

Abstract
Manufacturing of cell culture-derived virus particles for vaccination and gene therapy is a rapidly growing field in the biopharmaceutical industry. The process involves a number of complex tasks and unit operations ranging from selection of host cells and virus strains for the cultivation in bioreactors to the purification and formulation of the final product. For the majority of cell culture-derived products, efforts focused on maximization of bioreactor yields, whereas design and optimization of downstream processes were often neglected. Owing to this biased focus, downstream procedures today often constitute a bottleneck in various manufacturing processes and account for the majority of the overall production costs. For efficient production methods, particularly in sight of constantly increasing economic pressure within human healthcare systems, highly productive downstream schemes have to be developed. Here, we discuss unit operations and downstream trains to purify virus particles for use as vaccines and vectors for gene therapy.
Keywords: chromatography, downstream processing, filtration, gene therapy vector, membrane adsorbers, monoliths, vaccine, virus
Virus particles are currently used for medical [1–5], analytical and scientific applications [6–9], and as bioinsecticides [10,11]. Recently, medical applications are gaining an increasing interest owing to the growing markets for viral vaccines (Table 1) and the potential broad usage of viral gene therapy vectors. Vaccines, administered to prevent or treat viral diseases, are mainly based on attenuated or killed viruses, membrane fractions derived from purified virus particles, or recombinant viral proteins expressed in various hosts. Examples of successful attenuated or killed virus vaccines are influenza, measles, mumps, rubella, rotavirus, yellow fever and varicella [3,4,12,13]. Gene therapy involves the transfer of genetic information to cells or tissues of individuals to achieve a therapeutic effect [14]. Therefore, required genes are largely delivered by viral vector systems based, for example, on the herpes simplex virus, adenovirus, adeno-associated virus (AAV), retrovirus (e.g., lentivirus) and Vaccinia virus [1,15]. Currently, numerous clinical gene therapy trials are conducted to investigate the treatment of diseases such as cancer, cystic fibrosis, Alzheimer’s, Parkinson’s, hemophilia and HIV/AIDS [5,16,17].
Table 1. Human vaccines produced with animal and human cell technology†.
| Therapeutic area | Vaccine type | Vaccine (virus) strain | Cells | Company |
First approval |
Trade name | Notes | |
|---|---|---|---|---|---|---|---|---|
| FDA | EMA | |||||||
| Hepatitis A | Inactivated | GBM | MRC-5 diploid cell line | Sanofi-Pasteur | 1996 | Avaxim® | ||
| CR 326F | Merck | 1996 | 1997 | Vaqta® | ||||
| HM 175 | GSK | 1995 | 1993 | Havrix™ | Also part of Ambirix, Twinrix and Hepatyrix combo | |||
| |
|
RG-SB |
|
Berna Biotech |
|
1997 |
Epaxal® |
|
| Human papillomavirus |
Virus-like particle, adjuvanted |
Types 16 and 18 |
Insect cell line derived from trichoplusia |
GSK |
2009 |
2007 |
Cervarix® |
|
| Influenza | Inactivated | A H1N1 | Vero cells (African green monkey kidney cells) | Baxter | 2009 | Celvapan® | ||
| A H5N1 | 2009 | Pandemic influenza vaccine Baxter | ||||||
| |
|
A H1N1, A H3N2 and B |
Madine–Darby canine kidney cells |
Novartis |
|
2007 |
Optaflu® |
|
| Japanese encephalitis |
Inactivated |
S14-14-12 |
Vero cells (African green monkey kidney cells) |
Intercell/Novartis |
2009 |
2009 |
Ixiaro® |
|
| Measles | Live-attenuated | Ender Edmonston or Schwarz | Primary chicken embryo fibroblasts | Sanofi-Pasteur | 1986 | Rouvax® | Also in combination with measles and mumps | |
| Merck | 2006 | MMR VaxPro | Also in combination with measles, mumps, and varicella | |||||
| GSK | Attenuvax® | Also in combination with rubella, mumps, and varicella | ||||||
| |
|
|
|
Crucell |
|
|
Trivitaren |
|
| Mumps | Live-attenuated | Jeryl Lynn | Primary chicken embryo fibroblasts | Sanofi-Pasteur | 1985 | MMR vaccine | Also in combination with measles and rubella | |
| Merck | 2006 | Mumpsvax | Also in combination with rubella, mumps, and varicella | |||||
| GSK | Priorix® | Also in combination with measles and rubella | ||||||
| |
|
|
|
Crucell |
|
|
Trivitaren |
|
| Poliomyelitis | Inactivated | Type 1 Mahoney, type 2 MEF-2, and type 3 Sauket | Primary monkey kidney cells | Sanofi-Pasteur | 1966 | DTPolio | ||
| MRC-5 diploid cell line | Sanofi-Pasteur | 1987 | 1987 | Poliovax | Never marketed | |||
| Live-attenuated | Types 1, 2 and 3 | GSK | OPV oral Polio vaccine | |||||
| Live-attenuated | Types 1, 2 and 3 | Vero cells (African green monkey kidney cells) | Sanofi-Pasteur | 1988 | OPV oral Polio vaccine | |||
| Inactivated | Type 1 Mahoney, type 2 MEF-2, and type 3 Sauket | Sanofi-Pasteur | 1982 | Imovax Polio | ||||
| Sanofi-Pasteur | 1990 | Ipol | ||||||
| |
|
|
|
GSK |
2002 |
|
Poliorix™ |
Also part of Infanrix hexa, Infanrix penta, and pediatrix |
| Prostate cancer |
|
|
Autologous cells |
Dendreon |
|
|
Provenge® |
|
| Rabies | Inactivated | PM | MRC-5 diploid cell line | Sanofi-Pasteur | 1980 | Imovax® Rabies | ||
| PM | Vero cells (African green monkey kidney cells) | Sanofi-Pasteur | 1985 | Verorab | ||||
| |
|
PM-1503-3M |
|
Novartis |
|
|
Rabavert® |
|
| Rotavirus | Live-attenuated | G1, G2, G3, G4 and G6 capsid proteins | Vero cells (African green monkey kidney cells) | Merck | 2006 | 2006 | Rotateq® | |
| |
|
89-12 G1P[8] RIX 4414 |
|
GSK |
2008 |
2006 |
Rotarix® |
|
| Rubella | Live-attenuated | WI RA 27/3 | MRC-5 diploid cell line | Sanofi-Pasteur | 1988 | Rudivax | Also in combination with measles and mumps | |
| Merck | 2006 | MMR VaxPro | Also in combination with measles, mumps and varicella | |||||
| 1969 | Meruvax II | Also in combination with rubella, mumps and varicella | ||||||
| GSK | Priorix® | Also in combination with measles and mumps | ||||||
| |
|
|
|
Crucell |
|
|
Trivitaren |
|
| Varicella/chicken pox | Live-attenuated | Oka/Merck | MRC-5 diploid cell line | Merck | 1996 | 2001 | Varivax® | Also in combination with measles, mumps and rubella |
| |
|
Oka |
|
GSK |
|
|
Varilix® |
|
| Zoster Herpesvirus | Live-attenuated | Oka/Merck | MRC-5 diploid cell line | Merck | 2006 | 2006 | Zostavax® | |
The broad spectrum of these applications and the current expansions of medical markets underline the ongoing efforts to improve production procedures for viral vaccines and gene therapy vectors. One striking example is the development of cell culture-derived influenza vaccines. While conventional production processes rely on egg-based systems, optimized cell culture systems are currently being established to cope with sudden demands for pandemic vaccines and increasing supply of seasonal vaccines. With advances in upstream procedures to increase yields and harvest volumes for influenza vaccines, as well as for other vaccines and viral vectors, downstream processing (DSP) is becoming an important factor in the race for higher overall productivity and decreased cost of goods. The general aim of DSP is the recovery and purification of biological products from process- and product-related impurities. Process-related impurities might originate from cell culture reagents and additives (e.g., antibiotics, bovine serum albumin and Benzonase® [Merck KGaA, Darmstadt, Germany]), from the purification process (e.g., extractables and leachables in chromatography), or from the cell substrate (e.g., host cell protein, nucleic acids, proteoglycans and glycosaminoglycans). Examples for virus particle-related impurities include free envelope proteins, virus aggregates or empty capsids, and virus particles that contain nucleic acid sequences other than the intended genome [18–20]. Naturally, the requirements on product purity and product safety depend on the particular application. Vaccines and viral vectors need to meet the stringent guidelines of regulatory authorities such as the US FDA and EMA.
Viruses are complex bioparticles with varying size, shape, composition and surface structure. The virus surface defines their individual physicochemical characteristics including number and distribution of charges, hydrophobic residues and post-translational modifications (i.e., glycans) of surface proteins. Purification of virus particles based on these unique characteristics and removal of contaminants according to the regulatory guidelines can only be achieved by a combination of different unit operations.
This article provides an overview of individual unit operations currently used for DSP of viral vaccines and gene therapy vectors. This includes methods such as precipitation, flocculation, extraction, centrifugation, microfiltration, ultrafiltration, bead-based and membrane-based chromatography, and the use of monoliths. In addition, it addresses issues concerning the use of continuous chromatography methods, that is, simulated moving bed chromatography, and the utilization of kits for small- and medium-scale purifications and concentrations of virus particles and vectors for gene therapy. Finally, examples for complete purification trains are presented for DSP of virus particles for therapeutic applications and vaccine manufacturing.
Precipitation & flocculation
Owing to the complexity of large bioparticles and the necessity to maintain the specific immunogenicity and infectivity of virus particles and viral vectors, precipitation and flocculation are rarely used in DSP. Even for purification of recombinant proteins, use of these methods is considered problematic owing to potential losses in biological activity. However, the latest advances in upstream processes for recombinant proteins in terms of high cell density cultures and drastically improved product levels triggered the re-evaluation of these methods, particularly for purification of monoclonal antibodies [21,22] or viral proteins [23].
Small-scale precipitation of virus particles was demonstrated for AAV vectors [19], cowpea chlorotic mottle virus [24], turkey coronavirus [25], mycovirus OMIV [26] and rotaviruses [27]. Turkey coronavirus was precipitated with ammonium sulfate but with low recoveries compared with ultracentrifugation (60% sucrose cushion) [25]. Other virus particles were precipitated with varying combinations of polyethylene glycol (PEG) and sodium chloride. In particular, PEG precipitation of bovine rotavirus particles resulted in an approximately tenfold better yield than pelleting by high-speed centrifugation based on results determined by the tissue culture infective dose (TCID50) assay [27]. This could be owing to increased damage of virus particles during high-speed centrifugation [27] or an irreversible aggregation of virus particles leading to overall reduced TCID50 measurements. Furthermore, precipitation of AAV vectors via PEG can lead to an enhanced purity compared with the classically conducted cesium chloride (CsCl) gradient ultracentrifugation [19].
An alternative to the precipitation of the target virus is the removal of contaminating host cell DNA or protein from crude culture harvests. A recent study focusing on the precipitation of host cell DNA from a clarified cultivation broth of influenza virus particles has been conducted by Kröber et al.[28]. They investigated cationic reagents with respect to their ability to selectively precipitate host cell DNA and observed successful DNA reduction applying the cationic polymers protamine sulfate and polyethyleneimine [28]. However, as discussed by the authors, the acidic isoelectric point of influenza virus particles (Table 2) may have resulted in co-precipitation of virus particles with cationic polymers [28], and therefore product losses. In the past effective purification of encephalomyocarditis virus from cellular components [29], and the removal of cellular DNA from poliovirus produced in continuous cell lines [30] as well as from preparations of inactivated vaccines against tick-borne encephalitis [31] by protamine sulfate precipitations was demonstrated.
Table 2. Overview of viruses relevant for vaccination and gene therapy.
| Virus | Family (genus) | Size (nm) | Isoelectric points† | Virion morphology | Envelope | Genome | Ref. |
|---|---|---|---|---|---|---|---|
| Adenovirus |
Adenoviridae |
70–100 |
Human adenovirus 5: 4.5 |
Icosahedral |
No |
dsDNA |
[34,163] |
| Adeno-associated virus |
Parvoviridae |
18–26 |
AAV-4: 2.6 |
Icosahedral |
No |
ssDNA |
[163–165] |
| Baculovirus |
Baculoviridae |
D: 30–60 L: 250–300 |
AcMNPV: 5.4 |
Rod shaped |
Yes |
dsDNA |
[166–168] |
| Bromovirus |
Bromoviridae |
26 |
NA |
Icosahedral |
No |
(+)ssRNA |
[169] |
| Flavivirus |
Flaviviridae |
40–65 |
NA |
Spherical |
Yes |
(+)ssRNA |
[170,171] |
| Hepatitis A virus |
Picornaviridae |
30 |
Hepatitis A virus: 2.8 |
Icosahedral |
No |
(+)ssRNA |
[170,172,173] |
| Hepatitis B virus |
Hepadnaviridae |
a: 42–47 and b: D: 20–22 L: Variable |
NA |
a: Spherical, b: Filamentous |
Yes |
dsDNA and RNA |
[170,174] |
| Hepatitis C virus |
Flaviviridae |
30–60 |
NA |
Spherical |
Yes |
(+)ssRNA |
[175] |
| Herpes simplex virus |
Herpesviridae |
180–200 |
NA |
Spherical; tegument |
Yes |
dsDNA |
[170,176] |
| Influenza virus |
Orthomyxoviridae |
80–120 |
(H2N2) A2/Singapore/57: 5.0 (H3N1) A/Pol/L/71: 6.5–6.8 (H3N1) A/Pol/20/72: 6.5–6.8 (H3N1) A/Enfl/42/72: 6.5–6.8 (H3N1) A/Phil/2/82: 6.5–6.8 (H3N1) A/Pol/79/85: 6.5–6.8 (H1N1) A1/Lenigrad: 5.0 (H3N2) A3/Lenigrad): 5.0 (H1N1) A/PR/8: 5.3 |
Pleomorphic, spherical |
Yes |
(-)ssRNA |
[177–183] |
| Japanese encephalitis |
Flaviviridae |
50–60 |
NA |
Spherical |
Yes |
(+)ssRNA |
[184,185] |
| Lentivirus |
Retroviridae |
80–130 |
NA |
Spherical |
Yes |
(+)ssRNA |
[163,186] |
| Measles virus |
Paramyxoviridae (Morbillivirus) |
100–300 |
NA |
Pleomorphic |
Yes |
(-)ssRNA |
[170,187] |
| Mumps virus |
Paramyxoviridae (Rubulavirus) |
a: 150–350 b: L: up to 600 |
NA |
a: Spherical, b: Filamentous, pleomorphic |
Yes |
(-)ssRNA |
[170,188] |
| Murine leukemia virus |
Retroviridae |
80–120 |
NA |
Spherical |
Yes |
(+)ssRNA |
[189] |
| Papillomavirus |
Papillomaviridae |
52–55 |
Papillomavirus: 5.0 |
Icosahedral |
No |
dsDNA |
[170,190,191] |
| Poliovirus |
Picornaviridae |
28–30 |
PV-1: 7.4 and 4.0 PV1 Brunender: 7.4 and 3.8 PV-1 Brunhilde: 7.1 and 4.5 PV-1 LSc2ab: 6.75 and 4.5 PV-1 Mahoney: 8.3 |
Icosahedral |
No |
(+)ssRNA |
[170,173,192–196] |
| Rabies virus |
Rhabdoviridae |
D: 75–80 L: 100–300 |
NA |
Bullet shaped |
Yes |
(-)ssRNA |
[170,197] |
| Rotavirus |
Reoviridae |
70–85 |
Simian rotavirus A/SA11: 8.0 |
Quasi-spherical, icosahedral |
No |
dsRNA |
[170,195] |
| Rubella virus |
Togaviridae (Rubivirus) |
50–75 |
NA |
Quasi-spherical |
Yes |
(+)ssRNA |
[198,199] |
| Semliki forest virus |
Togaviridae (Alphavirus) |
65–70 |
NA |
Spherical |
Yes |
(+)ssRNA |
[200,201] |
| Vaccinia virus |
Poxviridae |
250–270–360‡ |
Chaumier: 5.0 Connaught: 4.9 Lister: 5.1 Lister: 3.9 Lister (egg): 3.7 Lister (rabbit): 3.0 WR: 4.8 |
Brick shaped |
Yes |
dsDNA |
[46,202–207] |
| Varicella-Zoster virus | Herpesviridae | 180–200 | NA | Spherical, tegument | Yes | dsDNA | [170] |
†Data on isoelectric points of different viruses taken from Michen and Graule [208].
‡This is a brick-shaped viral particle therefore the dimension are shown in terms of axis x–y–z.
D: Diameter; L: Length; NA: Not available.
Considering the continuous efforts to increase harvest volumes and cell concentration in vaccine and gene therapy vector production, precipitation represents an interesting approach for reduction of the high loads of host cell DNA and proteins. However, process robustness (i.e., the specificity of the applied precipitants) needs to be improved, and nontoxic compounds for precipitation processes need to be identified for economic applications in pharmaceutical production.
Extraction
As for precipitation, the system’s complexity, which is mainly dependent on the structural and physicochemical diversity of virus particles, constrains this technique to become widely used.
Further drawbacks of applying extraction methods for the purification of virus particles are the possible losses in viral infectivity, immunogenicity or transduction efficiency, high levels of co-extracted contaminants and the high cost for recycling of the utilized organic solvents [32,33]. Nevertheless, extractions via aqueous two-phase systems with PEG, dextran, salt or polyvenyl alcohol are used for the separation of virus particles from cultivation medium. Examples for these applications are the purification or concentration of adenovirus [34], bovine leukemia virus [35], feline leukemia virus [36], and human and simian immunodeficiency virus [37].
Centrifugation
The number of publications [24,38–46] and patents [301–303] describing the purification or concentration of virus particles by centrifugation methods demonstrates that these procedures are extensively used at industrial- and small-scale levels for viral vectors and vaccine production processes. For industrial scales only continuous flow centrifugation methods can be considered as they allow handling of larger volumes. However, these methods require high investment costs and suffer in some cases from losses of infectivity [33,38,47], leading to an increased usage of ultrafiltration techniques particularly outside of laboratory scales. A clear advantage of centrifigation methods is their potential to separate some assembled viral vectors from their empty capsids, which is commonly challenging with other separation techniques [38]. Another advantage is in the DSP of viruses with frequently changing strains (e.g., influenza) as these centrifugation methods are robust in relation to viral strain differences, which resulted in a rediscovery of this technology for a number of processes.
The most common centrifugation method for the purification of macromolecules and virus particles is density gradient centrifugation using CsCl, iodixanol, or sucrose gradients. These can be classified into two categories: rate-zonal separation and isopycnic separation. The latter is based on the buoyant density of the particle and the rate zonal separation on the particle size and mass. Examples of virus particles that were purified by gradient centrifugation techniques are feline leukemia virus [48], Gibbon ape lymphoma virus [49], hepatitis B virus [50], influenza virus [51], Japanese encephalitis virus [52], mammalian type C virus [53], mouse mammary tumor virus [54], mouse oncorna virus [55], mumps virus [56], murine leukemia virus [57], tick-borne encephalitis virus [58], turkey coronavirus [25], rabies virus [59] and Vaccinia virus [60]. An alternative centrifugation method is the differential centrifugation where pelleting (Epstein–Barr virus [61] and hepatitis A virus [62]) or simple sedimentation onto a sucrose cushion (Moloney murine sarcoma virus [63]) is done once or repeatedly at different centrifugation speeds.
Current examples for the concentration or purification of virus particles via centrifugation are: adenovirus and AAV [38,39], bromovirus [24], hepatitis C virus [40], herpes-like virus [41], retrovirus [42–44], rotavirus [45] and Vaccinia virus [46].
Microfiltration
Microfiltration is commonly used for clarification in biotechnological production processes as an alternative to centrifugation to remove microcarriers, producer cells, cell debris and organelles. Frequently, membranes with pore sizes in the range of 0.45–1 µm are applied, although this technique involves the risk of viral losses [44,64,65]. Recoveries depend on the initial membrane pore size, type of membrane (material), quality of the crude stock, virus (i.e., size), virus subtype and its aggregation behavior, applied buffer conditions, filtration rate, nature and level of protein concentration in the filtrate, and extent of pore obstruction as demonstrated for retroviral vectors [65]. Virus losses are mainly attributed to mechanical disruption, particularly for active virus particles (i.e., gene therapy vectors), exclusion of virus particles [64] and membrane entrapment as well as unspecific virus adsorption. Losses due to pore obstruction can be reduced by applying membrane cascades with decreasing pore sizes or filter capsules with dual membranes to minimize membrane fouling [50,66,67] and applying larger membrane areas (i.e., frequent changes of the filtration units or usage of larger units) to limit the volume of supernatant per membrane surface.
Ultrafiltration
Ultrafiltration is one of the preferred methods for virus removal in biological production methods [68–70] and for virus concentration and buffer exchanges in vaccine and viral gene transfer vector manufacturing processes (Table 3). Here, virus particles are usually enriched or maintained in the retentate while water and small-molecular-weight molecules are removed with the permeate. Although ultrafiltration has a low resolution, the size difference of virus particles (Table 2) compared with soluble biopolymers (i.e., nucleic acids and proteins) allows a relatively efficient and economic separation of the virus.
Table 3. Examples of ultrafiltration methods used for the purification and concentration of virus particles.
| Virus | Type of filtration | Membrane | Cutoff | Recovery (%) | Ref. |
|---|---|---|---|---|---|
| Adenoviridae (Adenovirus) | Tangential flow filtration (GE; Quix Stand, hollow-fiber cartridge) | Polysulfone; hollow-fiber | 750 kDa | 42 | [147] |
| 500 kDa | 71 | ||||
| 300 kDa | 96 | ||||
| Tangential flow filtration (Millipore; Pellicon II filter module) | Polyethersulfone; flat sheet | 500 kDa | Not provided | [209] | |
| Tangential flow filtration (Millipore; Ultra-15 centrifugal filter unit) | Regenerated cellulose; flat sheet | 100 kDa | 57 | [210] | |
| Tangential flow filtration (Millipore; Pellicon II filter module) | Polyethersulfone; flat sheet | 1000 kDa | Not provided | [82] | |
| 300 kDa | Not provided | ||||
| Tangential flow filtration (individual setup) | Polysulfone (GE-Healthcare) | 300 kDa | 83 | [211] | |
| Polysulfone (Spectrum Laboratories) | 400 kDa | 100 | |||
| |
|
|
0.05 µm |
75 |
|
|
Bromoviridae (Bromovirus) |
Tangential flow filtration (Millipore; Centricon Plus-20 centrifugal filter device) |
Polyethersulfone; flat sheet |
300 kDa |
Not provided |
[212] |
|
Herpesviridae (Anatid herpesvirus 1) |
Tangential flow filtration (not provided) |
Not provided |
50 kDa |
56† |
[213] |
|
Nimaviridae (white spot syndrome virus) |
Tangential flow filtration (Millipore; Ultra-15 centrifugal filter unit) |
Regenerated cellulose; flat sheet |
100 kDa |
Not provided |
[214] |
| Orthomyxoviridae (Influenza virus A) | Tangential flow filtration (GE-Healthcare; hollow-fiber cartridge) | Polysulfone; hollow fiber | 750 kDa | 106 | [67] |
| 0.1 µm | 54 | ||||
| 0.45 µm | 0 | ||||
| Tangential flow filtration (Sartorius; Sartocon cassette) | Polyethersulfone; flat sheet | 100 kDa | 95 | [215] | |
| Tangential flow filtration (Sartorius; Sartocon Slice 200 cassette) | Polyethersulfone; flat sheet | 100 kDa | 100 | [216] | |
| 300 kDa | ˜100 | ||||
| 0.1 µm | ˜80–10‡ | ||||
| 0.2 µm | ˜0–5 | ||||
| |
|
|
0.45 µm |
0 |
|
| Parvoviridae (Aedes Aegypti densonucleosis virus) | Tangential flow filtration (Sartorius; Sartocon Slice 200 cassette) | Polyethersulfone; flat sheet | 100 kDa | 100 | [72,78] |
| Tangential flow filtration (Sartorius; Sartocon Slice 200 cassette) | 300 kDa | <30‡ | |||
| High-performance tangential flow filtration (Sartorius; Sartocon Slice 200 cassette) | 100 kDa | <50 | |||
| High-performance tangential flow filtration (Sartorius; Sartocon Slice 200 cassette) | 300 kDa | <30 | |||
| Tangential flow filtration (Sartorius; Sartocon Slice 200 cassette) | 30 kDa | >95 | [72,163,217] | ||
| 50 kDa | >95 | ||||
| 100 kDa | >95 | ||||
| |
|
|
300 kDa |
<30‡ |
|
| Parvoviridae (minute virus of mice) | Tangential flow filtration (Sartorius; Sartocon Slice 200 cassette) | Polyethersulfone; flat sheet | 50 kDa | >90 | [218] |
| 100 kDa | >90 | ||||
| 300 kDa | <10‡ | ||||
| High performance tangential flow filtration (Sartorius; Sartocon Slice 200 cassette) | 50 kDa | >90 | |||
| 100 kDa | >90 | ||||
| |
|
|
300 kDa |
<10‡ |
|
| Retroviridae (vector; murine leukemia viral vector) | Tangential flow filtration (GE; Advanced MidJet system, hollow-fiber cartridge) | Polysulfone; hollow fiber | 500 kDa | 101 (6°C) | [152] |
| |
|
|
|
70 (21°C) |
|
|
Retroviridae (vector; tgLS[-f]HyTK retroviral vector) |
Tangential flow filtration (A.G. Technology, hollow-fiber cartridge) |
Polysulfone; hollow fiber |
500 kDa |
54–86 |
[219] |
|
Retroviridae (vector; GINa, GlNaSvAd and LASN retroviral vectors) |
Tangential flow filtration (Millipore; Pellicon filter module) |
Regenerated cellulose, flat sheet |
300 kDa |
91–96 |
[220] |
|
Retroviridae (vector; HIV type 1-derived vector and Moloney murine leukemia viral vector derived from LP-BM5 MoMLV) |
Tangential flow filtration (individual setup) |
Regenerated cellulose, hollow fiber |
35 and 75 nm§ |
˜50 |
[221] |
| Retroviridae (vector; HIV type 1 viral vector) | Tangential flow filtration (GE-Healthcare; Amersham Quickstand) | Polysulfone; hollow fiber | 750 kDa | Not provided¶ | [222] |
†Overall yield with subsequent sucrose gradient ultracentrifugation.
‡Internal fouling.
§Both pore sizes 35 and 75 nm were used, and with both pore sizes a comparable viral recovery was obtained.
¶In combination with ultracentrifugation.
MoMLV: Moloney murine leukemia virus.
The key parameters for an ultrafiltration process are the transmembrane pressure (TMP), feed (retentate) and permeate flux as well as the nominal molecular weight limit, also sometimes referred to as molecular weight cutoff (MWCO). Pore sizes defining the MWCO are normally distributed about the mean pore size, which varies depending on the production method for the membrane, and therefore also between the manufacturers. In order to minimize product losses via the permeate, the MWCO has to be significantly smaller than the virus particle. However, if the MWCO selected is too small, permeate fluxes are reduced,, which leads to longer filtration times, increased TMP and decreases the possibility of reducing the amount of contaminating proteins and nucleic acids. Furthermore, membrane fouling, which decreases the permeate flux, can often be greater for larger pore size membranes than for smaller ones. Here, small virus particles but also cell and virus debris are able to enter the pores where they are eventually trapped in constricted channels. Asymmetric ultrafiltration membranes with the finer filtering surface facing the feed suspension should be applied where possible to reduce internal fouling.
Important for concentration of active virus particles is a gentle processing for which TMP and retentate flux have to be optimized for low wall shear rates during filtration.
The filtration performance and in particular the permeate flux is heavily affected by the type or components of the cultivation medium, thereby underlining the importance of a tight interaction between the up- and down-stream development. Nowadays, serum containing growth media are rarely used for production processes. Serum-free media often contain defined proteins such as bovine albumin and insulin, but also protein hydrolysates. Protein-free media only include defined polypeptides and amino acids. The higher the concentration of proteins, target virus, and nucleic acids and the larger the size of contaminating DNA fragments in the retentate, the higher the sample viscosity, leading to a continuously decreasing permeate flux at constant TMP. Additionally, high retentate protein concentrations result in an increased degree of concentration polarization, which also reduces the permeate flux [71]. Finally, ionic strength, type of ions and the pH in the suspending buffer influence the sieving coefficient of proteins [72–75] and probably also macromolecular biomolecules and virus particles.
Ultrafiltration can be carried out by a wide variety of filtration devices (Table 3). For small scales (up to 10 ml) centrifugal filtration devices are well suited, and for small to medium volumes (10–300 ml) stirred cell tanks are ideal. Larger volumes are usually concentrated by tangential flow filtration. There are three types of membranes available: tubular membranes, flat sheets (cassettes) and hollow-fiber membranes from which the latter two are commonly used in the biopharmaceutical industry. Generally, flat-sheet and tubular tangential-flow membrane modules have higher mass transfer coefficients at low cross-flow rates compared with hollow-fiber modules. The latter have wide feed flow paths, providing laminar flow with low shear rates [76], which are crucial processing life vaccines and gene therapy vectors. High shear rates can significantly reduce their efficacy and overall yield. A general advantage of hollow-fiber membrane modules compared with flat sheets is the high membrane surface-to-module volume ratio. However, individual modules of flat-sheet tangential-flow membrane modules are easier to clean and replacement of defective membrane elements is uncomplicated. Overall, the type of membranes eventually used for a specific process depends largely on the viscosity, solid content and volume of the feed as well as the product stability (shear-sensitivity). Thus, for the concentration or diafiltration of active virus particles, hollow-fiber membranes are superior to flat-sheet membranes.
A potential improvement of the classical tangential-flow filtration is the high-performance tangential-flow filtration, which achieves a superior separation factor [71,77]. In high-performance tangential-flow filtration, part of the permeate is circulated co-current to the feed on the permeate side of the module, resulting in a more constant TMP throughout the module due to an axial pressure drop along the permeate flow channel [78]. This allows, by careful optimization of the operating conditions, a more stable transmembrane flux over time and in some cases leads to an improved selectivity for the purification of virus particles (Table 3).
In summary, the main advantages of ultrafiltration compared with other methods are their high-throughput and (for the concentration of active virus particles) the gentle processing at optimal operating conditions [43,47] that results in improved efficacies for purification of viral vectors for gene therapy. Another advantage of utilizing size differences of virus particles to other soluble components in the culture medium for their purification and concentration is its broad applicability among different types of viruses, virus subtypes or different recombinant forms of a virus. This opens the path for the establishment of generic platform technologies.
Considering a complete purification train for the production of vaccines or gene therapy vectors (Figure 1), current improvements of the dynamic binding capacities in chromatography media might facilitate the removal of the initial concentration step within the downstream process. However, later concentration steps and buffer exchanges for final formulations will continue to be done via ultrafiltration.
Figure 1. Generic flowchart of current downstream processes for gene therapy vectors and inactivated virus vaccines.
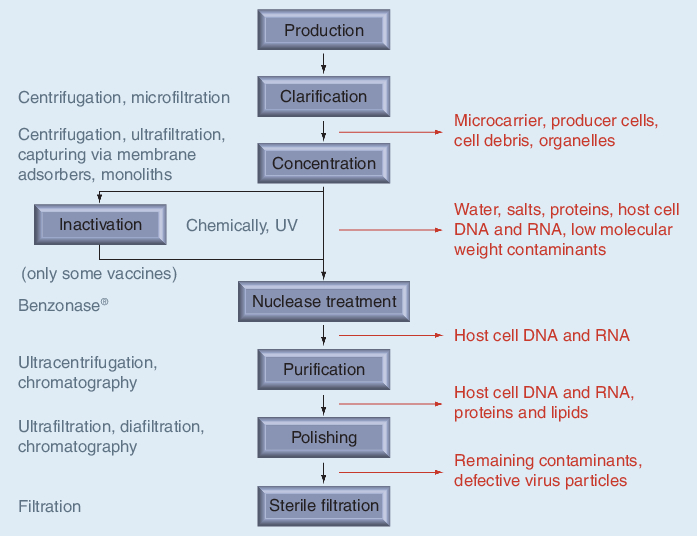
Chromatography using beads, membranes & monoliths
Chromatography is the most popular methodology for large-scale purification of vaccines and viral gene therapy vectors. Chromatographic separation is based on differences in the interaction of target virus and other components to the applied stationary phase. The specific characteristics of the virus particles exploited are described by the physicochemical properties of the outer particle surface, electrostatic properties of the whole particle, or their large hydrodynamic volume compared with other soluble process components. There are four main points of chromatography that have to be considered for a successful purification [79]:
• Physical structure of the stationary phase including the morphology (i.e., beads, membranes and monoliths), pore size, porosity, mechanical and chemical properties (e.g., resistance to pressure and surface charge of the mobile phase), and specificity for resin particle size and particle distribution;
• Surface chemistry of the stationary phase and the composition of the mobile phase;
• Mode of operation such as adsorption or partitioning, type of elution;
• Column/housing and system hardware.
For each application a balance between these individual aspects must be found to obtain an efficient chromatographic procedure. In the following sections, however, we will focus on the first two points.
Physical structure of the stationary phase
Chromatographic purification of virus particles is mainly achieved by use of three different types of stationary phases: packed beds, membrane adsorbers and monoliths.
In the case of packed beds the matrix consists of resins (beads), which are filled into a column. For bioseparations polymer and inorganic resin materials are commonly used. Polymer-based beads either consist of natural polymers (agarose, agarose-dextran composites, cellulose and dextran) or synthetic polymers (polyacrylamide, polystyrene divinylbenzene, derivatives of polyacrylamide derivatives and polymethacrylate) [80]. Examples for inorganic matrices used for the purification of virus particles are hydroxyapatite [81–83], silica [84] and controlled-pore glass [85,86]. Beads can be spherical and nonspherical shaped and are available as porous, nonporous and solid-core resins. Pore sizes of conventional porous beads range from 10 to 100 nm [79], although specific particles with pore diameters of up to 400 nm are available [33]. Virus particles vary generally from 30 nm to greater than 300 nm (Table 2). Hence, the material transport of virus particles is limited by pore-diffusion or pore-exclusion effects. Even small virus particles, which are able to enter large pores, diffuse through the intra-particular channels two- to 100-fold slower than proteins [87]. In fact, the majority of chromatography resins currently available are optimized for purification of proteins rather than virus particles, which primarily adsorb to the outer bead surface [88]. Furthermore, the inter-particle porosity for packed beds can reach values of maximally 40%, whereas for stationary phases such as monoliths this value can be up to 90% or more [89].
Monoliths are continuous stationary phases of homologous columns, where interconnecting channels allow for convective flow of the mobile phase through the entire matrix [90,91]. Commercially available monoliths are made of polymethacrylate copolymers (BIA Separations, Ljubljana, Slovenia; Dionex Sunnyvale, CA, USA), polyacrylamide (Bio-Rad, Hercules, CA, USA; Protista Biotechnology AB, Lund, Sweden), polystyrene (Dionex, Sunnyvale, CA, USA), polystyrene-divinylbenzene (Dionex, Sunnyvale, CA, USA), modified cellulose (Sepragen, Hayward, CA, USA) and silica (Merck, Darmstadt, Germany; Phenomenex, Torrence, CA, USA) [92,93]. Depending on the manufacturer they are sold as disks, tubes and rods with pore sizes ranging from 13 nm (silica, Merck) [94] to 6 µm (polymethacrylate, BIA Separations).
Membrane materials for chromatographic bioseparations include cellulose, polyamine, polysulfone, hydrazide and composite membranes such as polyethylene oxide and polyethersulfone coated with hydroxyethyl-cellulose [95,96]. There are three types of chromatographic membrane adsorbers used for the purification of bioproducts: flat sheets (e.g., Pall, Dreieich, Germany; Sartorius, Göttingen, Germany), hollow fibers (e.g., Kinetic Systems Inc., Boston, MA, USA) and radial-flow devices (e.g., 3M, Neuss, Germany) [96,97]. The advantages of hollow-fiber membrane adsorbers are the high surface area and the reduction in particle accumulation near the pore entrance owing to the cross-flow principle used during separation. However, the breakthrough of target compounds is generally broadened, leading to poor adsorber utilization compared with flat sheets [97]. Furthermore, eluted products are heavily diluted and internal mixing within the fiber housing results in low resolutions, and the application of gradient elution is very limited. Radial-flow adsorbers have flat-sheet membranes spirally wound over a cylindrical core [97] whereby the available surface area can be significantly increased. Thus, the volume-to-surface area for spiral-wound membrane adsorbers is better than for the majority of hollow-fiber adsorbers. Still, products are diluted and gradient elutions are accordingly challenging. Flat sheets are mainly used as stacks of numerous individual sheets providing a larger adsorbent volume. In flat-sheet membrane adsorbers the liquid is commonly introduced vertical to the membrane adsorber surface, allowing a minimum dead volume in an optimally designed membrane module. Thus, these membrane adsorbers do not suffer from diluted product fractions and can be used for gradient elution procedures.
The main advantages of membrane adsorbers and monoliths for the separation of virus particles are the predominance of convective material transport with nearly no diffusion limitations, [79,90,96–99] owing to large pores, and well-interconnected channels with small constrictions and high dynamic binding capacities [79,99,100], whereby the latter is sometimes lower for membrane adsorbers than for comparable monoliths [101]. The pore structure of membrane adsorbers and monoliths facilitates the accessibility of large virus particles to bound ligands in contrast to typical resins where they are excluded from the internal bead pores [98].
Except for monoliths, eddies are formed in the majority of chromatographic matrices including membrane adsorbers. As a result, labile virus particles can be deteriorated and separation impaired by peak broadening. Stacks of membrane adsorbers can be considered as very thin slices of monoliths. Pores between membrane layers are connected by a void space between individual membrane slices, leading potentially to eddy formations. Flow distributions in hollow-fiber and radial-flow membrane adsorbers are less well controlled compared with either monoliths or packed-bed columns. In addition to eddy formations this could result in reduced dynamic binding capacities and dispersions. The latter does not only affect negative-mode adsorption chromatography, but also reduce the resolution of positive-mode applications.
The majority of membrane adsorbers are designed for single-use applications, supporting current efforts in the pharmaceutical industry to reduce costs for validation, cleaning and sanitization. Monoliths are currently following this trend. For beads, however, it remains to be seen if disposable approaches can be realized economically. Nevertheless, a wider diversity of adsorptive surface chemistries is currently available for resins than for convective alternatives, but the product range for membrane-based separations constantly expands.
Surface chemistry of the stationary phase & the composition of the mobile phase
Currently, five different modes are used for the chromatographic separation of virus particles: size-exclusion chromatography (SEC), ion exchange chromatography (IEC), affinity chromatography (AC), hydrophobic-interaction chromatography (HIC) and mixed-mode chromatography (Table 4). The action principles and possibilities for process optimization are limited by the stability of the individual viruses. Extreme running conditions in terms of pH, osmolarity, ion compositions or certain organic solvents might influence the efficacy of vaccines or gene therapy vectors. Hence, normal or reversed-phase chromatography requiring organic solvents is usually not applied for whole-virus separations. However, for split vaccines or subunit vaccines, where virus membrane integrity does not necessarily have to be conserved, organic solvents and thus reversed- or normal-phase chromatography might be used depending on the solvent effects on the target proteins.
Table 4. Recent examples (since 2003) of chromatographic methods and purification schemes for the purification of virus particles.
| Virus | Main purification steps | Resin | Recovery (%) | Ref. |
|---|---|---|---|---|
| Adenoviridae (adenovirus) | DNAse, filtration, AEC, UF, SEC | Fractogel EMD DEAE-650 (M); Sephacryl S-400HR | 80 | [223] |
| AEC | Streamline Q XL | 35–65 | [224] | |
| EBA-AEC, UF | Streamline Q XL | 32 | ||
| AEC | Sartobind Q | 15–43 | [147] | |
| AEC | Sartobind anion direct Q | 60–62 | ||
| |
Filtration, UF, AEC, UF |
Sartobind anion direct Q |
52 |
|
|
Arteriviridae (porcine reproductive and respiratory syndrome virus) |
Filtration, UF AC |
HiTrap Heparin HP |
53 |
[97] |
|
Baculoviridae (baculovirus) |
Centrifugation, DNAse, SEC |
Sepharose CL-4B |
25 (24†) |
[225] |
| AC | Con A-Sepharose 4B | 29 (21†) | [110] | |
| AC | Heparin-agarose | 2 | [226] | |
| CEC | Sartobind S | 20 | ||
| |
CEC |
Mustang S |
78 |
|
| Hepadnaviridae (hepatitis B) and Flaviviridae (hepatitis C) | AC | HiTrap-Heparin | Not provided‡ | [227] |
| Orthomyxoviridae (influenza virus A and B) | SEC | Sepharose CL 2B | 38 | [215] |
| SEC | Sepharose 4 FF | 85 | [228] | |
| Filtration, UF, SEC, AEC | Sepharose 4FF; Sepharose Q XL | 53 | ||
| AEC | Sartobind Q | 86 | [102] | |
| Sartobind D | 38 | |||
| AC (Euonymus europaeus lectin) | Sartobind EEL-MA | 94§ | [229] | |
| AC (sulfated cellulose) | Sulfated cellulose MA | 73–94 | [105] | |
| AC | Sartobind Zn-IDA MA | 75 | [104] | |
| |
AC |
Different ligands immobilized to monoliths¶ |
Not provided |
[156] |
|
Parvoviridae (Aedes Aegypti densonucleosis virus) |
AEC CEC |
Sartobind Q, D Sartobind S, C |
Not provided |
[230,231] |
|
Parvoviridae (adeno-associated virus, AAV1-GFP) |
AEC |
Mini Q 4.6/50 PE |
42–72# |
[143] |
|
Parvoviridae (adeno-associated virus, AAV1, baculovirus-mediated production system) |
AC |
AVB Sepharose |
29–82 |
[232] |
|
Parvoviridae (adeno-associated virus, AAV2) |
AEC |
Q Sepharose XL |
74†† |
[20] |
|
Parvoviridae (adeno-associated virus, AAV5) |
Filtration, DNAase, AEC |
Mono Q HR |
30–50 |
[233] |
| Parvoviridae (adeno-associated virus, AAV2/8) | AEC | Mustang Q | 25–58 | [234] |
| |
CEC |
Mustang S |
|
|
|
Parvoviridae (adeno-associated virus, AAV8) |
AEC |
Mustang Q |
43 |
[234] |
| Poxviridae (Vaccinia virus, MVA-BN®) | AC, AEC | Sulfated cellulose and heparin membrane adsorber; Sartobind Q | 58–59 | [117] |
| |
AC, HIC |
Sulfated cellulose and heparin membrane adsorber; ToyoScreen Phenyl-650M |
52–56 (34–37†) |
[100] |
| Retroviridae (vector; MoMLV-derived VSV-G-pseudotyped retroviral vector) | AC | Fractogel EMD heparin (S) | 61 | [66] |
| |
Filtration, UF, DF, AC, SEC |
Fractogel EMD heparin (S) Sepharose CL-4B |
38 |
|
|
Retroviridae (vector; MoMLV-derived VSV-G-pseudotyped retroviral vector) |
AC |
Fractogel EMD heparin (S) |
61 |
[106] |
|
Retroviridae (vector; MoMLV-derived RD114-pseudotyped retroviral vector) |
AC |
Fractogel EMD heparin (S) |
43 |
[106] |
| Retroviridae (vector; VRX496 a VSV-G pseudotyped human immunodeficiency virus type 1-derived vector) | AEC | Mustang Q | 65 | [235] |
| |
SEC |
Sephacryl S500 |
70–80 |
|
| Retroviridae (vector; VSV-G-pseudotyped retroviral vector) | SEC | Sepharose CL-4B | 70 | [236] |
| |
UF, SEC, UF/DF |
Sepharose CL-4B |
19 |
|
|
Retroviridae (vector; His6-tagged retroviral vector) |
AC |
Ni-NTA agarose |
56 |
[114] |
| Retroviridae (vector; biotin-tagged retroviral vector) | AC | Fractogel EMD streptavidin‡‡ | 17 | [237] |
| |
|
Steptavidin-Monolith§§ |
8 |
[109] |
| Retroviridae (vector; MoMLV-derived retroviral vector) | AEC | DEAE FF HiTrap | 53–57 | [83] |
| Q XL HiTrap | 51–53 | |||
| |
|
Q FF HiTrap |
16–25 |
|
| Retroviridae (vector; Murine leukemia viral vector) | AEC | Fractogel DEAE | 56–77 | [152] |
| |
MF, UF/DF, AEC, UF/DF |
Fractogel DEAE |
62 |
|
| Rhabdoviridae (rabies virus) | SEC | Sepharose 4FF | 35–40 | [238] |
| AEC | Streamline Q XL | 2 | ||
| CEC | Streamline SP XL | 17 |
†Infectious virus particles.
‡Captured from infected plasma samples.
§Euonymus europaeus lectin immobilized onto Sartobind-Epoxy membrane adsorber.
¶Epoxy- or carbodiimide-activated monoliths with immobilized sialyllactosylamine, sialyllactose, ceruloplasmin, chitosan, heparin and remantadine.
#Recovery after removal of empty capsids.
††Separation of AAV2 empty particles from genome-containing vectors.
‡‡Streptavidin immobilized onto Fractogel EMD-Azlactone.
§§Epoxy-activated monoliths with immobilized streptavidin.
AC: Affinity chromatography; AEC: Anion-exchange chromatography; CEC: Cation-exchange chromatography; DF: Diafiltration; EBA: Expanded bed adsorption; MA: Membrane adsorber; MF: Microfiltration; MoMLV: Moloney murine leukemia virus; MVA-BN®: Modified Vaccinia Ankara – Bavarian Nordic; SEC: Size-exclusion chromatography; UF: Ultrafiltration; VSV-G: Vesicular stomatitis virus G protein.
Partition chromatography such as SEC can only be achieved by porous particles but not by membrane adsorbers or monoliths. For SEC, virus particles are generally recovered in the column void volume allowing the separation of bulk proteins and small molecular contaminants without buffer changes under gentle operating conditions, thereby maintaining virus infectivity and immunogenicity.
Owing to the large difference in hydrodynamic volume of intact virus particles and soluble contaminants, the SEC exclusion limit can easily be optimized not to distinguish between morphologically similar viruses, allowing robust process conditions and broad applicability where different strains are routinely required (e.g., for influenza vaccines). The main disadvantages of SEC are the low capacity, product dilution and the poor pressure resistance of the matrix, requiring low flow rates. Despite these disadvantages, SEC represents a valuable method in addition to adsorptive chromatography, but high-throughput options for ultrafiltration processes will challenge the application of this unit operation for the separation of virus particles in the future.
Industrial chromatography processes are usually optimized for high dynamic binding capacities, particularly for the primary capturing step, and the elimination of residual contaminants during the polishing step (Figure 1). High capacities are attained by large accessible surface areas, optimized ligand densities and high selectivity. The latter is important if highly contaminated loads (i.e., feedstocks) are applied. In addition to the dynamic binding capacity, the ligand density of adsorption matrices such as IEC, AC, and HIC also contributes to the overall adsorption strength. This is mainly due to the multivalent interactions of virus particles with the matrix ligands. Thus, ligand density needs to be carefully selected taking into account both of these aspects. Furthermore, the desorption conditions need to be considered, selecting optimal ligands and ligand densities, and allowing elution conditions that do not affect the virus immunogenicity or transfection efficiency of vectors. Strong adsorptions, requiring harsh desorption conditions for the target virus particles, are often observed for IEC or HIC [102].
Adsorption matrices are commonly used both in positive mode (i.e., virus particles adsorbing to the chromatography medium) and negative mode (i.e., contaminants adsorbing to the matrix and virus particles eluting). Positive-mode applications are usually used for capturing or intermediate purification and negative modes for the final polishing.
Ion exchange chromatography exploits the charge-to-charge interaction between the virus surface and immobilized ion exchange groups on the matrix. Depending on the overall isoelectric point (Table 2) of the individual virus particles, cation- or anion exchange chromatography (AEC) matrixes are utilized. Different variations of IEC have been applied for the purification of a variety of viruses (Table 4). Adsorbed virus particles are in general displaced by an increasing ionic strength. Alternatively, they are desorbed by a change in buffer pH, leading to an unfavorable overall charge of the virus surface or the matrix, at which the virus particles do not bind anymore. IEC has a relatively low specificity resulting in a reduced dynamic binding capacity for the target virus and often results in co-elutions of contaminants (e.g., host cell DNA for AEC). Differential elution between the target virus and contaminants can sometimes be achieved by linear- or step-gradient elutions. If high salt concentrations or nonphysiological pH conditions are required for purification, it is crucial to monitor immunogenicity or transfection efficiency of virus particles [47,304]. Hence, IEC matrices have to be carefully selected in terms of the type of charged groups, ligand density, and applied buffer conditions with respect to the virus type, product application and utilized assays for process characterization.
Affinity chromatography of virus particles relies on a specific and reversible adsorption and subsequent recovery of the active target virus from a ligand immobilized onto the chromatography matrix. The specific interactions between ligand and virus are based on individual structural properties, and the virus particles are commonly eluted by an altered conformation via a changed buffer pH or ionic strength or competitive displacement. However, the latter is generally not economic for large-scale production processes, but is frequently applied at smaller scales. Applied ligands or matrices for the purification of virus particles are immobilized metal ion affinity [103,104], sulfated carbohydrates (e.g., heparin) [100,105–107], specific antibodies [108] or antibody fragments, biotin streptavidine system for recombinant viral vectors [109], lectins [110,111], DNA aptamers [112] and peptides (Table 4). For the majority of viruses, no specific ligands are known, resulting in the application of less-specific ligands such as lectins or sulfated carbohydrates (i.e., heparin). Lectins interact with accessible carbohydrate moieties of the surface glycoproteins according to their specificity. Heparin is primarily a weak and strong cation exchanger containing sulfo and carboxyl groups as well as numerous hydroxyl groups [113]. Desorption of the virus particles from heparin matrices is done by increasing the ionic strength. However, it often requires a higher ionic strength than for classical cation exchangers owing to the multivalent interactions of the branched linear structure of heparin and the formation of hydrogen bonding between the target virus and heparin [113]. An alternative to laborious ligand screens or the application of less specific pseudo-affinity ligands are engineered viral vectors containing affinity tags on the virus surface [109,114]. Crucial for specific affinity ligands is the consideration of their dissociation constants. For recombinant proteins, dissociation constants of 10-4–10-10 M are usually considered a good working range [115]. Optimal affinity ligands allow fast binding kinetics for high flow rates but mild elution conditions to avoid reduction in immunogenicity or activity. Therefore, it is essential to consider the overall binding strength of large virus particles, which is due to the already discussed multivalent interactions. If proteins or larger biomolecules are used as ligands, their potential toxicity or immunogenicity has to be considered and, if applied, assays have to be developed for their detection and quantification. Furthermore, for many proteins, high costs are associated and the low stability of these ligands towards sanitizing agents in some cases prohibits their application for large-scale processes [116]. In some cases, additional drawbacks of specific ligands are their restricted application as platform technology and limited process robustness for vaccines with frequently varying subtypes (e.g., influenza vaccines). Furthermore, for viruses with varying progenies (e.g., Vaccinia virus) [117], some virus particles might be excluded, thereby affecting the overall yield. On the other hand, the applied specificity allows the separation of specific progenies. Ligands targeting post-translational modifications on the virus surface are heavily susceptible to the cultivation conditions and host cells [118]. Hence, target epitopes for AC matrices on the virus surface have to be carefully selected, considering their stability during the entire process. Nevertheless, matrices with highly specific affinity ligands and high dynamic binding capacities are very well suited to capture virus particles after clarification from low concentrated cultivation broths.
Hydrophobic-interaction chromatography separates biomolecules dependent on the differential interaction of these compounds with hydrophobic ligands on the surface of the stationary phase. It is routinely used for bioseparations of proteins [119–122], and has also been applied for virus purification [100,113] since it is an orthogonal separation technique to purification methods based on ionic interactions. However, many factors such as type of salt and ionic strength of buffer, pH, temperature, ligand and ligand density influence the performance of HIC, making its development and optimization a difficult and time-consuming endeavor. The two most commonly used HIC ligands are butyl and phenyl [113]. Interactions with these ligands are relatively strong and have been frequently cited to denature labile proteins, potentially reducing their efficacy. However, often proteins subjected to these methods revert upon elution to their native structure [113]. Even so, if used for purification of gene transfer vectors or vaccines, their transfection efficiency or immunogenicity has to be closely monitored, just as for the high salt applications for the IEC methods. Stronger HIC ligands such as hexyl and octyl are commonly destructive to proteins, and most likely also to virus particles or gene therapy vectors if applied in positive mode. Furthermore, HIC requires the application of high concentrations of kosmotropic salts, which potentially also reduces virus efficacies. Also, the application of high salt concentrations influences virus aggregation in the majority of cases, affecting further downstream processes and often process analytics. In addition, disaggregation of the formed aggregates usually impacts virus transfection efficiency and immunogenicity. Nevertheless, it was recently shown that HIC can be successfully applied to remove residual DNA after AC for purification of active Vaccinia virus particles (MVA-BN®; Bavarian Nordic A/S, Kvistgaard, Denmark) (Table 4)[100,305].
Mixed-mode chromatography exploits a multimodal functionality that allows virus particles or contaminants to adsorb to the stationary phase by a combination of ionic interactions, hydrogen bonds and/or hydrophobic interactions. A typical example for a mixed-mode chromatography media is hydroxyapatite, which supports metal affinity interactions through its hydroxyapatite calcium groups, and cation exchange interactions through its hydroxyapatite phosphate groups [81]. Successful applications of mixed-mode matrices for the purification of gene therapy vectors have been described for adenoviruses [82], AAV [123], Moloney murine leukemia viruses [124] and retroviruses (Table 4)[83].
Current topics to promote cell culture-derived viral vaccine & gene therapy vector production processes
Continuous chromatography: a potential future method for the purification of virus particles
Continuous chromatographic processes, such as the simulated moving bed (SMB) technology is today well established for the separation of binary mixtures of petrochemicals, sugars and small molecule pharmaceuticals [125,126]. SMB significantly improves the volumetric throughput as well as the purity and concentration factor relative to batch chromatography. However, purification of complex biological mixtures is still challenging because most systems are operated as a binary fractionator, providing only two exit streams [127]. However, new operation modes such as the three-fraction SMB [128,129], solvent gradient SMB, and cleaning in-place SMB are important steps leading the way for its successful application in bioseparations. Finally, the complexity of the equipment and the experimental setup – that is, the large number of valves required – poses a serious challenge for process validation.
Applying chromatographic models for process optimization
The design of chromatographic purification processes is challenging owing to the multitude of unit operations and process variables, that is, a constantly growing number of available matrices, different chromatography modes and diverse operating conditions. As a result, an extremely large amount of design freedom exists. So far, the selection of unit operation and the choice of process conditions relies mainly on practical experiences or on trial and error approaches. In order to save valuable time and resources, miniaturized parallel screening tools have been developed [130], that can be combined with additional kinetic studies (e.g., surface plasmon resonance [SPR] spectroscopy) [98,131], thermodynamic measurements and dynamic light-scattering analysis [132] to guide the early stage process development and optimization. For the selection of the most appropriate experiments and parameters, the screens are commonly planned via experimental design software tools (e.g., Design-Expert, MODDE, STATISTICA), implementing the concepts of ‘quality by design’. Furthermore, software tools for computer-aided process design, production scheduling and process debottlenecking is often used [133,134].
To date, high-throughput process developments have been mainly applied to production processes for recombinant proteins. However, these methods can also be applied for the DSP of viral vaccines and gene therapy vectors. Here, progress will largely depend on the establishment of reliable and robust analytical assays, which can cope with the high number of samples. A particular problem is the determination of product titers, which requires provision of suitable standards, biological and technical replicates, and assay validation on a logarithmic range. The latter typically results in comparatively large errors. Typically, standard deviation of methods for virus titer assays in the range of 0.3 to 0.5 log10 have to be accepted.
Screening and analysis of well-proven operating conditions are one point, to further facilitate efficient downstream development, good modeling tools have to be established and applied. For instance, Vicente et al. combined the steric mass action model of ion exchange [135] with a standard chromatographic column model to simulate and predict adsorption and elution conditions for virus-like particles on anion-exchange (AEX) membrane adsorbers [136]. The same research group recently described the effect of ligand density on AEX membrane adsorbers for the purification of recombinant baculoviruses by comparing model strategies based on SPR spectroscopy with diethylaminoethyl (DEAE) ligand densities of membrane adsorbers. Both methods indicated that a lower ligand density increased the overall yields by over 20% [98], demonstrating that SPR technology can be successfully used to model membrane adsorption processes. Their studies were further aided by a theoretical model to predict process conditions for DSP improvement [98].
Overall these studies demonstrate how chromatographic models combined with parallel screening techniques and rationally designed experiments can streamline the design and optimization of viral DSP in the future.
Use of disposables in DSP
Single-use components for individual unit operations in DSP have been used for years in the pharmaceutical industry, mainly for filtration and buffer/media storage. Over the last decade, the relevance of disposable concepts has been extended to other unit operations including chromatographic applications for which matrices such as membrane adsorbers, monoliths and more recently resins are used. The driving force for the increasing interest in single-use concepts for the production of viral vaccines is the fast and highly flexible set up of production processes, which is expected to improve the response time for increasing vaccine demands such as for pandemic preparations. In addition, disposable concepts for limited production campaigns are economically attractive considering that under current good manufacturing practice (cGMP) standards, raw material and equipment in direct contact with the product have to be dedicated to the particular production process [137]. Furthermore, single-use concepts commonly reduce or eliminate time-consuming and cost-intensive process steps such as cleaning and sanitization and their validation. However, the economic application depends heavily on the scale and requirements of the individual product, in addition to the available production facilities. Hence, the use of disposable concepts always has to be considered on a by-case basis, taking into account investment costs, the entire variable (running) costs of the particular manufacturing facility and individual processes, and the product requirements [137].
Purification of virus particles: from kits to complete downstream processes
Kits for virus purification
During recent years, kits for small- and medium-scale purifications and concentrations were introduced for adenoviruses (Adenoviridae), AAV (Parvoviridae) and lentiviruses (Retroviridae). In the ease of operation they are comparable to plasmid purification kits and require approximately 2 h for the entire process. The majority of manufacturers rely on adsorption membrane technology based on centrifugal devices (small scale), syringe capsules (small scale) or larger capsules for fast protein liquid chromatography systems. Purification of lentivirus and AAV particles is mainly achieved by the use of AEX and cation-exchange (CEX) membrane adsorbers. According to the manufacturer’s handbooks, the maximum capacities of the largest units currently available are approximately 1 × 1012 and 3 × 1013 total virus particles for adenovirus and AAV, respectively, and 2 × 108 infectious particles for lentivirus. The required culture volumes range from 20 to 500 ml, allowing recoveries of approximately 60–80% of the total particles depending on the culture conditions and virus. For the best results, it is important to consider the virus type for which the respective kit has been optimized. However, the kits can be used for other virus species after adapting the operating conditions. Importantly, nucleic acids (i.e., host cell DNA and proteoglycans) will also bind to AEX membrane adsorbers and potentially co-elute. Hence, kit procedures often include nuclease treatment of the harvest to reduce nucleic acid contaminations and to improve the flow rates and thus process time (i.e., kits for adenovirus purification). In addition, a digestion step using chondroitinase ABC, an enzyme that degrades glycosaminoglycan side chains of chondroitin sulfate proteoglycans [138], is sometimes included to remove proteoglycan contaminations. Certainly, the purity of the resulting virus fractions heavily depends on culture conditions, virus species, type and strain, as well as the applied kit. Therefore, no detailed information on virus purity can be provided by the manufacturers. It should also be pointed out, however, that viruses purified by this method can only be used for research purposes and are not intended for clinical studies or medical applications.
Downstream processes of virus particles
Current downstream processes aim to combine individual unit operations to an overall purification train taking into account the virus type (morphology, specific surface characteristics), the final product type (e.g., subunits vaccines, split vaccines, inactivated or active virus particles), application requirements (contamination levels), product yields and size of the production batch. For the development of new purification schemes it will be equally important to develop and optimize new technologies, as to design efficient DSP trains based on optimal combinations of these new technologies with currently used methods. Unfortunately, very few complete purification schemes, from culture supernatant to clinical grade virus product, have been described in detail with overall recoveries and degree of contaminations in the literature. Hence, a precise evaluation of the respective methods is nearly impossible. In general, however, overall viral recoveries of greater than 30% are currently considered acceptable for human pharmaceutical products.
A particularly challenging issue for the DSP of human vaccines and the purification of gene therapy vectors is the removal of host cell DNA. Current guidelines for newly licensed human inactive vaccine products from continuous cell lines stipulate that residual DNA levels exceeding 10 ng per dose are not acceptable [139,140]. Levels of contaminating host cell DNA for gene therapy applications are even lower. Here, levels of 10–100 pg of residual host cell DNA per parenterally administered dose are considered acceptable by most medical agencies [141]. For many applications these levels can only be achieved via nuclease treatments such as Benzonase. The clear advantage of such a treatment is the elimination of potential oncogenes or other functional DNA sequences [142] even in very low amounts of host cell DNA and the cleavage of nucleic acids adsorbed to the target virus. In addition, nuclease and condroitinase treatment frequently results in improved efficacies for gene therapy vectors.
For the purification of gene therapy vectors, the removal of empty capsids or virus particles containing nucleic acid sequences other than the intended genome is crucial for their efficacy. However, owing to the small differences in specific surface characteristics or in virus morphology this is fairly difficult. Successful separations were described via density centrifugation (adenovirus) [38], IEC (AAV) [20,143], and partial depletions by HIC and immobilized zinc-chelating interaction chromatography (adenovirus) [144]. However, the possible application of the methods and their optimization has to be considered on a by-case basis.
A further substantial challenge for DSP of viral vaccines, gene therapy vectors and other biotechnological products is batch-to-batch variations from the upstream processes. Cell culture harvests can vary considerably not only in product concentration and quality but also in the type and amount of contaminations. For efficient and successful process development, all the aforementioned issues have to be addressed within a tightly coupled up- and down-stream process. Therefore, critical parameters of the cultivation process effecting the DSP should be closely monitored and the subsequent purification methods adapted accordingly [145].
In the following paragraphs, we summarize unit operations and purification trains for different virus types that have been described in the last 5 years (Table 4).
Adenovirus
A thorough review has been published by Lusky et al. on the GMP production of adenoviral vectors for clinical trials [146]. Another overview by Burova and Loffe deals with the chromatographic purification of recombinant adenoviral and AAV vectors [38]. In general, AEC is the driving force for the described processes for adenoviruses [147,148]. Eglon described a purification via AEC in combination with SEC from clarified and benzonase-treated cell lysate, which yielded total virus recoveries of 36% compared with 27.5% via the classical CsCl-purification method [149]. Duffy et al. compared a novel membrane-based technology for the purification of adenovirus and AAV to conventional techniques [150] and Peixoto et al. described a downstream process for adenoviral vectors based on membrane technology [147]. This purification train comprising clarification, ultrafiltration, AEX membrane adsorber, ultrafiltration and sterile filtration resulted in a recovery of 52% of the infectious particles [147]. Another very important aspect, namely the virus aggregation and association of DNA to virus particles, was addressed by Konz et al.[82].
Adeno-associated virus
Wright reviewed, among other issues, vector quality characteristics such as AAV-related impurities (e.g., AAV-encapsidated DNA impurities) and their pharmacological impact as well as GMP considerations for clinical AAV vector production methods [18]. A chromatographic purification train for AAV using a combination of hydroxyapatite, DEAE-Sepharose and Cellufine® (Chisso America Incorporated, Rye, NY, USA) sulfate with an overall yield of 30% has been described by O’Riordan et al.[123]. More recently Okada et al. described the use of a combination of CEX and AEX membrane adsorbers for the purification of AAV serotype 1 and 8 with average recoveries of 17.4 and 49%, respectively [151].
Retroviral vectors (e.g., lentivirus)
Downstream processing strategies for purification of retroviral vectors are summarized in at least two excellent reviews from Segura et al.[47] and Andreadis et al.[116], the latter being published in 1999, ahead of the former. Rodrigues et al. described the purification of murine leukemia virus-derived gene therapy vectors by means of membrane separation and AEC with overall recoveries of 26% and a purity of greater than 99% relative to the protein concentration [152]. Moreover, Lesch et al. illustrated a scalable capture step for lentiviral vectors generated in 293T cells with baculoviral vectors [153]. Capturing based on DEAE monolithic columns reached a 65% recovery.
Oncolytic gene therapy vectors
Ausubel et al. described the cGMP large-scale production of an oncolytic recombinant vesicular stomatitis virus based on a downstream process composed of a primary clarification step (filtration), Benzonase treatment, AEC, dia-/ultra-filtration and sterile filtration. The obtained yields of individual unit operations for infectious virus particles ranged from 59% to approximately 100% after sterile filtration [5]. The fact that full recovery was obtained after the complete purification of some batches suggests that some of the titers, which were estimated via plaque forming assays, might be overestimated. However, the overall process is described comprehensively. Working et al. discussed critical points affecting the development and production of clinical grade oncolytic adenoviruses, describing the up- and downstream process in detail [154]. They illustrated a downstream process composed of cell lysis (Triton), clarification, Benzonase treatment, AEC capture, ultra-/dia-filtration, SEC, ultra-/dia-filtration (final formulation), followed by a final sterile filtration. However, the achievable purities and yields were not discussed in detail.
Vaccines: influenza
Downstream processes for purification of influenza virus particles were recently reviewed, summarizing applied purification trains for egg- and cell culture-derived influenza virus particles by Wolff and Reichl [155]. Motivated by the recent pandemic threats of influenza, numerous research laboratories worked on the optimization of influenza vaccine production processes. For the downstream side the center of attention was on specific chromatography media to substitute the frequently applied Cellufine Sulfate for the capturing of influenza virus particles [105,156] and on the use of chromatographic matrices with improved volumetric throughputs (i.e., monoliths [156,306]) and membrane adsorbers [102,104].
Vaccines: smallpox
Wolff et al. described a downstream process for Vaccinia virus based on a combination of pseudo-affinity and ion-exchange membrane adsorbers as well as pseudo-affinity and HIC matrices [100,117]. The most promising downstream train resulted in an overall yield of 34% (infectious virus particles). Furthermore, depletion of total DNA to 0.01% of the starting material and a total protein amount of less than 25 µg per dose was achieved [100]. However, batch variations of the starting material resulted in significant variations in the product yield and purity, and need to be further addressed.
Expert commentary
The success story of vaccination against major infectious diseases rests on a 200-year-old history [157]. Gene therapy in contrast is a relatively new but promising technology with the revolution of molecular genetics in the 1970s paving the way into successful medical applications [158]. Vaccines currently comprise a rapidly growing market within the biopharmaceutical industry and gene therapy vectors have the potential to follow this line. Nevertheless, for some vaccines, manufacturing capacities are still limited (i.e., for pandemic influenza vaccines). In general these problems are related to both, upsteam processing and DSP, and can only be efficiently solved if both are considered together. In the last 10 years, upstream production processes of some vaccines moved away from their respective classical systems to the usage of diploid and continuous cell lines. However, downstream purification trains were in many cases only adapted to the new requirements but still lack further optimization. Main improvements for vaccine purifications have to be governed by enhanced throughputs, capacities and potentially specificities of individual unit operations, as well as an enhanced linking of applied unit operations and the reduction of the number of purification steps. Process development for gene therapy vectors could significantly benefit from the experience accumulated in vaccine production, especially with vectors derived from vaccines. Nevertheless, there are still several technical problems to be solved for both product classes such as the Benzonase-free removal of host cell nucleic acids. Specific difficulties for the purification of gene therapy vectors are:
• High final vector titers;
• Removal of transduction inhibitors from producer cells (proteoglycans, glycosaminoglycans [159–161]);
• Elimination of free envelope proteins, defective-virus particles and empty capsids.
Removal of host cell proteins, medium proteins and peptides is usually less problematic but equally important owing to potential allergic reactions. Of note, the reduced starting levels of process-related protein contaminations are mainly due to increased usage of serum-free, protein-free or even fully defined media for upstream cultivations – an excellent example for improved overall process economics via a tighter coupling of up- and down-stream methods.
In summary, for the downstream processes described in Table 4, overall yields of virus particles of greater than 30% for production of human vaccines or gene therapy applications should be considered satisfactory. For other purposes, higher yields can be achieved depending on the requirements on product purity, the type and the amount of contaminants in the starting material, and the exertion.
Five-year view
Constant pressure towards improved product quality and safety as well as tighter timelines for the development of biopharmaceuticals will certainly push for more efficient process development. Current advances in the field indicate that increased application of high-throughput automated scaled-down models in combination with good mathematical models for experimental design and process development will support these efforts. Certainly, these advances will also require the establishment of high-throughput assay platforms for precise and reliable process characterization. As a result, unit operations should be available that have not only higher specificity and increased volumetric throughput but also enable design of purification trains with a reduced number of steps.
Advances in DSP will be most likely involve a broader use of filtration techniques (e.g., tangential flow filtration and diafiltration), the availability of new chromatography matrices such as membrane adsorbers, monoliths or bifunctional beads, and the identification of new, specific ligands. Furthermore, single use concepts and the introduction of continuous DSP methods will play an important role as well as tighter process integration in terms of the up- and downstream process. Furthermore, the establishment of platform technologies will have high priority whereas a large step towards platform technologies has already been accomplished by the development of downstream purification kits for gene therapy vectors.
For additional readings, Pedro et al. recently published a detailed summary dealing with the purification of bionanoparticles [162], Gagnon published a comprehensive description on the chromatographic purification of virus particles [113] and Segura et al. published an overview of current scalable methods for purification of viral vectors [43].
Key issues
• The application of parallel screening techniques, rationally designed experiments and the development of chromatographic models needs to be implemented to support downstream processing (DSP) development.
• Development and application of DSP platform technologies to improve the flexibility and response time of production processes is also required.
• Overall process economics need to be enhanced via a tighter coupling of upstream and downstream methods.
• Specific capturing of virus particles from bioreactor harvests via affinity chromatography can optimize the overall DSP by combining the primary concentration and purification step.
• Utilization of modern chromatography matrices such as membrane adsorbers and monoliths in place of classical resins enables an improved productivity in numerous DSP for viral particles.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest •• of considerable interest
- 1.Howarth JL, Lee YB, Uney JB. Using viral vectors as gene transfer tools (Cell Biology and Toxicology Special Issue: ETCS-UK 1 day meeting on genetic manipulation of cells). Cell Biol. Toxicol. 26(1), 1–20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett PN, Portsmouth D, Ehrlich HJ. Developing cell culture-derived pandemic vaccines. Curr. Opin. Mol. Ther. 12(1), 21–30 (2010). [PubMed] [Google Scholar]
- 3.Lauring AS, Jones JO, Andino R. Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 28(6), 573–579 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudek T, Knipe DM. Replication-defective viruses as vaccines and vaccine vectors. Virology 344(1), 230–239 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Ausubel LJ, Meseck M, Derecho I et al. cGMP production of an oncolytic rVSV vector for cancer treatment. Hum. Gene Ther. 22, 489–497 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes the current good manufacturing practice (GMP) large-scale production of a recombinant oncolytic virus including the complete downstream process.
- 6.Pelkmans L. Viruses as probes for systems analysis of cellular signalling, cytoskeleton reorganization and endocytosis. Curr. Opin. Microbiol. 8(3), 331–337 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Purkayastha A, Dasgupta I. Virus-induced gene silencing: a versatile tool for discovery of gene functions in plants. Plant Physiol. Biochem. 47(11–12), 967–976 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Gould B, Kramer E. Virus-induced gene silencing as a tool for functional analyses in the emerging model plant Aquilegia (columbine, Ranunculaceae). Plant Methods 3(1), 6 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnell MJ, McGettigan JP, Wirblich C, Papaneri A. The cell biology of rabies virus: using stealth to reach the brain. Nat. Rev. Micro. 8(1), 51–61 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Masetti A, De Luigi V, Burgio G. Effects of nucleopolyhedrovirus based product on Spodoptera littoralis. Bull. Insectol. 61(2), 299–302 (2008). [Google Scholar]
- 11.Berling M, Blachere-Lopez C, Soubabere O et al.Cydia pomonella granulovirus genotypes overcome virus resistance in the codling moth and improve virus efficiency by selection against resistant hosts. Appl. Environ. Microbiol. 75(4), 925–930 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandey A, Singh N, Sambhara S, Mittal SK. Egg-independent vaccine strategies for highly pathogenic H5N1 influenza viruses. Human Vaccines 6(2), 178–188 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genzel Y, Reichl U. Continuous cell lines as a production system for influenza vaccines. Expert Rev. Vaccines 8(12), 1681–1692 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Mulligan RC. The basic science of gene therapy. Science 260(5110), 926–932 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Collins SA, Guinn BA, Harrison PT, Scallan MF, O’Sullivan GC, Tangney M. Viral vectors in cancer immunotherapy: which vector for which strategy? Curr. Gene Ther. 8(2), 66–78 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Alton E. Progress and prospects: gene therapy clinical trials (part 1). Gene Ther. 14(20), 1439–1447 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Griesenbach U. Progress and prospects: gene therapy clinical trials (part 2). Gene Ther. 14(22), 1555–1563 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Wright JF. Transient transfection methods for clinical adeno-associated viral vector production. Hum. Gene Ther. 20(7), 698–706 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Concise summary on vector quality characteristics including product-related impurities and their pharmacological impact.
- 19.Ayuso E, Mingozzi F, Montane J et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 17(4), 503–510 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Qu G, Bahr-Davidson J, Prado J et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. J. Virol. Methods 140(1–2), 183–192 (2007). [DOI] [PubMed] [Google Scholar]
- 21.McDonald P, Victa C, Carter-Franklin JN, Fahrner R. Selective antibody precipitation using polyelectrolytes: a novel approach to the purification of monoclonal antibodies. Biotechnol. Bioeng. 102(4), 1141–1151 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Tscheliessnig A, Ong D, Lee J et al. Engineering of a two-step purification strategy for a panel of monoclonal immunoglobulin M directed against undifferentiated human embryonic stem cells. J. Chromatogr. A 1216(45), 7851–7864 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Kim HJ, Kim SY, Lim SJ, Kim JY, Lee SJ. One-step chromatographic purification of human papillomavirus type 16 L1 protein from Saccharomyces cerevisiae Protein Expr. Purif. 70(1), 68–74 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Ali A, Roossinck MJ. Rapid and efficient purification of Cowpea chlorotic mottle virus by sucrose cushion ultracentrifugation. J. Virol. Methods 141(1), 84–86 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Loa CC, Lin TL, Wu CC et al. Purification of turkey coronavirus by Sephacryl size-exclusion chromatography. J. Virol. Methods 104(2), 187–194 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ro HS, Lee NJ, Lee CW, Lee HS. Isolation of a novel mycovirus OMIV in Pleurotus ostreatus and its detection using a triple antibody sandwich-ELISA. J. Virol. Methods, 138(1–2), 24–29 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Fontes LVQ, Campos GS, Beck PA, Brandao CFL, Sardi SI. Precipitation of bovine rotavirus by polyethylen glycol (PEG) and its application to produce polyclonal and monoclonal antibodies. J. Virol. Methods 123(2), 147–153 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Kröber T, Knochlein A, Eisold K, Kalbfuss-Zimmermann B, Reichl U. DNA depletion by precipitation in the purification of cell culture-derived influenza vaccines. Chem. Eng. Technol. 33(6), 941–959 (2010). [Google Scholar]
- 29.Kerr IM, Martin EM. Simple method for isolation of encephalomyocarditis virus ribonucleic-acid J. Virol. 9(3), 559–561 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amosenko FA, Svitkin YV, Popova VD et al. Use of protamine sulphate for elimination of substrate DNA in poliovaccines produced on continuous cell lines. Vaccine 9(3), 207–209 (1991). [DOI] [PubMed] [Google Scholar]
- 31.Elbert LB, Terletskaya EN, Timofeev AV et al. Inactivated vaccine against tick-borne encephalitis (TBE) derived from heteroploid continuous monkey cell-line Vaccine 7(5), 477–477 (1989). [DOI] [PubMed] [Google Scholar]
- 32.Hammar L, Gilljam G. Extraction of HIV-1 in aqueous 2-phase systems to obtain a high-yield of gp120 AIDS Res. Hum. Retroviruses 6(12), 1379–1388 (1990). [DOI] [PubMed] [Google Scholar]
- 33.Morenweiser R. Downstream processing of viral vectors and vaccines. Gene Ther. 12, S103–S110 (2005). [DOI] [PubMed] [Google Scholar]; •• Review on general purification methods for viral vectors and vaccines providing a detailed overview on currently applied techniques.
- 34.Braas GMF, Walker SG, Lyddiatt A. Recovery in aqueous two-phase systems of nanoparticulates applied as surrogate mimics for viral gene therapy vectors. J. Chromatogr. B Biomed. Sci. Appl. 743(1–2), 409–419 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Hammar L, Merza M, Malm K, Eriksson S, Morein B. The use of aqueous 2-phase systems to concentrate and purify bovine leukemia-virus outer envelope protein gp51 Biotechnol. Appl. Biochem. 11(3), 296–306 (1989). [PubMed] [Google Scholar]
- 36.Hammar L, Eriksson S, Malm K, Morein B. Concentration and purification of feline leukemia-virus (FELV) and its outer envelope protein gp70 by aqueous 2 phase systems J. Virol. Methods 24(1–2), 91–102 (1989). [DOI] [PubMed] [Google Scholar]
- 37.Gilljam G, Siridewa K, Hammar L. Purification of simian immunodeficiency virus, SIVMAC251, and of its external envelope glycoprotein, gp148. J. Chromatogr. A 675(1–2), 89–100 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Burova E, Loffe E. Chromatographic purification of recombinant adenoviral and adeno-associated viral vectors: methods and implications. Gene Ther. 12, S5–S17 (2005). [DOI] [PubMed] [Google Scholar]; • Extensive overview on methods and implications on the chromatographic purification of adenoviral and adeno-associated viral vectors.
- 39.Yamada K, Morishita N, Katsuda T, Kubo S, Gotoh A, Yamaji H. Adenovirus vector production using low-multiplicity infection of 293 cells. Cytotechnology 59(3), 153–160 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo J, Yan R, Xu GD, Li WY, Zheng CY. Construction of the Vero cell culture system that can produce infectious HCV particles. Mol. Biol. Rep. 36(1), 111–120 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Tan JM, Lancaster M, Hyatt A, van Driel R, Wong F, Wamer S. Purification of a herpes-like virus from abalone (Haliotis spp.) with ganglioneuritis and detection by transmission electron microscopy. J. Virol. Methods 149(2), 338–341 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Reiser J. Production and concentration of pseudotyped HIV-1-based gene transfer vectors. Gene Ther. 7(11), 910–913 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Segura MM, Kamen AA, Garnier A. Overview of current scalable methods for purification of viral vectors. In: Viral Vectors for Gene Therapy (Volume 737) Merten O-W, Al-Rubeai M (Eds). Humana Press Inc., NJ, USA, 89–116 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Segura MdlM, Garnier A, Kamen A. Purification and characterization of retrovirus vector particles by rate zonal ultracentrifugation. J. Virol. Methods 133(1), 82–91 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Villegas GA, Arguelles MH, Castello AA, Mas NJ, Glikmann G. A rapid method to produce high yields of purified rotavirus particles. J. Virol. Methods 104(1), 9–19 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Manes NP, Estep RD, Mottaz HM et al. Comparative proteomics of human monkeypox and vaccinia intracellular mature and extracellular enveloped virions. J. Proteome Res. 7(3), 960–968 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Segura MD, Kamen A, Garnier A. Downstream processing of oncoretroviral and lentiviral gene therapy vectors. Biotechnol. Adv. 24(3), 321–337 (2006). [DOI] [PubMed] [Google Scholar]; •• Summarizes downstream processing of oncoretroviral and lentiviral vectors.
- 48.Hoekstra J, Deinhardt F. Simian sarcoma and feline leukemia virus antigens: isolation of species- and interspecies-specific proteins. Intervirology 2(4), 222–230 (1973). [DOI] [PubMed] [Google Scholar]
- 49.Harewood KR, Chang P, Higdon C, Larson D. The endogenous reverse transcriptase activity of gibbon ape lymphoma virus: characterization of the DNA product. Biochim. Biophys. Acta 407(1), 14–23 (1975). [DOI] [PubMed] [Google Scholar]
- 50.Tsuda F, Takahashi T, Takahashi K, Miyakawa Y, Mayumi M. Determination of antibody to hepatitis B core antigen by means of immune adherence hemagglutination. J. Immunol. 115(3), 834–838 (1975). [PubMed] [Google Scholar]
- 51.Iordan AG, Mazhul LA, Shcheglovitova ON, Ageeva ON. Comparative study of the biological properties if influenza virus preparations purified by methods of centrifugation and chromatography. Vopr. Virusol. 2, 150–154 (1983). [PubMed] [Google Scholar]
- 52.Okuda K, Ito K, Miyake K, Morita M, Ogonuki M. Purification of Japanese encephalitis virus vaccine by zonal centrifugation. J. Clin. Microbiol. 1(1), 96–101 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oroszlan S, Summers MR, Foreman C, Gilden RV. Murine type-c virus group-specific antigens – interstrain immunochemical, biophysical, and amino-acid sequence differences J. Virol. 14(6), 1559–1574 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drohan W, Kettmann R, Colcher D, Schlom J. Isolation of the mouse mammary tumor virus sequences not transmitted as germinal provirus in the C3H and RIII mouse strains. J. Virol. 21(3), 986–995 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burnette WN, Riggin CH, Mitchell WM. Physical and chemical properties of an oncornavirus associated with a murine adrenal carcinoma cell line. J. Virol. 14(1), 110–115 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hilfenhaus J, Köhler R, Gruschkau H. Large-scale purification of animal viruses in the RK-model zonal ultracentrifuge: III. Semliki Forest virus and vaccinia virus. J. Biol. Stand. 4(4), 285–293 (1976). [DOI] [PubMed] [Google Scholar]
- 57.Chattopadhyay SK, Hartley JW, Lander MR, Kramer BS, Rowe WP. Biochemical characterization of the amphotropic group of murine leukemia viruses. J. Virol. 26(1), 29–39 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinz FX, Kunz C, Fauma H. Preparation of a highly purified vaccine against tick-borne encephalitis by continuous flow zonal ultracentrifugation. J. Med. Virol. 6(3), 213–221 (1980). [DOI] [PubMed] [Google Scholar]
- 59.Kumar AAP, Mani KR, Palaniappan C, Bhau LNR, Swaminathan K. Purification, potency and immunogenicity analysis of Vero cell culture-derived rabies vaccine: a comparative study of single-step column chromatography and zonal centrifuge purification. Microb. Infect. 7(9–10), 1110–1116 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Smith GL, Moss B. Infectious poxvirus vectors have capacity for at least 25000 base pairs of foreign DNA. Gene 25(1), 21–28 (1983). [DOI] [PubMed] [Google Scholar]
- 61.Shibley GP, Manousos M, Munch K et al. New method for large-scale growth; and concentration of the Epstein–Barr viruses. Appl. Environ. Microbiol. 40(6), 1044–1048 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wheeler CM, Robertson BH, Van Nest G, Dina D, Bradley DW, Fields HA. Structure of the hepatitis A virion: peptide mapping of the capsid region. J. Virol. 58(2), 307–313 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gilden RV, Oroszlan S, Huebner RJ. Antigenic differentiation of M-MSV(O) from mouse, hamster, and cat C-type viruses. Virology 43(3), 722–724 (1971). [DOI] [PubMed] [Google Scholar]
- 64.Rodrigues T, Carrondo MJT, Alves PM, Cruz PE. Purification of retroviral vectors for clinical application: biological implications and technological challenges. J. Biotechnol. 127(3), 520–541 (2007). [DOI] [PubMed] [Google Scholar]; •• Comprehensive review on the purification of retroviral vectors.
- 65.Reeves L, Cornetta K. Clinical retroviral vector production: step filtration using clinically approved filters improves titers. Gene Ther. 7(23), 1993–1998 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Segura MD, Kamen A, Trudel P, Garnier A. A novel purification strategy for retrovirus gene therapy vectors using heparin affinity chromatography. Biotechnol. Bioeng. 90(4), 391–404 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Kalbfuss B, Genzel Y, Wolff M, Zimmermann A, Morenweiser R, Reichl U. Harvesting and concentration of human influenza A virus produced in serum-free mammalian cell culture for the production of vaccines. Biotechnol. Bioeng. 97(1), 73–85 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Birch JR, Racher AJ. Antibody production. Adv. Drug Del. Rev. 58(5–6), 671–685 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Kern G, Krishnan M. Virus removal by filtration: points to consider. BioPharm Int. 19(10), 32 (2006). [Google Scholar]
- 70.Roberts PL, Feldman P, Crombie D, Walker C, Lowery K. Virus removal from factor IX by filtration: validation of the integrity test and effect of manufacturing process conditions. Biologicals 38(2), 303–310 (2010). [DOI] [PubMed] [Google Scholar]
- 71.van Reis R, Brake JM, Charkoudian J, Burns DB, Zydney AL. High-performance tangential flow filtration using charged membranes. J. Membr. Sci. 159(1–2), 133–142 (1999). [Google Scholar]
- 72.Grzenia DL, Carlson JO, Wickramasinghe SR. Tangential flow filtration for virus purification. J. Membr. Sci. 321(2), 373–380 (2008). [Google Scholar]
- 73.Pujar NS, Zydney AL. Electrostatic and electrokinetic interactions during protein-transport through narrow pore membranes Ind. Eng. Chem. Res. 33(10), 2473–2482 (1994). [Google Scholar]
- 74.Saksena S, Zydney AL. Effect of solution pH and ionic-strength on the separation of albumin from immunoglobulins (IgG) by selective filtration Biotechnol. Bioeng. 43(10), 960–968 (1994). [DOI] [PubMed] [Google Scholar]
- 75.van Eijndhoven R, Saksena S, Zydney AL. Protein fractionation using electrostatic interactions in membrane filtration Biotechnol. Bioeng. 48(4), 406–414 (1995). [DOI] [PubMed] [Google Scholar]
- 76.van Reis R, Zydney A. Bioprocess membrane technology. J. Membr. Sci. 297(1–2), 16–50 (2007). [Google Scholar]
- 77.van Reis R, Gadam S, Frautschy LN et al. High performance tangential flow filtration. Biotechnol. Bioeng. 56(1), 71–82 (1997). [DOI] [PubMed] [Google Scholar]
- 78.Grzenia DL, Wickramasinghe SR, Carlson JO. Ultrafiltration of Parvovirus. Sep. Sci. Technol. 42(11), 2387–2403 (2007). [Google Scholar]
- 79.Jungbauer A, Hahn R. Polymethacrylate monoliths for preparative and industrial separation of biomolecular assemblies. J. Chromatogr. A 1184(1–2), 62–79 (2008). [DOI] [PubMed] [Google Scholar]; •• Compares conventional chromatography media and monoliths for industrial bioseparations.
- 80.Jungbauer A. Chromatographic media for bioseparation. J. Chromatogr. A 1065(1), 3–12 (2005). [DOI] [PubMed] [Google Scholar]; •• Detailed overview on chromatographic media used for bioseparations.
- 81.Gagnon P. Hydroxyapatite for biomolecule purification. Genetic Engineering & Biotechnology News 30(7), 28–29 (2010). [Google Scholar]
- 82.Konz JO, Lee AL, Lewis JA, Sagar SL. Development of a purification process for adenovirus: controlling virus aggregation to improve the clearance of host cell DNA. Biotechnol. Prog. 21(2), 466–472 (2005). [DOI] [PubMed] [Google Scholar]; • Describes the development of a purification process for adenoviruses (Ad5).
- 83.Rodrigues T, Carvalho A, Roldao A, Carrondo MJT, Alves PM, Cruz PE. Screening anion-exchange chromatographic matrices for isolation of onco-retroviral vectors. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 837(1–2), 59–68 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Chen Z, Hsu F-C, Battigelli D, Chang H-C. Capture and release of viruses using amino-functionalized silica particles. Anal. Chim. Acta 569(1–2), 76–82 (2006). [Google Scholar]
- 85.Heyward JT, Klimas RA, Stapp MD, Obijeski JF. The rapid concentration and purification of influenza virus from allantoic fluid. Arch. Virol. 55(1–2), 107–119 (1977). [DOI] [PubMed] [Google Scholar]
- 86.Schnabel R, Langer P. Controlled-pore glass as a stationary phase in chromatography. J. Chromatogr. A 544, 137–146 (1991). [Google Scholar]
- 87.Ljunglof A, Bergvall P, Bhikhabhai R, Hjorth R. Direct visualisation of plasmid DNA in individual chromatography adsorbent particles by confocal scanning laser microscopy. J. Chromatogr. A 844(1–2), 129–135 (1999). [DOI] [PubMed] [Google Scholar]
- 88.Ferreira GNM. Chromatographic approaches in the purification of plasmid DNA for therapy and vaccination. Chem. Eng. Technol. 28(11), 1285–1294 (2005). [Google Scholar]
- 89.Podgornik A, Vidic J, Jancar J, Lendero N, Frankovic V, Strancar A. Noninvasive methods for characterization of large-volume monolithic chromatographic columns. Chem. Eng. Technol. 28(11), 1435–1441 (2005). [Google Scholar]
- 90.Hahn R, Tscheliessnig A, Bauerhansl P, Jungbauer A. Dispersion effects in preparative polymethacrylate monoliths operated in radial-flow columns. J. Biochem. Biophys. Methods 70(1), 87–94 (2007). [DOI] [PubMed] [Google Scholar]
- 91.Podgornik A, Jancar J, Mihelic I, Barut M, Strancar A. Large volume monolithic stationary phases: preparation, properties, and applications. Acta Chimica Slovenica 57(1), 1–8 (2010). [PubMed] [Google Scholar]
- 92.Plieva FM, Seta ED, Galaev IY, Mattiasson B. Macroporous elastic polyacrylamide monolith columns: processing under compression and scale-up. Sep. Purif. Technol. 65(1), 110–116 (2009). [Google Scholar]
- 93.Trilisky EI, Lenhoff AM. Flow-dependent entrapment of large bioparticles in porous process media. Biotechnol. Bioeng. 104(1), 127–133 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jungbauer A, Hahn R. Monoliths for fast bioseparation and bioconversion and their applications in biotechnology. J. Sep. Sci. 27(10–11), 767–778 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Charcosset C. Review: purification of proteins by membrane chromatography. J. Chem. Technol. Biotechnol. 71(2), 95–110 (1998). [Google Scholar]
- 96.Charcosset C. Membrane processes in biotechnology: an overview. Biotechnol. Adv. 24(5), 482–492 (2006). [DOI] [PubMed] [Google Scholar]; • Comprehensive review on membrane processes in biotechnology.
- 97.Ghosh R. Protein separation using membrane chromatography: opportunities and challenges. J. Chromatogr. A 952(1–2), 13–27 (2002). [DOI] [PubMed] [Google Scholar]; • Detailed overview on protein separations via membrane adsorbers.
- 98.Vicente T, Fáber R, Alves PM, Carrondo MJT, Mota JPB. Impact of ligand density on the optimization of ion-exchange membrane chromatography for viral vector purification. Biotechnol. Bioeng. 108(6), 1347–1359 (2011). [DOI] [PubMed] [Google Scholar]
- 99.Gagnon P. The emerging generation of chromatography tools for virus purification. BioProcess Int. 6(Suppl. 6), 24, 26, 28–30 (2008). [Google Scholar]; •• Outlines the advantages of monoliths as chromatography tools for virus purification.
- 100.Wolff MW, Siewert C, Hansen SP, Faber R, Reichl U. Purification of cell culture-derived modified vaccinia ankara virus by pseudo-affinity membrane adsorbers and hydrophobic interaction chromatography. Biotechnol. Bioeng. 107(2), 312–320 (2010). [DOI] [PubMed] [Google Scholar]
- 101.Gagnon P. Monoliths emerge as key purification methodology. Genetic Engineering & Biotechnology News 28(14), 1 (2008). [Google Scholar]
- 102.Kalbfuss B, Wolff M, Geisler L et al. Direct capture of influenza A virus from cell culture supernatant with Sartobind anion-exchange membrane adsorbers. J. Membr. Sci. 299(1–2), 251–260 (2007). [Google Scholar]
- 103.Koerber JT, Jang JH, Yu JH, Kane RS, Schaffer DV. Engineering adeno-associated virus for one-step purification via immobilized metal affinity chromatography. Hum. Gene Ther. 18(4), 367–378 (2007). [DOI] [PubMed] [Google Scholar]
- 104.Opitz L, Hohlweg J, Reichl U, Wolff MW. Purification of cell culture-derived influenza virus A/Puerto Rico/8/34 by membrane-based immobilized metal affinity chromatography. J. Virol. Methods 161(2), 312–316 (2009). [DOI] [PubMed] [Google Scholar]
- 105.Opitz L, Lehmann S, Reichl U, Wolff MW. Sulfated membrane adsorbers for economic pseudo-affinity capture of influenza virus particles. Biotechnol. Bioeng. 103(6), 1144–1154 (2009). [DOI] [PubMed] [Google Scholar]
- 106.Segura MM, Kamen A, Lavoie MC, Garnier A. Exploiting heparin-binding properties of MoMLV-based retroviral vectors for affinity chromatography. J. Chromatogr. B 846(1–2), 124–131 (2007). [DOI] [PubMed] [Google Scholar]
- 107.Hu JZ, Ni YY, Dryman BA, Meng XJ, Zhang CM. Purification of porcine reproductive and respiratory syndrome virus from cell culture using ultrafiltration and heparin affinity chromatography. J. Chromatogr. A 1217(21), 3489–3493 (2010). [DOI] [PubMed] [Google Scholar]
- 108.Cameron-Smith R, Harbour C. Removal of poliovirus type 1 from a protein mixture using an immunoaffinity chromatography column. Biomed. Chromatogr. 15(7), 471–483 (2001). [DOI] [PubMed] [Google Scholar]
- 109.Williams SL, Eccleston ME, Slater NKH. Affinity capture of a biotinylated retrovirus on macroporous monolithic adsorbents: towards a rapid single-step purification process. Biotechnol. Bioeng. 89(7), 783–787 (2005). [DOI] [PubMed] [Google Scholar]
- 110.Chen GY, Chen CY, Chang MDT, Matsuura Y, Hu YC. Concanavalin A affinity chromatography for efficient baculovirus purification. Biotechnol. Prog. 25(6), 1669–1677 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Opitz L, Salaklang J, Buttner H, Reichl U, Wolff MW. Lectin-affinity chromatography for downstream processing of MDCK cell culture derived human influenza A viruses. Vaccine 25(5), 939–947 (2007). [DOI] [PubMed] [Google Scholar]
- 112.Nitsche A, Kurth A, Dunkhorst A et al. One-step selection of Vaccinia virus-binding DNA aptamers by MonoLEX. BMC Biotechnol. 7, 48 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gagnon P. Chromatographic purification of virus particles. In: Encyclopedia of Industrial Biotechnology (Volume 3) Flickinger MC (Ed.). John Wiley & Sons, Inc., NY, USA, 1591–1611 (2010). [Google Scholar]
- 114.Ye KM, Jin S, Ataai MM, Schultz JS, Ibeh J. Tagging retrovirus vectors with a metal binding peptide and one-step purification by immobilized metal affinity chromatography. J. Virol. 78(18), 9820–9827 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Patel DC, Luo RG. Protein adsorption dissociation constants in various types of biochromatography. In: Adsorption and its Application in Industry and Environmental Protection (Volume 1: Applications in Industry) Dabrowski A (Ed.). Elsevier, Amsterdam, The Netherlands (1999). [Google Scholar]
- 116.Andreadis ST, Roth CM, Le Doux JM, Morgan JR, Yarmush ML. Large-scale processing of recombinant retroviruses for gene therapy. Biotechnol. Prog. 15(1), 1–11 (1999). [DOI] [PubMed] [Google Scholar]; • Outlines critical issues on design strategies for the production, concentration, purification and formulation of recombinant retroviruses.
- 117.Wolff MW, Siewert C, Lehmann S et al. Capturing of cell culture-derived modified Vaccinia Ankara virus by ion exchange and pseudo-affinity membrane adsorbers. Biotechnol. Bioeng. 105(4), 761–769 (2010). [DOI] [PubMed] [Google Scholar]
- 118.Opitz L, Zimmermann A, Lehmann S et al. Capture of cell culture-derived influenza virus by lectins: strain independent, but host cell dependent. J. Virol. Methods 154(1–2), 61–68 (2008). [DOI] [PubMed] [Google Scholar]
- 119.Graumann K, Ebenbichler AA. Development and scale up of preparative HIC for the purification of a recombinant therapeutic protein. Chem. Eng. Technol. 28(11), 1398–1407 (2005). [Google Scholar]
- 120.Kramarczyk JF, Kelley BD, Coffman JL. High-throughput screening of chromatographic separations: II. Hydrophobic interaction. Biotechnol. Bioeng. 100(4), 707–720 (2008). [DOI] [PubMed] [Google Scholar]
- 121.Lu Y, Williamson B, Gillespie R. Recent advancement in application of hydrophobic interaction chromatography for aggregate removal in industrial purification process. Curr. Pharm. Biotechnol. 10(4), 427–433 (2009). [DOI] [PubMed] [Google Scholar]
- 122.Wang FH, Dai XJ, Gong BL. New method for preparation of hydrophobic interaction chromatographic stationary phases based on polymer-grafted silica and their chromatographic properties. J. Appl. Polym. Sci. 118(3), 1513–1519 (2010). [Google Scholar]
- 123.O’Riordan CR, Lachapelle AL, Vincent KA, Wadsworth SC. Scaleable chromatographic purification process for recombinant adeno-associated virus (rAAV). J. Gene Med. 2(6), 444–454 (2000). [DOI] [PubMed] [Google Scholar]
- 124.Kuiper M, Sanches RM, Walford JA, Slater NKH. Purification of a functional gene therapy vector derived from Moloney murine leukaemia virus using membrane filtration and ceramic hydroxyapatite chromatography. Biotechnol. Bioeng. 80(4), 445–453 (2002). [DOI] [PubMed] [Google Scholar]
- 125.Kessler LC, Gueorguieva L, Rinas U, Seidel-Morgenstern A. Step gradients in 3-zone simulated moving bed chromatography: application to the purification of antibodies and bone morphogenetic protein-2. J. Chromatogr. A 1176(1–2), 69–78 (2007). [DOI] [PubMed] [Google Scholar]
- 126.Juza M, Mazzotti M, Morbidelli M. Simulated moving-bed chromatography and its application to chirotechnology. Trends Biotechnol. 18(3), 108–118 (2000). [DOI] [PubMed] [Google Scholar]
- 127.Kessler LC, Seidel-Morgenstern A. Theoretical study of multicomponent continuous countercurrent chromatography based on connected 4-zone units. J. Chromatogr. A 1126(1–2), 323–337 (2006). [DOI] [PubMed] [Google Scholar]
- 128.Abel S, Babler MU, Arpagaus C, Mazzotti M, Stadler J. Two-fraction and three-fraction continuous simulated moving bed separation of nucleosides. J. Chromatogr. A 1043(2), 201–210 (2004). [DOI] [PubMed] [Google Scholar]
- 129.Paredes G, Abel S, Mazzotti M, Morbidelli M, Stadler J. Analysis of a simulated moving bed operation for three-fraction separations (3F-SMB). Ind. Eng. Chem. Res. 43(19), 6157–6167 (2004). [Google Scholar]
- 130.Coffman JL, Kramarczyk JF, Kelley BD. High-throughput screening of chromatographic separations: I. Method development and column modeling. Biotechnol. Bioeng. 100(4), 605–618 (2008). [DOI] [PubMed] [Google Scholar]; •• One article of a series (four publications) covering in detail high-throughput screening of chromatographic separations.
- 131.Vicente T, Mota JPB, Peixoto C, Alves PM, Carrondo MJT. Modeling protein binding and elution over a chromatographic surface probed by surface plasmon resonance. J. Chromatogr. A 1217(13), 2032–2041 (2010). [DOI] [PubMed] [Google Scholar]
- 132.Vicente T, Peixoto C, Alves PM, Carrondo MJT. Modeling electrostatic interactions of baculovirus vectors for ion-exchange process development. J. Chromatogr. A 1217(24), 3754–3764 (2010). [DOI] [PubMed] [Google Scholar]
- 133.Petrides DP, Koulouris A, Lagonikos PT. The role of process simulation in pharmaceutical process development and product commercialization. Pharmaceutical Engineering 22(1), 1–8 (2002). [Google Scholar]; • Describes the application of batch process simulators to expedite the development of pharmaceutical products.
- 134.Neumann K, Schwindt C, Trautmann N. Advanced production scheduling for batch plants in process industries. Or Spectrum 24(3), 251–279 (2002). [Google Scholar]
- 135.Brooks CA, Cramer SM. Steric mass-action ion exchange: displacement profiles and induced salt gradients. AICHE J. 38(12), 1969–1978 (1992). [Google Scholar]
- 136.Vicente T, Sousa MFQ, Peixoto C, Mota JPB, Alves PM, Carrondo MJT. Anion-exchange membrane chromatography for purification of rotavirus-like particles. J. Membr. Sci. 311(1–2), 270–283 (2008). [Google Scholar]
- 137.Gottschalk U. Disposables in downstream processing. In: Disposable Bioreactors (Volume 115) Eibl R, Eibl D (Eds). Springer-Verlag, Heidelberg, Germany, 171–183 (2009). [DOI] [PubMed] [Google Scholar]
- 138.Nakamae T, Tanaka N, Nakanishi K et al. Chondroitinase ABC promotes corticospinal axon growth in organotypic cocultures. Spinal Cord 47(2), 161–165 (2009). [DOI] [PubMed] [Google Scholar]
- 139.World Health Organization. WHO Expert Commitee on Biological Standardization. World Health Organ. Tech. Rep. Ser. 878(i–vi), 1–101 (1998). [PubMed] [Google Scholar]
- 140.Sheng-Fowler L, Lewis Jr AM, Peden K. Issues associated with residual cell-substrate DNA in viral vaccines. Biologicals 37(3), 190–195 (2009). [DOI] [PubMed] [Google Scholar]
- 141.Iznaga AB, Frontera MC, Uramis JR, Gonzalez JBT, Parra YC, Gonzalez YM. DNA removal from a purification process of recombinant hepatitis B surface antigen. Electron. J. Biotechnol. 10(1), 37–47 (2007). [Google Scholar]
- 142.Knezevic I, Stacey G, Petricciani J. WHO Study Group on cell substrates for production of biologicals, Geneva, Switzerland, 11–12 June 2007. Biologicals 36(3), 203–211 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Urabe M, Xin KQ, Obara Y et al. Removal of empty capsids from type 1 adeno-associated virus vector stocks by anion-exchange chromatography potentiates transgene expression. Mol. Ther. 13(4), 823–828 (2006). [DOI] [PubMed] [Google Scholar]
- 144.Vellekamp G, Porter FW, Sutjipto S et al. Empty capsids in column-purified recombinant adenovirus preparations. Hum. Gene Ther. 12(15), 1923–1936 (2001). [DOI] [PubMed] [Google Scholar]
- 145.Whitfield RJ, Battom SE, Barut M, Gilham DE, Ball PD. Rapid high-performance liquid chromatographic analysis of adenovirus type 5 particles with a prototype anion-exchange analytical monolith column. J. Chromatogr. A 1216(13), 2725–2729 (2009). [DOI] [PubMed] [Google Scholar]
- 146.Lusky M. Good manufacturing practice production of adenoviral vectors for clinical trials. Hum. Gene Ther. 16(3), 281–291 (2005). [DOI] [PubMed] [Google Scholar]; • Overview on GMP production processes for adenoviral vectors including a section focusing on GMP regulations in the USA and Europe.
- 147.Peixoto C, Ferreira TB, Sousa MFQ, Carrondo MJT, Alves PM. Towards purification of adenoviral vectors based on membrane technology. Biotechnol. Prog. 24(6), 1290–1296 (2008). [DOI] [PubMed] [Google Scholar]; • Describes the purification of adenoviral vectors based on a combination of filtration techniques and membrane adsorbers.
- 148.Altaras NE, Aunins JG, Evans RK, Kamen A, Konz JO, Wolf JJ. Production and formulation of adenovirus vectors. In: Gene Therapy and Gene Delivery Systems (Volume 99) Scheper T (Ed.). Springer-Verlag, Berlin, Germany, 193–260 (2005). [DOI] [PubMed] [Google Scholar]
- 149.Eglon MN, Duffy AM, O’Brien T, Strappe PM. Purification of adenoviral vectors by combined anion exchange and gel filtration chromatography. J. Gene Med. 11(11), 978–989 (2009). [DOI] [PubMed] [Google Scholar]
- 150.Duffy AM, O’Doherty AM, O’Brien T, Strappe PM. Purification of adenovirus and adeno-associated virus: comparison of novel membrane-based technology to conventional techniques. Gene Ther. 12, S62–S72 (2005). [DOI] [PubMed] [Google Scholar]
- 151.Okada T, Nonaka-Sarukawa M, Uchibori R et al. Scalable purification of adeno-associated virus serotype 1 (AAV1) and AAV8 vectors, using dual ion-exchange adsorptive membranes. Hum. Gene Ther. 20(9), 1013–1021 (2009). [DOI] [PubMed] [Google Scholar]
- 152.Rodrigues T, Carvalho A, Carmo M, Carrondo MJT, Alves PM, Cruz PE. Scateabte purification process for gene therapy retroviral vectors. J. Gene Med. 9(4), 233–243 (2007). [DOI] [PubMed] [Google Scholar]
- 153.Lesch HP, Laitinen A, Peixoto C et al. Production and purification of lentiviral vectors generated in 293T suspension cells with baculoviral vectors. Gene Ther. 18(6), 531–538 (2011). [DOI] [PubMed] [Google Scholar]
- 154.Working PK, Lin A, Borellini F. Meeting product development challenges in manufacturing clinical grade oncolytic adenoviruses. Oncogene 24(52), 7792–7801 (2005). [DOI] [PubMed] [Google Scholar]; • Describes the up- and down-stream process in detail of clinical grade oncolytic adenoviruses.
- 155.Wolff MW, Reichl U. Downstream processing: from egg to cell culture-derived influenza virus particles. Chem. Eng. Technol. 31(6), 846–857 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Summary on downstream processing methods and purification trains for egg-derived and cell culture-derived influenza viruses.
- 156.Kalashnikova I, Ivanova N, Tennikova T. Development of a strategy of influenza virus separation based on pseudoaffinity chromatography on short monolithic columns. Anal. Chem. 80(6), 2188–2198 (2008). [DOI] [PubMed] [Google Scholar]
- 157.Plotkin SA. Vaccination against the major infectious diseases. Comptes Rendus De L Academie Des Sciences Serie Iii-Sciences De La Vie-Life Sciences 322(11), 943–951 (1999). [DOI] [PubMed] [Google Scholar]
- 158.Marcum JA. From the molecular genetics revolution to gene therapy: translating basic research into medicine. J. Lab. Clin. Med. 146(6), 312–316 (2005). [DOI] [PubMed] [Google Scholar]
- 159.Le Doux JM, Morgan JR, Yarmush ML. Removal of proteoglycans increases efficiency of retroviral gene transfer. Biotechnol. Bioeng. 58(1), 23–34 (1998). [DOI] [PubMed] [Google Scholar]
- 160.Le Doux JM, Morgan JR, Snow RG, Yarmush ML. Proteoglycans secreted by packaging cell lines inhibit retrovirus infection. J. Virol. 70(9), 6468–6473 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Slingsby JH, Baban D, Sutton J et al. Analysis of 4070A envelope levels in retroviral preparations and effect on target cell transduction efficiency. Hum. Gene Ther. 11(10), 1439–1451 (2000). [DOI] [PubMed] [Google Scholar]
- 162.Pedro L, Soares SS, Ferreira GNM. Purification of bionanoparticles. Chem. Eng. Technol. 31(6), 815–825 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Grzenia DL, Carlson JO, Czermak P, Han BB, Specht RK, Wickramasinghe SR. Purification of densonucleosis virus by tangential flow ultrafiltration. Biotechnol. Prog. 22(5), 1346–1353 (2006). [DOI] [PubMed] [Google Scholar]
- 164.Pfeifer A, Verma IM. Gene therapy: promises and problems. Annu. Rev. Genomics Hum. Genet. 2(1), 177–211 (2001). [DOI] [PubMed] [Google Scholar]
- 165.Salo RJ, Mayor HD. Isoelectric-focusing of parvoviruses. Intervirology 10(2), 87–93 (1978). [DOI] [PubMed] [Google Scholar]
- 166.Harrap KA. The structure of nuclear polyhedrosis viruses: II. The virus particle. Virology 50(1), 124–132 (1972). [DOI] [PubMed] [Google Scholar]
- 167.Ayres MD, Howard SC, Kuzio J, Lopezferber M, Possee RD. The complete DNA-sequence of Autographa-Californica nuclear polyhedrosis-virus. Virology 202(2), 586–605 (1994). [DOI] [PubMed] [Google Scholar]
- 168.Yang Y, Lo S-L, Yang J et al. Polyethylenimine coating to produce serum-resistant baculoviral vectors for in vivo gene delivery. Biomaterials 30(29), 5767–5774 (2009). [DOI] [PubMed] [Google Scholar]
- 169.Bujarski JJ. Bromovirus Isolation and RNA Extraction In: Methods in Molecular Biology: Plant Virology Protocols: From Virus Isolation to Transgenic Resistance (Volume 81) Foster GD, Taylor SC (Eds). Humana Press Inc., NJ, USA, 183–188 (1998). [DOI] [PubMed] [Google Scholar]
- 170.Condit R. Principles of Virology. In: Fields Virology (Volume 1) Knipe D, Howley P (Eds). Lippincott Williams & Wilkins, PA, USA, 21 (2001). [Google Scholar]
- 171.Westaway EG, Brinton MA, Gaidamovich SY et al.Flaviviridae Intervirology 24(4), 183–192 (1985). [DOI] [PubMed] [Google Scholar]
- 172.Melnick JL. Properties and classification of hepatitis-A virus Vaccine 10, S24–S26 (1992). [DOI] [PubMed] [Google Scholar]
- 173.Nasser A, Battagelli D, Sobsey M. Isoelectric focusing of hepatitis A virus in sucrose gradients. Israel J. Med. Sci. 28, 73 (1992). [Google Scholar]
- 174.Liang TJ. Hepatitis B: the virus and disease. Hepatology 49(S5), S13–S21 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Purcell R. The hepatitis C virus: overview. Hepatology 26(S3), 11S–14S (1997). [DOI] [PubMed] [Google Scholar]
- 176.Chang JT, Schmid MF, Rixon FJ, Chiu W. Electron cryotomography reveals the portal in the herpesvirus capsid. J. Virol. 81(4), 2065–2068 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Granter A, Wille M, Whitney H, Robertson G, Ojkic D, Lang A. The genome sequence of an H11N2 avian influenza virus from a thick-billed murre (Uria lomvia) shows marine-specific and regional patterns of relationships to other viruses. Virus Genes 41(2), 224–230 (2010). [DOI] [PubMed] [Google Scholar]
- 178.Dea S, Elazhary MA, Roy RS. Les virus influenza chez l’homme et les animaux. Une revue de la littérature. Can. Vet. J. 21, 171–178 (1980). [PMC free article] [PubMed] [Google Scholar]
- 179.Bruce EA, Digard P, Stuart AD. The Rab11 pathway is required for influenza A virus budding and filament formation. J. Virol. 84(12), 5848–5859 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhilinskaya IN, El-Saed LH, Ivanova NA, Golubev DE, Sokolov NN. Isolation of A2-Singapore-57 influenza virus V and S antigens by isoelectric focusing. Acta Virol. 16(5), 436–439 (1972). [PubMed] [Google Scholar]
- 181.Brydak L. Studies on the adaptation of influenza-virus replication at low temperature. 5. Isoelectric-focusing studies Acta Microbiol. Pol. 42(1), 29–33 (1993). [PubMed] [Google Scholar]
- 182.Molodkina LM, Molodkin VM, Vostryukhina OA, Kolikov VM, Golikova EV, Chernoberezhskii YM. Study of the electrophoretic mobility of A1 (Leningrad) and A3 (Leningrad) influenza-viruses Colloid Journal of the USSR 48(1), 66–70 (1986). [Google Scholar]
- 183.Miller G, Lauffer M, Stanley W. Electrophoretic studies on PR8 influenza virus. J. Exp. Med. 80(6), 549–559 (1944). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Misra UK, Kalita J. Overview: Japanese encephalitis. Prog. Neurobiol. 91(2), 108–120 (2010). [DOI] [PubMed] [Google Scholar]
- 185.Kitano T, Suzuki K, Yamaguchi T. Morphological, chemical, and biological characterization of Japanese encephalitis virus virion and its hemagglutinin. J. Virol. 14(3), 631–639 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kafri T. Gene delivery by lentivirus vectors an overview. Methods Mol. Biol. 246, 367–390 (2004). [DOI] [PubMed] [Google Scholar]
- 187.Berghäll H, Sirén J, Sarkar D et al. The interferon-inducible RNA helicase, MDA-5, is involved in measles virus-induced expression of antiviral cytokines. Microb. Infect. 8(8), 2138–2144 (2006). [DOI] [PubMed] [Google Scholar]
- 188.Kim HS, Jung SH, Kim SH et al. High-throughput analysis of mumps virus and the virus-specific monoclonal antibody on the arrays of a cationic polyelectrolyte with a spectral SPR biosensor. Proteomics 6(24), 6426–6432 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Corbin A, Darlix JL. Functions of the 5´ leader of murine leukemia virus genomic RNA in virion structure, viral replication and pathogenesis, and MLV-derived vectors. Biochimie 78(7), 632–638 (1996). [DOI] [PubMed] [Google Scholar]
- 190.Cobo F, Concha Á, Marta Ortiz M. Human papillomavirus (HPV) type distribution in females with abnormal cervical cytology. A correlation with histological study. Open Virol. J. 3, 60–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Beard JW, Wyckoff RWG. The pH stability of the papilloma virus protein. J. Biol. Chem. 123(2), 461–470 (1938). [Google Scholar]
- 192.Oshima KH, Evansstrickfaden TT, Highsmith AK, Ades EW. The removal of phages T1 and PP7, and poliovirus from fluids with hollow-fiber ultrafilters with molecular-weight cutoffs of 50000, 13000, and 6000. Can. J. Microbiol. 41(4–5), 316–322 (1995). [DOI] [PubMed] [Google Scholar]
- 193.La Colia P, Marcialis MA, Mereu GP, Loddo B. Behaviour of a guanidine-dependent strain of poliovirus 1 in sucrose density and pH gradients. Cell. Mol. Life Sci. 28(9), 1115–1117 (1972). [DOI] [PubMed] [Google Scholar]
- 194.Mandel B. Characterization of type 1 poliovirus by electrophoretic analysis. Virology 44(3), 554–568 (1971). [DOI] [PubMed] [Google Scholar]
- 195.Butler M, Medlen AR, Taylor GR. Electrofocusing of viruses and sensitivity to desinfection Water Sci. Technol. 17(10), 201–210 (1985). [Google Scholar]
- 196.Floyd R, Sharp DG. Viral aggregation – effects of salts on aggregation of poliovirus and reovirus at low-pH Appl. Environ. Microbiol. 35(6), 1084–1094 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Simani S, Gholami A, Farahtaj F, Yousefi-Behzadi M, Fayaz A. Epidemiological survay of different rabies virus strains in Iran. J. Sci. IR Iran 12(4), 315–319 (2001). [Google Scholar]
- 198.Best J, Banatvala JE, Almeida J, Waterson AP. Morphological characteristics of rubella virus. Lancet 290(7509), 237–239 (1967). [Google Scholar]
- 199.Tzeng W-P, Frey TK. Mapping the rubella virus subgenomic promoter. J. Virol. 76(7), 3189–3201 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lundstrom K. Biology and application of alphaviruses in gene therapy. Gene Ther. 12, S92–S97 (2005). [DOI] [PubMed] [Google Scholar]
- 201.Ehrengruber MU, Lundstrom K. Alphaviruses: semliki forest virus and sindbis virus vectors for gene transfer into neurons. In: Current Protocols in Neuroscience Gerfen C, Holmes A, Sibley D, Skolnick P, Wray S (Eds). John Wiley & Sons, Inc., NY, USA (2001). [Google Scholar]
- 202.Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia ankara strain: comparison with other orthopoxviruses. Virology 244(2), 365–396 (1998). [DOI] [PubMed] [Google Scholar]
- 203.Carrascosa JL, Chichon FJ, Pereiro E et al. Cryo-x-ray tomography of vaccinia virus membranes and inner compartments. J. Struct. Biol. 168(2), 234–239 (2009). [DOI] [PubMed] [Google Scholar]
- 204.Mouillot L, Netter R. Identification of orthopox virus by isoelectrofocusing in a granulated gel. Ann. Microbiol. (Paris) B128(3), 417–419 (1977). [PubMed] [Google Scholar]
- 205.Douglas HW, Williams BL, Rondle CJM. Micro-electrophoresis of pox viruses in molar sucrose. J. Gen. Virol. 5(3), 391–396 (1969). [DOI] [PubMed] [Google Scholar]
- 206.Douglas HW, Rondle CJM, Williams BL. Micro-electrophoresis of cowpox and Vaccinia viruses in molar sucrose. J. Gen. Microbiol. 42(1), 107–113 (1966). [DOI] [PubMed] [Google Scholar]
- 207.Taylor DH, Bosmann HB. Measurement of the electrokinetic properties of Vaccinia and Reovirus by laser-illuminated whole-particle micro-electrophoresis J. Virol. Methods 2(5), 251–260 (1981). [DOI] [PubMed] [Google Scholar]
- 208.Michen B, Graule T. Isoelectric points of viruses. J. Appl. Microbiol. 109(2), 388–397 (2010). [DOI] [PubMed] [Google Scholar]; • Detailed collection on isoelectric points of viruses.
- 209.Goerke AR, To BC, Lee AL, Sagar SL, Konz JO. Development of a novel adenovirus purification process utilizing selective precipitation of cellular DNA. Biotechnol. Bioeng. 91(1), 12–21 (2005). [DOI] [PubMed] [Google Scholar]
- 210.Ugai H, Yamasaki T, Hirose M et al. Purification of infectious adenovirus in two hours by ultracentrifugation and tangential flow filtration. Biochem. Biophys. Res. Commun. 331(4), 1053–1060 (2005). [DOI] [PubMed] [Google Scholar]
- 211.Subramanian S, Altaras GM, Chen J, Hughes BS, Zhou W, Altaras NE. Pilot-scale adenovirus seed production through concurrent virus release and concentration by hollow fiber filtration. Biotechnol. Prog. 21(3), 851–859 (2005). [DOI] [PubMed] [Google Scholar]
- 212.Michel JP, Gingery M, Lavelle L. Efficient purification of bromoviruses by ultrafiltration. J. Virol. Methods 122(2), 195–198 (2004). [DOI] [PubMed] [Google Scholar]
- 213.Guo YF, Cheng AC, Wang MS, Zhou Y. Purification of anatid herpesvirus 1 particles by tangential-flow ultrafiltration and sucrose gradient ultracentrifugation. J. Virol. Methods 161(1), 1–6 (2009). [DOI] [PubMed] [Google Scholar]
- 214.Gracia-Valenzuela MH, Coronado-Molina D, Hernandez-Lopez J, Gollas-Galvan T. A simple method for purifying the white spot syndrome virus using ultrafiltration. Aquacult. Res. 40(6), 737–743 (2009). [Google Scholar]
- 215.Nayak DP, Lehmann S, Reichl U. Downstream processing of MDCK cell-derived equine influenza virus. J. Chromatogr. B 823(2), 75–81 (2005). [DOI] [PubMed] [Google Scholar]
- 216.Wickramasinghe SR, Kalbfuss B, Zimmermann A, Thom V, Reichl U. Tangential flow microfiltration and ultrafiltration for human influenza A virus concentration and purification. Biotechnol. Bioeng. 92(2), 199–208 (2005). [DOI] [PubMed] [Google Scholar]
- 217.Czermak P, Grzenia DL, Wolf A et al. Purification of the densonucleosis virus by tangential flow ultrafiltration and by ion exchange membranes. Desalination 224(1–3), 23–27 (2008). [Google Scholar]
- 218.Hensgen MI, Czermak P, Carlson JO, Wickramasinghe SR. Purification of minute virus of mice using high performance tangential flow filtration. Desalination 250(3), 1121–1124 (2010). [Google Scholar]
- 219.Paul RW, Morris D, Hess BW, Dunn J, Overell RW. Increased viral titer through concentration of viral harvests from retroviral packaging lines. Hum. Gene Ther. 4(5), 609–615 (1993). [DOI] [PubMed] [Google Scholar]
- 220.Kotani H, Newton PB, Zhang SY et al. Improved methods of retrovrral vector transduction and production for gene-therapy. Hum. Gene Ther. 5(1), 19–28 (1994). [DOI] [PubMed] [Google Scholar]
- 221.Makino M, Ishikawa G, Yamaguchi K et al. Concentration of live retrovirus with a regenerated cellulose hollow-fiber, BMM. Arch. Virol. 139(1–2), 87–96 (1994). [DOI] [PubMed] [Google Scholar]
- 222.Koldej R, Cmielewski P, Stocker A, Parsons DW, Anson DS. Optimisation of a multipartite human immunodeficiency virus based vector system; control of virus infectivity and large-scale production. J. Gene Med. 7(11), 1390–1399 (2005). [DOI] [PubMed] [Google Scholar]
- 223.Kamen A, Henry O. Development and optimization of an adenovirus production process. J. Gene Med. 6, S184–S192 (2004). [DOI] [PubMed] [Google Scholar]
- 224.Peixoto C, Ferreira TB, Carrondo MJT, Cruz PE, Alves PM. Purification of adenoviral vectors using expanded bed chromatography. J. Virol. Methods 132(1–2), 121–126 (2006). [DOI] [PubMed] [Google Scholar]
- 225.Transfiguracion J, Jorio H, Meghrous J, Jacob D, Kamen A. High yield purification of functional baculovirus vectors by size exclusion chromatography. J. Virol. Methods 142(1–2), 21–28 (2007). [DOI] [PubMed] [Google Scholar]
- 226.Wu CX, Soh KY, Wang S. Ion-exchange membrane chromatography method for rapid and efficient purification of recombinant baculovirus and baculovirus gp64 protein. Hum. Gene Ther. 18(7), 665–672 (2007). [DOI] [PubMed] [Google Scholar]
- 227.Zahn A, Allain JP. Hepatitis C virus and hepatitis B virus bind to heparin: purification of largely IgG-free virions from infected plasma by heparin chromatography. J. Gen. Virol. 86, 677–685 (2005). [DOI] [PubMed] [Google Scholar]
- 228.Kalbfuss B, Wolff M, Morenweiser R, Reichl U. Purification of cell culture-derived human influenza A virus by size-exclusion and anion-exchange chromatography. Biotechnol. Bioeng. 96(5), 932–944 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Opitz L, Lehmann S, Zimmermann A, Reichl U, Wolff MW. Impact of adsorbents selection on capture efficiency of cell culture derived human influenza viruses. J. Biotechnol. 131(3), 309–317 (2007). [DOI] [PubMed] [Google Scholar]
- 230.Han BB, Specht R, Wickramasinghe SR, Carlson JO. Binding Aedes aegypti densonucleosis virus to ion exchange membranes. J. Chromatogr. A 1092(1), 114–124 (2005). [DOI] [PubMed] [Google Scholar]
- 231.Specht R, Han BB, Wickramasinghe SR et al. Densonucleosis virus purification by ion exchange membranes. Biotechnol. Bioeng. 88(4), 465–473 (2004). [DOI] [PubMed] [Google Scholar]
- 232.Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther. 17(11), 1888–1896 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Smith RH, Ding CT, Kotin RM. Serum-free production and column purification of adeno-associated virus type 5. J. Virol. Methods 114(2), 115–124 (2003). [DOI] [PubMed] [Google Scholar]
- 234.Davidoff AM, Ng CYC, Sleep S et al. Purification of recombinant adeno-associated virus type 8 vectors by ion exchange chromatography generates clinical grade vector stock. J. Virol. Methods 121(2), 209–215 (2004). [DOI] [PubMed] [Google Scholar]
- 235.Slepushkin V, Chang N, Cohen R et al. Large-scale purification of a lentiviral vector by size exclusion chromatography or mustang q ion exchange capsule. BioProcess. J. 2, 89–95 (2003). [Google Scholar]
- 236.Transfiguracion J, Jaalouk DE, Ghani K, Galipeau J, Kamen A. Size-exclusion chromatography purification of high-titer vesicular stomatitis virus G glycoprotein-pseudotyped retrovectors for cell and gene therapy applications. Hum. Gene Ther. 14(12), 1139–1153 (2003). [DOI] [PubMed] [Google Scholar]
- 237.Williams SL, Nesbeth D, Darling DC, Farzaneh F, Slater NKH. Affinity recovery of Moloney murine leukaemia virus. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 820(1), 111–119 (2005). [DOI] [PubMed] [Google Scholar]
- 238.Chtioui M, Trabelsi K, Kallel H. Purification of rabies virus produced on vero cells using chromatography techniques. In: Purification of Rabies Virus Produced on Vero Cells Using Chromatography Techniques Smith R (Ed.). Springer-Verlag, NY, USA (2007). [Google Scholar]
Patents
- 301.GlaxoSmithKline Biologicals SA. WO2010089339-A1 (2010).
- 302.Merck & Co., Inc. WO2003097797-A2 (2003).
- 303.Baxter Healthcare SA. WO2003049766-A2 (2003).
- 304.Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. WO2008125361- A1 (2008).
- 305.Bavarian Nordic. A/S US 20100119552-A1 (2010).
- 306.BIA-Separations D.O.O. and Greenhills Biotechnology GmbH. WO2008006780-A1 (2008).
Websites
- 401.Human vaccines produced with animal cell technology (status 2010) www.actip.org/pages/vaccinestable.html (Accessed March 2011)
- 402.Complete List of Vaccines Licensed for Immunization and Distribution in the US www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm093833.htm (Accessed March 2011)
- 403.European Medicines Agency: human medicines www.ema.europa.eu/ema/index.jsp?curl=pages/includes/medicines/medicines_landing_page.jsp&murl=menus/medicines/medicines.jsp&mid=WC0b01ac058001d124 (Accessed March 2011)