

Abstract
Many positive-strand RNA viruses remodel the endomembrane to form specialized replication organelles. However, knowledge regarding whether negative-strand RNA viruses take advantage of intracellular membranes for replication is limited. Here we show that a negative-strand RNA virus, human parainfluenza virus type 3 (HPIV3), remodels the endoplasmic reticulum (ER) membrane to form inclusion bodies (IBs), whereby the phosphoprotein (P) of HPIV3 recruits phosphatidylinositol 4-kinase beta (PI4KB) to IBs to generate PI4P, creating a PI4P-enriched microenvironment to promote HPIV3 replication. In addition, we find that human respiratory syncytial virus (HRSV) also takes advantage of the ER to form IBs and that these IBs are also enriched with PI4P. The nucleoprotein of HRSV recruits PI4KB to IBs. These results suggest that paramyxoviruses also exploit the host endomembrane to form IBs and that PI4KB is recruited by viral proteins to enrich IBs with PI4P to facilitate viral replication.
Keywords: PI4KB, IBs, ER membrane, HPIV3
Graphical Abstract
Highlights
-
•
Inclusion bodies (IBs) of HPIV3 induce membrane rearrangement of ER
-
•
PI4P generated by PI4KB on IBs facilitates replication of HPIV3
-
•
PI4KB is recruited to IBs via interaction with the HPIV3 phosphoprotein, P
-
•
Remodeling ER is a general mechanism for IBs of negative-strand RNA viruses
Many positive-strand RNA viruses reorganize the endomembrane to form specialized replication organelles. However, whether negative-strand RNA viruses use intracellular membranes for replication is largely unknown. Li et al. demonstrate that inclusion bodies of HPIV3 induce ER membrane rearrangement. PI4KB is recruited to IBs to generate PI4P, which facilitates viral replication.
Introduction
Viruses rely on their host for viability and replication, and the host cell provides not only nucleotides and amino acids for viral metabolism but also a structural platform for viral replication and assembly (Belov and van Kuppeveld, 2012, Salonen et al., 2005). Positive-strand RNA viruses and even some DNA viruses, such as vaccinia virus, remodel intracellular membranes to form their replication organelles (ROs) (Miller and Krijnse-Locker, 2008, Salonen et al., 2005, Schwartz et al., 2004). Different viruses rely on different organelles, and the appearance of ROs is manifold. For example, during infection, hepatitis C virus (HCV), dengue virus, severe acute respiratory syndrome (SARS) coronavirus, and arterivirus induce positive curvature of the endoplasmic reticulum (ER) membrane, forming discrete membrane vesicles (Knoops et al., 2008, Knoops et al., 2012, Romero-Brey et al., 2012, Welsch et al., 2009). Cells infected with poliovirus, coxsackie virus B3 (CVB3), and rhinovirus contain many complex and dynamic structures that originate from the cis-Golgi membrane (Belov and Sztul, 2014, Roulin et al., 2014). Flock House virus (FHV) induces formation of spherules with negative curvature on the mitochondrial membrane (Kopek et al., 2007). Electron microscopy of the replication structure of tomato bushy stunt virus showed that ROs were generated from the peroxisome membrane (McCartney et al., 2005).
Positive-strand RNA viruses remodel intracellular membranes to form replication sites that contain unique lipids and proteins distinct from the original organelles, in which phosphatidylinositol 4-kinase (PI4K) is a common component. PI4K contains four isoforms: PI4KA, PI4KB, PI4K2A, and PI4K2B (Roulin et al., 2014). In general, PI4K is recruited to replication sites via its interaction with viral proteins directly or with the help of cytokines, such as GBF1/ARF1 or ACBD3 (Hsu et al., 2010, Lei et al., 2017, Sasaki et al., 2012, Xiao et al., 2017). Then, PI4K yields phosphatidylinositol-4-phosphate (PI4P) on ROs to bind and induce conformational changes in the RNA-dependent RNA polymerase (RdRp) to modulate RdRp activity (Hsu et al., 2010). Moreover, PI4P locally changes the RO membrane curvature during viral infection to generate a high-curvature membrane pocket to shield viral components from host defense (Belov and van Kuppeveld, 2012, Miller and Krijnse-Locker, 2008). In addition, PI4P is exchanged with cholesterol to maintain essential membrane properties that determine the shape and function of ROs (Roulin et al., 2014). However, whether PI4K is required for negative-strand RNA viral replication has not been explored, possibly because the existence of ROs in negative-strand RNA viruses has not been confirmed.
A variety of animal viruses, such as DNA viruses (e.g., poxviruses and herpesviruses) and some RNA viruses (e.g., reovirus, togavirus, bunyavirus), form inclusion bodies (IBs) or viroplasms, which have been speculated to be viral replication factories (Netherton et al., 2007, Novoa et al., 2005a), but we know little about the organization and composition of IBs. Recent studies of double-strand RNA viruses (e.g., wheat yellow mosaic virus and reovirus) showed that these viruses take advantage of the ER membrane to form IBs, and IBs are thought to derive from the ER (Fernández de Castro et al., 2014, Sun et al., 2014). In addition, a few reports have suggested that negative-strand RNA viruses may also remodel the endomembrane to form IBs. For example, ribosomes have been found close to Ebola virus IBs, suggesting that IBs of Ebola virus may form near rough ER, although the ER structure was not clearly detected (Nelson et al., 2016). Negri body-like structures (NBLs) are sites of rabies virus RNA synthesis, and electron microscopy observations have indicated that NBLs are progressively associated with a double membrane that may have been derived from the rough ER (Nikolic et al., 2017). Bunyamwera virus infection induces fragmented Golgi stacks, where assembly and maturation take place (Novoa et al., 2005b, Salanueva et al., 2003). Oropouche virus attracts endosomal sorting complex required for transport (ESCRT) machinery components to modify Golgi cisternae, forming viral factory units (Barbosa et al., 2018). However, compared with positive-strand RNA viruses, the IB formation mechanisms of many negative-strand RNA viruses remain unexplored.
Human parainfluenza virus type 3 (HPIV3), a classical enveloped negative-strand RNA virus, is in the Paramyxoviridae family. HPIV3 infection can also induce formation of IBs. Its genome encodes six structural proteins: nucleoprotein (N), phosphoprotein (P), polymerase (L), matrix protein (M), and two spike glycoproteins consisting of hemagglutinin-neuraminidase protein (HN) and fusion protein (F) (Moscona, 2005). Our previous study showed that the association of N and P is the minimum requirement for formation of IBs. A mutant of N, NL478A, was unable to interact with P and failed to induce formation of IBs when co-expressed with P (Zhang et al., 2013). HPIV3 can cause serious respiratory diseases in children and infants. However, no effective treatments exist; thus, further exploration of IB organization and the cytokines involved in executing IB function can enhance our understanding of HPIV3 replication mechanisms so that we can better develop antiviral therapies or vaccines.
In the current study, we focused on the in situ properties of the viral RNA replication structures in cells infected with paramyxoviruses. We demonstrate that HPIV3 remodels the ER membrane to form IBs. P recruits PI4KB to IBs, and PI4KB on IBs produces a PI4P lipid microenvironment, which reinforces IB structures and facilitates HPIV3 replication. Finally, we found that another paramyxovirus, human respiratory syncytial virus (HRSV), also takes advantage of the ER membrane to modulate the formation of IBs and that PI4P is also rich in HRSV IBs. The N of HRSV interacts with PI4KB and recruits PI4KB to IBs.
Results
HPIV3 IBs Change the Distribution of ER Proteins
Our previous results showed that expression of HPIV3 N and P is the minimum requirement for viral IB formation and that IBs are replication sites for HPIV3 (Zhang et al., 2013, Zhang et al., 2017). To determine whether the IBs of HPIV3 bear membrane structures and to assess the relationship between HPIV3 IBs and the endomembrane, we first sought to determine localization changes of organelle marker proteins during IB formation. After HeLa cells were transfected with plasmids encoding N and P for 24 h, we found that the distribution of the Golgi marker protein TGN46 and the mitochondrial marker protein Tom20 remained unchanged (Figures 1A and 1B). However, the ER marker protein Calnexin formed aggregates and colocalized with viral IBs (Figure 1C), and the expression of N or P alone failed to induce formation of aggregates by Calnexin (Figure S1A), suggesting that re-localization of Calnexin was induced only during IB formation.
Figure 1.

HPIV3 IBs Change the Distribution of ER Proteins
(A–D) HeLa cells grown in 24-well plates were transfected with plasmids encoding N-Myc (0.1 μg) and HA-P (0.4 μg) for 24 h to form IBs and analyzed for colocalization of the Golgi protein TGN46 (A), the mitochondrial protein Tom20 (B), the ER protein Calnexin (C), the ER protein PDI (D), and IBs. The fluorescence intensity profile of IBs (green) and organelle proteins (red) was measured along the line drawn on a 2× zoom panel by Leica Application Suite Advanced Fluorescence Lite.
(E and F) HeLa cells stably expressing GFP-tagged P were infected with HPIV3 (MOI = 0.1) for consecutive times (0 h, 12 h, 24 h, 30 h, and 36 h) and analyzed for distribution of the ER proteins Calnexin (E) and PDI (F). Yellow arrows indicate representative colocalization of ER proteins and IBs.
(G) Kinetic process of the ER protein Calnexin fusing into IBs. HeLa cells expressing GFP-tagged P were seeded into 20-mm dishes for 24 h, infected with HPIV3 (MOI = 0.1), transfected with mCherry-Calnexin (0.5 μg), and visualized by live-cell imaging. The fusion event is marked with white arrows and numbers. In (a) and (b), white arrows indicate Calnexin protein (red) attached to small IBs (green) fused into small IBs to form a homogeneous structure. In (b) and (c), 1 and 2 fused into 3. In (d) and (e), arrow 4 indicates that Calnexin was absorbed into small IBs. In (f)–(h), 3 and 4 fused into 7 and 5 and 6 fused into 8. In (i) and (j), 7 and 8 fused into 9.
Scale bars, 10 μm. See also Figure S1.
Next we assessed another ER protein, PDI, and a similar conclusion was reached (Figures 1D and S1B). To exclude bleed-through from the green channel into the red channel, HeLa cells were transfected with N-Myc and HA-P and only stained for cellular marker proteins, using the same acquisition parameters for the red channel as used for imaging of TGN46, Tom20, Calnexin, and PDI. We found that Calnexin and PDI still formed aggregates but that the distribution of TGN46 and Tom20 remained unchanged (Figures S1C–S1F). Next we sought to determine how ER marker proteins are recruited to IBs, and we were not able to show interaction of either N or P with Calnexin or PDI (Figures S1G–S1J), suggesting that the distribution changes and colocalization of Calnexin or PDI with IBs were not achieved through their interaction with N or P. To determine whether HPIV3 infection induces ER marker proteins to form aggregates, we infected HeLa cells stably expressing GFP-tagged P with HPIV3 (MOI = 0.1) for up to 36 h and analyzed the distribution of Calnexin and PDI at different time points post-infection (p.i.). As infection progressed, small IBs were observed at the early stage of infection (24 h p.i.) and gradually became larger at 30 h and 36 h p.i. At the same time, the aggregates of Calnexin and PDI also gradually became larger and colocalized with IBs (Figures 1E and 1F). Of note, the expression levels of Calnexin and PDI remained unchanged (Figures S1K and S1L), suggesting that IBs only change the distribution of ER proteins, not the amount of protein.
To gain insight into the dynamic fusion of ER proteins with IBs, live-cell imaging was performed. For this, HeLa cells stably expressing GFP-tagged P were infected with HPIV3 (MOI = 0.1) and transfected with a plasmid that expressed the mCherry-tagged Calnexin protein. Upon IB fusion, Calnexin aggregates also became bigger. We found that Calnexin could be detected on the surface of small IBs and fused with IBs, which reflected the gradual absorption and integration of Calnexin by IBs (Figure 1G; Video S1). Taken together, these results suggest that HPIV3 changes the distribution of ER components and recruits them to IBs; thus, the ER membrane might be remodeled and serve as a source of IB components for viral replication.
HPIV3 IBs Are Surrounded by a Fragmented ER Membrane, and IB Formation Depends on the ER
To confirm the relationship between HPIV3 IBs and the ER, we used transmission electron microscopy to observe the ultrastructure of the ER and IBs. When HeLa cells were transfected with an empty vector or a vector encoding NL478 and P, HeLa cells were unable to form IBs and were enriched with integrated ER structures. In contrast, when HeLa cells were co-transfected with plasmids encoding N and P to form IBs, the number of classical ER structures was significantly reduced, and we observed many fragmented ER structures around IBs. Moreover, we detected fragmented ER structures that closely surrounded IBs in HPIV3-infected HeLa cells (Figure 2 A), suggesting that HPIV3 IBs induced ER fragmentation, and debris from the ER surrounded and was attached to IBs. Next, HeLa cells stably expressing GFP-tagged P were infected with HPIV3 (MOI = 0.1) for 36 h. Through Airyscan super-resolution microscopy, we found that Calnexin colocalized with IBs, and surface projections of three-dimensional reconstructions showed that ER membrane fragments existed near IBs (Figure 2B), which suggests that IBs were surrounded by a fragmented ER membrane. To confirm whether IBs contained membrane structures derived from the ER, HeLa cells were treated with a hypotonic solution and lysed with a Dounce homogenizer. The homogenate was left untreated or treated with Triton X-100, trypsin, or Triton X-100 plus trypsin. In the homogenate treated with trypsin, N and P were not completely digested (Figures 2C, lane 3, and 2D), but NL478A and P were completely digested (Figures 2C, lane 7, and 2D), suggesting that the fragmented ER membrane around IBs protected N and P from degradation by trypsin.
Figure 2.

HPIV3 IBs Are Surrounded by a Fragmented ER Membrane, and IB Formation Depends on the ER
(A) HeLa cells were transfected with vector (a), NL478A-Myc and HA-P (b), or N-Myc and HA-P (c) for 24 h or infected with HPIV3 (MOI = 0.1) for 36 h and analyzed via electron microscopy to assess the ultrastructure of the ER and IBs. Black arrows indicate the fragmented ER membrane. The black spheroid structure indicates an IB.
(B) Surface projections of a three-dimensional reconstruction with accumulations of fragmented ER membranes around IBs. HeLa cells expressing GFP-tagged P were mock-treated or infected with HPIV3 (MOI = 0.1), transfected with mcherry-Calnexin for 36 h, and analyzed via Zeiss Airyscan Super-resolution microscopy. Representative Airyscan images were acquired without pixel saturation. Boxed regions were magnified and processed to a three-dimensional surface reconstruction.
(C) HeLa cells were transfected with the indicated plasmids for 24 h. Cell lysates were harvested and treated with a hypotonic solution for 20 min and broken with a Dounce homogenizer. The homogenate was left untreated or treated with Triton X-100, trypsin, or Triton X-100 plus trypsin and analyzed via WB.
(D) HeLa cells are treated in the way described above, and the contents of N and P protein were analyzed to determine quantity by Quantity one software.
(E–G) Effects of the ER stress inducer Tu on IB formation. HeLa cells were transfected with N-Myc and HA-P for 12 h, treated with Tu (2 μg/mL) for 6 h, and analyzed for the location of Calnexin and the size and number of IBs. Representative images are shown (E). Also shown are quantitative analysis of the average number of IBs per cell treated with DMSO or Tu (F; n = 3, 50 cells were counted) and quantitative analysis of the average diameter of IBs treated with DMSO or Tu (G; n = 3, 100 IBs were counted).
(H–J) Effects of the ER stress inducers Tg and Tu on HPIV3 replication. HeLa cells were infected with HPIV3 (MOI = 0.1) for 12 h and treated with Tg (1 μM) for 3 h or Tu (2 μg/mL) for 6 h. The HPIV3 protein HN was analyzed via western blot (WB) (H), extracellular viral production was analyzed via plaque assay (I), and HPIV3 RNA was analyzed by qPCR (J).
(K–M) The IB components N and P were rich in rough ER. HeLa cells were transfected with plasmids encoding N-Myc (0.5 μg) and HA-P (2 μg) or NL478A-Myc (0.5 μg) and HA-P (2 μg) for 24 h, and the RER was extracted to detect IB composition. Representative images are shown (K). Also shown is quantitative analysis of the N or NL478A and P content in rough endoplasmic reticulum (RER) when expressing IBs (N and P) or non-IBs (NL478A and P) (L and M).
Images are representative of experiments carried out at least three times. Error bars, mean ± SD of three experiments (n = 3). Student’s t test, ★p < 0.05, ★★p < 0.01, ★★★p < 0.001. Scale bars in (A), 500 nm; scale bars in (B), 3 μm; All other scale bars, 10 μm. See also Figure S2.
Next, we used the ER stress inducers thapsigargin (Tg) and tunicamycin (Tu) to disrupt the structure of the ER. When HeLa cells were treated with Tg for 3 h or Tu for 6 h, proteolytic cleavage of ATF6 (an ER stress-regulated transmembrane transcription factor) was induced, and XBP1s protein was expressed (Figure S2A), which demonstrates that Tg and Tu can induce classic ER stress. Of note, Tg and Tu had no effects on the metabolic activity of cells (Figure S2B). HeLa cells were transfected with plasmids encoding N-Myc and HA-P and then treated with Tu. We calculated the average number of IBs per cell and the average diameter of IBs and found that, although Calnexin and PDI were still distributed to IBs, the size of IBs in Tu-treated cells was significantly smaller and the number of IBs was lower than that in untreated cells (Figures 2E–2G and S2C), and a comparable result was reached when cells were treated with Tg (data not shown). HPIV3 replication was also significantly suppressed (Figures 2H–2J), suggesting that formation of HPIV3 IBs and HPIV3 replication rely on the ER. Of note, expression of N and P was not influenced by treatment with Tg and Tu (Figure S2D), which demonstrates that the inhibition of IB formation by Tg and Tu is not due to expression of N and P. In contrast, EV71 replication was not inhibited by Tg or Tu treatment (Figures S2E and S2F), which indicates that EV71 replication site formation does not mainly rely on the ER. Furthermore, we found that the components of IBs, N and P, were enriched in purified ER (Figures 2K, lane 3, 2L, and 2M). Because of their inability to induce IB formation, NL478A and P were not enriched in purified ER (Figures 2K, lane 4, 2L, and 2M). In addition, N and P were not enriched in purified mitochondria (Figure S2G). Taken together, these findings strongly suggest that HPIV3 IBs might be derived from the ER by inducing ER fragmentation and recruiting ER ingredients.
HPIV3 IBs Are Enriched with PI4P
Positive-strand RNA viruses remodel the endomembrane to form ROs rather than directly replicating on original organelles, primarily because these ROs possess unique characteristics, such as abundant PI4P, which is critical for viral replication. Thus, we sought to determine whether and how HPIV3 IBs become enriched with PI4P to facilitate replication. First, we found that abundant PI4P localized on IBs in transfected and HPIV3-infected cells (Figures 3A and 3B). In addition, to exclude bleed-through from the green channel into the red channel, we only stained for PI4P, using the same acquisition parameters for the red channel as used for imaging of PI4P, and we found that PI4P still formed aggregates (Figure S3), suggesting that HPIV3 also takes advantage of lipid synthesis pathways to replenish ROs with PI4P.
Figure 3.

HPIV3 IBs Are Enriched with PI4P
(A) HeLa cells were transfected with N-Myc (0.1 μg) and HA-P (0.4 μg) for 24 h and then analyzed to detect PI4P on IBs. The fluorescence intensity profile of IBs (green) and PI4P (red) was measured along the line drawn on a 2× zoom panel by Leica Application Suite Advanced Fluorescence Lite.
(B) HeLa cells stably expressing GFP-P were infected with HPIV3 (MOI = 0.1) for consecutive times (0 h, 12 h, 24 h, 30 h, and 36 h) and then analyzed to detect PI4P on IBs. Yellow arrows indicate representative colocalization of PI4P and IBs.
Scale bars, 10 μm. See also Figure S3.
PI4P on IBs Is Generated by PI4KB
PI4P is generated by PI4K, which comprises four isoforms: PI4KA, PI4KB, PI4K2A, and PI4K2B. To determine which PI4K was involved in the generation of PI4P on IBs, we first assessed the proximity of the four PI4K isoforms to IBs via immunofluorescence and found that neither PI4KA nor PI4K2A was recruited to IBs (Figures S4A and S4E). Furthermore, using a pool of small interfering RNAs directed against the known PI4Ks, we found that HPIV3 replication was not significantly affected in cells treated with short hairpin RNA (shRNA) targeting PI4KA or PI4K2A (Figures S4B–S4D and S4F–S4H), suggesting that neither PI4KA nor PI4K2A was involved in generating PI4P on IBs and was not required for HPIV3 replication. Although PI4KB and PI4K2B colocalized with IBs (Figures 4 A and S4I), viral replication was significantly reduced only in cells treated with shRNA targeting PI4KB (Figures 4B–4D) but not PI4K2B (Figures S4J–S4L), suggesting that PI4KB was involved in generation of PI4P on IBs.
Figure 4.

PI4P on IBs Was Generated by PI4KB
(A) HeLa cells were transfected with plasmids encoding N-Myc (0.1 μg), HA-P (0.4 μg), and FLAG-PI4KB (0.5 μg) for 24 h and analyzed for colocalization of FLAG-PI4KB and IBs. The fluorescence intensity profile of IBs (green) and PI4KB (red) was measured along the line drawn on a 2× zoom panel by Leica Application Suite Advanced Fluorescence Lite.
(B–D) Effects of PI4KB knockdown on HPIV3 replication. HeLa cells were infected with HPIV3 (MOI = 0.1) and transfected with shRNA targeting PI4KB (3 μg) for 24 h. The HPIV3 protein HN was analyzed via WB (B), extracellular viral production was analyzed via plaque assay (C), and HPIV3 RNA was analyzed by qPCR (D).
(E) HeLa cells were transfected with the indicated plasmids for 24 h and analyzed for colocalization of endogenous PI4KB and IBs.
(F) HeLa cells stably expressing GFP-P were infected with HPIV3 (MOI = 0.1) for up to 36 h and analyzed for colocalization of endogenous PI4KB and IBs. Yellow arrows indicate representative colocalization of PI4KB and IBs.
(G and H) HeLa cells were transfected with N-Myc (0.5 μg) and HA-P (2 μg) for 24 h, and the ER was extracted to detect PI4KB in RER when expressing IBs (N and P) or non-IBs (NL478A and P) via WB. Representative images are shown (G). Also shown is quantitative analysis of the PI4KB content in RER when expressing IBs (N and P) or non-IBs (NL478A and P) (H).
Images are representative of experiments carried out at least three times. Error bars, mean ± SD of three experiments (n = 3). Student’s t test; ★p < 0.05, ★★p < 0.01, ★★★p < 0.001; ns, non-significant. Scale bars, 10 μm. See also Figure S4.
To confirm the colocalization of PI4KB and IBs, we also examined endogenous PI4KB and found that endogenous PI4KB aggregated and colocalized with IBs in transfected and HPIV3-infected cells (Figures 4E and 4F), whereas neither N nor P alone could induce PI4KB accumulation on IBs (Figures S4M and S4N). To exclude the bleed-through from the green channel into the red channel, HeLa cells were transfected with N-Myc, HA-P, and FLAG-PI4KB or N-Myc and HA-P, and stained for only PI4KB, using the same acquisition parameters for the red channel as used for imaging of FLAG and PI4KB, and we found that PI4KB still formed aggregates (Figures S4O and S4P). Moreover, PI4KB was present in purified ER of HeLa cells expressing IBs but not in purified ER of HeLa cells expressing NL478A and P (Figures 4G, lanes 3 and 4, and 4H). Taken together, these results show that HPIV3 recruits PI4KB to IBs to generate PI4P, resulting in enrichment of PI4P on IBs derived from the ER.
PI4KB Facilitates HPIV3 Replication
To confirm the relationship between PI4KB and HPIV3 replication, we employed the PI4KB-specific inhibitor PIK93 at a concentration of 2–4 μM (Figure S5A), which had minimal effects on the metabolic activity of cells (Figure S5B). When cells were treated with PIK93, PI4P generation on IBs declined significantly compared with DMSO-treated cells (Figure 5 A). Simultaneously, HPIV3 replication was apparently decreased (Figures 5B–5D). Of note, PIK93 did not influence the expression of PI4KB and its colocalization on IBs (Figures S5C and S5D). Furthermore, we carried out a recovery experiment and found that HPIV3 replication was strongly inhibited when cells were treated with shRNA targeting PI4KB and was rescued by expression of wild-type PI4KB, but HPIV3 replication was not rescued by expression of a functionally inactivated mutant of PI4KB, KD-PI4KB (Figures 5E–5G). Correspondingly, generation of PI4P on IBs was also reduced, and expression of wild-type PI4KB, but not KD-PI4KB, restored the levels of PI4P on IBs in PI4KB knockdown cells (Figure S5E). Of note, KD-PI4KB was also recruited to IBs but lost the ability to generate PI4P (Figure S5F). Next, to determine whether PI4KB or PI4P directly supports viral genome replication, an HPIV3 minigenome assay was performed with increasing amounts of FLAG-tagged PI4KB. The results showed that PI4KB increased the minigenome-encoded reporter activity in a dose-dependent manner (Figure 5H). In our experiment, HPIV3 minigenome-encoded reporter expression was driven by vTF7-3 (a recombinant vaccinia virus expressing T7 RNA polymerase), whereas it has been reported that the H7 protein of vaccinia virus binds PI4P, which is essential for poxvirus replication (Kolli et al., 2015). To exclude the possibility that the increase in minigenome reporter activity was due to the effect of PI4KB on vTF7-3, a control plasmid that encodes Renilla luciferase under control of the T7 promoter was constructed. The activity of Renilla luciferase was adjusted to a level comparable with that of the minigenome firefly luciferase (Figure 5I, lanes 1 and 3). Then, plasmids expressing PI4KB were co-transfected with pGBKT7-Renilla luciferase and the minigenome assay components, respectively. We found a significant increase in the activity of minigenome-encoded firefly luciferase and almost no increase in the activity of Renilla luciferase (Figure 5I, lanes 2 and 4). Taken together, these results show that PI4KB facilitates HPIV3 genome replication directly by producing PI4P on IBs.
Figure 5.

PI4KB Facilitates HPIV3 Replication
(A) PIK93 reduced PI4P on IBs. HeLa cells were treated with PIK93 (2 μM and 4 μM) for 1 h and transfected with plasmids encoding N-Myc (0.1 μg) and HA-P (0.4 μg) for 24 h. PI4P on IBs was analyzed by immunofluorescence.
(B–D) Effects of the PI4KB-specific inhibitor PIK93 on HPIV3 replication. HeLa cells were treated with PIK93 up to 4 μM for 1 h and infected with HPIV3 (MOI = 0.1) for 36 h. Cell lysates were harvested, and HPIV3 protein HN was analyzed via WB (B), extracellular viral production was analyzed via plaque assay (C), and HPIV3 RNA was analyzed by qPCR (D).
(E–G) HeLa cells were infected with HPIV3 (MOI = 0.1) and transfected with the indicated plasmids. The HPIV3 protein HN was analyzed via WB (E), extracellular viral production was analyzed via plaque assay (F), and HPIV3 RNA was analyzed via qPCR (G).
(H and I) PI4KB increases minigenome reporter activity.
(H) The effect of PI4KB on minigenome reporter RNA synthesis. vTF7-3-infected HeLa cells were transfected with pGEM4-N (125 ng), pGEM4-P (62.5 ng), pGEM4-L (100 ng), pOCUS-HPIV3-MG (50 ng), and increasing amounts of plasmid encoding PI4KB (200 ng, 400 ng, 800 ng, and 1600 ng). Relative luciferase activity was measured according to the manufacturer’s instructions. Expression of N, P, and PI4KB was detected via WB, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control.
(I) PI4KB has no effect on vTF7-3. Plasmids encoding Renilla luciferase (5 ng) and plasmids encoding N, P, L, and the minigenome were transfected alone or together with FLAG-PI4KB (1600 ng) in vTF7-3 infected HeLa cells to exclude nonspecific effects of PI4KB on vTF7-3. Relative luciferase activity was measured, and expression of PI4KB was detected via WB.
Images are representative of experiments carried out at least three times. Error bars, mean ± SD of three experiments (n = 3). Student’s t test; ★p < 0.05, ★★p < 0.01, ★★★p < 0.001. Scale bars, 10 μm. See also Figure S5.
PI4KB Is Recruited to IBs via Its Interaction with P
Next, we sought to determine how PI4KB is recruited to IBs. Previous studies of positive-strand RNA viruses showed that multiple picornaviruses utilize viral proteins, particularly 3A, to recruit PI4KB to ROs, and that 3A colocalizes with ROs (Greninger et al., 2012, Xiao et al., 2017). Given that the HPIV3 proteins N and P are major components of IBs, we first determined whether N or P was involved in PI4KB recruitment, and we found that FLAG-PI4KB strongly interacted with HA-P (Figure 6 A). Likewise, HA-P significantly interacted with FLAG-PI4KB (Figure 6B), and HA-P interacted with endogenous PI4KB (Figure 6C). These results confirm that P and PI4KB physically interact on IBs. However, we found no interaction between N and PI4KB (Figures 6D and 6E).
Figure 6.

PI4KB Is Recruited to IBs via Its Interaction with P
(A) HEK293T cells were transfected with empty vector (2 μg) and HA-P (1 μg) or with FLAG-PI4KB (2 μg) and HA-P (1 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
(B) HEK293T cells were transfected with empty vector (1 μg) and FLAG-PI4KB (2 μg) or with HA-P (1 μg) and FLAG-PI4KB (2 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
(C) HEK293T cells were transfected with HA-P (1 μg) or empty vector (1 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
(D) HEK293T cells were transfected with empty vector (2 μg) and N-Myc (1 μg) or with FLAG-PI4KB (2 μg) and N-Myc (1 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
(E) HEK293T cells were transfected with empty vector (1 μg) and FLAG-PI4KB (2 μg) or with N-Myc (1 μg) and FLAG-PI4KB (2 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
(F) HEK293T cells were transfected with empty vector (2 μg), N-Myc (1 μg), and HA-P (1 μg) or with FLAG-PI4KB (2 μg), N-Myc (1 μg), and HA-P (1 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
(G) HEK293T cells were transfected with empty vector (1 μg), HA-P (1 μg), and FLAG-PI4KB (2 μg); with N-Myc (1 μg), HA-P (1 μg), and FLAG-PI4KB (2 μg); or with NL478A-Myc (1 μg), HA-P (1 μg), and FLAG-PI4KB (2 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
Next, N-Myc, HA-P, and FLAG-PI4KB were co-expressed in HEK293T cells, and, surprisingly, we found that FLAG-PI4KB interacted with N-Myc (Figure 6F), suggesting that N interacts with PI4KB via the N-P complex. Finally, we employed a mutant, NL478A, which fails to interact with P, and found that N, but not NL478A, was able to interact with FLAG-PI4KB in the presence of P (Figure 6G). To confirm that P but not N was associated with PI4KB, we performed co-immunoprecipitation (coIP) in HeLa cells, and a comparable conclusion was reached (Figures S6A and S6B). Taken together, these results show that P is responsible for PI4KB recruitment to IBs via its interaction with PI4KB.
Remodeling the ER Structure and Recruiting PI4K Are General Mechanisms of IBs of Paramyxoviruses
Based on the aforementioned results, we concluded that HPIV3 reorganizes ER membranes to form IBs and that P recruits PI4KB to IBs to generate PI4P to facilitate HPIV3 replication. Next, we sought to determine whether other paramyxoviruses, such as HRSV, use similar mechanisms to generate PI4P on IBs. First, we found that the ER marker protein Calnexin neither aggregated nor colocalized with HRSV IBs (Figure 7 A). However, another ER marker protein, PDI, formed aggregates and colocalized with IBs (Figure 7B). Furthermore, PDI did not aggregate when N or P of HRSV was expressed alone (Figure S7A), suggesting that HRSV may also take advantage of the ER membrane to form IBs; however, the components absorbed and utilized are different from those utilized by HPIV3. Second, we found that PI4P lipids and PI4KB were also rich in HRSV IBs (Figures 7C and 7D) and that PI4KB did not aggregate when N or P of HRSV was expressed alone (Figure S7B), suggesting that HRSV also recruited PI4KB to produce PI4P on IBs. Finally, we found that N, but not P, of HRSV strongly interacted with FLAG-PI4KB (Figures 7E and 7F). Furthermore, PI4KB interacted with P of HRSV via the N-P complex (Figure 7G). Taken together, these data demonstrate that HRSV also takes advantage of the ER and that PI4P lipids are enriched in IBs. The HRSV N protein recruits PI4KB to generate PI4P on IBs.
Figure 7.

Remodeling the ER Structure Is a General Mechanism of IBs of Paramyxoviruses
(A and B) HeLa cells expressing GFP-tagged HRSV-P were transfected with HRSV-N-Myc (0.5 μg) for 24 h and analyzed for colocalization of Calnexin (A) and PDI (B) and HRSV IBs. The fluorescence intensity profile of IBs (green) and ER proteins (red) was measured along the line drawn on a 2× zoom panel by Leica Application Suite Advanced Fluorescence Lite.
(C) HeLa cells expressing GFP-tagged HRSV-P were transfected with HRSV-N-Myc (0.5 μg) for 24 h and analyzed for colocalization of PI4P and HRSV IBs. The fluorescence intensity profile of IBs (green) and PI4P (red) was measured.
(D) HeLa cells expressing GFP-tagged HRSV-P were transfected with HRSV-N-Myc (0.5 μg) and FLAG-PI4KB (0.5 μg) for 24 h and analyzed for colocalization of FLAG-PI4KB and HRSV IBs. The fluorescence intensity profile of IBs (green) and PI4KB (red) was measured.
(E) HEK293T cells were transfected with empty vector (2 μg) and HRSV-N-Myc (1 μg) or with FLAG-PI4KB (2 μg) and HRSV-N-Myc (1 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
(F) HEK293T cells were transfected with empty vector (2 μg) and HRSV-HA-P (1 μg) or with FLAG-PI4KB (2 μg) and HRSV-HA-P (1 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
(G) HEK293T cells were transfected with empty vector (2 μg), HRSV-N-Myc (1 μg), and HRSV-HA-P (1 μg) or with FLAG-PI4KB (2 μg), HRSV-N-Myc (1 μg), and HRSV-HA-P (1 μg) for 36 h. Cell lysates were subjected to IP and analyzed via WB.
Images are representative of experiments carried out at least three times; Scale bars, 10 μm. See also Figure S7.
Discussion
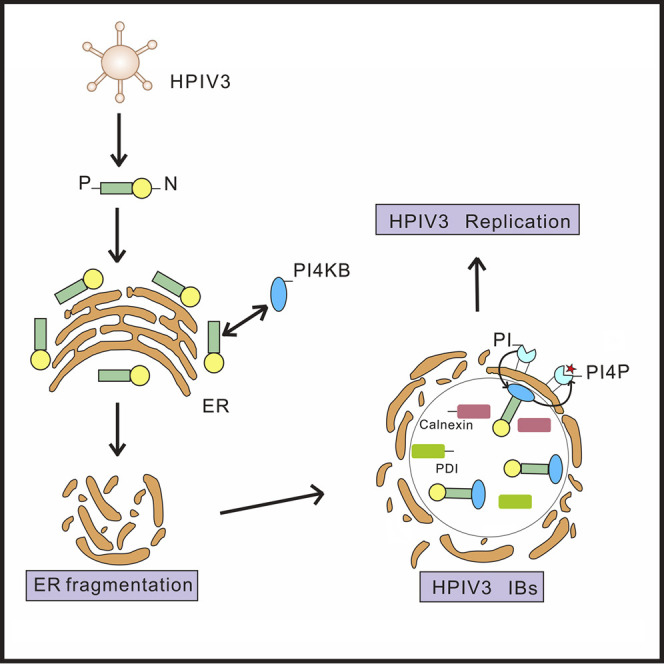
In summary, we show that, similar to positive-strand RNA viruses, the negative-strand RNA viruses HPIV3 and HRSV also remodel the endomembrane to form their replication factories (Figures 1, 2, 7A, and 7B), namely IBs, which are enriched with PI4P lipids (Figures 3 and 7C). The PI4P lipid microenvironment is an important facilitator of HPIV3 replication, and cellular PI4KB is a key player in this process (Figure 5). These findings highlight the dependence on the endomembrane and PI4P among negative-strand RNA viruses and should prompt related studies to determine the mechanism by which PI4P lipids modulate viral RNA replication. Furthermore, our findings may provide a basis for the design of a class of therapeutic agents that target the negative-strand RNA viral replication platform.
Based on our findings, we propose the following model for reorganization of the ER membrane in HPIV3 infection, which generates PI4P lipid-enriched IBs. In infected cells, HPIV3 translates mRNA into N and P, and increasing amounts of N and P result in ER membrane fragmentation. ER ingredients (such as Calnexin and PDI) and N and P form IBs together. Meanwhile, ER debris is loosely distributed around IBs. Subsequently, P recruits PI4KB to IBs, where it catalyzes the production of PI4P lipids, leading to biogenesis of a PI4P-enriched microenvironment, which facilitates HPIV3 replication.
EV71 replication was not inhibited by Tg or Tu treatment, which indicates that EV71 replication site formation did not rely on the ER (Figures S2E and S2F). Consistent with this finding, previous studies have reported that the ROs of EV71, CVB3, and poliovirus are often located close to the cis-Golgi and that the secretory vesicle budding machinery might be involved in RO formation (Belov et al., 2012, Belov and Sztul, 2014, Greninger et al., 2012, Hsu et al., 2010, Limpens et al., 2011). Furthermore, we found that PI4P was enriched on the IBs of paramyxoviruses just as it was enriched in the ROs of positive-strand RNA viruses. Therefore, we concluded that both positive- and negative-strand RNA viruses can remodel the endomembrane to form ROs and that they take advantage of lipid synthesis pathways to replenish ROs with PI4P to facilitate replication. In addition, we have a deeper understanding of paramyxovirus IB organization. IBs are not only the protein aggregates but also structures composed of ER ingredients and surrounded by a fragmented ER membrane.
We found that the ER marker proteins Calnexin and PDI both colocalized with HPIV3 IBs (Figures 1C and 1D); however, only PDI colocalized with HRSV IBs (Figures 7A and 7B). In other words, although HPIV3 and HRSV both take advantage of the ER membrane to form IBs, different viruses absorb different components of the ER, which implies the complexity of paramyxovirus IB organization. Similarly, previous studies considered the ER membrane to be the main membrane source of HCV ROs (Romero-Brey et al., 2012, Welsch et al., 2009). However, a recent study showed that the Golgi and autophagosome membrane are important for HCV replication and highlighted the complexity of the cellular membrane structures of positive-strand viruses (Hansen et al., 2017). Interestingly, the autophagy marker LC3 was also found on HPIV3 IBs (data not shown), suggesting that autophagy may also play a role in IB formation. Consistent with this, our previous results showed that HPIV3 P blocks autophagosome-lysosome fusion to increase virus production (Ding et al., 2014). Thus, we assume that P blocks autophagosome-lysosome fusion to increase the abundance of autophagosomes, and then HPIV3 takes advantage of autophagosomes other than the ER to form IBs. Moreover, a recent report showed that HRSV IBs contain sub-compartments that were termed “IB-associated granules” (IBAGs), and that newly synthetized viral mRNA and the viral transcription anti-terminator M2-1 concentrate in IBAGs. In contrast, viral genome RNA, N, L, and its cofactor P are excluded from IBAGs (Rincheval et al., 2017). Whether IBAGs are separated with other space of IBs by membrane structures is worth exploring. These findings provide substantial insights into the organization of IBs, which were previously thought to be aggregates of proteins. These findings suggest that IBs are extremely complex and elaborate structures, and understanding their composition could greatly help us understand the mechanisms of viral replication.
ROs of positive-strand RNA viruses promote efficient viral replication by anchoring the replication machinery in membrane compartments or by shielding it from host defense mechanisms (Netherton et al., 2007, Novoa et al., 2005a). Electron microscopy showed that dengue virus- and HCV-induced membrane structures contain vesicle pores, which could enable release of newly synthesized viral RNA, and revealed budding of viral particles on ROs directly opposed to vesicle pores (Miyanari et al., 2007, Paul et al., 2014, Romero-Brey et al., 2012, Welsch et al., 2009). Our results strongly suggest that the IBs of paramyxoviruses consist of membranous ingredients and that fragmented ER membranes loosely surround the core of HPIV3 IBs. On one hand, this membrane structure protects IBs from degradation to ensure viral replication (Figures 2C and 2D). On the other hand, many cytokines are involved in viral replication, and the gaps between incompact ER debris ensure communication between IBs and the cytosol. Most importantly, our previous results showed that HPIV3 IBs escape the antiviral effect of stress granules by concealing their own newly synthesized viral RNAs (Hu et al., 2018). Subsequently, loose ER membranes around IBs may create a favorable environment to transport synthesized viral RNA to the cytoplasm and escape the alarm system of host cells in a sustained-release manner.
During positive-strand RNA virus replication, PI4P can recruit cellular proteins that have a PI4P-binding pleckstrin homology domain, such as the oxysterol-binding protein (OSBP). OSBP exchanges cholesterol with PI4P on membrane contact sites between ROs and other organelles (Nchoutmboube et al., 2015, Roulin et al., 2014), which generate and maintain essential membrane properties that drive the shape and function of ROs. However, we did not detect enriched cholesterol around IBs (data not shown), which suggests that HPIV3 replication and transcription do not rely on cholesterol. In addition, RdRp 3Dpol of enterovirus preferentially binds PI4P lipids over other cellular lipid components (Hsu et al., 2010), indicating that PI4P serves as a protein docking site on the RO membrane. Furthermore, PI4P lipid binding may also induce conformational changes and modulate RdRp enzymatic activity (Hsu et al., 2010). The mechanism whereby PI4P facilitates HPIV3 replication is not clear.
Several protein partners have been investigated for their involvement in PI4K recruitment to produce PI4P. PI4K is recruited to replication sites via its interaction with viral proteins directly or with the help of cytokines, such as GBF1/ARF1 or ACBD3 (Greninger et al., 2012, Hong et al., 2014, Sasaki et al., 2012, Xiao et al., 2017). Here we found that recruitment of PI4KB to HPIV3 IBs depended on P (Figures 6A–6C and S6B). So far, we have not found any intermediate proteins involved in the process of recruitment.
HCV nonstructural protein 5A (NS5A) has been found to interact with PI4KA, and direct activation of a lipid kinase by HCV NS5A contributes critically to the integrity of the membranous ROs of HCV (Reiss et al., 2011). However, when we inhibited PI4KB activity via PIK93 or reduced its expression via shRNA targeting PI4KB, the morphological characteristics of IBs were almost unaffected (Figure 5A; Figures S5D and S5E). That is, PI4KB is involved in HPIV3 IB function but not IB formation. However, we are not sure whether PI4KB is recruited to IBs before or after fragmentation of the ER.
In conclusion, our work focused on describing the relationship between paramyxovirus IBs and the endomembrane, and we found that negative-strand viruses recruited PI4Ks to ROs. Research in these areas may offer fascinating insight into IBs of negative-strand RNA viruses and may also yield therapeutic strategies in negative-strand RNA virus-induced disease.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti HPIV3 HN | Abcam | Cat. #: ab49756; RRID: AB_2313773 |
| Mouse monoclonal anti-TGN46 | Sigma-Aldrich | Cat. #: SAB4200235; RRID: AB_10762671 |
| Mouse monoclonal anti-TOM20 | BD Biosciences | Cat. #: 612278; RRID: AB_399595 |
| Rabbit polyclonal anti-calnexin | Sigma-Aldrich | Cat. #: C4731; RRID: AB_476845 |
| Rabbit polyclonal anti-PDI | Abcam | Cat. #: ab3672; RRID: AB_303990 |
| Mouse monoclonal anti-GAPDH | Santa Cruz | Cat. #: sc32233; RRID: AB_627679 |
| Rabbit polyclonal anti-PI42A | ABclonal | Cat. #: A7168; RRID: AB_10683078 |
| Rabbit polyclonal anti-PI4K2B | Abcam | Cat. #: ab37812; RRID: AB_777261 |
| Rabbit polyclonal anti-PI4 Kinase | Cell Signaling Technology | Cat. #: 4902; RRID: AB_2164029 |
| Rabbit polyclonal anti-PI4 Kinase beta | Abcam | Cat. #: ab134756; RRID: AB_2313773 |
| Mouse monoclonal anti-PI4P | Echelon Biosciences | Cat. #: Z-P004; RRID: AB_11127796 |
| Bacterial and Virus Strains | ||
| Human parainfluenza virus type 3 | Lab stored | N/A |
| Enterovirus 71 | Lab stored | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Thapsigargin | Y.Liu lab, Wuhan university | N/A |
| Tunicamycin | Med Chem Express (MCE) | Cat. #: 11089-65-9 |
| PIK93 | Sigma-Aldrich | Cat. #: SML0546 |
| Lipofectamine 2000 | Invitrogen | Cat. #: 11668 |
| Critical Commercial Assays | ||
| Endoplasmic reticulum isolation kit | Sigma-Aldrich | Cat. #: ER0100 |
| In vitro toxicology assay kit | Sigma-Aldrich | Cat. #: TOX8 |
| Luciferase assay system kit | Promega | Cat. #: E1501 |
| Experimental Models: Cell Lines | ||
| 293T cells | CCTCC | GDC187 |
| HeLa cells | CCTCC | GDC009 |
| LLC-MK2 cells | CCTCC | GDC156 |
| Oligonucleotides | ||
| shRNA targeting sequences | N/A | |
| PI4KA | This paper | N/A |
| GCGTCTCATCACATGGTACAA | ||
| PI4KB | This paper | N/A |
| GCAAGAAACACGAAGGATCAT | ||
| PI4K2A | This paper | N/A |
| CAATGACAACTGGCTGATTAA | ||
| PI4K2B | This paper |
N/A |
| GCTGCAATTGATAATGGTCTA | ||
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Lead Contact and Materials Availability
Further information and requests for resources and regents should be directed to and will be fulfilled by the Lead Contact, Mingzhou Chen (chenmz@whu.edu.cn). This study did not generate new unique reagents.
Experimental Model and Subject Details
Experimental cells (HeLa cells, HEK293T cells, LLC-MK2 cells) obtained from ACTCC were cultured in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum (FBS; GIBCO) and 100U/ml penicillin/streptomycin (GIBCO) at 37°C with 5% CO2.
Method Details
Antibodies and reagents
Mouse anti-TGN46 monoclonal Ab was purchased from Sigma-Aldrich. Mouse anti-Tom20 monoclonal Ab was purchased from BD Biosciences. Rabbit anti-Calnexin polyclonal antibodies (Abs) was purchased from Sigma-Aldrich. Rabbit anti-PDI polyclonal Ab was purchased from Abcam. Rabbit anti-LMNB1 Ab was purchased from ABclonal. Rabbit anti-PI4KA polyclonal Ab was purchased from ABclonal Technology. Rabbit anti-PI4B and anti-PI4K2B polyclonal Abs were purchased from Abcam. Rabbit anti-PI42A polyclonal Ab was purchased from Cell Signaling Technology. Mouse anti-HPIV3 HN monoclonal Ab was purchased from Abcam. Mouse anti-PI4P monoclonal Ab was purchased from Echelon Biosciences. Mouse anti-Flag monoclonal Ab, rabbit anti-Flag polyclonal Ab, and mouse anti-HA monoclonal Ab were purchased from Sigma-Aldrich. Mouse anti-Myc and anti-GAPDH monoclonal Abs were purchased from Santa Cruz Biotechnology, Inc. Goat anti-rabbit IgG Rhodamine and goat anti-mouse IgG Fluorescein were purchased from Thermo. DAPI was purchased from Sigma-Aldrich. PIK93 was purchased from Sigma-Aldrich and dissolved in dimethylsulfoxide (DMSO). Thapsigargin (Tg) was a gift from Y. Liu Lab (Wuhan University). Tunicamycin (Tu) was purchased from Med Chem Express (MCE) and dissolved in DMSO.
Infection and transfection
For infection, HeLa or LLC-MK2 cells were infected with DMEM containing viruses with a multiplicity of infection (MOI) as indicated in the figure legends. 2 h later, the medium was replaced with fresh DMEM with 10% FBS. For transfection, plasmids were transfected by using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, cells were harvested or further treatment as indicated.
SDS-PAGE and Western Blot
Cells were harvested and lysed with lysis buffer (150 nM NaCL, 50 nM Tris-HCL [PH 7.4], 1% Triton X-100, 1mM EDTA [PH 8.0], 0.1% SDS with a protease inhibitor cocktail) for 30 min at 4°C. The supernatants were collected by centrifugation at 12000 g for 25 min at 4°C. Protein concentrations were determined based on the Bradford method. Equal amounts of proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane (GE Healthcare). After blocking with 5% non-fat milk dissolved in PBST (phosphate-buffered saline with 0.1% Tween 20), the membrane was incubated with the primary Abs, followed by horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Thermo Fisher Scientific). The proteins were detected on a Fujifilm LAS-4000 imaging system by using immobilon western HRP substrate (Millipore). The primary antibodies used were as follows: mouse anti-HN (1:2500), mouse anti-GAPDH (1:10000), mouse anti-HA (1:10000), mouse anti-Flag (1:5000), mouse anti-Myc (1:5000), rabbit anti-LMNB1 (1:500), mouse anti-Tom20 (1:5000), rabbit anti-calnexin (1:5000), rabbit anti-PDI (1:5000), rabbit anti-GM130 (1:5000), rabbit anti-PI4KA (1:1000), rabbit anti-PI42A (1:500), rabbit anti-PI4KB (1:5000), rabbit anti-PI42B (1:100), HRP-conjugated goat anti-mouse immunoglobulin (IgG) (1:5000) and goat anti-rabbit IgG (1:5000) were used as secondary antibodies.
Immunofluorescence Analysis
HeLa cells were cultured in 24-well plates with coverslips and transfected with the indicated plasmids or infected with HPIV3 (MOI = 0.1). Cells were washed with phosphate-buffered saline (PBS) three times (5 min each time) and fixed with 4% (wt/vol) paraformaldehyde/PBS for 20 min at room temperature. Then Cells were washed with PBS three times (5 min each time) and incubated with 50 μg/ml digitonin diluted with PBS for 5 min and washed with PBS three times (5 min each time). Next, cells were blocked with 3% (wt/vol) bovine serum albumin (BSA)/PBS at room temperature for 30 min and incubated with primary Abs diluted in 1% (wt/vol) BSA/PBS for 1 h at 4°C. Cells were washed with PBS three times (5 min each time) and incubated with secondary antibodies diluted in 1% (wt/vol) BSA/PBS for 1 h. After being stained with DAPI (sigma) for 5 min, cells were then observed with confocal microscope.
The primary antibodies used were as follows: mouse anti-TGN46 (1:100), rabbit mouse anti-Tom20 (1:200), anti-Calnexin (1:500), rabbit anti-PDI (1:500), mouse anti-HA (1:1000), rabbit anti-HA (1:1000), mouse anti-PI4P (1:500), rabbit anti-PI4KB (1:500), rabbit anti-Flag (1:1000), mouse anti-Myc (1:1000). All images were captured through Leica confocal microscopy unless otherwise marked.
Live cell imaging
For live cell imaging, HeLa cells stably expressing GFP tagged P were seeded into 20 mm dishes 24 h prior to infection and infected with HPIV3 (MOI = 0.1), and transfected with the mcherry-Calnexin. 30 h later, cells incubated in a 37°C and 5% CO2 environment were visualized on a Zeiss LSM 800 fluorescence microscope with a temporal resolution of 30 s per picture.
Co-IP using cell lysates
HEK293T cells or HeLa cells were transfected with the indicated plasmids for 24 h. Cells were harvested and lysed with 400 μL lysis buffer (150 mM NaCl, 50 mM Tris-HCl [pH,7.4], 1%Triton X-100, 1 mM EDTA [pH,8.0], and 0.1% SDS and protease inhibitor cocktail) for 30 min on ice. The supernatants were collected via centrifugation at 12000 g for 30 min at 4°C. Next, 50 μL supernatants were added with loading buffer and boiled at 100°C for 10 min. Additionally, 350 μL supernatants were precleared by adding 20 μL protein G Sepharose 4 Fast Flow beads at 4°C with rotation for 2 h. Supernatants were collected via centrifugation at 5000 g 4°C for 2 min. The indicated primary Abs and 30 μL beads were added and incubated at 4°C with rotation overnight. Beads were collected via centrifugation at 5000 g for 2 min and washed with lysis buffer three times (2 min each time). Next, beads were boiled with loading buffer at 100°C for 10 min, and the proteins were analyzed via WB.
plaque assay
For the plaque assay, MK2 cells were cultured in 24-well plates at a density of 70%–80% at 37°C overnight, and virus stocks were successively diluted 10-fold up to 105 with DMEM. Next, 400 μL dilutions were added to wells for 2 h at 37°C and 5% CO2. Then, the medium was replaced with methylcellulose, and cells were cultured at 37°C and 5% CO2 for 3-4 days. Cells were stained with crystal violet, and the viral titers were calculated.
Transmission Electron Microscopy
HeLa cells were transfected with the indicated plasmids for 24 h or infected with HPIV3 (MOI = 0.1) for 36 h, and the cells were fixed with fixative liquid (3% paraformaldehyde and 1.5% glutaraldehyde in 0.1M sodium phosphate buffer [pH,7.4]) for 1 h at room temperature, harvested and subjected to gradient centrifugation (1000 g, 5 min; 3000 g, 5 min; 6000 g, 5 min; 12000 g, 5 min) at 4°C, post-fixed with 1% osmium tetroxide for 1 h at 4°C under dark conditions, incubated with 2% uranyl acetate overnight, dehydrated in increasing concentrations of ethanol (50%, 75%, 95%, and 100%), and processed for embedding in epoxy resin. Ultrathin (∼70 nm) sections were collected on uncoated 200-mesh copper grids and stained with uranyl acetate and lead citrate, and were evaluated via transmission electron microscopy (JEOL, JEM-1400 plus) operating at 100kV.
Subcellular Fractionation
HeLa cells were transfected with the indicated plasmids; 24 h later, the cells were harvested and subjected according to the protocol for the ER isolation kit (Sigma). For mitochondrial isolation, cells were harvested and resuspended with 500 μL isolation buffer (10 mM Tris-HCl [pH,7.4], 2 mM MgCl2, 10 mM KCl, and 250 mM sucrose). Cells were homogenized with 30 strokes of a Dounce homogenizer on ice. The supernatants were collected after centrifugation at 500 g for 10 min at 4°C, transferred into another tip and subjected to centrifugation for 10 min at 5000 g. Precipitates were resuspended with 1 mL isolation buffer and collected via centrifugation at 5000 g for 10 min at 4°C. Then the precipitates were resuspended in protein loading buffer and boiled at 100°C for 10 min. Equivalent amounts of protein from each fraction were analyzed by western blotting (WB).
Plasmids and shRNA Oligonucleotides
The coding regions of PI4KA, PI4K2A, PI4KB and PI4K2B were obtained from HeLa cells via RNA extraction and subsequent reverse transcription polymerase chain reaction (RT-PCR) and were cloned into pCAGGS with a N-terminal Flag tag. The plasmids HPIV3-N-Myc, HPIV3-NL478A-Myc, HPIV3-HA-P, pGEM4-N, pGEM4-P, pGEM4-L, pOCUS-HPIV3-MG have been described previously (Zhang et al., 2013). The construct encoding Calnexin with an mCherry epitope tag at N terminus was generated by PCR-based cloning techniques and cloned into pCAGGS. Double-strand oligonucleotides corresponding to the target sequences were cloned into pLKO.1. The target sequences for PI4KA were as follows: GCGTCTCATCACATGGTACAA. The target sequences for PI4KB were as follows: GCAAGAAACACGAAGGATCAT. The target sequences for PI4K2A were as follows: CAATGACAACTGGCTGATTAA. The target sequences of PI4K2B were as follows: GCTGCAATTGATAATGGTCTA.
In vitro Minigenome Assay of HPIV3
HeLa cells grown in 12-well-plates were infected with vTF7-3 at an MOI of 1 for 1 h, and transfected with pGEM4-N (125 ng), pGEM4-P (62.5 ng), pGEM4-L (100 ng), pOCUS-HPIV3-MG (50 ng) alone or together with pCAGGS-Flag-PI4KB by using Lipofectamine 2000 (Invitrogen), 4 h later, the transfection medium was replaced by fresh DMEM with 10% FBS. 24 h post-transfection, the cells were harvested and lysed with 150 μl lysis buffer, and an aliquot of 20 μl was used for luciferase activity analysis according to the manufacturer’s instructions. Assays were repeated three times for accuracy.
RNA quantification
Total RNA from mock- or HPIV3-infected cells was isolated using TRIZOL reagent (Ambion, Invitrogen) according to the manufacturer’s instructions, and 1μg total RNA of each sample was used for reverse transcription (RT) using a reverse transcriptase (Fermentas, Waltham, MA, USA), followed by qPCR analysis to measure the indicated RNA abundance. Data shown are the relative abundance of the indicated RNA normalized to that of GAPDH. The following Primers were used: Human GAPDH forward: 5′- GAGTCAACGGATTTGGTCGT-3′; Human GAPDH reverse: 5′- GACAAGCTTCCCGTTCTCAG-3′; HPIV3 N forward: 5′-ATCAGATTGGGTCAATCAT; HPIV3 N reverse: 5′- AATTCCATACCTGATTGTATT-3′.
Quantification and Statistical Analysis
Statistical parameters including the definition and exact values of n, distribution and deviation are reported in the figure legends. Data are expressed as mean ± standard deviation (SD). The significance of the variability between different groups was determined by two-way analyses of variance using GraphPad Prism software (version 5.0). A p value of < 0.05 was considered statistically significant and a p value of > 0.05 was considered statistically non-significant.
Data and Code Availability
All data are available upon request to the lead contact. No proprietary software was used in the data analysis.
Acknowledgments
We acknowledge Yong Liu (Wuhan University, China) for providing thapsigargin. This research is supported by grants from the National Natural Science Foundation of China (81825015), National Key R&D Program of China (2017YFA0505801), the National Natural Science Foundation of China (81871650 and 31630086), National Science and Technology Major Project (2018ZX10101004), and The Natural Science Foundation of Hubei Province Innovation Group (2017CFA022).
Author Contributions
Z.L., D.G., Y.Q., and M.C. designed the experiments, analyzed and organized data, and wrote the paper. Z.L., Y.Q., and M.C. performed the experiments. D.G. helped construct plasmids and designed shRNAs. All authors discussed the results and commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 19, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.052.
Supplemental Information
References
- Barbosa N.S., Mendonça L.R., Dias M.V.S., Pontelli M.C., da Silva E.Z.M., Criado M.F., da Silva-Januário M.E., Schindler M., Jamur M.C., Oliver C. ESCRT machinery components are required for Orthobunyavirus particle production in Golgi compartments. PLoS Pathog. 2018;14:e1007047. doi: 10.1371/journal.ppat.1007047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., Sztul E. Rewiring of cellular membrane homeostasis by picornaviruses. J. Virol. 2014;88:9478–9489. doi: 10.1128/JVI.00922-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., van Kuppeveld F.J. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr. Opin. Virol. 2012;2:740–747. doi: 10.1016/j.coviro.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., Nair V., Hansen B.T., Hoyt F.H., Fischer E.R., Ehrenfeld E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012;86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B., Zhang G., Yang X., Zhang S., Chen L., Yan Q., Xu M., Banerjee A.K., Chen M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe. 2014;15:564–577. doi: 10.1016/j.chom.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Fernández de Castro I., Zamora P.F., Ooms L., Fernández J.J., Lai C.M., Mainou B.A., Dermody T.S., Risco C. Reovirus forms neo-organelles for progeny particle assembly within reorganized cell membranes. MBio. 2014;5:e00931-13. doi: 10.1128/mBio.00931-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greninger A.L., Knudsen G.M., Betegon M., Burlingame A.L., Derisi J.L. The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIβ. J. Virol. 2012;86:3605–3616. doi: 10.1128/JVI.06778-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M.D., Johnsen I.B., Stiberg K.A., Sherstova T., Wakita T., Richard G.M., Kandasamy R.K., Meurs E.F., Anthonsen M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. U.S.A. 2017;114:E3462–E3471. doi: 10.1073/pnas.1616683114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z., Yang X., Yang G., Zhang L. Hepatitis C virus NS5A competes with PI4KB for binding to ACBD3 in a genotype-dependent manner. Antiviral Res. 2014;107:50–55. doi: 10.1016/j.antiviral.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Hsu N.Y., Ilnytska O., Belov G., Santiana M., Chen Y.H., Takvorian P.M., Pau C., van der Schaar H., Kaushik-Basu N., Balla T. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z., Wang Y., Tang Q., Yang X., Qin Y., Chen M. Inclusion bodies of human parainfluenza virus type 3 inhibit antiviral stress granule formation by shielding viral RNAs. PLoS Pathog. 2018;14:e1006948. doi: 10.1371/journal.ppat.1006948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., Kikkert M., Worm S.H., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., Bárcena M., Limpens R.W., Koster A.J., Mommaas A.M., Snijder E.J. Ultrastructural characterization of arterivirus replication structures: reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 2012;86:2474–2487. doi: 10.1128/JVI.06677-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolli S., Meng X., Wu X., Shengjuler D., Cameron C.E., Xiang Y., Deng J. Structure-function analysis of vaccinia virus H7 protein reveals a novel phosphoinositide binding fold essential for poxvirus replication. J. Virol. 2015;89:2209–2219. doi: 10.1128/JVI.03073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopek B.G., Perkins G., Miller D.J., Ellisman M.H., Ahlquist P. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 2007;5:e220. doi: 10.1371/journal.pbio.0050220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X., Xiao X., Zhang Z., Ma Y., Qi J., Wu C., Xiao Y., Zhou Z., He B., Wang J. The Golgi protein ACBD3 facilitates Enterovirus 71 replication by interacting with 3A. Sci. Rep. 2017;7:44592. doi: 10.1038/srep44592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpens R.W., van der Schaar H.M., Kumar D., Koster A.J., Snijder E.J., van Kuppeveld F.J., Bárcena M. The transformation of enterovirus replication structures: a three-dimensional study of single- and double-membrane compartments. MBio. 2011;2 doi: 10.1128/mBio.00166-11. e00166–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney A.W., Greenwood J.S., Fabian M.R., White K.A., Mullen R.T. Localization of the tomato bushy stunt virus replication protein p33 reveals a peroxisome-to-endoplasmic reticulum sorting pathway. Plant Cell. 2005;17:3513–3531. doi: 10.1105/tpc.105.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S., Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008;6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanari Y., Atsuzawa K., Usuda N., Watashi K., Hishiki T., Zayas M., Bartenschlager R., Wakita T., Hijikata M., Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- Moscona A. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J. Clin. Invest. 2005;115:1688–1698. doi: 10.1172/JCI25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nchoutmboube J., Ford-Siltz L.A., Belov G.A. Enterovirus replication: go with the (counter)flow. Trends Microbiol. 2015;23:183–184. doi: 10.1016/j.tim.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Nelson E.V., Schmidt K.M., Deflubé L.R., Doğanay S., Banadyga L., Olejnik J., Hume A.J., Ryabchikova E., Ebihara H., Kedersha N. Ebola Virus Does Not Induce Stress Granule Formation during Infection and Sequesters Stress Granule Proteins within Viral Inclusions. J. Virol. 2016;90:7268–7284. doi: 10.1128/JVI.00459-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netherton C., Moffat K., Brooks E., Wileman T. A guide to viral inclusions, membrane rearrangements, factories, and viroplasm produced during virus replication. Adv. Virus Res. 2007;70:101–182. doi: 10.1016/S0065-3527(07)70004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic J., Le Bars R., Lama Z., Scrima N., Lagaudrière-Gesbert C., Gaudin Y., Blondel D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017;8:58. doi: 10.1038/s41467-017-00102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa R.R., Calderita G., Arranz R., Fontana J., Granzow H., Risco C. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol. Cell. 2005;97:147–172. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa R.R., Calderita G., Cabezas P., Elliott R.M., Risco C. Key Golgi factors for structural and functional maturation of bunyamwera virus. J. Virol. 2005;79:10852–10863. doi: 10.1128/JVI.79.17.10852-10863.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D., Madan V., Bartenschlager R. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe. 2014;16:569–579. doi: 10.1016/j.chom.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Kaderali L., Poenisch M., Blankenburg H., Hiet M.S. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 2011;9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincheval V., Lelek M., Gault E., Bouillier C., Sitterlin D., Blouquit-Laye S., Galloux M., Zimmer C., Eleouet J.F., Rameix-Welti M.A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017;8:563. doi: 10.1038/s41467-017-00655-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Brey I., Merz A., Chiramel A., Lee J.Y., Chlanda P., Haselman U., Santarella-Mellwig R., Habermann A., Hoppe S., Kallis S. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012;8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulin P.S., Lötzerich M., Torta F., Tanner L.B., van Kuppeveld F.J., Wenk M.R., Greber U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe. 2014;16:677–690. doi: 10.1016/j.chom.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Salanueva I.J., Novoa R.R., Cabezas P., López-Iglesias C., Carrascosa J.L., Elliott R.M., Risco C. Polymorphism and structural maturation of bunyamwera virus in Golgi and post-Golgi compartments. J. Virol. 2003;77:1368–1381. doi: 10.1128/JVI.77.2.1368-1381.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen A., Ahola T., Kääriäinen L. Viral RNA Replication in Association with Cellular Membranes. Curr. Top. Microbiol. Immunol. 2005;285:139–173. doi: 10.1007/3-540-26764-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki J., Ishikawa K., Arita M., Taniguchi K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2012;31:754–766. doi: 10.1038/emboj.2011.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M., Chen J., Lee W.M., Janda M., Ahlquist P. Alternate, virus-induced membrane rearrangements support positive-strand RNA virus genome replication. Proc. Natl. Acad. Sci. USA. 2004;101:11263–11268. doi: 10.1073/pnas.0404157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Andika I.B., Shen J., Yang D., Chen J. The P2 of Wheat yellow mosaic virus rearranges the endoplasmic reticulum and recruits other viral proteins into replication-associated inclusion bodies. Mol. Plant Pathol. 2014;15:466–478. doi: 10.1111/mpp.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K., Walther P., Fuller S.D., Antony C., Krijnse-Locker J., Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X., Lei X., Zhang Z., Ma Y., Qi J., Wu C., Xiao Y., Li L., He B., Wang J. Enterovirus 3A facilitates viral replication by promoting PI4KB-ACBD3 interaction. J. Virol. 2017;91 doi: 10.1128/JVI.00791-17. e00791–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Chen L., Zhang G., Yan Q., Yang X., Ding B., Tang Q., Sun S., Hu Z., Chen M. An amino acid of human parainfluenza virus type 3 nucleoprotein is critical for template function and cytoplasmic inclusion body formation. J. Virol. 2013;87:12457–12470. doi: 10.1128/JVI.01565-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Jiang Y., Cheng Q., Zhong Y., Qin Y., Chen M. Inclusion Body Fusion of Human Parainfluenza Virus Type 3 Regulated by Acetylated α-Tubulin Enhances Viral Replication. J. Virol. 2017;91 doi: 10.1128/JVI.01802-16. e01802–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available upon request to the lead contact. No proprietary software was used in the data analysis.