

Summary
RIG-I and MDA5 are cytosolic RNA sensors that play a critical role in innate antiviral responses. Major advances have been made in identifying RIG-I ligands, but our knowledge of the ligands for MDA5 remains restricted to data from transfection experiments mostly using poly(I:C), a synthetic dsRNA mimic. Here, we dissected the IFN-α/β-stimulatory activity of different viral RNA species produced during picornavirus infection, both by RNA transfection and in infected cells in which specific steps of viral RNA replication were inhibited. Our results show that the incoming genomic plus-strand RNA does not activate MDA5, but minus-strand RNA synthesis and production of the 7.5 kbp replicative form trigger a strong IFN-α/β response. IFN-α/β production does not rely on plus-strand RNA synthesis and thus generation of the partially double-stranded replicative intermediate. This study reports MDA5 activation by a natural RNA ligand under physiological conditions.
Graphical Abstract
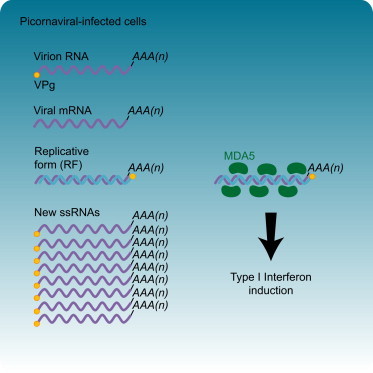
Highlights
► Viral ssRNA, with or without the 5′ VPg peptide, does not induce IFN-α/β ► Synthesis of minus-strand—but not plus-strand—RNA is crucial for MDA5 activation ► The picornavirus RF activates MDA5 both in vitro and in vivo ► MDA5 activation by viral replicative form is independent of the terminal groups of this RNA
MDA5 is a cytosolic RNA sensor and a crucial player in innate antiviral responses. However, our knowledge of its ligands, especially physiological ligands, remains restricted. Here, van Kuppeveld and colleagues dissected the MDA5-stimulatory activity of different picornavirus RNA species produced during infection. They show that the picornavirus double-stranded replicative form directly binds to and activates MDA5. This study shows MDA5 activation by a natural RNA ligand.
Introduction
RIG-I-like receptors (RLRs) are ubiquitously expressed cytoplasmic pathogen recognition receptors. The RLR family of proteins are DExD/H box RNA helicases, and two of its members, namely retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), specialize in detecting viral RNAs in infected cells (reviewed in Kato et al., 2011). Upon ligand recognition, RIG-I and MDA5 interact with a mitochondrion-anchored adaptor molecule, MAVS, which activates kinase complexes eventually leading to the transcription of type I interferons (IFN-α/β) and other proinflammatory cytokine genes (Kato et al., 2011).
RIG-I and MDA5 play differential roles in virus recognition. RIG-I recognizes most single-stranded (ss) RNA viruses investigated to date, including all minus-strand RNA [(−)RNA] viruses (e.g., influenza virus, Sendai virus, and vesicular stomatitis virus) and some plus-strand RNA [(+)RNA] viruses (e.g., hepatitis C virus and Japanese encephalitis virus) (Kato et al., 2011). MDA5 recognizes some other (+)RNA viruses, namely encephalomyocarditis virus (EMCV) (Gitlin et al., 2006; Kato et al., 2006), Theiler’s murine encephalomyelitis virus (TMEV) (Pichlmair et al., 2009), coxsackievirus B3 (CVB3) (Wang et al., 2010)—all of which are members of the Picornavirus family—and mouse norovirus (McCartney et al., 2008). Other ssRNA viruses, such as dengue virus, West Nile virus, mouse hepatitis virus, and several paramyxoviruses, are recognized by both RIG-I and MDA5 (Kato et al., 2011). Recognition of a double-stranded (ds) RNA virus, reovirus, has also been shown to involve both of these RLRs (Kato et al., 2008).
The differential recognition of viruses by RIG-I and MDA5 has been attributed to their distinct preferences for RNA ligands. RIG-I can be activated by 5′-triphosphate (ppp)-containing RNAs as well as short (<2 kbp) dsRNAs (Kato et al., 2011). The ligand specificity of RIG-I provides an explanation for how it differentiates viral RNAs from cellular RNAs. Many RNA viruses carry genomes that contain 5′ppp or produce 5′ppp-containing RNAs during their replication cycle in the cytoplasm of infected cells, whereas cytoplasmic cellular RNAs generally lack 5′ppp. The ligands of MDA5 are as yet poorly defined. MDA5 is activated by transfection of a long (>2 kbp), synthetic dsRNA analog of artificial sequence, namely polyinosinic:polycytidylic [poly(I:C)]. Additionally, transfection-based experiments showed that the L segments of the reovirus genome (∼3.9 kbp dsRNAs) induce an IFN-β response that is partially dependent on MDA5 (Kato et al., 2006). Based on these findings, it is believed that MDA5 recognizes long dsRNAs, which would be intrinsically “nonself.” However, little is known about the identities and characteristics of physiological ligands of MDA5 in infected cells.
Many (+)RNA viruses, including picornaviruses, produce dsRNAs during infection. Picornaviridae is a large, highly diverse family of human and animal viruses, including many pathogens of great medical and/or economical significance. The picornavirus genome is a 7.5–8.5 kb ssRNA molecule that harbors a single open reading frame flanked by structured 5′ and 3′ nontranslated regions and a poly(A) tail at the 3′ end. The 5′ terminus contains a small (20–24 aa) viral peptide, VPg, linked via an unusual tyrosine-RNA phosphodiester bond. Upon virus entry and uncoating, the virion RNA (vRNA) is released into the cytoplasm where it is directly translated. Viral RNA templates used for protein synthesis no longer contain VPg due to the activity of a yet unknown cellular enzyme (further referred to as unlinkase) (Rozovics et al., 2011). The viral polyprotein is proteolytically processed to release the viral proteins that engage in genomic RNA replication. During this complex process, several species of viral RNAs with unique features are generated (Figure 2A). First, the viral genomic ssRNA is transcribed into a complementary (−)RNA by the virally encoded RNA-dependent RNA polymerase using VPg as primer, yielding a 7.5 kbp dsRNA named the replicative form (RF). The (−)RNA is subsequently used as a template for VPg-primed synthesis of large amounts of new (+)RNAs, which are identical to vRNA. Newly synthesized (+)RNAs either enter a new cycle of translation and RNA replication or are encapsidated to form new virion particles. During the process of (+)RNA synthesis, a partially ds replicative intermediate (RI)—consisting of multiple incomplete (+)RNAs undergoing active transcription along the full-length (−)RNA—is formed.
Figure 2.
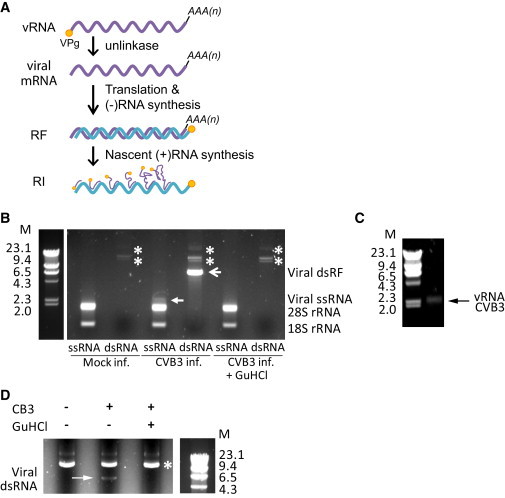
Enrichment of ssRNA and dsRNA Fractions of CVB3-Infected Cells
(A) Viral RNA species produced during picornavirus infection. RF, replicative form. RI, replication intermediate. Purple line, (+)RNA; blue line, (−)RNA; orange circle, VPg.
(B) HeLa cells were infected with CVB3 in the absence or presence of replication inhibitor GuHCl (2 mM) for 5 hr and total RNA was extracted. ssRNA and dsRNA fractions were separated by LiCl differential precipitation and examined on agarose gel. Note that the ssRNA fraction was diluted 400-fold relative to the dsRNA fraction to load comparable amounts of RNAs. M, dsDNA marker with indicated size in kbp; closed arrow, viral ssRNA; arrowhead, viral dsRNA; asterisk, unknown bands.
(C) vRNA purified from pelleted virus particles was visualized on agarose gel next to the same DNA marker as used in (A).
(D) HeLa cells were infected with CVB3 (MOI 100) in the absence or presence of GuHCl (2 mM) for 2.5 hr and the dsRNA fraction was purified from total RNA extract by LiCl precipitation and examined on gel. M, dsDNA marker; arrow, viral dsRNA; asterisk, unknown bands.
See also Figure S2.
It is generally assumed that viral dsRNAs produced in cells infected with picornaviruses, as well as other (+)RNA viruses, activate MDA5. However, experimental evidence supporting such a hypothesis is lacking. In this study, we sought to identify physiological ligand(s) of MDA5 by studying the recognition of distinct picornavirus RNA species in transfected cells as well as during infection. Here, we identify a naturally occurring RNA from infected cells, the picornavirus RF, as a potent and specific activator of MDA5 during infection.
Results
MDA5 Recognizes Viruses from Various Picornavirus Genera
The Picornavirus family contains hundreds of highly diverse members that are subcategorized in 12 genera. Based on the observation that MDA5 recognizes EMCV and TMEV, both members of the Cardiovirus genus, and CVB3, an Enterovirus (Kato et al., 2011), it is generally assumed that all picornaviruses are recognized by MDA5, but experimental proof for this is lacking. Here, we set out to investigate the role of RLRs in the recognition of representative picornaviruses from various genera using wild-type (WT) and RIG-I−/−, MDA5−/−, or MAVS−/− mouse embryonic fibroblasts (MEFs). To bypass the requirement for specific entry receptors, we introduced vRNAs into the cytoplasm by transfection rather than infection. Figure 1 A shows IFN-β responses in WT and knockout MEFs triggered by transfection of vRNA of WT mengovirus (a strain of EMCV) or infection of a mutant mengovirus (mengo-Zn). This virus generates high levels of IFN-β because its IFN antagonist has been compromised (Hato et al., 2007). Both methods resulted in a MDA5- and MAVS-dependent, but RIG-I-independent, IFN-β induction, demonstrating the suitability of the transfection method to identify the RLR(s) responsible for detecting picornaviruses.
Figure 1.
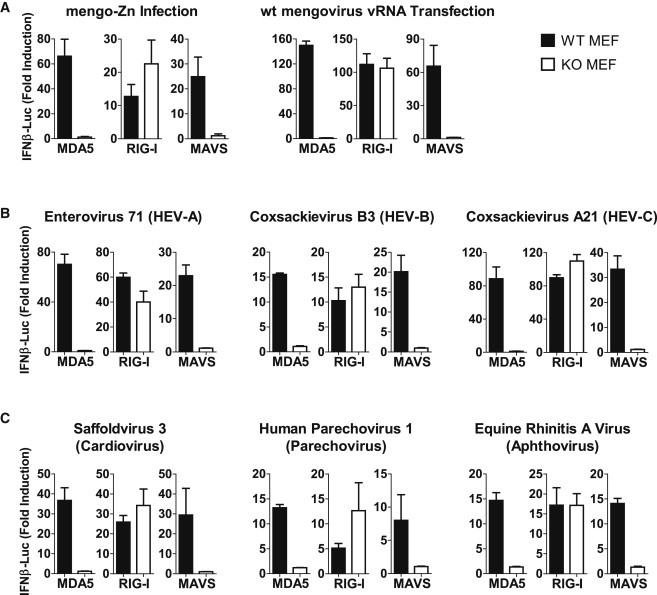
MDA5 Recognizes Picornaviruses across Various Genera
(A) MEFs of indicated genotypes were either infected with mengo-Zn (MOI 50) or transfected with virion RNA (vRNA) from WT mengovirus. IFN-β promoter activation at 8 hr.p.t. was determined by an IFN reporter assay.
(B) MEFs of indicated genotypes were transfected with vRNAs from three human enteroviruses (HEVs), namely enterovirus 71, coxsackievirus B3, or coxsackievirus A21, a close relative to poliovirus. IFN-β promoter activation at 8 hr.p.t. was determined by IFN reporter assay.
(C) Same assay as (B) using vRNAs from Saffold virus 3, human parechovirus 1, and equine rhinitis A virus. Data presented as mean ± SD.
See also Figure S1.
Using this vRNA transfection assay, we studied recognition of members of three human Enterovirus species. In addition, we included Saffold virus (a recently identified human Cardiovirus), human parechovirus (Parechovirus genus), and equine rhinitis A virus (a member of the Aphthovirus genus that also includes foot-and-mouth disease virus). Infectious viruses were produced upon transfection of all vRNAs, indicating efficient RNA replication in MEFs (data not shown). Replication of all viruses induced an MDA5- and MAVS-dependent, but RIG-I-independent, IFN-β response (Figures 1B and 1C). These data provide experimental evidence that a broad spectrum of picornaviruses is indeed recognized by MDA5.
Two other cellular proteins, namely PKR and RNaseL, have also been implicated in recognition of some viruses (Kato et al., 2011). However, we found no difference in IFN-α/β responses to mengo-Zn in cells deficient in RNaseL or PKR (Figure S1 ), arguing against a major role of these proteins in detecting picornavirus RNA (see Extended Results and Discussion).
Figure S1.
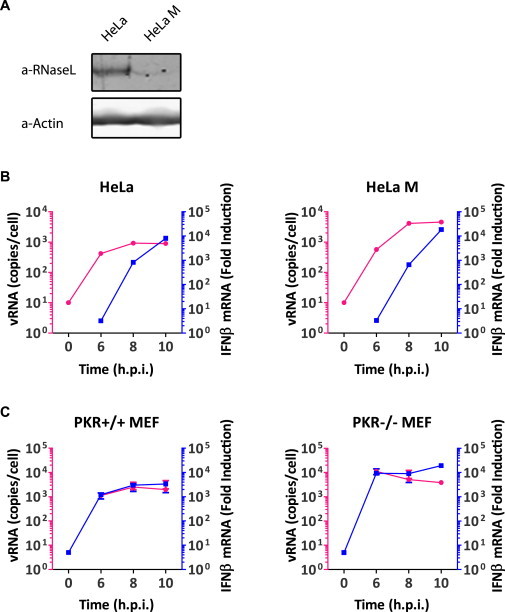
Picornavirus RF-Induced IFN-β Upregulation Is Independent of RNaseL and PKR, Related to Figure 1
(A) Lysates of HeLa and HeLa-M cells were subjected to immunoblotting with RNaseL-specific antibody. Actin was taken as loading control.
(B) HeLa and HeLa-M cells were infected with mengo-Zn (MOI 50) and viral RNA and host IFN-β mRNA levels in total RNA extracts at indicated hours postinfection (h.p.i.) were determined by real-time qPCR.
(C) Same experiment as B was performed with WT or PKR−/− MEFs.
Extended Results and Discussion.
RNaseL and PKR Are Not Essential for Recognition of Picornaviruses
Our data showed that picornavirus-induced IFN-β response is dependent on MDA5, but not RIG-I (Figures 1B and 1C). Two other cellular proteins, namely RNaseL and PKR, have also been implicated in virus recognition (Kato et al., 2011). RNaseL has been suggested to process RNAs to generate ligands for RIG-I (Malathi et al., 2010) and MDA5 (Luthra et al., 2011), while PKR has been implicated in IFN-α/β protein synthesis in response to a number of RNA viruses (Carpentier et al., 2007; Nallagatla et al., 2011). To investigate whether RNaseL and/or PKR also play a role in IFN-α/β induction in response to picornaviruses, we infected WT cells or cells deficient in RNaseL or PKR with mengo-Zn virus and examined their abilities to induce IFN-β transcription. Instead of RNaseL−/− MEFs we made use of a commonly used RNase L-deficient cell line, HeLa M (Dong et al., 2001; Xiang et al., 2003). These cells express virtually no RNaseL (Figure S1A) and no RNaseL activity, as demonstrated by rRNA degradation, was detectable in HeLa M cells after transfection of poly(I:C) or 2′,5′-oligoadenylates (Data not shown). Neither deficient cell line showed any major defect in triggering IFN-β gene transcription upon infection as compared to their WT control cells (Figures S1B and S1C), arguing against significant involvement of PKR or RNaseL in detecting picornavirus RNA.
The lack of a role of PKR in picornavirus recognition seems to be at odds with a previous report of reduced IFN-α/β protein levels in TMEV-infected astrocytes in the absence of PKR (Carpentier et al., 2007). This apparent discrepancy can be explained by the recently identified role of PKR in controlling IFN-α/β responses at the post-transcriptional level by regulating the integrity of the poly(A) tail of IFN-α/β mRNA (Schulz et al., 2010). Together with our data that the level of IFN-β mRNA is unaffected in picornavirus-infected PKR−/− MEFs, it seems that PKR does not participate in the initial detection of picornavirus RNA in MEFs.
Enrichment of ssRNA and dsRNA Fractions from Picornavirus-Infected Cells
To gain more insight into the identity of the RNA species detected by MDA5 in picornavirus-infected cells, we separately enriched for ssRNAs and dsRNAs by LiCl differential precipitation from mock- and CVB3-infected HeLa cells (Figure 2 B). The ssRNA pool of infected cells contained one additional RNA species of the same electrophoretic mobility as purified CVB3 vRNA (Figure 2C), suggesting that it represents viral genomic ssRNA. Although partially ds, the viral RI is known to precipitate by 2M LiCl (Richards et al., 1984), the same condition used here to prepare ssRNA fractions. However, the amount of RI is most likely too low to be detected on gel. The dsRNA pool of CVB3-infected cells contained an RNA species of approximately 7.5 kbp that was absent in the dsRNA pool of mock-infected cells (Figure 2B). Both the viral ssRNA and dsRNA bands were absent in HeLa cells infected with CVB3 in the presence of GuHCl (Figure 2B), a well-known inhibitor of enterovirus (−)RNA synthesis (Barton and Flanegan, 1997) (Figures 2D and S2 ), demonstrating that these RNAs were of viral origin.
Figure S2.
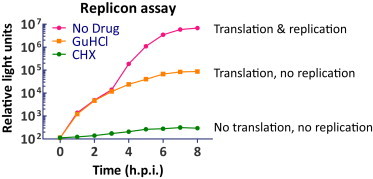
Effect of Translation and RNA Replication Inhibitors, Related to Figure 2
Wt MEFs were transfected with CVB3 replicon RNA (p53CB3-LUC), which contains the CVB3 cDNA in which the capsid-coding region is replaced by the firefly luciferase gene (Wessels et al., 2005), and incubated in the absence or presence of GuHCl (2mM) or CHX (10 μg/ml). Cells were collected every hour and the luciferase activity in the lysates was measured using a luminescent detector.
The Picornavirus 7.5 kbp RF Is a Potent MDA5 Ligand upon Transfection
The dsRNA fraction from CVB3-infected cells (Figure 2B) was transfected into MEFs and IFN-β mRNA levels were measured at 2 hr posttransfection (hr.p.t.), a time point preceding RNA replication (Figure S2), thereby excluding the possible role of other, newly produced viral RNA species. The dsRNA fraction triggered a MDA5- and MAVS-dependent IFN-β response (Figure 3 A). We next set out to identify the IFN-stimulatory RNA species in this fraction. In the dsRNA fractions, we observed two bands on gel; an infection-specific, 7.5 kbp species, and a larger fragment that was present in both mock- and CVB3-infected samples. To determine the nature of the higher band, we treated the dsRNA fractions with nucleases specific for ssRNA (RNase A), dsRNA (RNase III), and DNA. The nonspecific larger band was completely digested upon DNase treatment, indicating that it was DNA (Figure 3B). The 7.5 kbp CVB3 RF was specifically digested by RNase III, confirming that this is a dsRNA molecule (Figure 3B).
Figure 3.
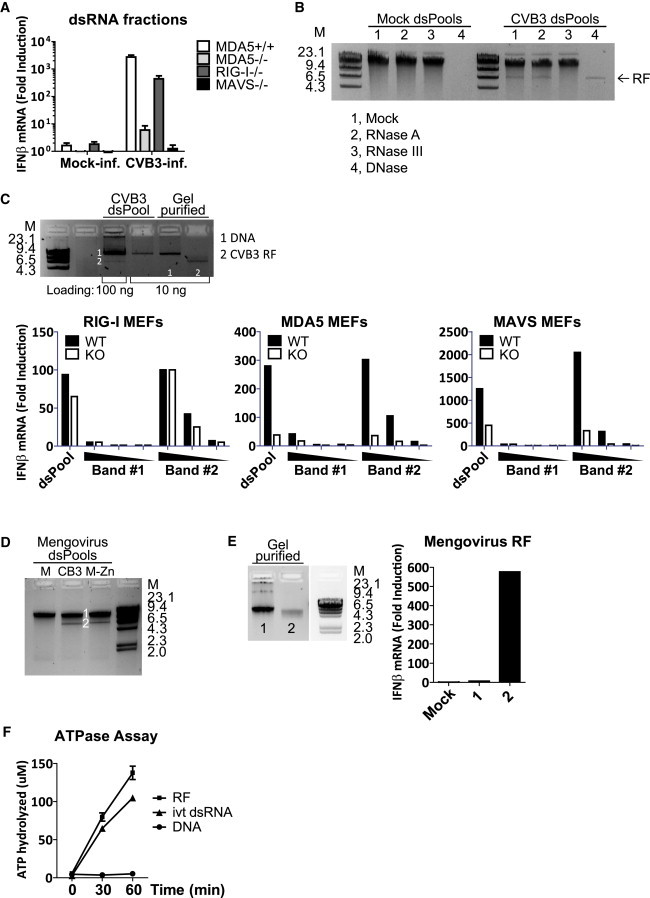
Picornavirus RF Is a Potent MDA5 Agonist
(A) dsRNA fractions from mock- or CVB3-infected HeLa cells were transfected-into MEFs of indicated genotypes, and IFN-β mRNA level at 2 hr.p.t. was measured by real-time qPCR. Data presented as mean ± SD.
(B) dsRNA fractions (dsPool) of mock- and CVB3-infected cells were treated with 10 ng/μl RNase A, 10 mU RNase III, or 10 mU DNase I at 37°C for 15 min, and analyzed on agarose gel.
(C) DNA and RF bands observed in CVB3 dsRNA fraction were gel purified and analyzed on agarose gel. CVB3 dsRNA fraction (10 ng per well in 24-well format), and the gel-purified RNAs (10, 2, or 0.4 ng/well) were transfected into WT, RIG-I−/−, MDA5−/−, or MAVS−/− MEFs in the presence of CHX (10 μg/ml). IFN-β mRNA induction was determined by real-time qPCR at 8 hr.p.t.
(D) dsRNA fractions of mock-, CVB3-, or mengovirus-infected cells were analyzed on agarose gel.
(E) DNA and RF bands from mengovirus dsRNA fraction were gel purified and transfected into RIG-I−/− MEFs in the presence of CHX (10 μg/ml). IFN-β response at 8 hr.p.t. was measured by real-time qPCR.
(F) Gel-purified RF and DNA bands from CVB3 dsRNA fraction, as well as a in vitro transcribed dsRNA of CVB3 sequence (ivt dsRNA) (0.3 μg/ml), were incubated with recombinant MDA5 (0.3 μM) at 37°C in the presence of 2mM ATP, and free Pi was measured using Green Reagent at 0, 30, and 60 min after reaction was started. M, dsDNA marker with indicated size in kbp. Bands number 1 and 2 on gel corresponds to the samples used in RNA transfection. Data presented as mean ± SD.
Next, we examined the ability of purified CVB3 RF to activate MDA5. When gel-extracted and transfected into MEFs, the RF, but not the DNA molecule, induced high levels of IFN-β transcription in an MDA5- and MAVS-specific manner (Figure 3C). Similar experiments were performed with nucleic acids extracted from cells infected with mengovirus. The dsRNA fraction from mengovirus-infected cells appeared indistinguishable on gel from that from CVB3-infected cells (Figure 3D), and the gel-purified mengovirus RF also proved to be a potent MDA5 agonist (Figure 3E). These data show that the RF of prototype members of at least two genera of picornaviruses can specifically and potently induce an MDA5-dependent IFN-α/β response.
To investigate whether the RF can directly activate MDA5, we performed in vitro ATPase assays where recombinant MDA5 was incubated with gel-purified RF or the nonspecific DNA band in the presence of ATP. ATP hydrolysis was followed at different time points by measuring free phosphate. CVB3 RF induced high levels of MDA5 ATPase activity—hydrolyzing nearly 150 μM ATP in 60 min—while the DNA band did not induce any ATP hydrolysis during this time period (Figure 3F). These results are in line with our transfection data (Figures 3C and 3E) and confirm that RF can be directly recognized by MDA5 and potently induce its activation.
Picornavirus ssRNAs Do Not Activate MDA5
We also tested the ssRNA fraction from infected cells (Figure 2B) for its IFN-β-stimulatory activity. To our surprise, transfection of this fraction also led to an MDA5- and MAVS-dependent IFN-β response (Figure 4 A). However, subsequent nuclease digestion experiment revealed that the IFN-β-stimulatory activity is sensitive to RNase III (dsRNA-specific) treatment, but not RNase A (ssRNA-specific) (Figures 4B and 4C). Indeed, after removal of rRNAs—the most abundant RNA species present in the ssRNA fractions—we observed a RNA species on gel that is specifically present in infected cells and has the same electrophoretic mobility as the RF band found in the dsRNA fraction from infected cells (Figure S3 A). These results suggest that residue amounts of RF are still present in our ssRNA fraction preparations, and the RF, but not viral ssRNAs, is responsible for the observed IFN-β induction upon transfection of this fraction. In line with this conclusion, we found that VPg-containing vRNA (Figures 4D and 4E), VPg-unlinked viral mRNAs (Figure 4E), and in vitro transcribed ssRNAs of viral sequence (Figures S3B and S3C) all failed to induce any IFN-β response when purified and transfected into cells.
Figure 4.
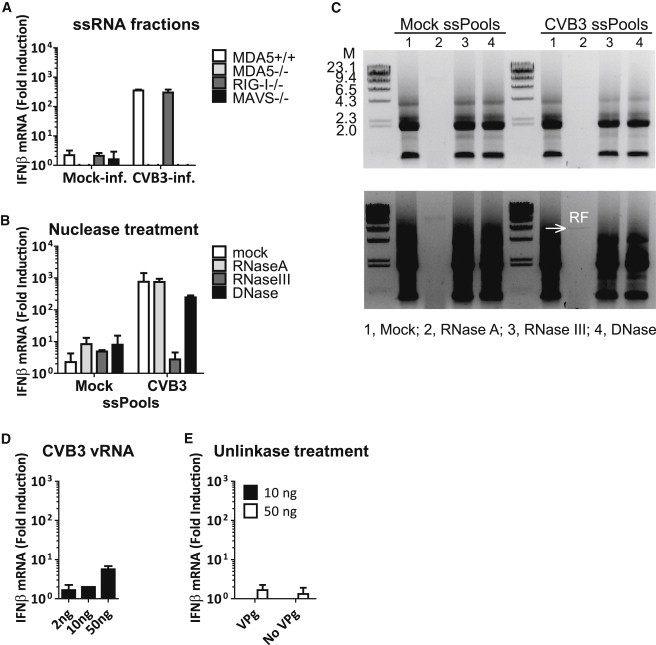
ssRNAs from CVB3-Infected Cells Do Not Activate MDA5
(A) ssRNA fraction from mock- or CVB3-infected HeLa cells were transfected into MEFs of indicated genotypes, and IFN-β mRNA level at 2 hr.p.t. was measured by real-time qPCR. Data presented as mean ± SD.
(B) ssRNA fractions (ssPools) of mock- and CVB3-infected cells were treated with 10 ng/μl RNase A, 10 mU RNase III, or 10 mU DNase I at 37°C for 15 min. Resulting RNA samples equivalent to 500 ng starting material were transfected into approximately 200,000 MAVS+/+ MEFs and IFN-β mRNA induction was determined by real-time qPCR at 8 hr.p.t. Data presented as mean ± SD.
(C) The same samples as in (B) were analyzed on agarose gel.
(D) CVB3 vRNA was isolated from pelleted viral particles. Indicated amounts of vRNA were transfected into RIG-I−/− MEFs in the presence of CHX (10 μg/ml), and IFN-β mRNA induction was determined by real-time qPCR at 8 hr.p.t.. Data presented as mean ± SD.
(E) Indicated amounts of mock-treated or unlinkase-treated poliovirus vRNAs were transfected into RIG-I−/− MEFs in the presence of CHX (10 μg/ml). Total RNA was extracted at 8 hr.p.t. and IFN-b mRNA induction was determined by real-time qPCR. Data presented as mean ± SD.
See also Figure S3.
Figure S3.
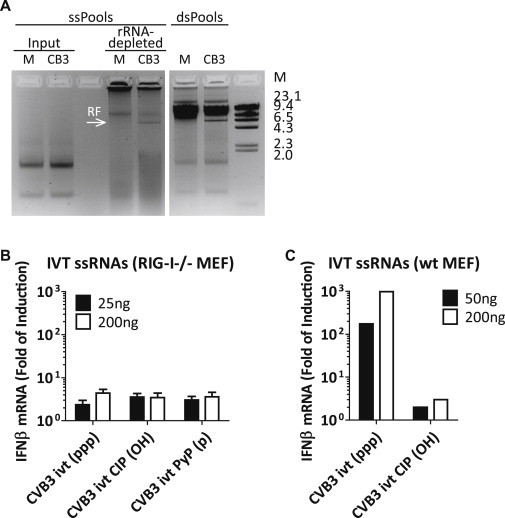
Picornavirus ssRNAs Do Not Exert IFN-β-Stimulatory Activity, Related to Figure 4
(A) ssRNA fractions (ssPools) of mock- and CVB3-infected cells were subjected to rRNA removal. The input (2%) and resulting materials were analyzed by agarose gel electrophoresis. The dsRNA fractions were loaded in parallel as a comparison.
(B) In vitro transcribed ssRNA was prepared from CVB3 cDNA clone (CVB3 ivt) and treated with either calf intestinal alkaline phosphatase (CIP), which removes all three phosphate groups from the 5′ end of the RNA, or 5′ polyphosphatase (PyP), which removes the beta- and gamma- phosphate groups. Remaining 5′ groups of these RNAs are indicated in brackets. RIG-I−/− MEFs were transfected with indicated amounts of these CVB3 IVT RNAs in the presence of CHX (10 μg/ml), and IFN-β induction was measured at 8 hr.p.t. by real-time qPCR. Data presented as mean ± SD.
(C) Mock- and CIP-treated CVB3 IVT RNAs as on the left were transfected into WT MEFs in the presence of CHX (10 μg/ml), and IFN-β induction at 8 hr.p.t. was measured by real-time qPCR. The 5′ppp-containing RNA, but not the 5′OH-containing RNA, induced high levels of IFN-β, confirming the quality of the in vitro transcribed RNAs produced and the efficiency of the CIP treatment.
The viral RI was also reported to segregate in the ssRNA fraction according to the LiCl precipitation method (Richards et al., 1984). To study the effect of RI on IFN-α/β induction without complications from RF, we attempted to remove the remaining RF from CVB3 ssRNA fraction by repeating the 2M LiCl precipitation procedure. However, even after two additional rounds of precipitation, we did not observe significant loss of RF from the CVB3 ssRNA fraction, nor did we observe a reduced IFN-β response upon transfection (data not shown). Hence, we were unable to determine whether RI also exerts IFN-inducing activity.
Formation of Viral RF, but Not RI, Is Required for IFN-β Induction in Infected Cells
Our results indicated that the picornavirus RF, and possibly also the RI, is a potent MDA5 activator when delivered into the cytoplasm by lipid-based transfection. However, the amount of RF/RI and their accessibility to MDA5 may differ greatly from the situation in infected cells, where viral RNA is covered with replication enzymes and localized in virus-induced, membrane-associated replication organelles (reviewed in Belov and van Kuppeveld, 2012). Therefore, we set out to dissect the IFN-stimulatory abilities of different viral RNAs produced in cells infected with mengo-Zn, which induces strong IFN-β responses.
We first generated a comprehensive profile of the kinetics of viral RNA replication as well as host IFN-β response to mengo-Zn in HeLa cells by measuring viral RNA and IFN-β mRNA levels by real-time quantitative PCR (qPCR) every hour up until 10 hr postinfection (hr.p.i.) (Figure 5 A). Viral RNA level remained at input levels in the first 3 hr of infection, then rapidly increased (i.e., active viral RNA replication) to reach a maximum at 6 hr.p.i., and stabilized at this level through 10 hr p.i. The host IFN-β response showed a slight delay as IFN-β mRNA levels in infected cells remained at background levels through the first 6 hr, then started to increase from 7 hr.p.i., and reached their maximum at 8–9 hr p.i.
Figure 5.
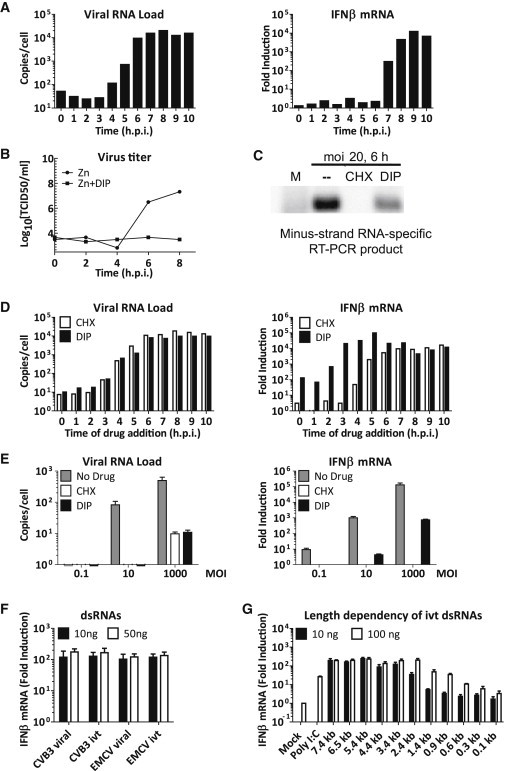
Viral RNA Recognition in Infected Cells and Characterization of Determinant for MDA5 Activation in RF
(A) HeLa cells were infected with mengo-Zn (MOI 10) and samples were taken every hour postinfection. Total RNA was extracted and viral RNA and host IFN-β mRNA levels were assayed by real-time qPCR.
(B) Cells were infected with mengo-Zn in the absence or presence of DIP (100 μM). Production of infectious particles was determined by end-point titration on BHK-21 cells.
(C) HeLa cells were mock-infected or infected with mengo-Zn (MOI 20) in the absence or presence of CHX (10 μg/ml) or DIP (100 μM) and total RNA was extracted at 6 hr.p.i. dsRNA fraction was purified by LiCl precipitation and subjected to RT-PCR using primers specific to viral (−) RNA.
(D) HeLa cells were infected with mengo-Zn at MOI 10. CHX (10 μg/ml) or DIP (100 μM) was added to infected cells either at time of infection or at indicated times postinfection. All samples were harvested at 12 hr.p.i. Total RNA was extracted and viral RNA and host IFN-β mRNA levels were analyzed by real-time qPCR.
(E) HeLa cells were infected with mengo-Zn at indicated MOI’s in the absence or presence of CHX (10 μg/ml) or DIP (100 μM). Total RNA was extracted at 10 hr.p.i. and viral RNA and host IFN-β mRNA levels were measured by real-time qPCR. Data presented as mean ± SD.
(F) RFs of CVB3 and mengovirus as well as in vitro transcribed dsRNAs of viral sequences were transfected into RIG-I−/− MEFs in the presence of CHX (10 μg/ml) at indicated amounts per well in 24-well format. IFN-β mRNA levels were determined at 8 hr.p.t. by real-time qPCR. Data presented as mean ± SD.
(G) polyIC or in vitro transcribed dsRNAs of indicated length were transfected into RIG-I−/− MEFs in the presence of CHX (10 μg/ml) at indicated amounts. IFN-β mRNA levels were determined at 8 hr.p.t. by real-time qPCR. Data presented as mean ± SD.
See also Figures S4 and S5.
To study the separate contribution of viral ssRNA and dsRNA in inducing IFN-α/β response during infection, we employed inhibitors of mengovirus replication, cycloheximide (CHX) and dipyridamole (DIP) (Figure 5B), which affect viral replication at different stages. CHX prevents protein synthesis and thereby RNA replication. DIP has little effect on (−)RNA synthesis but strongly inhibits (+)RNA synthesis in cell-free extract (Fata-Hartley and Palmenberg, 2005). Indeed, (−)RNAs were detected in cells infected in the presence of DIP, but not CHX, using a strand-specific PCR (Figure 5C). The finding that the signal obtained from DIP-treated cells was lower than that from no-drug-treated cells likely reflects a reduced amount of (−)RNA as DIP only allows (−)RNA synthesis from incoming genomic (+)RNA, whereas in the absence of DIP newly synthesized (+)RNAs can enter a new round of RNA replication, and therefore lead to the production of more RF.
To determine the IFN-β response to different viral RNA species formed in infected cells, we used DIP and CHX in a time-of-drug-addition experiment. HeLa cells were infected with mengo-Zn, and DIP or CHX was added to cells at the indicated times (Figure 5D). Cells were incubated until 12 hr.p.i., harvested, and viral RNA and IFN-β mRNA levels were determined by real-time qPCR. As shown in Figure 5D, both DIP and CHX were effective in inhibiting viral RNA replication when added at early stages (0–2 hr.p.i.) of infection. Later addition of the drugs resulted in partial, and eventually loss, of effect on RNA replication.
Under conditions where CHX and DIP completely inhibited viral RNA replication (i.e., added at 0–2 hr.p.i., Figure 5D), no IFN-β induction was detected in CHX-treated cells, whereas a 100-fold, MDA5-dependent (Figure S4 ) induction of IFN-β mRNA level was observed in DIP-treated cells. Additionally, when CHX or DIP was added at 3 hr.p.i., a maximal IFN-β response was observed in DIP-treated cells, but virtually no IFN-β was induced in the CHX-treated cells (Figure 5D). Analysis of viral RNA levels indicate that only a few new (+)RNAs had been made because addition of either CHX or DIP at this time point led to only a slight increase in viral RNA copy number at 12 hr.p.i. These newly synthesized (+)RNAs could serve as template to make additional RFs in the DIP-treated cells, but not in the CHX-treated cells, and result in the increase of IFN-β induction observed at 3 hr p.i. It is theoretically possible that the observed differences in IFN-β levels in CHX- and DIP-treated cells were due to nonspecific blockade or stimulation of the RLR signaling pathway by CHX or DIP, respectively. However, this is unlikely because these drugs had no such effects on poly(I:C)-induced IFN-β responses (Figure S5 ).
Figure S4.
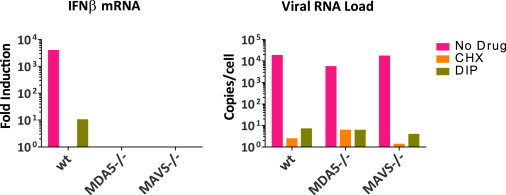
Mengo-Zn Infection Induces a MDA5-Dependent IFN-α/β Response in MEFs, Related to Figure 5
MEFs derived from RIG-I-, MDA5- or MAVS-deficient mice were infected with mengo-Zn at a MOI of 10 in the presence or absence of DIP (100 uM) or CHX (10 μg/ml). Total RNA was isolated at 10 hr.p.i. and IFN-β mRNA levels were determined by real-time qPCR.
Figure S5.
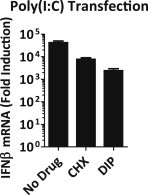
Poly(I:C)-Induced IFN-β mRNA Upregulation in the Absence or Presence of CHX and DIP, Related to Figure 5
HeLa cells were transfected with 0.5 μg/ml poly(I:C) in the presence or absence of CHX (10 μg/ml) or DIP (100 μM). IFN-β mRNA levels were determined at 10 hr.p.t. by real-time qPCR. Both CHX and DIP treatments resulted in a reduced IFN-β mRNA induction in response to poly(I:C). Importantly, these results show that the difference in IFN-β induction we observed in CHX- and DIP-treated samples in Figures 5D and 5E could not have been artifacts caused by these inhibitors themselves. Data presented as mean ± SD.
In the experiment described above, the incoming vRNA alone (i.e., in the presence of CHX) induced no IFN-β response. To exclude that the amount of incoming vRNA at the used MOI was too low to induce a detectable IFN-β mRNA upregulation, we increased the infection dose from MOI 10 to 1,000. This resulted in increased levels of IFN-β in no-drug- and DIP-treated samples, but in CHX-treated cells the IFN-β level remained undetectable (Figure 5E). This result clearly shows that the incoming vRNA, even when present at very high quantities, does not activate MDA5. Taken together, our results demonstrate that (−)RNA synthesis (i.e., formation of RF), but not (+)RNA synthesis (i.e., formation of RI), is required for achieving high levels of IFN-α/β induction.
Termini and Length Dependency of MDA5 Activation by Long dsRNAs
We next investigated whether the termini of picornavirus RF are important for MDA5 activation. The viral RF contains a VPg peptide at the 5′ end of the (−)RNA, but not at the 5′ end of the (+)RNA, because of the activity of the unlinking enzyme. Furthermore, the RF contains a large unpaired poly(A) tail at the 3′ end of the (+)RNA (van Ooij et al., 2006). To investigate whether these features are important for MDA5 activation, we produced CVB3 and mengovirus dsRNAs with 5′ppp and perfectly paired termini by in vitro transcription (ivt dsRNA) and compared them with viral RF purified from infected cells for their ability to activate MDA5. Strikingly, all dsRNAs triggered a similar IFN-β response upon transfection into RIG-I−/− MEFs (Figure 5F). Additionally, CVB3 ivt dsRNA activated MDA5 to a similar extent as CVB3 RF in our in vitro ATPase assay (Figure 3F). Thus, the recognition of long dsRNAs by MDA5 is likely independent of the termini of the RNA molecule.
Lastly, we investigated whether MDA5 activation by long dsRNAs is length dependent as proposed by Kato et al., who showed that only long polyIC molecules (>2 kb) are efficiently recognized by MDA5 (Kato et al., 2008). To this end we generated ivt dsRNAs of various lengths (0.1–7.4 kbp) and determined IFN-β activation levels by real-time qPCR upon transfection of different amounts of these fragments. At relatively low quantities (10 ng), ivt dsRNAs shorter than 2.4 kbp induced only low levels of IFN-β mRNA. However, when larger amounts of dsRNA (100 ng) were transfected, smaller dsRNAs were also able to trigger an IFN-β response (Figure 5G), indicating that there may not be a strict length cut-off value for MDA5 ligands.
Discussion
Since the initial discovery of RLRs, significant progress has been made in identifying ligands for RIG-I, but the identity and molecular characteristics of viral MDA5 ligands remain largely unknown. Based on transfection experiments using poly(I:C), MDA5 is believed to recognize long dsRNAs. However, viral ssRNAs should not be excluded because base-paired regions in ssRNAs, as those in the picornavirus genomic RNA, could potentially be sufficient for MDA5 recognition. Furthermore, a virally encoded peptide, VPg, is covalently attached to the 5′ terminus of picornavirus RNAs via a unique tyrosine-RNA bond, providing a potential “nonself” signature at the 5′ end. Bearing this information in mind, we followed an unbiased approach to determine the IFN-stimulatory activities of different viral RNA species produced in picornavirus-infected cells.
The viral dsRNA, RF, has been assumed to be the MDA5 ligand produced by picornaviruses based on the fact that all known activators of MDA5 are dsRNAs (mimics). Here, we provide experimental evidence that purified picornavirus RF can not only induce an MDA5-dependent IFN-α/β response upon transfection, but also directly bind to, and activate, MDA5 in vitro. Viral ssRNAs, on the other hand, proved poor MDA5 stimuli. Neither vRNA (VPg-containing) nor viral mRNA (VPg-lacking) induced an IFN-β response when transfected into cells. This is in line with the observation that the full-length EMCV RNA failed to activate MDA5 ATPase activity in vitro (Peisley et al., 2011). Although we could not test IFN-stimulatory activity of purified RI, this molecule does not seem to play a crucial role in inducing IFN-α/β because treatment of the CVB3 ssRNA fraction with RNase A did not affect its ability to induce IFN-α/β. This observation also speaks against the hypothesis that other (viral) ssRNAs—which might have been produced by the action of cellular RNases like RNaseL—may be recognized by MDA5.
Collectively, our transfection experiments clearly identify the RF as an MDA5 agonist. Previously, Pichlmair and colleagues identified two virus-induced RNA species in their total RNA extract from EMCV-infected cells: one dsRNA of approximately 12 kbp and one high-molecular-weight RNA (HMW-RNA) that consisted of both ssRNAs and dsRNAs (Pichlmair et al., 2009). The HMW-RNA, but not the 12 kbp dsRNA, was found to be IFN-α/β stimulatory upon transfection. We never observed either of these RNAs in our total RNA extracts or the fractionated RNA preparations. The identity of the HMW-RNA and the IFN-stimulatory element(s) within their mixed RNA population remains unclear.
Although transfection may be a convenient method to study aspects of MDA5 ligand recognition, it may not faithfully recapitulate physiological conditions for several reasons. First, transfection delivers large amounts of viral RNA directly into the cytoplasm at once. This is different from the RNA release process during infectious entry as well as the gradual accumulation of viral RNAs during virus replication. Second, purified, and therefore “naked,” RNAs are transfected into cells whereas viral RNAs are heavily coated with viral and host factors during infection. Third, (+)RNA viruses induce the formation of membranous replication organelles, which may restrict access of MDA5 to replication intermediates such as RF and RI. To gain insight into MDA5 activation in a more physiologically relevant situation, we studied MDA5 activation in infected cells. Our data show that the incoming genomic ssRNA alone cannot induce any detectable IFN-β response even when large amounts were delivered by high MOI infection. When (−)RNA synthesis (i.e., formation of RF) was permitted but (+)RNA synthesis (i.e., formation of RI) was inhibited, a significant upregulation of IFN-β mRNA was observed. These data further support the idea that picornavirus RF is the essential MDA5 agonist in the course of infection. Of note, a much higher IFN-β response was observed when RNA replication was allowed to proceed fully (i.e., in the absence of inhibitor). Although this finding may be interpreted as that RIs also activate MDA5, this increase can be attributed, at least partly, to the increased amount of RF produced from newly synthesized (+)RNAs that enter new rounds of RNA replication.
Recently, Züst et al. suggested that MDA5 may also recognize RNA molecules with a “nonself” 5′ termini, as a mutant coronavirus deficient in fully capping its mRNAs activated MDA5 in infected cells (Züst et al., 2011). Because picornavirus RF harbors distinctive termini, we investigated whether this terminal composition of the RF is essential for MDA5 activation. Changing the physiological terminal groups of RF into 5′ppp and fully complemented 3′ end did not change its ability to induce IFN-β upregulation. Other features such as secondary structures are common requirements for ligand recognition; however, the picornavirus RF is not known to contain distinct higher structures. Thus, it is likely that the long ds nature of the RF, and not its unique termini, renders this RNA a potent MDA5 activator.
We also investigated the length dependency of MDA5 ligand recognition because it has been suggested that only dsRNAs longer than 2 kbp can induce MDA5 activation (Kato et al., 2008). We tested dsRNAs of viral sequence in vitro that span a wide range of sizes (0.1–7.4 kbp), and found that although longer dsRNAs activate MDA5 more efficiently when provided at equal mass, short dsRNAs could also induce some levels of MDA5 activation when present in large quantities. These data suggest that MDA5 may not require its substrate to be longer than a strict cut-off size. In line with this, others have demonstrated efficient activation of MDA5 ATPase activity by dsRNAs as short as 112 bp (Peisley et al., 2011). Authors of this paper also showed that MDA5 ATPase activity, which is triggered by ligand binding, induces disassembly of MDA5 from the RNA ligand, the rate of which is inversely related to the length of the bound RNA molecule (Peisley et al., 2011). This provides some molecular explanation why longer dsRNAs are more efficient in activating MDA5 than shorter ones.
While this paper was in preparation, another study reported that enterovirus dsRNAs, but not ssRNAs, can activate MDA5-dependent IFN-β responses upon transfection (Triantafilou et al., 2012). Also shown in this study is colocalization of MDA5 with dsRNA in enterovirus-infected cells. Although not providing evidence of direct binding or functional activation, these data nicely show that MDA5 and viral dsRNA can be found in physical vicinity, and therefore support our results that the RF can activate MDA5 in infected cells.
Collectively, our data suggest that the 7.5 kbp RF of picornaviruses is a highly potent MDA5 activator and is directly recognized by MDA5. Moreover, this study shows that manipulating specific steps of virus replication is a powerful approach to study recognition of specific viral RNA species by RLRs under physiological conditions.
Experimental Procedures
Cells, Viruses, Infectious cDNA Clones, and Reagents
MEFs and HeLa cells were maintained in DMEM supplemented with 10% FBS and cyproxin (1 mg/ml). Viruses are described in Extended Experimental Procedures. Infectious cDNA clones p53CB3/T7 (Wessels et al., 2005) and pM16.1 (Hato et al., 2007) were used to produce in vitro transcribed RNAs with CVB3 or mengovirus sequences, respectively. CHX and DIP were purchased from Sigma-Aldrich, and RNase A, RNase III, and DNase I from Ambion.
Extended Experimental Procedures.
Cells
HeLa-M cells were provided by Prof. Robert Silvermann (Lerner Research Institute, Cleveland, USA) through Thomas Michiels (Université de Louvain, Belgium). RIG-I+/−, RIG-I−/−, MDA5+/+ and MDA5−/− MEFs were provided by Prof. S. Akira (Osaka University, Japan), MAVS+/+ and MAVS−/− MEFs were provided by Z.J. Chen (University of Texas, Dallas, USA), and PKR+/+ and PKR−/− MEFs (Abraham et al., 1999) were provided by Prof. John Bell (Ottawa Regional Cancer Center, Ontario, Canada) through Thomas Michiels.
Viruses
Both WT mengovirus and mengo-Zn were obtained by transfection of in vitro RNA transcripts of infectious cDNA clone based on pM16.1, which contains a copy of mengovirus with a shortened poly C tract (Zoll et al., 1996). Coxsackievirus B3 (CVB3, strain Nancy) was obtained by transfection of in vitro RNA transcripts of p53CB3/T7, an infectious clone that contains a full-length cDNA of CVB3 (Wessels et al., 2005). Saffold virus 3 (SAFV3, strain NL2007) and human parechovirus type 1 (HPeV, strain 755532) have been described previously (Zoll et al., 2009a; Zoll et al., 2009b). Enterovirus 71 (EV71, strain Br-Cr) and Coxsackievirus A21 (CVA21, strain Kuykendall) were obtained from the National Institute for Public Health and the Environment (RIVM, Bilthoven, the Netherlands). Equine rhinitis A virus (ERAV, strain NM11/67) was a kind gift from T. Tuthill and D. Rowlands (University of Leeds, United Kingdom). Mengoviruses were propagated on BHK-21 cells. CVB3, ERAV, EV71 were propagated on BGM cells. CVA21 and SAFV3 were propagated on HeLa cells. HPeV1 was propagated on Vero cells. Virus titers were determined by endpoint titration as described previously (van Kuppeveld et al., 1995).
IFN Reporter Assays
1 × 105 HeLa cells were transfected with 200 ng of pIFN-β-luc (Fitzgerald et al., 2003) and 50 ng pRL-TK (Promega; an internal control that constitutively expresses Renilla luciferase) using Fugene-6 (Roche). Cells were incubated for 24 hr and were either mock treated, infected with virus, or transfected with RNA using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Luciferase activity was measured at the indicated time points using the Dual-luciferase Reporter assay system (Promega) according to the manufacturer’s instructions. Firefly luciferase values were divided by Renilla luciferase values to normalize for transfection efficiency and are shown as fold induction over mock.
Real-Time qPCR Analysis
2 × 105 cells were transfected with indicated amounts of RNA using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. At the indicated time points, total cellular RNA was harvested using GenElute mammalian total RNA miniprep Kit (Sigma-Aldrich) according to the manufacturer’s instructions. RNA was treated with DNase I (Invitrogen) prior to reverse transcription, which was performed using TaqMan reverse transcription reagents kit (Applied Biosystems) according to the manufacturer’s instructions. Quantitative analysis of gene expression was performed using qPCR, using LightCycler 480 SYBR Green I master mix (Roche). Reactions were carried out by a LightCycler 480 (Roche) and primary data analysis was done with the provided software. The following primers were used. Murine IFN-β forward (fw) and reverse (rv) primers: 5′-CAGCTCCAAGAAAGGACGAAC-3′ and 5′-GGCAGTGTAACTCTTCTGCAT-3′, respectively. Primer sequences were acquired from the Primerbank (Wang and Seed, 2003) (Primerbank ID 6754304a1). Murine HPRT fw and rv primers: 5′-GTAATGATCAGTCAACGGGGGAC-3′ and 5′-CCAGCAAGCTTGCAACCTTAACCA-3′, respectively. Human IFN-β fw and rv primers: 5′-ATGACCAACAAGTGTCTCCTCC-3′ and 5′-GCTCATGGAAAGAGCTGTAGTG-3′, respectively. Human Actin fw and rv primers: 5′-CCTTCCTGGGCATGGAGTCCTG-3′ and 5′-GGAGCAATGATCTTGATCTTC-3′, respectively.
Preparation of Full-Length IVT RNAs
The in vitro transcribed CVB3 RNA was generated by using cDNA clones p53CB3/T7 (Wessels et al., 2005) or pRibCB3/T7 (van Ooij et al., 2006) as templates. Uncapped, in vitro transcribed RNAs from linearized plasmids were synthesized using Riboprobe in vitro transcription systems (Promega) following the manufacturer’s instructions. Clean-up reactions were performed using TRIzol reagent (Invitrogen) or GenElute mammalian total RNA miniprep Kit (Sigma-Aldrich) according to manufacturer’s instructions. RNAs were treated with DNase I (Invitrogen) and again cleaned up. RNA integrity was checked on agarose gel. To produce IVT dsRNAs of CVB3 and mengovirus the full-length genomes were amplified by PCR using primers with flanking T7 and Phi6 promoter sites and the cDNA clones p53CB3/T7 and pM16.1 as templates, respectively. dsRNAs were in vitro transcribed using Replicator RNAi Kit (Finnzyme) according to manufacturer’s instructions.
Generation of Partially Purified VPg Unlinkase
Approximately 70 ml of packed HeLa cells were homogenized by cryogenic grinding (Retsch) then solubilized in TDE buffer (10 mM Tris-HCl, pH 7.5, 5 mM DTT, 1 mM EDTA). The homogenate was centrifuged at 70,000 x g for 1 hr and the supernatant was subjected to ammonium sulfate fractionation followed by successive chromatography on Q-Sepharose (Pharmacia Biotech), Heparin-Sepharose (GE Healthcare), SP-Sepharose (GE Healthcare), Sephacryl S200 (GE Healthcare), and Mono Q (Amersham) to generate partially purified unlinkase. To generate VPg-depleted ssRNA, 5 μl of partially purified unlinkase or buffer was incubated with 0.4 pmol of vRNA in a 20 μl reaction volume containing PDGM buffer (20 mM phosphate buffer, pH 7.0, 5 mM DTT, 10% glycerol, 2 mM MgCl2) for 30 min at 30°C. To assay for the removal of VPg from vRNA, parallel reactions were spiked with 700 CPM 35S-methionine labeled vRNA and quantified as described in (Rozovics et al., 2011).
Isolation of 35S-methionine Labeled vRNA
A mutant poliovirus carrying a VPg that can be radioactively labeled with [35S]methionine and the method for isolating 35S-methionine labeled vRNA have been described previously (Rozovics et al., 2011). Briefly, 1 mCi of 35S-methionine was added to HeLa cells in suspension culture following poliovirus W1-VPg31 infection and methionine starvation for three hours at 37°C. At six hours post infection, the infected cells were pelleted and subjected to five cycles of freezing and thawing in RSB+MgCl2 buffer (10 mM NaCl, 10 mM Tris-HCl, pH 7.5, 1.5 mM MgCl2) to release the virions. The virions in the supernatant were pelleted by centrifugation at 29,700 rpm for 3.5 hr in a Ti 70.1 rotor at 24°C. The pellet was resuspended in 0.1 buffer (0.1 M NaCl, 10 mM Tris-HCl, pH 7.5, 5 mM EDTA, 0.5% SDS) and then applied to a 15%–30% sucrose gradient (5 mM EDTA, 0.1 M NaCl, 10 mM Tris-HCl, pH 7.5, 0.5% SDS) and centrifuged at 27,700 rpm for 2.5 hr in an SW41 at 24°C. Fractions containing 35S-methionine labeled virions were identified by scintillation counting, pooled and then subjected to two cycles of phenol/chloroform extraction and ethanol precipitation. The final volume of 35S-methionine labeled vRNA was adjusted to 700 cpm/μl (∼0.04 pmol/μl).
In Vitro MDA5 ATPase Assay
Recombinant MDA5 protein was purified as previously described (PNAS, 2011, 108(52):21010-5). Following a 3 min pre-incubation of 0.3 uM MDA5 with 0.3 μg/ml nucleic acid in 20 mM HEPES, pH 7.5, 0.1 M NaCl, 1 mM MgCl2 at 37 C the reaction was initiated by addition of 2 mM ATP. Aliquots were withdrawn at time 0, 30 and 60 min, and were quenched with 50 mM EDTA. The amount of ATP hydrolyzed was determined by measuring released phosphate using BIOMOL Green Reagent (Enzo), which was added to the quenched reaction at a ratio of 9:1. OD650 was measured using a Synergy2 plate reader (BioTek).
Isolation of Viral RNA from Virions
Cells were infected with virus and grown until cytopathic effect was complete. This lysate was subjected to three freeze-thaw cycles, cleared, and centrifuged at 25,000 rpm for 6 hr in an SW28 rotor at 4°C. The virus pellet was suspended in either TRIzol reagent (Invitrogen) or the lysis buffer of GenElute mammalian total RNA Miniprep Kit (Sigma-Aldrich), and RNA was isolated according to the manufacturers' instructions.
Isolation of ssRNA and dsRNA Fractions from Control and Infected Cells
Total RNA was isolated using TRIzol (Invitrogen) according to manufacturer’s protocol. LiCl was added to the total RNA extract to an end concentration of 2 M. This was incubated at 4°C for 2–16 hr, and ssRNAs were precipitated by centrifugation at 16,000 × g for 30 min at 4°C. The supernatant was subjected to ethanol precipitate in the presence of 0.8M LiCl to precipitate dsRNAs. RNA pellets were washed with 75% ethanol, air-dried, and dissolved in RNase-free water.
IFN reporter assay and real-time qPCR analysis were carried out as described previously (Hato et al., 2007).
Preparation of Full-Length IVT RNAs
ssRNAs and dsRNAs were transcribed using Riboprobe in vitro transcription systems (Promega) and Replicator RNAi Kit (Finnzyme), respectively, following the manufacturers' instructions. RNAs were treated with DNaseI and purified prior to transfection.
Generation of partially purified VPg unlinkase, isolation of 35S-methionine-labeled vRNA, and in vitro unlinking reactions were conducted as described previously (Rozovics et al., 2011).
In vitro MDA5 ATPase assay was performed as described previously (Peisley et al., 2011).
Ribosomal RNA removal was performed using Ribo-Zero rRNA Removal Kit (Human.Mouse.Rat) (Epicenter RZH1046) according to the manufacturer’s protocol.
Additional Information
More detailed descriptions of experimental procedures and materials are provided in Extended Experimental Procedures.
Acknowledgments
We wish to thank Drs. Shizuo Akira and James Chen for the gifts of MEFs, Thomas Michiels and Robert Silverman for the gift of HeLa-M cells, and Toby Tuthill and Dave Rowlands for the gift of ERAV. S.H. and Q.F. are supported by Mosaic grants (NWO-017.002.025 and NWO-017.006.043, respectively), M.L. by a Rubicon grant (NWO-825.11.022), and F.K. by an ECHO grant (NWO-CW-700.59.007), all from the Netherlands Organisation for Scientific Research (NWO). B.L.S. is a Senior Fellow of the American Asthma Foundation.
Published: November 8, 2012
Footnotes
Supplemental Information includes Extended Results and Discussion, Extended Experimental Procedures, and five figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2012.10.005.
Licensing Information
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplemental Information
References
- Barton D.J., Flanegan J.B. Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J. Virol. 1997;71:8482–8489. doi: 10.1128/jvi.71.11.8482-8489.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., van Kuppeveld F.J.M. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr. Opin. Virol. 2012 doi: 10.1016/j.coviro.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata-Hartley C.L., Palmenberg A.C. Dipyridamole reversibly inhibits mengovirus RNA replication. J. Virol. 2005;79:11062–11070. doi: 10.1128/JVI.79.17.11062-11070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L., Barchet W., Gilfillan S., Cella M., Beutler B., Flavell R.A., Diamond M.S., Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hato S.V., Ricour C., Schulte B.M., Lanke K.H., de Bruijni M., Zoll J., Melchers W.J., Michiels T., van Kuppeveld F.J. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 2007;9:2921–2930. doi: 10.1111/j.1462-5822.2007.01006.x. [DOI] [PubMed] [Google Scholar]
- Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K., Hiiragi A., Dermody T.S., Fujita T., Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H., Takahasi K., Fujita T. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol. Rev. 2011;243:91–98. doi: 10.1111/j.1600-065X.2011.01052.x. [DOI] [PubMed] [Google Scholar]
- McCartney S.A., Thackray L.B., Gitlin L., Gilfillan S., Virgin H.W., Colonna M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008;4:e1000108. doi: 10.1371/journal.ppat.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A., Lin C., Wu B., Orme-Johnson M., Liu M., Walz T., Hur S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA. 2011;108:21010–21015. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A., Schulz O., Tan C.P., Rehwinkel J., Kato H., Takeuchi O., Akira S., Way M., Schiavo G., Reis e Sousa C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards O.C., Martin S.C., Jense H.G., Ehrenfeld E. Structure of poliovirus replicative intermediate RNA. Electron microscope analysis of RNA cross-linked in vivo with psoralen derivative. J. Mol. Biol. 1984;173:325–340. doi: 10.1016/0022-2836(84)90124-4. [DOI] [PubMed] [Google Scholar]
- Rozovics J.M., Virgen-Slane R., Semler B.L. Engineered picornavirus VPg-RNA substrates: analysis of a tyrosyl-RNA phosphodiesterase activity. PLoS ONE. 2011;6:e16559. doi: 10.1371/journal.pone.0016559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou K., Vakakis E., Kar S., Richer E., Evans G.L., Triantafilou M. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci. 2012 doi: 10.1242/jcs.103887. Published online July 13, 2012. [DOI] [PubMed] [Google Scholar]
- van Ooij M.J., Polacek C., Glaudemans D.H., Kuijpers J., van Kuppeveld F.J., Andino R., Agol V.I., Melchers W.J. Polyadenylation of genomic RNA and initiation of antigenomic RNA in a positive-strand RNA virus are controlled by the same cis-element. Nucleic Acids Res. 2006;34:2953–2965. doi: 10.1093/nar/gkl349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.P., Cerny A., Asher D.R., Kurt-Jones E.A., Bronson R.T., Finberg R.W. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 2010;84:254–260. doi: 10.1128/JVI.00631-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels E., Duijsings D., Notebaart R.A., Melchers W.J., van Kuppeveld F.J. A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-golgi transport. J. Virol. 2005;79:5163–5173. doi: 10.1128/JVI.79.8.5163-5173.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züst R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B.W., Ziebuhr J., Szretter K.J., Baker S.C., Barchet W., Diamond M.S. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplemental References
- Abraham, N., Stojdl, D.F., Duncan, P.I., Méthot, N., Ishii, T., Dubé, M., Vanderhyden, B.C., Atkins, H.L., Gray, D.A., McBurney, M.W., et al. (1999). Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J. Biol. Chem. 274, 5953–5962. [DOI] [PubMed]
- Carpentier, P.A., Williams, B.R., and Miller, S.D. (2007). Distinct roles of protein kinase R and toll-like receptor 3 in the activation of astrocytes by viral stimuli. Glia 55, 239–252. [DOI] [PubMed]
- Dong, B., Niwa, M., Walter, P., and Silverman, R.H. (2001). Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. RNA 7, 361–373. [DOI] [PMC free article] [PubMed]
- Fitzgerald, K.A., McWhirter, S.M., Faia, K.L., Rowe, D.C., Latz, E., Golenbock, D.T., Coyle, A.J., Liao, S.M., and Maniatis, T. (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496. [DOI] [PubMed]
- Luthra, P., Sun, D., Silverman, R.H., and He, B. (2011). Activation of IFN-β expression by a viral mRNA through RNase L and MDA5. Proc. Natl. Acad. Sci. USA 108, 2118–2123. [DOI] [PMC free article] [PubMed]
- Malathi, K., Saito, T., Crochet, N., Barton, D.J., Gale, M., Jr., and Silverman, R.H. (2010). RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 16, 2108–2119. [DOI] [PMC free article] [PubMed]
- Nallagatla, S.R., Toroney, R., and Bevilacqua, P.C. (2011). Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr. Opin. Struct. Biol. 21, 119–127. [DOI] [PMC free article] [PubMed]
- Schulz, O., Pichlmair, A., Rehwinkel, J., Rogers, N.C., Scheuner, D., Kato, H., Takeuchi, O., Akira, S., Kaufman, R.J., and Reis e Sousa, C. (2010). Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 7, 354–361. [DOI] [PMC free article] [PubMed]
- van Kuppeveld, F.J., Galama, J.M., Zoll, J., and Melchers, W.J. (1995). Genetic analysis of a hydrophobic domain of coxsackie B3 virus protein 2B: a moderate degree of hydrophobicity is required for a cis-acting function in viral RNA synthesis. J. Virol. 69, 7782–7790. [DOI] [PMC free article] [PubMed]
- van Ooij, M.J., Vogt, D.A., Paul, A., Castro, C., Kuijpers, J., van Kuppeveld, F.J., Cameron, C.E., Wimmer, E., Andino, R., and Melchers, W.J. (2006). Structural and functional characterization of the coxsackievirus B3 CRE(2C): role of CRE(2C) in negative- and positive-strand RNA synthesis. J. Gen. Virol. 87, 103–113. [DOI] [PubMed]
- Wang, X., and Seed, B. (2003). A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 31, e154. [DOI] [PMC free article] [PubMed]
- Xiang, Y., Wang, Z., Murakami, J., Plummer, S., Klein, E.A., Carpten, J.D., Trent, J.M., Isaacs, W.B., Casey, G., and Silverman, R.H. (2003). Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res. 63, 6795–6801. [PubMed]
- Zoll, J., Galama, J.M., van Kuppeveld, F.J., and Melchers, W.J. (1996). Mengovirus leader is involved in the inhibition of host cell protein synthesis. J. Virol. 70, 4948–4952. [DOI] [PMC free article] [PubMed]
- Zoll, J., Erkens Hulshof, S., Lanke, K., Verduyn Lunel, F., Melchers, W.J., Schoondermark-van de Ven, E., Roivainen, M., Galama, J.M., and van Kuppeveld, F.J. (2009a). Saffold virus, a human Theiler’s-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog. 5, e1000416. [DOI] [PMC free article] [PubMed]
- Zoll, J., Galama, J.M., and van Kuppeveld, F.J. (2009b). Identification of potential recombination breakpoints in human parechoviruses. J. Virol. 83, 3379–3383. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.