups. We compensated for the losses of control animals and analyzed 10 rats per group in order to achieve statistical significance.
Table 1. Summary of Hemodynamic Data.
at baseline and at 10 weeks post-MI. Gene therapy treatment groups, especially the self-complementary genome group, demonstrated superior left ventricular performance and preserved left ventricular geometry compared to control groups.
CO, cardiac index; SV, stroke volume; EDVI, end-diastolic volume index; ESVI, end-systolic volume index; EF, ejection fraction; FS, fraction shortening;
| Baseline | 10 Weeks Post MI | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Measure | Sham | Saline | AAV Null | ssAAV/S100A1 | scAAV/S100A1 | Sham | Saline | AAV Null | ssAAV/S100A1 | scAAV/S100A1 |
| CO index (ml/min/cm2) | .18 (.16,.21) | .18 (.17,.22) | .15 (.13,.18) | .175 (.14,.2) | .16 (.14,.2) | .215 (.18,.26) | .16 (.15,.19) | .15 (.13,.17) | .18 (.17,.2) | .215 (. 19,.23) |
| SV index (ml/min/cm2) | .47±.07 | .39±.08 | 0.41±.07 | .46±.09 | .44±.09 | .57±.08 | .31±.04★ | .29±.04★ | .45±.07 | .54±.08§□ |
| EDVI (ml/cm2) | .93±.14 | .83±.20 | 0.83±.14 | .73±.17 | .80±.18 | 1.08±. 18 | 1.59±.15★ | 1.49±.12★ | 1.33±.13★ | 1. 14±. 14§□ |
| ESVI (ml/cm2) | .335 (.32,.36) | .31 (.26,.35) | .345 (.32,.4) | .305 (.26,.34) | .27 (.25,.31) | .435 (.38,.48) | .825 (.79,.96)★ | .915 (.88,.99)★ | .785 (.73,.84)★ | .63 (.58,.71)★§□¶ |
| EF% | 69.2±2.6 | 71.5±3.4 | 74.21±4.1 | 72.4±3.8 | 73.7±3.1 | 67.8±3.8 | 32.3±3.1★ | 31.7±3.4★ | 44.6±3.5★§□ | 54.8±5.9★§□¶ |
| FS% | 45±4 | 45±4 | 43±4 | 46±4 | 45±4 | 41±4 | 26±5★ | 27±4★ | 31±3★ | 38±4§□¶ |
| Myocardial thickening (mm) | 1.6±.7 | 1.7±.1 | 1.6±.1 | 1.5±.2 | 1.6±.2 | 1.6±.1 | 1.2±. 1★ | 1.2±. 1★ | 1.4±.01§ | 1.4±. 1§□ |
| Weight (g) | 308 (287,317) | 326 (318,330) | 325 (312,330) | 272 (267,281) | 315 (306,317) | 525 (518,539) | 510.5 (490,520) ★ | 491 (480,502)★ | 518 (513,524)§□ | 480 (471, 492)★¶ |
| Heart rate (bpm) | 381.5 (371,385) | 350 (343,353) | 340.5 (330,345) | 340.5 (339,350) | 362.5 (352,370) | 350 (340,360) | 356.5 (346,365) ★ | 380 (369,380)★§ | 386.5 (380,390)★§ | 362.5 (355,374) ★□¶ |
P <0.05 versus sham
P<0.05 versus saline
P<0.05 versus null
P<0.05 versus ss
Myocardial gene delivery and S100A1 expression
IM delivery of AAV9/S100A1 resulted in robust infection as assessed by qPCR data and demonstrated increased expression of S100A1 in both the anterior and lateral walls of the LV and liver (Figure 1A–C). However, quantification results indicated a significant difference in S100A1 expression level between self-complimentary and single stranded groups in the anterior LV wall (SC group, 5.68±0.44 log10 gc per 100 ng DNA versus SS group, 4.09±0.25 log10 gc per 100 ng DNA; p<0.0001) (Figure 1A). The same trend was seen in the LV lateral wall, which also showed a significant difference between the SC and SS groups (SC group, 5.88±0.63 log10 gc per 100 ng DNA versus SS group, 3.61±0.16 log10 gc per 100 ng DNA; p<0.001) (Figure 1B). There was no significant difference in collateral (liver) expression in both treated groups (Figure 1C, F). Moreover, the number of genome copies per cell in the anterior wall of LV using scAAV was 1.46±0.68 versus 0.22±0.29 using ssAAV (p<0.0001) (Figure 1D). The data followed the same trend in the LV lateral wall (p<0.0001) (Figure 1E).
Figure 1. AAV9/S100A1 Genome Copy Detection.
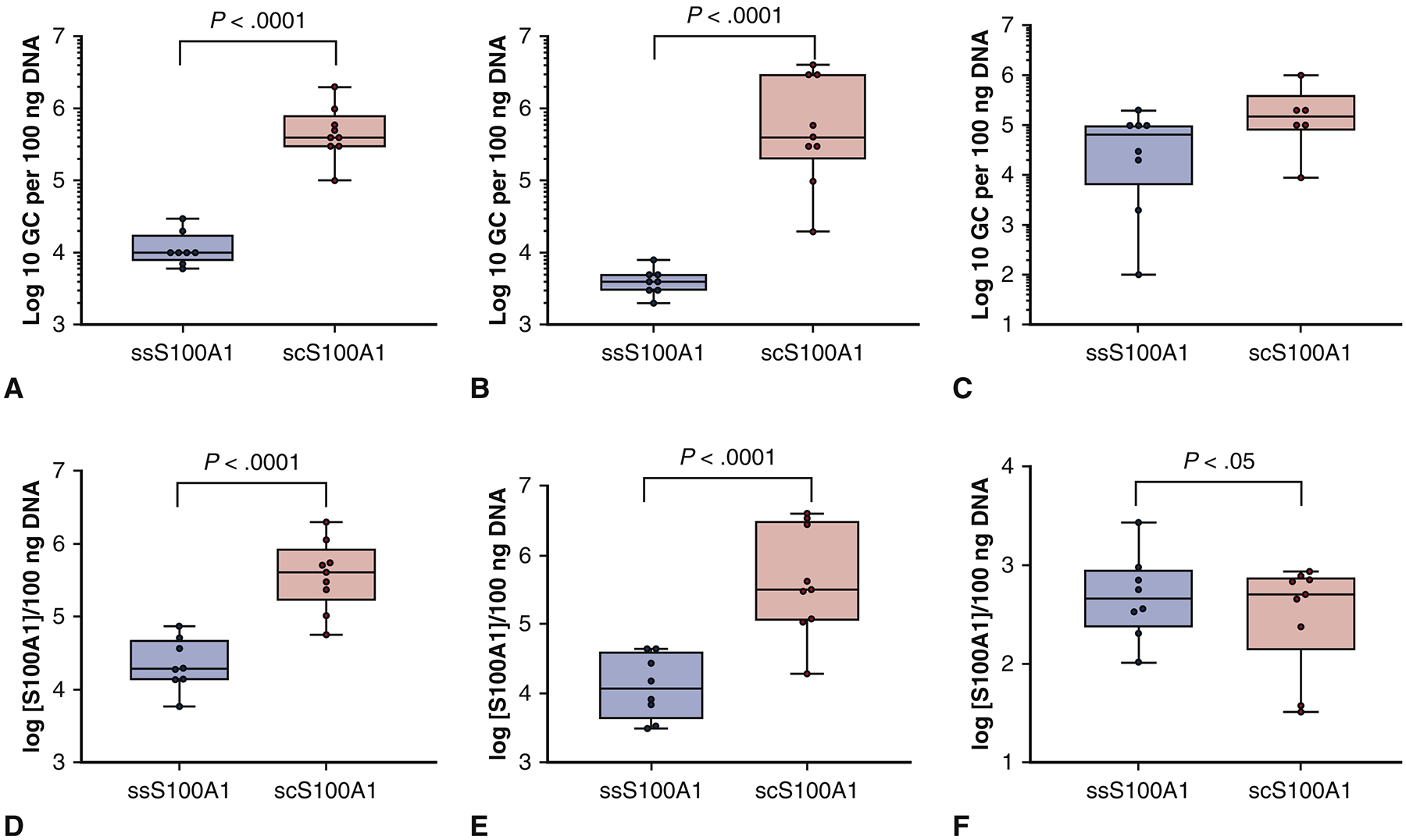
QPCR was performed to determine the S100A1 gene bio-distribution in (A) the left ventricle anterior wall, (B) the left ventricle lateral wall, and (C) the liver as a control (genome copies per 100 ng DNA). QPCR was also performed to assess the genome copies of S100A1 gene bio-distribution per cell in (D) the left ventricle anterior wall, (E) the left ventricle lateral wall, and (F) in the liver.
Myocardial infarct size and ventricular fibrosis assessment
Morphometric analyses of the histopathology of the heart sections revealed that in all groups besides Sham, the area of infarct was located in the anterior wall of left ventricle (Figure 2A). MI size was significantly reduced in the SC group (29.2±3.26%) as compared to the Saline (39.6±1.79%) and Null (39.6±3.64%) groups (p<0.0001) (Figure 2B). The percentage of ventricular fibrosis was also lower in the transfected groups: SS (21.6±4.78%) and SC (15.9±3.16%) compared to the Saline and Null groups (p<0.0001) (Figure 2C).
Figure 2. Myocardial infarct size and ventricular fibrosis assessment.
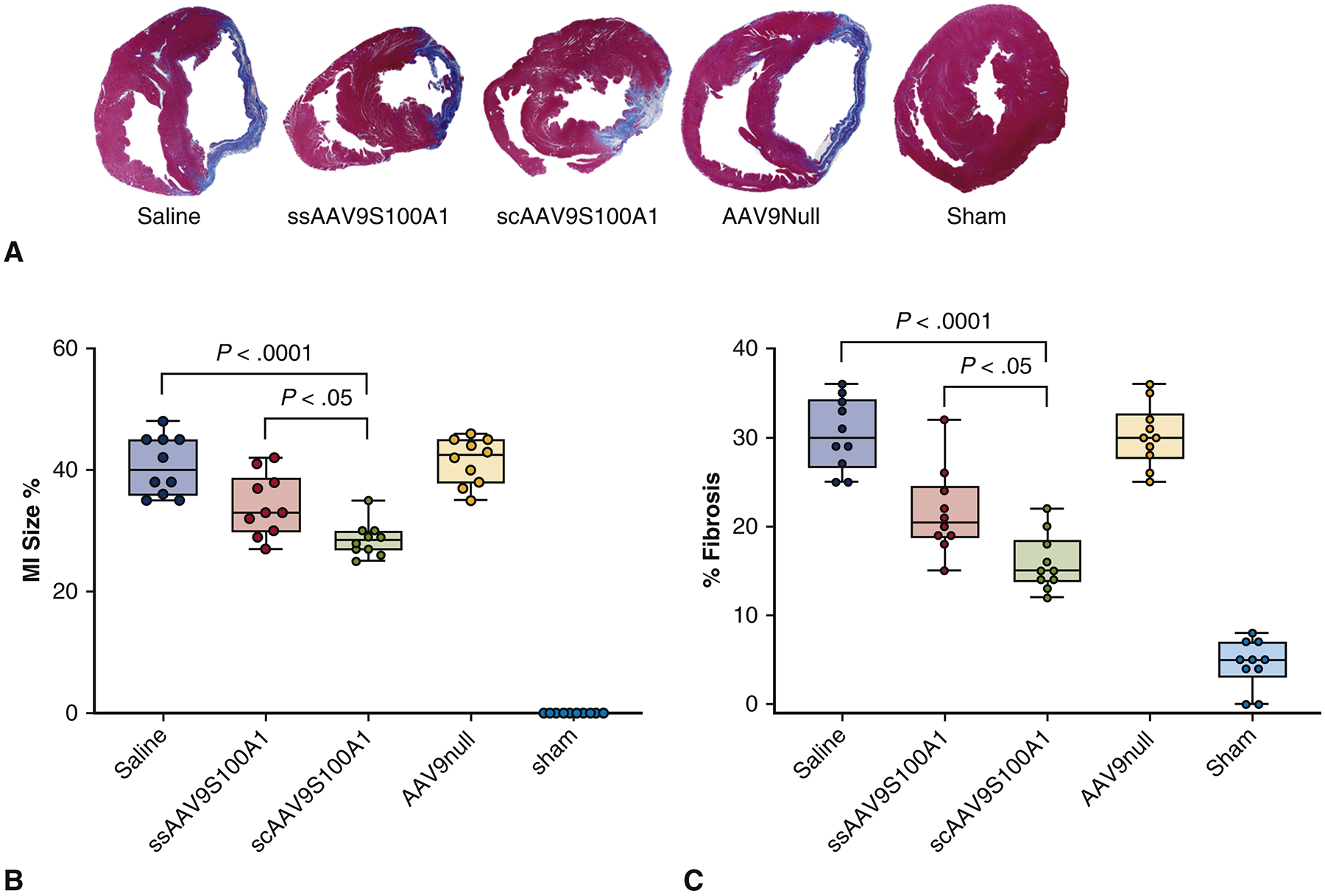
(A) Masson’s trichrome staining of midventricular cardiac sections 10 weeks after infarction in five different groups. Scar tissue stains blue and muscle fibers stain red. (B) Quantitative assessment of MI size and (C) quantitative assessment of fibrosis were performed and compared across the treatment groups.
Cytokine Expression Analysis
Distributions of serum levels of the main inflammatory cytokines and anti-inflammatory cytokines differed significantly among the groups. Overall, among the 23 cytokine analytes investigated, most showed significant differences at 10-week follow-up between the Saline and SS and SC groups (p<0.05) (Figure 3A). Mean concentrations of cytokines EP0, GLP-1, and IL-10 were significantly elevated in rats after administration of ssAAV9/S100A1 as compared to scAAV9/S100A1 (p<0.05). Significant changes in two pro-inflammatory and five anti-inflammatory cytokines were observed between the Null group and the SC and SS groups together (p<0.05) (Figure 3B) (Table 2).
Figure 3. Changes in pro-inflammatory and anti-inflammatory cytokines expression.
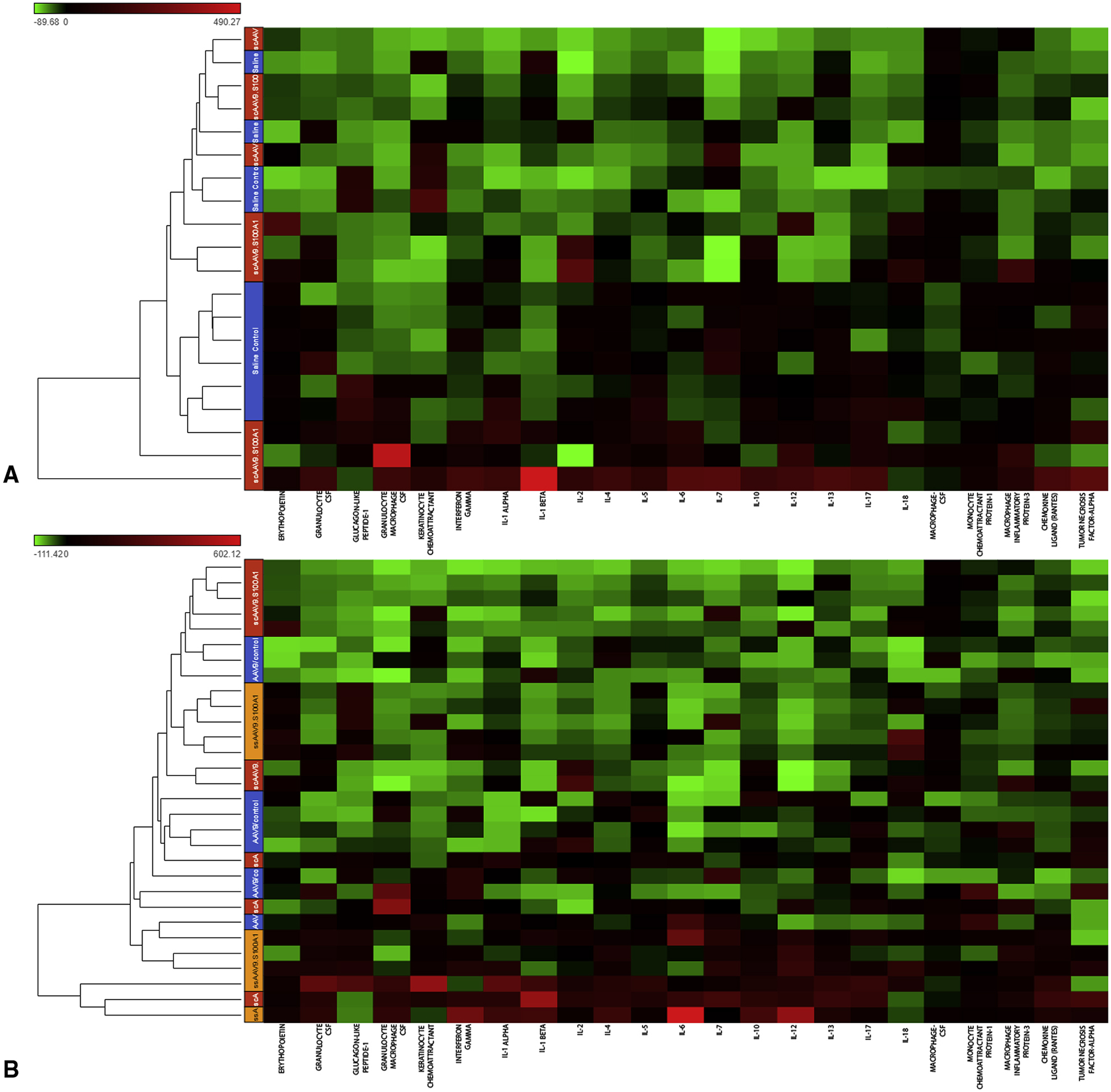
Hierarchical cluster dendrograms (heat maps) were made to compare cytokine levels in the serum of the groups. The lower dendrogram shows the clustering of cytokines. The colors on the left side represent cytokines expression, scaled to a mean of zero and unit variance. The expression analysis revealed that cytokines remain elevated in the empty virus group and that administration of the self-complementary vector mediated S100A1 gene resulted in less inflammation than with administration of the single-stranded vector.
CSF, colony stimulating factor; IL, interleukin
Table 2. Summary of changes in pro-inflammatory and anti-inflammatory cytokines expression in different treatment groups at 10 weeks post-MI.
CSF, colony stimulating factor; IL, interleukin
| Cytokines (pg/ml) | |||||||
|---|---|---|---|---|---|---|---|
| Groups | Erythopoietin | Granulocyte CSF | Glucagon-like Peptide-1 | Granulocyte-Macrophage-CSF | Keratinocyte Chemoattractant | Interferon-gamma | IL-1 alpha |
| Sham | 584.15 (205.75, 673.11) |
17.62±14.29 | 933.25 (576.8, 3932.6) |
107.15 (56.32, 151.1) |
97.36 (51.1, 169.98) |
70.715 (41.32, 117.44) |
107.93±81.25 |
| Saline | 736.2 (653.5, 912.9)★ |
46.29±18.59★ | 3426.05 (2983.4, 4562.3)★ |
619.85 (402.3, 780.3)★ |
315.3 (156.4, 567.4)★ |
299.7 (177.3, 388.9)★ |
192.30±94.64 |
| AAV9/null | 551.15 (407.9, 578.5)§ |
29.80±30.35 | 1698 (657, 3425) |
334.4 (275.8, 432)★ |
153.65 (86.7, 234.5)§ |
260.25 (67.9, 326.5)★ |
211.60±165.46 |
| SS | 954.71 (828.13, 1072.52)★□ |
34.78±27.38 | 4607.65 (3456, 4923.6)★□ |
212.7 (152.9, 288.2)§ |
122.2 (45.25, 189.2)§ |
159.11 (102.5, 257.92)★§ |
237.18±161.98 |
| 481.58 (383.82, 856.66)¶ |
21.31±14.98 | 609.45 (570.7, 938.6)§¶ |
103.185 (47.39, 434.4)§□ |
50.12 (21.07, 132.5)§□ |
77.37 (52.79, 165.9)§□ |
134.76±115.68 | |
| IL-1 beta | IL-2 | IL-4 | IL-5 | IL-6 | IL-7 | IL-10 | IL-12 | |
|---|---|---|---|---|---|---|---|---|
| Sham | 118.9 (85.75,142.2) |
333.83±183.14 | 64.525 (25.7, 84.9) |
200.76 (124.34, 217.8) |
47.63 (35.3, 62.2) |
272.225 (322.9, 174.83) |
605.95 (330.3, 801.2) |
79.91±58.76 |
| Saline | 382.8 (238.4, 603.4)★ |
340.89±156.16 | 158.85 (143.5, 187.6)★ |
287.9 (244.3, 382.1) |
122.2 (67.7, 188.3)★ |
274.8 (208.4, 377.4) |
789 (566.5, 923.4) |
184.76±83.70 |
| AAV9/null | 361.1 (219.4, 473.8)★ |
415.69±293.55 | 113.95 (67.5, 145.4)★ |
250 (200.3, 443.5) |
47.25 (43.7, 106.9) |
195.3 (78.5, 342.2) |
570.8 (438.9, 946.8) |
156.91±104.57 |
| SS | 172.14 (103.69, 474.8) |
399.06±207.01 | 82.855 (33.71, 156.8)§ |
253.85 (201.9, 288.2) |
91.95 (34.46, 135.7) |
321.615 (83.54, 583.9) |
658.235 (395.2, 1023) |
272.55±268.29 |
| SC | 160.935 (68.06, 244.67)§ |
441.57±460.77 | 48.445 (34.33, 89.77)§□ |
152.415 (108.76, 275.9) |
62.5 (38.3, 159.8) |
120.965 (9.9, 259.8)§ |
344.45 (294, 731.24) §¶ |
139.86±142.76 |
| IL-13 | IL-17 | IL-18 | Macrophage-CSF | Monocyte Chemoattractant Protein-1 | Macrophage Inflammatory Protein-3 | Chemokine ligand (RANTES) | Tumor Necrosis Factor-alpha | |
| Sham | 85.25 (77.3, 117.4) |
61.02 (29.9, 107.47) |
2997.87±1712.14 | 237.35±58.93 | 725.12±179.54 | 54.945 (40.77, 72.9) |
329.6±39.7 | 256.9±43.0 |
| Saline | 172 (102.3, 236.7)★ |
143.1 (78.4, 203.4)★ |
4024.00±2126.39 | 219.35±97.12 | 1641.84± 875.7★ | 160.3 (129.7, 255.6)★ |
340.9±52.9 | 272.5±40.1 |
| AAV9/null | 102.4 (83.4, 143.5) |
93.5 (43.6, 145.3) |
3571.90±2552.83 | 209.14±130.96 | 966.77±874.71 | 96.7 (56.8, 187.5)★ |
279.7±60.3 | 113.4±33.7 |
| SS | 100.8 (49.7, 164.3) |
70.7 (55.58, 129.9) |
6916.78±4362.81★ | 270.07±69.21 | 851.22±261.92§ | 80.625 (44.49, 113.57)§ |
294.1±43.3 | 215.7±50.8 |
| SC | 63.9 (29.9, 106.9)§□ |
56.575 (32.94, 134.5)§ |
3820.35±2266.95 | 284.64±38.78 | 845.27±177.46§ | 59.985 (34.13, 146.23)§ |
240.6±31.8 | 128.7±22.4 |
P <0.05 versus sham
P<0.05 versus saline
P<0.05 versus null
P<0.05 versus ss
Reactive oxygen species: lipid peroxidation analysis
Creation of MI with saline administration was associated with an increased production of malodialdehyde (MDA) (2.66±1.49 pmole.mg−1 protein) and 4-hydroxynonenal (HNE) (2.30±0.85 pmole.mg−1 protein) compared to Sham (MDA 0.64±0.54; HNE 0.72±0.57 pmole.mg−1 protein; p<0.05) (Figure 4A–B). Thus, saline injection did not stop adverse remodeling and resulted in significant enhancement of lipid peroxidation levels. Injection of scAAV9/S100A1 significantly decreased the degree of lipid peroxidation as compared to SS and Null groups. Although these values were higher than in the Sham group, the differences between SC and Sham were not statistically significant. An increase in MDA and HNE concentrations after ssAAV9/S100A1 administration was also observed (2.54±1.55 and 2.36±1.11 pmole.mg−1 protein), but these concentrations were not significantly lower than in Null group (2.55±1.23 and 2.61±1.45 pmole.mg−1 protein). From these observations it can be concluded that administration of scAAV9/S100A1 decreased the level of lipid peroxidation, which most likely plays an important role in reversal of the post-MI remodeling cascade. Levels of HNE in the Saline and Null groups were significantly higher when compared with treatment groups.
Figure 4. Changes in lipid peroxidation markers.
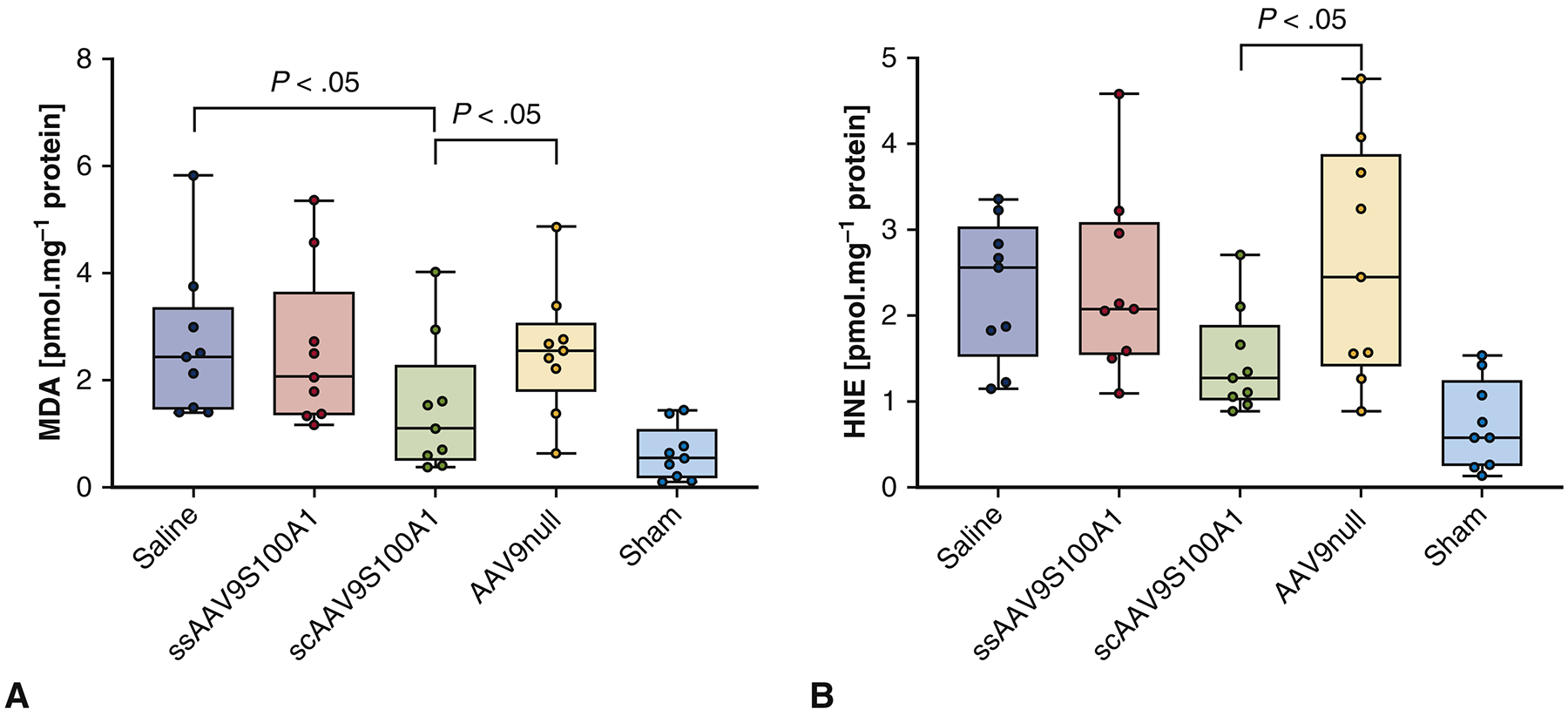
(A) Malodialdehyde (MDA) and (B) 4-hydroxynonenal (HNE). MI creation with administration of saline or empty virus was associated with an increased production of both MDA and HNE. Injection of self-complimentary AAV9/S100A1 decreased the degree of lipid peroxidation compared to other groups,..
Changes in mitochondrial function and structure
Mitochondrial respiration data demonstrates the compromised function in an infarcted heart without treatment. Administration of Saline and AAV/Null was associated with a decrease in mitochondrial efficiency, specifically a reduction in state 3 respiration to 40.26±19.48 and 38.08±11.42 nmolO2/ml/min, respectively, and state 4 respiration to 16.80±5.76 and 18.38±7.27 nmolO2/ml/min, respectively, compared to Sham (70.01±15.15 (state 3) and 32.77±6.23 (state 4) nmolO2/ml/min) (Figure 5A–B). The SC group demonstrated similar levels of mitochondrial function (61.6±8.95 (state 3) and 28.9±5.91 (state 4) nmolO2/ml/min) compared to Sham. While the administration of SS vector also demonstrated this trend but to a lesser extent. Mitochondrial fractional volume increased in the SC group compared to the Saline and Null groups (p<0.05) (Figure 5C). Mitochondrial fractional volume trended similarly in the SS group as in the SC group but to a smaller extent. The same trend was observed with mitochondrial density (p<0.05) (Figure 5D). Electron microscopic assessment of mitochondria in different groups demonstrated that in the Sham group, mitochondria were well organized and compacted between contractile filaments. In both control untreated groups, there was significant mitochondrial fragmentation and disintegration of ultrastructure with areas of autophagolysis. In gene-treated groups, mitochondrial ultrastructure was better organized and appeared normal. However, some areas of swelling and vacuolization, as well as nuclear and cytoplasmic degeneration persisted (Figure 6).
Figure 5. Mitochondrial respiration, mitochondrial density, and fractional volume data across the groups.
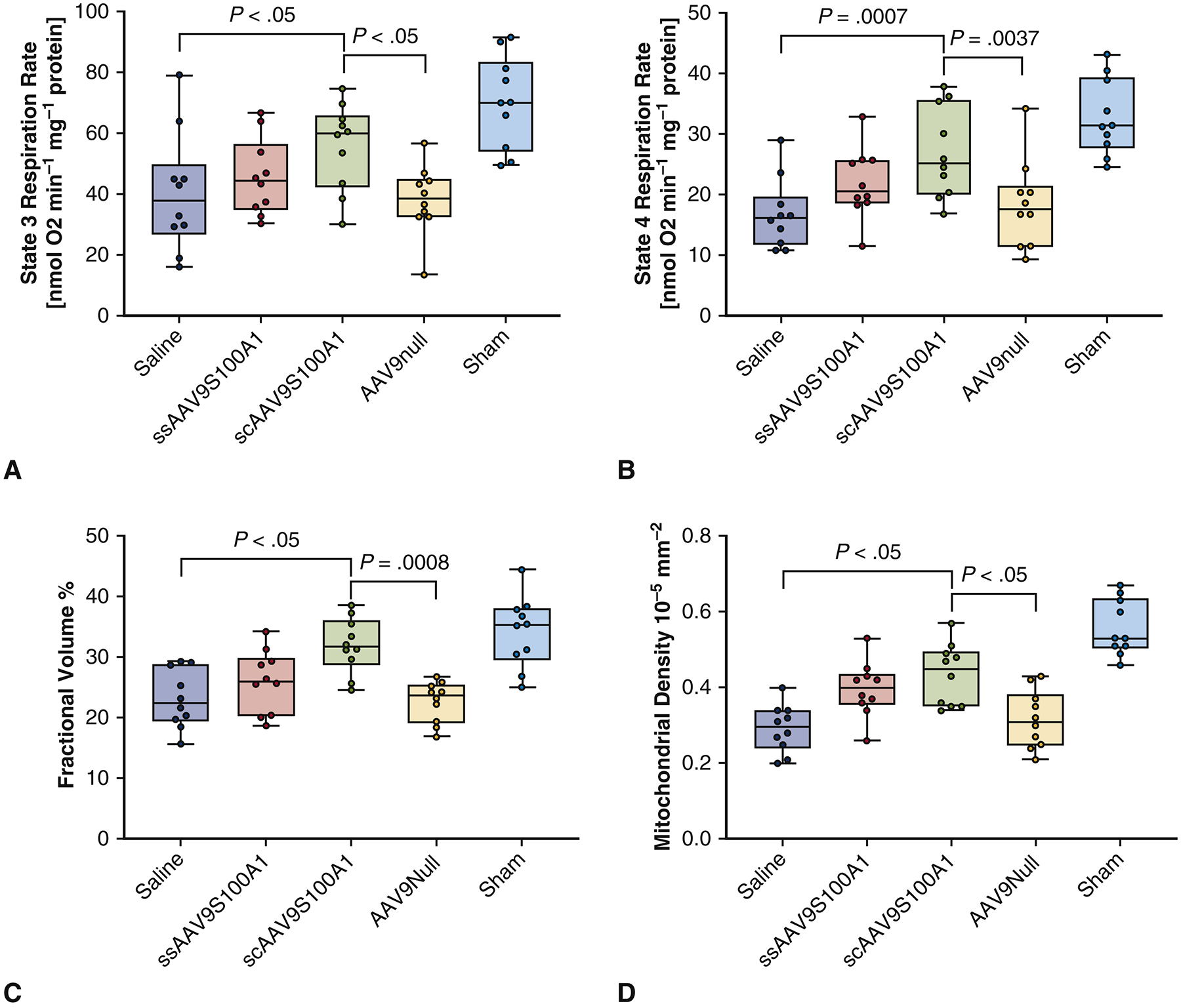
(A, B) Mitochondrial respiration state 3 represents the active phase of mitochondrial function with conversion of adenosine diphosphate into adenosine triphosphate. State 4 mitochondrial respiration is linked to the resting state of mitochondria. Administration of saline and empty virus after myocardial infarction was associated with a reduction in states 3 and 4. Using self-complimentary vector improved states 3 and 4 of respiration. (C). Mitochondrial fractional volume increased in the treated groups compared to control. (D) Mitochondrial density increased in the treated groups compared to control.
Figure 6. Representative Transmission Electron Microscopy of Mitochondrial Ultrastructure.
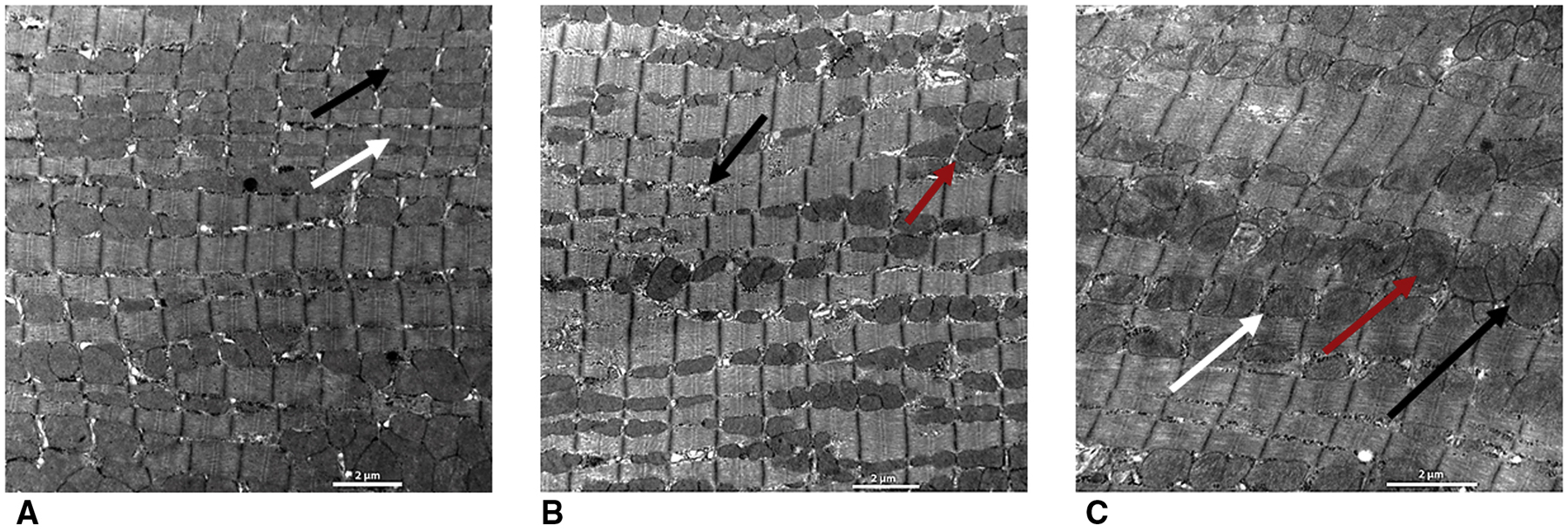
(A) Sham group. Mitochondria are well organized (black arrow) and compacted between contractile filaments (white arrow). (B) Control untreated group. There is significant mitochondrial fragmentation and disintegration of mitochondrial ultrastructure (red arrow) with areas of autophagolysis and decreases in average mitochondrial density and volume as evidenced by areas lacking mitochondria (black arrow). (C) Gene-treated groups. Mitochondrial ultrastructure is better organized and appears normal (black arrow), but areas of swelling and vacuolization (white arrow), as well as nuclear and cytoplasmic degeneration persist (red arrow). Overexpression of double stranded adeno-associated virus mediated S100A1 attenuated mitochondrial injury as compared to the single-stranded group.
Discussion
Successful gene therapy requires three key steps: identifying the molecular defect, developing a correcting gene, and determining the best approach to introduce and deliver the gene of interest into the appropriate host cells4,5. One of the major hurdles in gene transfer is the development and selection of adequate vectors. Among vector systems, AAVs are used in more than 100 of on-going clinical trials worldwide. Recent clinical trials using AAVs demonstrated sustained therapeutic effects in patients with Leber’s congenital amaurosis, hemophilia B, spinal muscular atrophy, alpha-1 antitrypsin and lipoprotein lipase deficiency6. Inherent cardiotropism adds to the profile of AAVs as a gene vehicle of choice7. AAV belongs to the family Parvoviridae and packages a linear single-stranded DNA (ssDNA) genome of 4.7 kb. However, there are limitations to using a ssDNA virus. Prior to expression, the ssDNA genome has to be converted into double-stranded, self-complimentary DNA (scDNA). It is accepted that second-strand DNA synthesis is a rate-limiting step for AAV transduction, based on the existing of amount of ssDNA after AAV infection and the deficiency of host enzymes in the nucleus. Other drawbacks of the ssDNA genome include long transport to the nucleus, uncoating from the capsid, and a period of vector genome instability after conversion. Consequentially, these limitations lead to a significant reduction of gene expression8,9. The results of cardiac gene therapy clinical trials with single-stranded genome have reported a low level of transgene efficacy10. The inability to deliver genome copies into a substantial percentage of cardiac cells decreases the chances of achieving therapeutic efficacy. In theory the use of scAAV vector can ameliorate these problems, and recent hematological clinical trials using self-complimentary AAV confirmed this hypothesis11.
A number of animal studies have demonstrated that calcium-regulated proteins like Serca2A and S100A1 are depleted as HF progresses2,3,12,13. Restoration of these protein levels normalizes the decreased gene expression and main cell processes in failing myocardium2,3. However, the detailed intracellular and extracellular functions of S100A1 have not been explored yet.
The scAAV9.S100A1 vector restored gene expression significantly better than the ssAAV9.S100A1 vector. In our model of ischemic heart failure, qPCR analysis showed that the SC vector resulted in detection of a significantly higher number of genome copies per 100 ng DNA and genome copies per cell of myocardium as compared with SS vector. Overexpression of scAAV9.S100A1 resulted in an enhancement of contractility, as evidenced by improvement in cardiac mechanics, and the substantial arrest of pathological remodeling, as revealed by significantly decreased LV dimensions. This study demonstrated that the scAAV9.S100A1 exhibits high levels of transduction and subsequent increases in genome copies within transfected cardiomyocytes and establishes a significant relationship between genome-copy transfer and the level of improvement of heart contractility.
Beyond improving hemodynamics, delivery of scAAV.S100A1 gene significantly decreased production of ROS, especially lipid peroxides that are closely associated with extensive myocardial damage after ischemia14,15. In humans, increased MDA and HNE concentrations have been reported in ischemic myocardial tissue and serum as compared to control14. This process triggers cytokine release. In this study, we documented the role of oxidative stress and pro-inflammatory cytokine response following acute myocardial infarction and explored the inflammatory mechanisms of cardiac injury.
Mitochondria are the main source of cellular ROS. During myocardial ischemia, transport of mitochondria is changed and causes elevation of nuclear ROS. Furthermore, mitochondrial uncoupling proteins, which should serve to reduce the production of ROS, stopped working effectively16. A primary function of mitochondria is to generate adenosine triphosphate (ATP)-energy through oxidative phosphorylation. Mitochondrial respiration state 3 represents the active phase of oxygen capacity with conversion of adenosine diphosphate (ADP) into ATP. State 4 respiration is linked to the resting state of mitochondria17. The infarcted heart is highly dependent on mitochondrial oxidative energy and mitochondrial defects can lead to the pathological remodeling18. A number of reports have shown a direct correlation between mitochondrial phosphorylation oxygen capacity and mitochondrial structure19,20. Previous evidence indicates that S100A1 interacts with the α- and β-chains of the mitochondrial ATPase21. Our data demonstrates that viral-based S100A1 augments mitochondrial function in both active and resting phases and prevents disorders of mitochondrial organization and that this effect is more pronounced with the self-complimentary genome.
Our next aim was to reveal the inflammatory reaction after gene transfer. Persistent cardiac inflammation plays a pathogenic role in maladaptive cardiac remodeling. The opposing actions of pro- and anti-inflammatory cytokines are important for the maintenance of homeostasis, and cytokine imbalance is implicated in the development of HF22. Not only does expression of these inflammatory cytokines increase in failing myocardium, but it also remains elevated in circulation for extended periods of time23. MI triggers a protective repair response featuring three overlapping phases: inflammatory, proliferative, and maturation. The duration of each phase varies in different myocardial zones after infarction24. Initially, during the acute phase of MI the production of cytokines induces a cardioprotective signaling cascade25,26. At the end of the proliferative phase, and the beginning of the maturation, cytokines begin to decrease. However, in cases of large transmural MI, cytokine expression levels may remain elevated for a prolonged period of time or may even induce a second wave of cytokine activation27,28. In the present study, plasma level expression of inflammatory and anti-inflammatory cytokines appeared to be changed in direct proportion with post-MI remodeling, depending on the use of gene therapy. Future studies should clarify the balance between adaptive and maladaptive effects of these mediators after MI as well as the specific mechanism of gene therapy that influences inflammation and cardiac remodeling.
Gene therapy is a complex process and many factors should be considered in its application. The main factors limiting the introduction of cardiac gene therapy in clinical practice are: (i) lack of methods to extend vector residence time and transient therapeutic gene expression in myocytes, (ii) lack of gene delivery techniques that limit collateral expression in other organs, (iii) limited number of clinical trials with the correct selection of patients and clinical end-points. We believe that cardiac gene delivery could be clinically applicable for patients with LV dysfunction who would normally undergo cardiac operations for coronary revascularization or other reasons, patients in whom percutaneous coronary interventions is contraindicated, and patients with dilated cardiomyopathy, particularly those with an X-linked or autosomal recessive inheritance pattern.
Conclusion
Our results demonstrate that restoring S100A1 through the gene transfer of scAAV9.S100A1 consistently promotes more robust transduction efficiency than ssAAV9.S100A1 and improves cardiac function in a failing heart (Figure 7, Video 1). Moreover, scAAV9.S100A1 reverses pathological post-MI remodeling by means of reducing both ROS production and pro- and anti-inflammatory cytokine concentrations.
Figure 7.
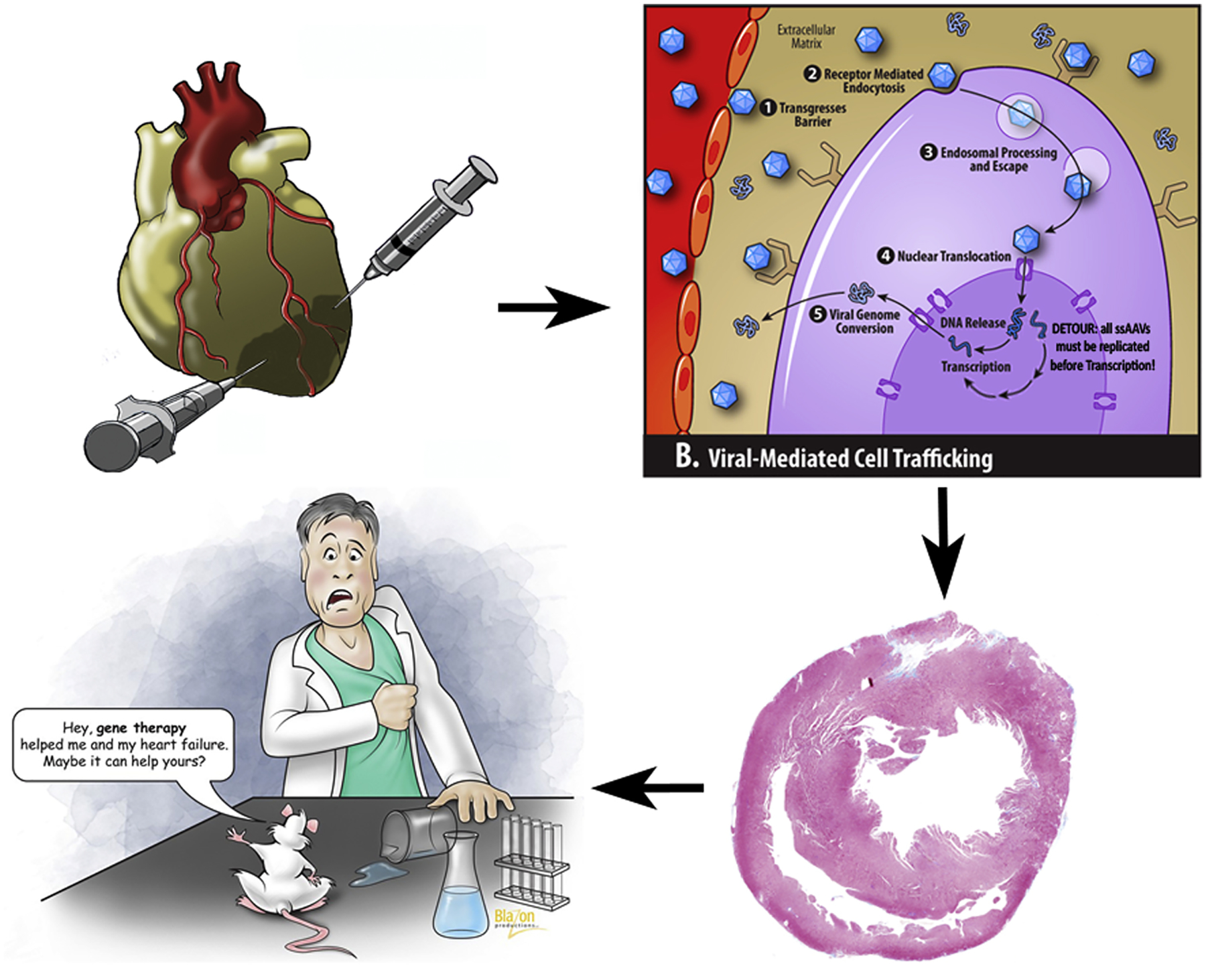
After intramyocardial injection of adeno-associated virus-mediated S100A1, the key trafficking steps include binding to myocyte cell surface receptors, endocytosis and endosomal processing, nuclear translocation and viral genome conversion from a single-stranded to a double-stranded DNA. Using the double-stranded vector bypasses the last step and provides much more effective transfection, leading to better heart function.
Limitations
This study was performed using a rodent model at the time of acute ischemia, and the results may not be reproducible in humans. We did not incorporate a group with ischemia/reperfusion model and chronic MI, which would be more similar to the clinical scenario. Inter-observer variability for determining the echo analysis data and dose response study were not performed as well.
Supplementary Material
Video 1. Creation of myocardial infarction via ligation of the left anterior descending artery & left heart catheterization in a rat.
Acknowledgements
We would like to acknowledge the Gene Therapy Resource Program, Richard D Williams, Andrew Parker Kendle, Inna M Sokolova, Anna V Ivanina, Nury M Steuerwald, Judy Parsons, Daisy M Ridings, Kim Michalko, and Karen. Fay.
Sources of Funding
This study was supported by NIH grant 5-R01 HL083078-08.
Abbreviations and Acronyms
- S100A1
the EF-hand Ca2+ sensor protein S100A1
- AAV
adeno-associated virus
- ss
single stranded DNA genome
- sc
self-complimentary(double-stranded) DNA genome
- ROS
reactive oxygen species
- MDA
malondialdehyde, lipid peroxide
- 4HNE
4-hydroxynonenal, lipid peroxide
- RT-qPCR
real-time quantitative polymerase chain reaction
- IL-1β, TNFα, IFNγ, IL-6
main pro-inflammatory cytokines
- IL-2, IL-4, IL-7, IL-10, EP0, GLP-1
main anti-inflammatory cytokines
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest
Disclosures
None.
References
- 1.Katz MG, Fargnoli AS, Williams RD, Bridges CR. Cell and Gene Therapies for Cardiovascular Disease: In Templeton NS, eds. Gene and Cell Therapy. 4th ed. New York: CRC Press; 2015:34861–901. [Google Scholar]
- 2.Most P, T Pleger S, Völkers M, Heidt B, Boerries M, Weichenhan D, et al. Cardiac Adenoviral S100a1 Gene Delivery Rescues Failing Myocardium. J Clin Invest. 2004;114:1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pleger Sven T, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, et al. Stable Myocardial-Specific AAV6-S100A1 Gene Therapy Results in Chronic Functional Heart Failure Rescue. Circulation. 2007;115:2506–2515. [DOI] [PubMed] [Google Scholar]
- 4.Sellke FW. Gene therapy in cardiac surgery: is there a role? J. Thorac. Cardiovasc. Surg 2003;125:994–997. [DOI] [PubMed] [Google Scholar]
- 5.Katz MG, Fargnoli AS, Kendle AP, Hajjar RJ, Bridges CR. Gene Therapy in Cardiac Surgery: Clinical Trials, Challenges, and Perspectives. Ann. Thorac. Surg 2016;101:2407–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vandamme C, Adjali O, Mingozzi F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial After Trial. Hum. Gene Ther 2017;11:1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katz MG, Fargnoli AS, Williams RD, Bridges CR. Gene therapy delivery systems for enhancing viral and nonviral vectors for cardiac diseases: current concepts and future applications. Hum. Gene Ther 2013;24:914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarty DM. Self-complementary AAV Vectors; Advances and Applications. Mol. Ther 2008;16:1648–1656. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z, Ma HI, Li J, Sun L, Zhang J, Xiao X. Rapid and highly efficient transduction by - double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003;10:2105–2011. [DOI] [PubMed] [Google Scholar]
- 10.Hulot J-S, Ishikawa K, Hajjar RJ. Gene therapy for the treatment of heart failure: promise postponed. Eur. Heart J 2016;37:1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med 2014; 371:1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katz MG, Fargnoli AS, Williams RD, Steuerwald NM, Isidro A, Ivanina AV, et al. Safety and efficacy of high-dose adeno-associated virus 9 encoding sarcoplasmic reticulum Ca2+ adenosine triphosphatase delivered by molecular cardiac surgery with recirculating delivery in ovine ischemic cardiomyopathy. J. Thorac. and Cardiovasc. Surg 2014;148:1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katz MG, Brandon-Warner E, Fargnoli AS, Williams RD, Kendle AP, Hajjar RJ, et al. Mitigation of myocardial fibrosis by molecular cardiac surgery-mediated gene overexpression. J. Thorac. Cardiovasc. Surg 2016;151:1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dominguez-Rodriguez A, Abreu-Gonzalez P, de la Rosa A, Vargas M, Ferrer J, Garcia Mn. Role of endogenous interleukin-10 production and lipid peroxidation in patients with acute myocardial infarction treated with primary percutaneous transluminal coronary angioplasty interleukin-10 and primary angioplasty. Int. J. Cardiol 2005;99:77–81. [DOI] [PubMed] [Google Scholar]
- 15.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res 2008;81:457–464. [DOI] [PubMed] [Google Scholar]
- 16.Lee H-L, Chen C-L, Yeh ST, Zweier JL, Chen Y-R. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol 2012;302:H1410–H1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brand MD, Nicholls DG. Assessing Mitochondrial Dysfunction In Cells. Biochem. J 2011;435:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Knowlton AA. Mitochondrial dynamics in heart failure. Congest. Heart Fail 2011;17:257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One. 2008;3:e3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem 2005;280:26185–26192. [DOI] [PubMed] [Google Scholar]
- 21.Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, et al. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol. Cell. Biol 2007;27:4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aukrust P, Gullestad L, Ueland T, Damås JK, Yndestad A. Inflammatory and anti-inflammatory cytokines in chronic heart failure: potential therapeutic implications. Ann. Med 2005;37:74–85. [DOI] [PubMed] [Google Scholar]
- 23.Müller F, Yndestad A, Holm AM, Simonsen S, Frøland SS, Gullestad L, et al. Enhanced expression of inflammatory cytokines and activation markers in T-cells from patients with chronic heart failure. Cardiovasc. Res 2003;60:141–146. [DOI] [PubMed] [Google Scholar]
- 24.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol 2014;11:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lecour S, James RW. When are pro-inflammatory cytokines SAFE in heart failure? Eur. Heart J 2011;32:680–685. [DOI] [PubMed] [Google Scholar]
- 26.Feldman AM, Combes A, Wagner D, Kadakomi T, Kubota T, Li YY, et al. The role of tumor necrosis factor in the pathophysiology of heart failure. J. Am. Coll. Cardiol 2000;35:537–544. [DOI] [PubMed] [Google Scholar]
- 27.Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S. Cytokine gene expression after myocardial infarction in rat hearts. Circulation. 1998;98:149–156. [DOI] [PubMed] [Google Scholar]
- 28.Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the studies of left ventricular dysfunction (SOLVD). J. Am. Coll. Cardiol 1996;27:1201–1206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Creation of myocardial infarction via ligation of the left anterior descending artery & left heart catheterization in a rat.