

Significance
MYC is a transcriptional regulator that controls much of the coding and noncoding transcriptome. It is also an oncoprotein that functions as a driver in numerous human cancers. The mechanism of this oncogenic activity is not known, but it probably involves specific MYC-regulated target genes. Here we systematically identify and characterize MYC-regulated, long noncoding RNAs (lncRNAs) that are required for MYC-driven cellular proliferation in human lymphoid cells. We use targeted transcriptional repression, adding new tools to this technology that will facilitate the functional analysis of any transcriptional regulator.
Keywords: CRISPRi, MYC, oncogenesis, transcriptional regulation, lncRNAs
Abstract
MYC controls the transcription of large numbers of long noncoding RNAs (lncRNAs). Since MYC is a ubiquitous oncoprotein, some of these lncRNAs probably play a significant role in cancer. We applied CRISPR interference (CRISPRi) to the identification of MYC-regulated lncRNAs that are required for MYC-driven cell proliferation in the P493-6 and RAMOS human lymphoid cell lines. We identified 320 noncoding loci that play positive roles in cell growth. Transcriptional repression of any one of these lncRNAs reduces the proliferative capacity of the cells. Selected hits were validated by RT-qPCR and in CRISPRi competition assays with individual GFP-expressing sgRNA constructs. We also showed binding of MYC to the promoter of two candidate genes by chromatin immunoprecipitation. In the course of our studies, we discovered that the repressor domain SID (SIN3-interacting domain) derived from the MXD1 protein is highly effective in P493-6 and RAMOS cells in terms of the number of guides depleted in library screening and the extent of the induced transcriptional repression. In the cell lines used, SID is superior to the KRAB repressor domain, which serves routinely as a transcriptional repressor domain in CRISPRi. The SID transcriptional repressor domain is effective as a fusion to the MS2 aptamer binding protein MCP, allowing the construction of a doxycycline-regulatable CRISPRi system that allows controlled repression of targeted genes and will facilitate the functional analysis of growth-promoting lncRNAs.
Members of the MYC family of oncogenes (c-Myc [MYC], N-Myc, and L-Myc) are ubiquitous transcriptional regulators that are deregulated, translocated, or amplified in most human cancers (1–4). Elevated MYC levels are often associated with cancer aggressiveness and poor prognosis; however, therapies directed against this important oncoprotein have yet to reach the clinic (5–7). MYC plays a central role in the pathogenesis of Burkitt’s lymphoma (BL), an aggressive type of non-Hodgkin’s lymphoma that arises from germinal center B cells. A hallmark of BL is the t(8,14) chromosomal translocation that brings the MYC coding sequence under the control of Ig enhancers, leading to uncontrolled MYC overexpression (8–10).
MYC is a basic helix-loop-helix leucine zipper (bHLH-LZ) protein that forms dimers with the small bHLH-LZ protein MAX, which enables sequence-specific binding to the E-box DNA element CACGTG or variants thereof (3, 11). Along with transcriptional activators, the proximal MYC network also includes bHLH-LZ proteins that function as strong transcriptional repressors (e.g., MXD family members). MXD-MAX dimers compete with MYC for binding to the same DNA elements and have the potential to counteract MYC activity on the same transcriptional targets (3, 12, 13).
In addition to protein coding genes, MYC pervasively regulates the expression of noncoding transcripts, including long noncoding RNAs (lncRNAs) (14, 15). In mammalian genomes, the number of lncRNA genes range in the tens of thousands, and their expression is highly cell type-specific (16). There is mounting evidence that deregulation of lncRNA expression is directly involved in the pathogenesis of cancer (17). Various molecular functions have been ascribed to lncRNAs, including gene regulation in cis, regulation of mRNA stability, and modulation of protein function (18–21); however, the functions of the vast majority of these enigmatic transcripts are unknown.
We have previously shown that in the BL model cell line P493-6, doxycycline (dox)-mediated inhibition of MYC expression from a tet-OFF controlled transgene leads to significant changes in the transcription of hundreds of noncoding RNAs (ncRNAs) (14). Some of these (e.g., MIR17HG and DANCR) have been shown to contribute to the oncogenic phenotypes induced by MYC overexpression (22–24); however, most of the MYC-regulated noncoding genes remain to be investigated. Modern gene engineering technologies provide tools for systematic functional analysis of genes, and such an analysis could help to select the most important lncRNAs that mediate the effects of MYC.
The type II prokaryotic CRISPR/Cas9 adaptive immune system is a powerful tool for RNA-guided site-specific DNA cleavage (25). A single guide RNA (sgRNA) directs the nuclease Cas9 to efficiently cleave targeted loci, which results in indels (insertion/deletions) upon repair by nonhomologous end-joining. While induced indels within open reading frames can disrupt translation of functional proteins, indels are less likely to affect the function of most noncoding genes (26).
Paired guide RNAs along with Cas9 have been used in CRISPR screens to delete lncRNAs; however, cloning of such libraries is laborious, and large genomic deletions potentially affect nearby or overlapping genes (26, 27).
An alternative is catalytically inactive Cas9 (dCas9) fused to effector domains that can be used as RNA-guided transcriptional regulators to activate (CRISPRa) or inhibit (CRISPRi) the expression of endogenous genes. These CRISPR tools have been successfully used in large-scale functional genomic screens to interrogate noncoding loci (28, 29).
Here we designed an sgRNA library to target the transcription start sites (TSSs) of MYC-regulated noncoding genes in human B cells. Pooled library screens using dCas9 fused to the highly effective transcriptional repressor domain of MXD1 identified noncoding loci that are required for robust growth of the human lymphoid cell line P493-6 (30) and the BL cell line RAMOS (31). This study also provides additional tools based on CRISPRi to facilitate genetic perturbation of noncoding targets. The investigation of lncRNAs that mediate oncogenic MYC functions will deepen our understanding of the roles of MYC in cancer.
Results
Effective CRISPRi for Noncoding MYC Targets.
For functional knockout of lncRNAs, we first tested whether gene expression can be prevented by deleting sequences surrounding the TSS of a noncoding target. Expression of Cas9 along with paired sgRNAs eliminated portions of the MALAT1 proximal promoter but had little effect on RNA expression (SI Appendix, Fig. S1 A–C). Lack of robustness, low deletion efficiency, and challenges in library preparation of paired sgRNA vectors prompted us to abandon this approach and instead use CRISPRi for the systematic identification of essential MYC-regulated lncRNAs. We designed and produced a CRISPR/sgRNA library to target sequences surrounding the TSSs of 508 noncoding genes whose expression levels respond to changing MYC levels in the human lymphoid cell line P493-6 (14, 32). As controls, we included 100 MYC-regulated coding genes, as well as 14 genes known to code for proteins that are essential for cell survival and proliferation (essential control genes). We also included 100 nontargeting (NT) guide sequences as negative controls. Stringent selection criteria, detailed in Materials and Methods, were applied to select up to 10 guides per gene. The resulting MYCncLibrary consisted of 5,708 individual sgRNAs (Dataset S1), and the guide sequences were cloned into lentiviral vectors for pooled expression in human cells. Pooled library screens were used to interrogate individual loci for their impact on MYC-mediated cell proliferation.
For these experiments, we used the BL cell line RAMOS in addition to P493-6. RNA-sequencing (RNA-seq) analysis showed that in these two lymphoid cell lines, the expression of coding and noncoding genes strongly overlaps, and that 83% of the genes targeted by the MYCncLibrary were also expressed in RAMOS cells (SI Appendix, Fig. S2A and Dataset S2) (32–34).
For the initial library screen, RAMOS cells stably expressing dCas9 fused to the transcriptional repressor domain KRAB of ZNF10 (dCas9-KRAB) along with parental RAMOS cells (no dCas9 expression) were transduced with a lentiviral pool of the MYCncLibrary. The cells were selected for their ability to survive and proliferate for 14 d, after which the genomic DNA was isolated and the PCR-amplified libraries were subjected to next-generation sequencing, a competition assay comparing sgRNA abundance in dCas9-KRAB–expressing cells with nonexpressing control cells (35). Since CRISPRi resulted in the down-regulation of the targeted loci, sgRNAs targeting genes involved in cell proliferation and survival were depleted. Comparing the dCas9-KRAB–expressing cells with controls revealed remarkably few depleted sgRNAs. The greatest changes were obtained for sgRNAs that target the protein-coding control genes EIF4A3 and DNM2 and the noncoding loci CKMT2-AS1 and ENSG00000263013 (Fig. 1A and Dataset S3).
Fig. 1.

Improved CRISPRi for MYC-regulated noncoding genes. (A) Pooled MYCncLibrary screen using the ZNF10 KRAB domain fused to dCas9 for CRISPRi. CPM, counts per million of CRISPRi sample; CPM0, counts per million of control sample (RAMOS cells, no dCas9). (B) Schematic depiction of KRAB-mediated recruitment of the corepressor TRIM28 and histone-modifying enzymes (HMEs) to induce transcriptional repression (Upper), and co-IP experiments to confirm TRIM28 binding to dCas9-KRAB and dCas9-kPOGO (Lower). Two lanes of the blot were removed (position indicated by dotted line); the original blot is shown in the SI Appendix, Fig. S2D. IB, immunoblot; IP, immunoprecipitation; kPOGO, KRAB domain of POGK. (C) Pooled MYCncLibrary screen using kPOGO fused to dCas9 for CRISPRi. (D) Schematic depiction of SID-mediated recruitment of the corepressor SIN3A and HMEs to induce transcriptional repression (Upper), and co-IP experiments to confirm SIN3A binding to dCas9-SID (Lower). SID, SIN3A-interacting domain. (E) Pooled MYCncLibrary screen using dCas9-SID for CRISPRi in RAMOS and P493-6 cells.
The lack of effective depletion of sgRNAs that target essential control genes led us to hypothesize that dCas9-KRAB–mediated transcriptional repression was inefficient, likely due to low protein expression (SI Appendix, Fig. S2B), and that identification of a better suited repressor domain could lead to more effective repression. KRAB zinc finger proteins constitute the largest group of transcriptional repressors encoded by the human genome. KRAB recruits TRIM28/KAP1, which acts as a scaffold for various heterochromatin-inducing factors (Fig. 1B) (36–38). Based on this knowledge, we used a proteomics approach, immunoprecipitating TRIM28 from P493-6 cell extracts and identifying associated proteins by mass spectrometry (SI Appendix, Fig. S2C). When ranked by numbers of identified peptides over control, the KRAB domain-containing proteins POGK and ZNF595 were atop the list of TRIM28-binding proteins (Dataset S4). Their KRAB domains (kPOGO and k595) were fused to dCas9. Because of the high expression and strong TRIM28 binding of dCas9-kPOGO (Fig. 1B and SI Appendix, Fig. S2 B and D), this fusion construct was used for CRISPRi library screens in RAMOS and P493-6 (35). Compared with control cells, dCas9-kPOGO led to the depletion of essentially the same sgRNAs as dCas9-KRAB, also failing to perturb most essential control genes (Fig. 1C; SI Appendix, Fig. S2E; and Dataset S3).
Because of the modest success from using KRAB domains in CRISPRi, we decided to test an unrelated small repressor peptide, the SIN3 interacting domain (SID) of MXD1 (Fig. 1D) (12, 13). We fused the 31-aa SID to the C terminus of dCas9 and obtained robust dCas9-SID expression, as analyzed by immunoblotting (SI Appendix, Fig. S2B). Co-immunoprecipitation (co-IP) experiments confirmed dCas9-SID binding to the corepressor SIN3A in both RAMOS and P493-6 cells (Fig. 1D). dCas9-SID–expressing cells were subjected to CRISPRi library screens as described above (35). The use of dCas9-SID in these screens caused significant depletion of guides targeting 12 of the 14 essential control genes in the MYCncLibrary, suggesting that in this experimental setting SID is more effective in repressing gene expression than KRAB. In addition, a large number of guides targeting MYC-regulated ncRNAs were depleted with dCas9-SID in both cell lines (Fig. 1E and Dataset S3).
The SID Repressor Domain Is an Important Addition to the CRISPRi Toolbox.
We compared the performance of the two repressor domains SID and KRAB in the CRISPRi screens. Fold changes (FCs) of the most depleted genes with dCas9-KRAB were plotted against those of dCas9-SID (Fig. 2A). Setting a two FC depletion cutoff, five genes were affected by both repressor domains, three were specific for KRAB, and 219 guides were depleted by SID only. The absolute FC values in the dCas9-SID library screen were significantly higher than those achieved with dCas9-KRAB (Fig. 2A). We then validated the CRISPRi-mediated knockdown of selected genes—DNM2, MALAT1, SNHG17, SNORA71A, SNHG26 and ZNF433-AS1—in RAMOS with both repressor domains by RT-qPCR. dCas9-KRAB–induced repression varied among the different genes, from strong for MALAT1, to ∼30% compared with an NT guide, to not significant for the small nucleolar host gene SNHG17 and its encoded small nucleolar RNA SNORA71A. In this analysis, SID significantly affected the expression of all tested genes, including SNHG17 and SNORA71. KRAB led to stronger perturbation of MALAT1 expression compared with SID.
Fig. 2.
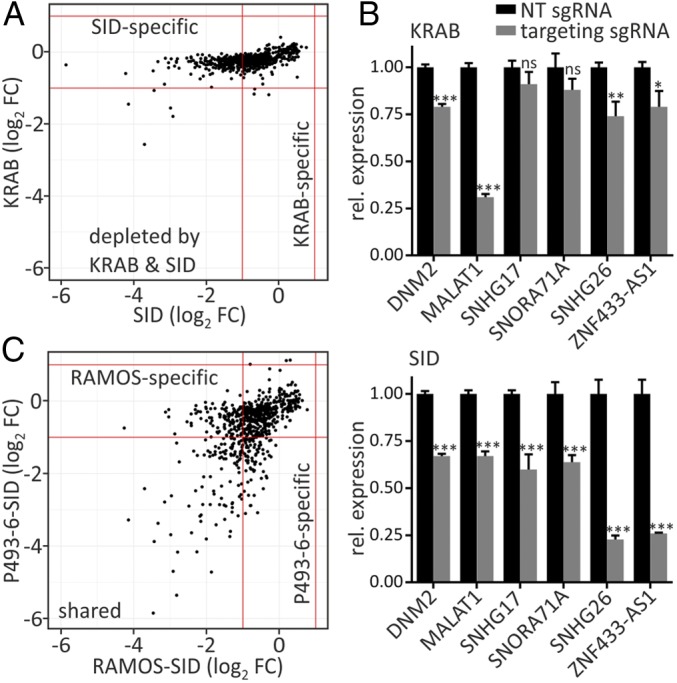
Comparison of repressor domains and CRISPRi screens. (A) Screening results of dCas9-KRAB and dCas9-SID in RAMOS were compared based on best-performing guides per gene. (B) RT-qPCR experiments to test CRISPRi-mediated transcriptional inhibition of selected candidate genes in RAMOS stably expressing dCas9-KRAB or -SID fusion proteins along with gene-specific sgRNAs or nontargeting (NT) guides. (C) Comparison of dCas9-SID screening results between P493-6 and RAMOS based on the best-performing guides per gene. Mean ± SD of three replicates. ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant. FC, fold change.
Another interesting finding of this expression analysis was the modest transcriptional inhibition of some essential control genes. For example, guides targeting the protein-coding gene DNM2 were highly depleted in all CRISPRi screens, yet according to RT-qPCR data, CRISPRi-mediated inhibition with both KRAB and SID was relatively weak (Fig. 2B). One possible explanation for this phenomenon could be selection against cells with strong transcriptional repression occurring between lentiviral transduction and RNA isolation.
We compared the CRISPRi results of the P493-6 and RAMOS cell lines. Of the genes whose targeting guides showed more than two FCs in the CRISPRi screens, 109 were depleted in both cell lines, 105 were depleted in RAMOS only, and 77 were affected only in P493-6. Since the MYCncLibrary was designed to target lncRNAs of P493-6, the genes specifically depleted in RAMOS would ipso facto be expressed in P493-6. Of the P493-6–specific depletions, the major fraction (84%) was expressed in RAMOS. These data show that the effectiveness of dCas9 repression constructs is influenced not only by the repression domains, but also by the cellular environment. Comparing sequences of the best-performing guides from the SID-CRISPRi screen with all guides in the library showed a strong sequence preference toward the 3′ end of the sgRNA next to the protospacer adjacent motif (PAM), where purines (especially Gs) were favored over pyrimidines (especially Ts) (SI Appendix, Fig. S2F). This finding could be helpful in the design of future libraries using dCas9-SID CRISPRi.
Identification of MYC-Regulated Noncoding Loci with Oncogenic Functions.
The sgRNAs significantly altered in abundance in our CRISPRi screens are MYC-regulated ncRNAs that have a role in cell proliferation. In both cell lines, the CRISPRi screen with dCas9-SID primarily caused depletion of sgRNAs, with only a few showing enrichment (Fig. 1E). The MYCncLibrary includes positive controls targeting essential coding genes and negative control sgRNAs that do not have corresponding sequences in the human genome and remained unchanged in abundance (Dataset S3). A selection of the genes targeted by most depleted sgRNAs in the screen is presented in Table 1. Among the candidates putatively necessary for cell survival or proliferation are known cancer-relevant lncRNAs, such as MIR17HG, NEAT1, and DANCR (22, 24, 39). Most of the top hits are identical in the P493-6 and RAMOS screens; exceptions are SNHG26 and SNHG5, whose sgRNAs were strongly depleted in the P493-6 screen but not in the RAMOS screen, and ERVK13-1, which was specific for RAMOS (Table 1).
Table 1.
Selected hits from the CRISPRi screen with SID
| Gene | P493-6 | RAMOS | ||
| log2FC | P value | log2FC | P value | |
| Protein coding | ||||
| DNM2 | −8.35 | 4.64E-186 | −4.15 | 2.92E-138 |
| EIF4A3 | −5.88 | 1.03E-235 | −3.70 | 5.54E-133 |
| RPL18A | −4.17 | 8.17E-94 | −2.79 | 1.49E-42 |
| Noncoding | ||||
| ENSG00000267169 | −5.85 | 1.70E-85 | −3.46 | 6.10E-60 |
| RAD51-AS1 | −5.36 | 1.23E-90 | −2.82 | 3.74E-49 |
| TPTEP2 | −4.72 | 4.35E-69 | −1.86 | 3.21E-25 |
| CKMT2-AS1 | −4.70 | 3.69E-110 | −2.92 | 1.56E-59 |
| SNHG17 | −4.16 | 7.75E-88 | −2.26 | 2.41E-56 |
| SNAI3-AS1 | −3.87 | 3.66E-47 | −3.44 | 2.07E-55 |
| ENSG00000254887 | −3.80 | 3.19E-42 | −2.01 | 4.07E-20 |
| ENSG00000263013 | −3.69 | 2.60E-51 | −2.98 | 1.19E-24 |
| ZNF433-AS1 | −3.63 | 1.34E-39 | −2.41 | 1.13E-35 |
| MIR17HG | −3.41 | 5.76E-99 | −1.74 | 3.60E-24 |
| SLC7A5P1 | −3.37 | 3.87E-34 | −3.33 | 2.45E-43 |
| LINC00476 | −3.32 | 1.72E-101 | −2.16 | 1.66E-43 |
| NEAT1 | −3.14 | 3.73E-61 | −2.15 | 3.87E-46 |
| SNHG26 | −3.10 | 9.84E-132 | −0.93 | 7.96E-21 |
| SNHG5 | −3.08 | 3.84E-135 | −0.60 | 9.53E-06 |
| ENSG00000226944 | −2.90 | 2.39E-26 | −2.48 | 7.97E-36 |
| ENSG00000245904 | −2.86 | 6.69E-130 | −1.64 | 1.71E-56 |
| KMT2E-AS1 | −2.85 | 9.94E-39 | −1.90 | 2.66E-33 |
| ENSG00000272831 | −2.85 | 6.48E-59 | −2.10 | 2.61E-27 |
| TTN-AS1 | −2.81 | 2.35E-82 | −1.29 | 3.30E-23 |
| KRT8P46 | −2.79 | 6.96E-15 | −3.15 | 7.07E-30 |
| ENSG00000273451 | −2.61 | 9.26E-56 | −2.96 | 1.05E-23 |
| DANCR | −1.50 | 4.18E-60 | −2.25 | 7.29E-32 |
| ERVK13-1 | −0.57 | 1.98E-03 | −2.53 | 6.12E-38 |
Numbers refer to the most-depleted guide per gene.
To confirm the role of top hits in cell proliferation, selected guide sequences were cloned into GFP-expressing lentiviral guide vectors, thereby marking the guide sequence for fluorescent detection (40). dCas9-SID–expressing P493-6 and RAMOS cells were transduced at low multiplicity of infection with such GFP lentiviral guide vectors targeting a specific gene candidate and grown for 14 d. Guides that repress genes important for cell proliferation cause transduced cells to proliferate at a reduced rate, limiting the fraction of GFP-expressing cells over time. As a positive control, a guide targeting the essential control gene DNM2 was included in this experiment, and a nontargeting guide served as a negative control. Guides targeting lncRNA hits from the library screen reduced the fraction of GFP-expressing cells over time, as did the guide targeting DNM2 (Fig. 3; additional targets shown in SI Appendix, Fig. S3). These data support the conclusion that the sgRNAs depleted in the CRISPRi screens target genes required for proliferation in these MYC-driven cell lines.
Fig. 3.

Validation of selected candidates from the library screen. Competition-based CRISPRi assays using GFP-expressing sgRNA vectors in P493-6 and RAMOS. GFP-positive cells were quantified by flow cytometry at the indicated time points. Data are mean ± SD of three replicates. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant, unpaired t test.
The CRISPRi hits include several antisense RNAs. This raises the question of whether corresponding sense RNA modulation is a consequence of the down-regulation of the antisense RNA (41–43). We have examined several examples of such sense-antisense pairs. One of the top CRISPRi hits, RAD51-AS1, shares its promoter with the sense gene RAD51, and guides targeting the RAD51-AS1 promoter also affect the expression of RAD51 (SI Appendix, Fig. S4 A and B). Published genome-wide CRISPR/Cas9 screens suggest that RAD51 is an essential gene in many cell types, including the BL cell lines Raji and Jiyoye (44, 45). In contrast, ZNF433-AS1 and TTN-AS1 are in a tail-to-tail orientation relative to their corresponding sense genes ZNF433 and TTN with their 3′ ends partially overlapping with the coding gene. In this orientation, the sense and antisense genes have independent promoters (SI Appendix, Fig. S4A). RNA-seq data revealed that ZNF433 is not expressed in P493-6 or in RAMOS (Dataset S2), and published genome-wide CRISPR/Cas9 screens suggest that this gene is dispensable for cell proliferation and survival (44, 45). In P493-6 cells expressing dCas9-SID, an sgRNA targeting TTN-AS1 strongly reduced expression of the noncoding gene but had no impact on expression of the protein-coding TTN gene (SI Appendix, Fig. S4B). These data suggest that although ZNF433-AS1 and TTN-AS1 overlap with coding genes, it is unlikely that the sgRNA-induced growth inhibitory phenotype is caused by perturbation of the overlapping coding genes.
Characterization of Selected Candidate Genes.
Guides targeting the small nucleolar RNA host gene SNHG17 were consistently depleted from the library on phenotypic selection in P493-6 and RAMOS cells. The importance of SNHG17 for proliferation or survival was also validated in CRISPRi competition assays using flow cytometry. At its genomic locus, SNHG17 is well separated from other genes; the closest neighbor is SNHG11. The TSSs of SNHG17 and SNHG11 are separated by 10 kb, making it unlikely that SNHG17-targeting guides directly affect the expression of SNHG11 or any other genes. Furthermore, guides directed against SNHG11 were not significantly depleted in the library screens (Dataset S3), suggesting that the growth-inhibitory effect induced by sgRNAs targeting SNHG17 in CRISPRi are unlikely to be caused by distal regulatory effects on its genomic neighborhood. Similar considerations apply to SNHG26, another small nucleolar RNA host gene and top candidate in the P493-6 CRISPRi screen.
To determine whether SNHG17 and SNHG26 are direct transcriptional targets of MYC, we performed chromatin immunoprecipitation (ChIP) assays for MYC binding. Both genes have a canonical (5′-CACGTG-3′) and a noncanonical (5′-CGCGTG-3′) MYC E-box directly adjacent to their TSSs (SI Appendix, Fig. S4C). ChIP-qPCR experiments with MYC-specific antibodies revealed strong enrichment of SNHG17 and SNHG26 promoter sequences over IgG control in P493-6 and RAMOS cells (Fig. 4A). No ChIP enrichment over IgG was obtained at 24 h after dox-mediated MYC repression in P493-6 cells. Cellular RNA levels of both host genes were dramatically decreased in P493-6 cells at 24 h after the addition of dox, as quantified by RT-qPCR (Fig. 4B). Taken together, these results indicate that SNHG17 and SNHG26 are directly bound by MYC in their promoter regions and are direct transcriptional targets of MYC.
Fig. 4.

Characterization of SNHG17, SNHG26, and ZNF433-AS1. (A) ChIP-qPCR analysis of MYC binding to proximal promoters of indicated small nucleolar host genes in P493-6 high (−dox) or low (+dox) MYC and in RAMOS cells. Dox was added 24 h before cross-linking. Control represents ChIP signals from an unrelated intergenic locus. Data are mean ± SD of three replicates. (B) RT-qPCR analysis of indicated transcripts at high and low MYC in P493-6 with 24 h of dox treatment. Data are mean ± SD of three replicates. (C) Cytoplasmic/nuclear distribution of indicated transcripts determined by RT-qPCR after cell fractionation. (Left) RNAs that are mainly cytoplasmic. (Right) Primarily nuclear transcripts. GAPDH and MALAT1 served as cytoplasmic and nuclear markers, respectively. Data are mean ± SD of three replicates. (D) GFP-expressing shRNA vectors targeting the indicated transcripts used in growth competition assays. (Left) GFP-positive cells were quantified by flow cytometry at the indicated time points. (Right) shRNA-mediated knockdown was confirmed by RT-qPCR and compared with a control (ctrl) hairpin. Data are mean ± SD of three replicates. Statistical significance for day 10 was tested for targeting shRNAs relative to ctrl-shRNA. **P < 0.01; ***P < 0.001, unpaired t test.
In addition, we analyzed MYC ChIP-seq data of P493-6 at low and high MYC levels and found MYC enrichment at the TSSs of most library target genes. Strong ChIP-seq signals were obtained for genes that are up-regulated by MYC in P493-6. When genes were ranked by sgRNA depletion on library screens in P493-6, we found that many of the target loci that led to strong guide depletion have stronger MYC binding to their proximal promoters than genes with low sgRNA depletion (SI Appendix, Fig. S4D).
We then determined the subcellular localization of these candidate transcripts. To this end, RNA was isolated from the cytoplasmic and nuclear fractions of P493-6 and RAMOS and subjected to RT-qPCR analyses. The primarily cytoplasmic GAPDH mRNA and the nuclear lncRNA MALAT1 were included in the analysis to confirm successful cell fractionation. The SNHG17-encoded small nucleolar RNA SNORA71A was highly enriched in the nucleus (Fig. 4C). Nuclear enrichment was also found for the transcript of its host gene and for SNHG26.
An orthogonal approach to depleting lncRNAs is RNA interference (RNAi). RNAi-mediated knockdown is especially effective against transcripts that localize to the cytoplasm (46), which is the case for ZNF433-AS1 (Fig. 4C). We designed short hairpin RNAs (shRNAs) directed against this transcript and prepared lentiviral constructs for stable expression. To test their impact on proliferation and survival in P493-6, we used GFP-expressing shRNA vectors and followed the fate of fluorescent cells during proliferation by flow cytometry, similar to the CRISPRi approach depicted in Fig. 3. Both ZNF433-AS1–targeting shRNAs and a positive-control hairpin targeting the MYC transcript were significantly depleted at 10 d after lentiviral transduction compared with control shRNA. Knockdown of the ncRNA and the MYC mRNA was independently confirmed by RT-qPCR (Fig. 4D).
MS2 Aptamer-Mediated Recruitment of SID for Regulatable Transcriptional Control.
Modified sgRNAs with hairpin aptamers attached to the tetraloop and stem loop 2 of the sgRNA have been used as scaffolding molecules to anchor effector domains for modulation of endogenous gene expression (28, 47). The MS2 coat protein (MCP) binds with high selectivity and high affinity to the MS2 RNA aptamer (48), and MCP fused to transcriptional activator domains have been shown to effectively up-regulate gene expression (28, 47). In contrast, MCP fused to the KRAB repressor domain was ineffective at mediating transcriptional inhibition, presumably due to MCP dimerization (49). We hypothesized that SID would not be affected by this restriction and would be capable of acting as a repressor when fused to the MCP protein. To test this possibility, we prepared a lentiviral vector for expression of MCP-SID (Fig. 5A).
Fig. 5.
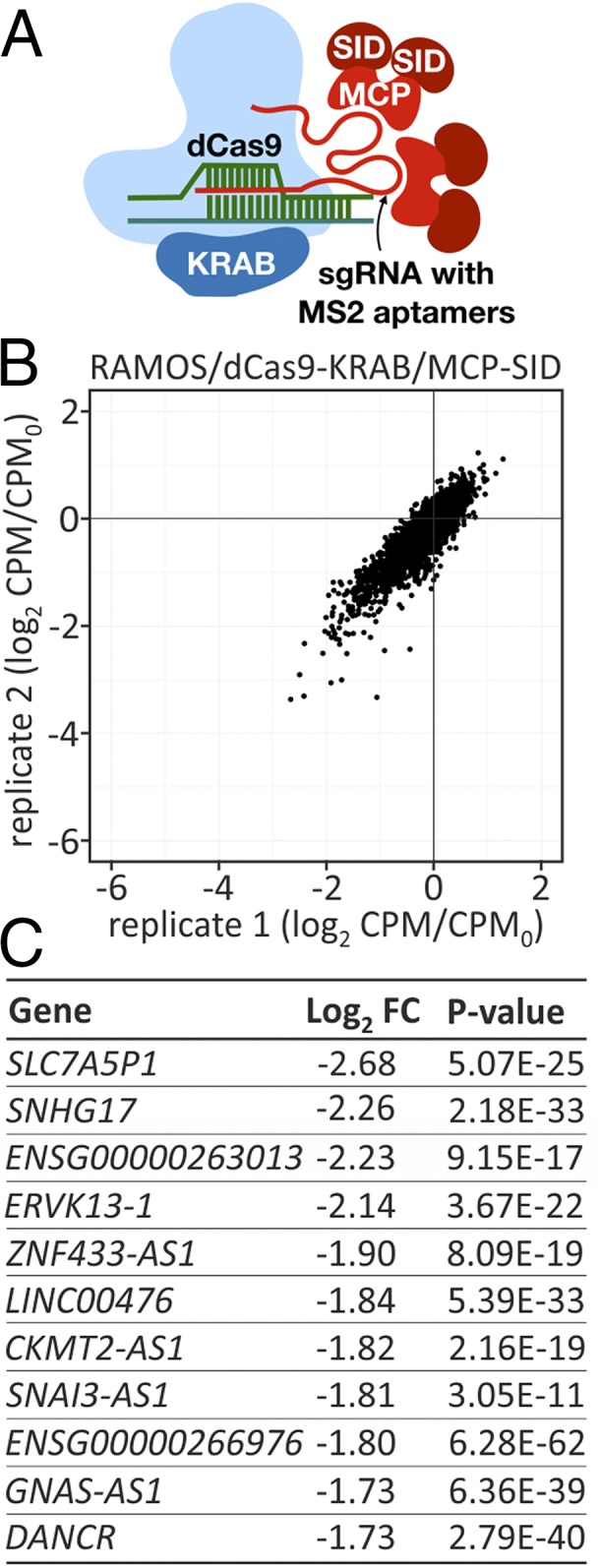
Aptamer-mediated recruitment of SID for CRISPRi. (A) Schematic depiction of the sgRNA-dCas9-MCP-SID complex. MCP, MS2 capsid protein dimer. (B) Phenotypic pooled sgRNA library screens. CPM, counts per million of CRISPRi sample; CPM0, counts per million of control sample. (C) List of selected top noncoding candidates from the library screen in B. Values refer to the most-depleted guides per gene.
The capacity of MCP-SID to mediate transcriptional repression was tested in a library screen using RAMOS cells stably expressing dCas9-KRAB and MCP-SID. This was possible because the MYCncLibrary consists of lentiviral constructs that express sgRNAs with the necessary MS2 aptamers in their scaffold. The depletion efficiency of sgRNAs from the library was comparable to that of the dCas9-SID CRISPRi screen (Fig. 5B) (35). In this experiment, the sgRNA-dCas9 ribonucleoprotein complex contained both repressor domains, KRAB and SID. As discussed previously, using dCas9-KRAB alone leads to the depletion of only a few sgRNAs in the CRISPRi library screen (Fig. 1A); therefore, further increases in sgRNA depletion are attributable to MCP-SID. This screen identified many of the essential coding and noncoding genes seen in the RAMOS/dCas9-SID screen (Fig. 5C and Dataset S3), suggesting that MCP-SID recruited via MS2 aptamers on the sgRNA is a near-functional equivalent of the dCas9-SID fusion protein.
Encouraged by these results, we decided to test whether this system can also be used for conditional CRISPRi. To this end, we cloned the MCP-SID coding region into the pLVX-TetOne-Puro vector for dox-inducible expression (SI Appendix, Fig. S5A). This vector was transduced into RAMOS cells, and subsequent immunoblotting experiments confirmed the induction of MCP-SID expression at 24 h after dox addition, while in the absence of dox, no fusion protein was detected with antibodies directed against MCP (SI Appendix, Fig. S5B). Stable expression of dCas9 and sgRNAs directed against the TSS of SNHG17 or ZNF433-AS1, along with dox-induced expression of MCP-SID, led to transcriptional inhibition of the targeted genes, causing inhibition of cell proliferation. No significant inhibitory effect on transcription and proliferation was obtained in the no-dox control compared with an NT guide (SI Appendix, Fig. S5 C and D).
Discussion
Oncogenic alterations that cause increased MYC activity can be found in numerous human cancers. A recent study found that across the 33 tumor types included in The Cancer Genome Atlas, 28% have amplifications of at least one of the MYC paralogs (4). Transcriptional regulation by MYC extends beyond the coding transcriptome. In fact, mounting evidence suggests that MYC controls the expression of a large segment of the noncoding transcriptome, and that transcripts derived from these loci are likely contributors to MYC oncogenicity (14, 15, 22–24). The results of the present study indicate that many of the MYC-regulated lncRNAs are essential to the proliferation and survival of MYC-driven growth in P493-6 and in the BL cell line RAMOS.
In the different experiments presented, a total of 320 individual noncoding genes led to the depletion of sgRNAs by more than two FCs when targeted by CRISPRi. Among these are noncoding genes previously identified as MYC-targeted functional RNAs, including DANCR and MIR17HG. In addition, we found that SNHG17, which was also previously identified as a MYC-regulated noncoding RNA with broad expression in various cancer types (24, 50, 51), has a proproliferative function in P493-6 and RAMOS cells. Among the ncRNAs depleted, many are directly adjacent to or share a promoter with a coding gene. CRISPRi library screening does not provide direct evidence for function of the noncoding RNA, but rather indicates that the particular targeted locus is important for proliferation or survival. CRISPRi does not discriminate between the two transcripts emanating from a bidirectional promoter. In these cases, further experiments are needed to elucidate the underlying mechanism leading to guide depletion.
In applying CRISPRi in a systematic investigation of MYC-controlled long noncoding genes, we discovered that the repressor domain SID, derived from the MXD1 protein, is highly effective in perturbing the transcription of MYC-regulated noncoding loci. dCas9 fused to the SID of MXI (MXD2) has previously been used for CRISPRi in yeast (52, 53), but not in mammalian cells to our knowledge. MXD1 is a member of the MXD family of transcriptional repressors. Their highly conserved amino terminal SID directly interacts with one of four paired amphipathic α-helical domains of SIN3 proteins. SIN3 forms large multiprotein complexes with chromatin-modifying enzymes, including class I histone deacetylases (HDAC1 and HDAC2). These enzymes catalyze the deacetylation of histones H3 and H4 in active chromatin, which is frequently associated with transcriptional silencing. The exact molecular functions of other SIN3-interacting proteins are not well understood, but many of them have also been linked to transcriptional repression and might be involved in MXD-mediated functions (3, 12, 13, 54–57). MXD1 acts as an endogenous antagonist of MYC by repressing MYC-regulated genes. In the present study, dCas9-SID–induced transcriptional inhibition occurred in the presence of high MYC levels in P493-6 and RAMOS, suggesting that SID exerts a dominant repressive effect, even on direct transcriptional targets of MYC such as MIR17HG, DANCR, SNHG17, and SNHG26. ChIP experiments and expression analysis carried out in this study indicate that SNHG17 and SNHG26 are directly regulated by MYC.
In a surprising contrast, the standard CRISPRi repressor peptide KRAB of ZNF10 did not cause effective depletion of most targets in our screens. Initially, we had supposed that this was a result of poor expression of the fusion protein, combined with weak binding to the corepressor TRIM28. The KRAB domain of POGK (kPOGO) fused to dCas9 was expressed at high levels, and co-IP experiments confirmed that it bound TRIM28 more efficiently than dCas9-KRAB. However, its performance in CRISPRi screens was comparable to that of dCas9-KRAB.
SIN3 and TRIM28 protein complexes may cause transcriptional repression through different mechanisms. Many genes directly bound and regulated by MYC are also targets of MXD family proteins, and these genes might be more susceptible to SID-mediated repression (58). Whether the high effectiveness of SID is general or unique to the selected targets or cell lines used in this study remains an open question. Our work supports the conclusion that CRISPRi screens need to use a diverse set of transcriptional repressors, because a single repressor might not provide a comprehensive set of results.
One of the main challenges in studying noncoding RNAs is the lack of efficient loss-of-function strategies, especially for transcripts that primarily localize to the cell nucleus (26, 59). Our study adds useful tools to the CRISPRi arsenal of techniques, applying MS2 aptamers to allow SID to function as a synergistic repressor with other repressor domains (e.g., KRAB) and placing MCP-SID expression under the control of dox for conditional repression of target genes. The latter technique in particular will facilitate functional analysis of lncRNAs. On a general scale, the methods applied and developed in this study open the door to the investigation of coding and noncoding RNAs identified by RNA-seq analysis as controlled by a specific transcription factor.
By applying these cutting-edge technologies, we have identified several noncoding loci that not only are transcriptionally controlled by MYC, but also are critically involved in mediating oncogenic phenotypes. Additional studies are needed for in-depth characterization of candidate genes to learn more about their molecular and cellular functions and how they impact MYC-driven phenotypes.
Materials and Methods
Design and Cloning of sgRNA Library.
All noncoding RNA targets that are differentially regulated by MYC (log2FC >0.5, P < 0.01) in P493-6 (14, 32) were selected as potential targets (a total of 563 genes). In addition, we included the 100 protein-coding genes most differentially regulated by MYC in P493-6 and the following genes as controls: RPL14, RPL5, RPL8, RPL18A, RPS26, CDK1, DNM2, EIF4A3, IARS, TFRC, TXNL4A, SF3B3, MYC, and JAG2. For the set of all 677 target genes, isoform abundance was determined from our RNA-seq data using RSEM (60). All the potential isoforms that map to the gene were considered, and the dominant isoform was selected by majority voting.
The genomic coordinates of the TSSs were obtained from Gencode v25 (61, 62), and sgRNAs were designed to target within 400 bases upstream or downstream of this site. All PAM sequences on either strand were identified as potential targets. Potential sgRNAs were searched for in the genome (63–65), and those with exact matches elsewhere were removed. A second alignment allowing for up to four mismatches was performed for each sgRNA, and an off-target score was assigned for each sgRNA using previously described procedures (66). SNPs in the sgRNA sequences were determined from 1000 Genomes phase 1 high-confidence SNPs (67–69). A score was assigned based on the population major allele frequency or a product of these allele frequencies in cases where there were more than one SNP in the area targeted by the guide. The guides were sorted based on these scores. The top guide was selected, and guides in the vicinity of this guide had a penalty added to their score. The guides were then sorted again by score, and the selection process was continued until 10 guides were chosen or the pool was exhausted. This procedure yielded a selection of guides that were distributed over the region while selecting those with the highest possible score. An additional 100 guides that are nontargeting in the human genome were generated. Six additional guides were included that target the tetracycline response element sequence bound by the tetracycline-controlled transactivator, which controls the exogenous expression of MYC in P493-6 cells. A total of 5,598 guides were generated against a total of 594 genes.
Oligonucleotide pools were designed with flanking cloning sites and synthesized by CustomArray. PCR amplicons were Gibson assembly-cloned into the sgRNA expression vector lenti-sgRNA(MS2)_Puro (Addgene; 73797) (47). Electrocompetent Escherichia coli (Lucigen) was transformed with assembly reactions, and bacterial colonies (∼100× library coverage) were pooled to prepare a liquid culture for plasmid DNA isolation using the QIAfilter Plasmid Midi Kit (Qiagen).
DNA Constructs.
LentiCas9_Blast (Addgene; 52962) (70) was used as template for mutagenesis PCR to introduce D10A and H840A point mutations into Streptococcus pyogenes Cas9 to obtain lenti-dCas9_Blast. The coding regions of human ZNF10-KRAB (amino acids 2 to 72), ZNF595-KRAB (amino acids 2 to 65), POGK-KRAB (amino acids 34 to 115), and SID (amino acids 2 to 32 of MXD1) were cloned into the BamHI-site of lenti-dCas9-Blast. Lenti-guide(MS2)_PuroT2AGFP was prepared by cloning the T2A-GFP coding region downstream of and in frame with the puromycin resistance gene of lenti-sgRNA(MS2)_Puro. The GFP-coding sequence was ligated into the BamHI/KpnI sites of pLKO.1_Blast (Addgene; 26655) (71) to obtain pLKO.1_GFP. shRNA sequences (SI Appendix, Table S1) were cloned into the AgeI/EcoRI sites of the pLKO.1_Blast and pLKO.1_GFP vectors. The SID coding region (amino acids 2 to 32 of MXD1) was cloned into the BamHI/BsrGI sites of lenti MS2-P65-HSF1_Hygro (Addgene; 61426) (47) to obtain lenti-MCP-SID_Hygro.
Lentivirus Production and Transduction.
293-T cells were cultured in DMEM (Gibco), and P493-6 and RAMOS cells were cultured in RPMI 1640 (Gibco). Complete cell culture media were obtained by supplementing with 100 U/mL penicillin, 0.1 mg/mL streptomycin, 2 mM glutamine (Gibco), and 10% (vol/vol) fetal bovine serum (FBS; Omega Scientific). For P493-6 cells, tetracycline-free FBS (Omega Scientific) was used.
Lentiviral vectors were transfected along with packaging plasmids (Addgene; pRSV-Rev, 12253; pMDLg/pRRE, 12251; and pMD2.G, 12259) (72) into 293-T cells using Lipofectamine 3000 reagent (Invitrogen), and lentiviruses were harvested at 2 d after transfection. P493-6 and RAMOS cells were transduced with lentiviruses, and antibiotic selection was started 2 d later and continued until cells appeared to be resistant. Blasticidin (Thermo Fisher Scientific), hygromycin B (Thermo Fisher Scientific), puromycin (Sigma-Aldrich), and Zeocin (Thermo Fisher Scientific) were used at a final concentration of 7.5 μg/mL, 100 μg/mL, 1 μg/mL, and 75 μg/mL, respectively.
CRISPRi Screens.
Pooled lentiviral library stocks were prepared in 293-T cells as described above, and P493-6 or RAMOS cells with or without (library control) stable dCas9 fusion protein expression were transduced to obtain an infection rate of 10 to 15%. After infection of three replicates, cells were cultured for 2 d, treated with puromycin for 3 d, allowed to recover for 2 d, and then cultured for 14 d while maintaining at least 5.5 × 106 cells at all times to maintain the 1,000× library representation.
For each condition and replicate, genomic DNA was isolated from 6 × 106 cells (∼1,000× coverage) using the DNeasy Blood and Tissue Kit (Qiagen). The DNA was then used as a template for PCR amplification of guide sequences using primers with Illumina adaptors, sequencing primer binding sites, and barcodes (SI Appendix, Table S2). A total of 23 PCR cycles at an annealing temperature of 61 °C were performed using Q5 Hot Start High-Fidelity 2X Master Mix (New England BioLabs) according to the manufacturer’s instructions. PCR reactions were subjected to TAE agarose gel electrophoresis (1.5% wt/vol), and amplicons were recovered using the QIAquick Gel Extraction Kit (Qiagen). Barcode multiplexed samples were pooled and loaded onto an Illumina NextSeq 500 instrument together with unrelated sequencing samples to obtain sequence diversity. Read lengths were 1 × 75 bp, and read depth per multiplexed library was 20 million. Raw data were processed by the CASSAVA pipeline version 1.8 (Illumina). The FASTQ files were processed using count_spacers.py (60) and GNU parallel (73). Differential representation of the guides within the library was determined using the negative binomial method implemented in edgeR version 3.28.0 (74) in R 3.6.1 (75), with additional graphics produced with ggplot2 (76). Plots of replicates with log2 (CPM/CPM0) were based on Weissman CRISPRi gamma plots (29). Raw count data were normalized by total read number to CPM. The CPM of treated samples were divided by the mean of the control samples and then log2-transformed (34). Sequence logograms were produced using the methods detailed previously (77) but reimplemented to use log2 (odds ratio).
For CRISPRi-based growth competition assays, candidate guides were synthesized (IDT) and cloned into lenti-guide(MS2)_PuroT2AGFP. dCas9-SID–expressing cells were incubated with lentiviruses produced from these GFP/guide vectors to achieve 10 to 15% transduction efficiency. P493-6 and RAMOS cells were grown in 24-well plates, and the number of GFP-positive cells present in the pool was determined by flow cytometry (Novocyte; ACEA Biosciences) at days 3, 7, and 14 after transduction. Data were processed in FlowJo (78). shRNA-based competition assays were carried out in the same way.
Protein–Protein Interaction Studies.
For co-IP experiments, 1 × 107 cells per sample were washed with ice-cold PBS and resuspended in IP lysis buffer (20 mM Hepes pH 7.5, 200 mM NaCl, 5% glycerol, and 0.75% Triton X-100) supplemented with protease inhibitors (Thermo Fisher Scientific). After sonication, cell debris was removed by centrifugation, and lysates were precleared using protein A Sepharose beads. Cleared lysates were incubated with antibodies overnight at 4 °C, and immunocomplexes were immobilized on protein G Sepharose beads and washed with IP lysis buffer. For Flag-IP, anti-Flag magnetic beads (Sigma-Aldrich) were used. IP samples along with input samples were subjected to SDS-PGE and immunoblotting. Anti-Cas9 (14697), SIN3A (8056), and TRIM28 (4123) antibodies were from Cell Signaling Technology, and MCP (MS2 coat protein) antibody (ABE76-I) was from EMD Millipore.
ChIP.
P493-6 were grown with or without 0.1 μg/mL dox (Sigma-Aldrich) for 24 h. A total of 1 × 107 cells per sample were cross-linked in PBS containing 1% formaldehyde for 10 min at room temperature and then quenched with glycine (125 mM final concentration) for 5 min at room temperature. Nuclei were extracted using hypotonic buffer (50 mM Tris⋅HCl pH 8.0, 5 mM EDTA pH 8.0, and 10% glycerol) and collected by centrifugation. Cell nuclei were resuspended in high-SDS RIPA buffer (50 mM Tris⋅HCl pH 8.0, 5 mM EDTA pH 8.0, 150 mM NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, and 0.5% SDS) supplemented with protease inhibitor mixture (Cell Signaling Technology), and lysates were sonicated to shear DNA to an average fragment size of 200 to 1,000 bp. Sheared chromatin was diluted 1:5 with RIPA buffer without SDS (50 mM Tris⋅HCl pH 8.0, 5 mM EDTA pH 8.0, 150 mM NaCl, 1% Igepal CA-630, and 0.5% sodium deoxycholate) supplemented with protease inhibitor mixture, and input samples (2%) were prepared. The rest of the diluted chromatin was incubated overnight with Rabbit IgG (Cell Signaling Technology; 2729) or anti-MYC antibodies (Cell Signaling Technology; 9402). Subsequently, reagents of the Chromatin IP Kit (Cell Signaling Technology; 9005) were used for IP, chromatin elution, and DNA purification according to the manufacturer’s instructions. A 2-μL DNA sample per reaction was used for qPCR using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific). The following formula was used to calculate signal relative to input: Percent Input = 2% × 2(C[T] 2% Input Sample – C[T] IP Sample); C[T] = CT = threshold cycle of PCR.
RT-qPCR.
Total RNA was isolated from cells using the Maxwell RSC simplyRNA Cells Kit (Promega) according to the manufacturer’s instructions. RNA samples were reverse-transcribed using the High-Capacity cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions, using random priming. qPCR was performed on a LightCycler 96 real-time PCR instrument (Roche). Reactions were prepared using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) and universal cycling conditions (95 °C for 10 min followed by 45 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 10 s). Reaction specificity was confirmed by melting curve analysis. qPCR primer sequences are listed in SI Appendix, Table S1.
Cellular Fractionation.
Cell nuclei were extracted as described previously (79). Cell pellets were resuspended in ice-cold Nonidet P-40 lysis buffer (10 mM Tris⋅HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, and 0.5% Nonidet P-40) and incubated on ice for 5 min. Nuclei were separated by centrifugation at 300 × g for 4 min, the supernatant (cytoplasmic fraction) was removed, and nuclei were washed once with Nonidet P-40 lysis buffer. RNA from the cytoplasmic fraction and from nuclei was isolated using the Maxwell RSC simplyRNA Cells Kit (Promega).
Supplementary Material
Acknowledgments
We thank Chi Van Dang for generously providing the P493-6 cells, Benjamin F. Cravatt for providing mass spectrometry instruments and expertise, Lynn Ueno for technical assistance, and Anja Zembrzycki for assistance with organizing and producing the manuscript. Funding was provided by the National Cancer Institute (Grants R35 CA197582, to P.K.V.; R50 CA243899, to J.R.H.; and K99 CA215249, to L.B.-P.) and the George E. Hewitt Foundation for Biomedical Research (P.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This is manuscript 29922 of The Scripps Research Institute.
Footnotes
The authors declare no competing interest.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo (accession no. GSE141615).
See online for related content such as Commentaries.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1921786117/-/DCSupplemental.
References
- 1.Eilers M., Eisenman R. N., Myc’s broad reach. Genes Dev. 22, 2755–2766 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dang C. V., MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conacci-Sorrell M., McFerrin L., Eisenman R. N., An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 4, a014357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaub F. X., et al. , Pan-cancer alterations of the MYC oncogene and its proximal network across the Cancer Genome Atlas. Cell Syst. 6, 282–300. e2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prochownik E. V., Vogt P. K., Therapeutic targeting of Myc. Genes Cancer 1, 650–659 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabay M., Li Y., Felsher D. W., MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 4, a014241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKeown M. R., Bradner J. E., Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med. 4, a014266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalla-Favera R., et al. , Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 79, 7824–7827 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klapproth K., Wirth T., Advances in the understanding of MYC-induced lymphomagenesis. Br. J. Haematol. 149, 484–497 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Calado D. P., et al. , The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat. Immunol. 13, 1092–1100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blackwood E. M., Eisenman R. N., Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211–1217 (1991). [DOI] [PubMed] [Google Scholar]
- 12.Ayer D. E., Lawrence Q. A., Eisenman R. N., Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell 80, 767–776 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Carroll P. A., Freie B. W., Mathsyaraja H., Eisenman R. N., The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 12, 412–425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hart J. R., Roberts T. C., Weinberg M. S., Morris K. V., Vogt P. K., MYC regulates the non-coding transcriptome. Oncotarget 5, 12543–12554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim T., et al. , Role of MYC-regulated long noncoding RNAs in cell cycle regulation and tumorigenesis. J. Natl. Cancer Inst. 107, dju505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris K. V., Mattick J. S., The rise of regulatory RNA. Nat. Rev. Genet. 15, 423–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anastasiadou E., Jacob L. S., Slack F. J., Non-coding RNA networks in cancer. Nat. Rev. Cancer 18, 5–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khalil A. M., et al. , Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malecová B., Morris K. V., Transcriptional gene silencing through epigenetic changes mediated by non-coding RNAs. Curr. Opin. Mol. Ther. 12, 214–222 (2010). [PMC free article] [PubMed] [Google Scholar]
- 20.Tay Y., Rinn J., Pandolfi P. P., The multilayered complexity of ceRNA crosstalk and competition. Nature 505, 344–352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercer T. R., Dinger M. E., Mattick J. S., Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 10, 155–159 (2009). [DOI] [PubMed] [Google Scholar]
- 22.O’Donnell K. A., Wentzel E. A., Zeller K. I., Dang C. V., Mendell J. T., c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Li Y., Choi P. S., Casey S. C., Dill D. L., Felsher D. W., MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell 26, 262–272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Y., et al. , MYC-targeted long noncoding RNA DANCR promotes cancer in part by reducing p21 levels. Cancer Res. 78, 64–74 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong L., et al. , Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goyal A., et al. , Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 45, e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu S., et al. , Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 34, 1279–1286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joung J., et al. , Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 548, 343–346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu S. J., et al. , CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355, eaah7111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pajic A., et al. , Cell cycle activation by c-myc in a Burkitt lymphoma model cell line. Int. J. Cancer 87, 787–793 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Maesako Y., Uchiyama T., Ohno H., Comparison of gene expression profiles of lymphoma cell lines from transformed follicular lymphoma, Burkitt’s lymphoma and de novo diffuse large B-cell lymphoma. Cancer Sci. 94, 774–781 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart J. R., et al. , P493-6 treated with KJ-Pyr-9 and/or doxycycline. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58168. Deposited 2 June 2014.
- 33.Raffeiner P., et al. , An MXD1-derived repressor peptide identifies non-coding mediators of MYCdriven cell proliferation. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141482. Deposited 4 December 2019.
- 34.Raffeiner P., et al. , An MXD1-derived repressor peptide identifies non-coding mediators of MYCdriven cell proliferation. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141615. Deposited 4 December 2019.
- 35.Raffeiner P., et al. , An MXD1-derived repressor peptide identifies non-coding mediators of MYCdriven cell proliferation. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141491. Deposited 4 December 2019.
- 36.Margolin J. F., et al. , Krüppel-associated boxes are potent transcriptional repression domains. Proc. Natl. Acad. Sci. U.S.A. 91, 4509–4513 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moosmann P., Georgiev O., Le Douarin B., Bourquin J. P., Schaffner W., Transcriptional repression by RING finger protein TIF1 beta that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res. 24, 4859–4867 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ecco G., Imbeault M., Trono D., KRAB zinc finger proteins. Development 144, 2719–2729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakravarty D., et al. , The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat. Commun. 5, 5383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erb M. A., et al. , Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guttman M., Rinn J. L., Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beiter T., Reich E., Williams R. W., Simon P., Antisense transcription: A critical look in both directions. Cell. Mol. Life Sci. 66, 94–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goyal A., et al. , A cautionary tale of sense-antisense gene pairs: Independent regulation despite inverse correlation of expression. Nucleic Acids Res. 45, 12496–12508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang T., et al. , Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsherniak A., et al. , Defining a cancer dependency map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lennox K. A., Behlke M. A., Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 44, 863–877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konermann S., et al. , Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peabody D. S., The RNA binding site of bacteriophage MS2 coat protein. EMBO J. 12, 595–600 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zalatan J. G., et al. , Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 160, 339–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J. X., Li W., Li J. T., Liu F., Zhou L., Screening key long non-coding RNAs in early-stage colon adenocarcinoma by RNA-sequencing. Epigenomics 10, 1215–1228 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Xu T., et al. , Gene amplification-driven long noncoding RNA SNHG17 regulates cell proliferation and migration in human non-small-cell lung cancer. Mol. Ther. Nucleic Acids 17, 405–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilbert L. A., et al. , CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gander M. W., Vrana J. D., Voje W. E., Carothers J. M., Klavins E., Digital logic circuits in yeast with CRISPR-dCas9 NOR gates. Nat. Commun. 8, 15459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ayer D. E., Laherty C. D., Lawrence Q. A., Armstrong A. P., Eisenman R. N., Mad proteins contain a dominant transcription repression domain. Mol. Cell. Biol. 16, 5772–5781 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brubaker K., et al. , Solution structure of the interacting domains of the Mad-Sin3 complex: Implications for recruitment of a chromatin-modifying complex. Cell 103, 655–665 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Cowley S. M., et al. , Functional analysis of the Mad1-mSin3A repressor-corepressor interaction reveals determinants of specificity, affinity, and transcriptional response. Mol. Cell. Biol. 24, 2698–2709 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adams G. E., Chandru A., Cowley S. M., Co-repressor, co-activator and general transcription factor: The many faces of the Sin3 histone deacetylase (HDAC) complex. Biochem. J. 475, 3921–3932 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mathsyaraja H., et al. , Max deletion destabilizes MYC protein and abrogates Eµ-Myc lymphomagenesis. Genes Dev. 33, 1252–1264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leone S., Santoro R., Challenges in the analysis of long noncoding RNA functionality. FEBS Lett. 590, 2342–2353 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frankish A., et al. , GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gencode , Release 25. https://www.gencodegenes.org/human/release_25.html. Accessed 11 December 2016.
- 63.Langmead B., Wilks C., Antonescu V., Charles R., Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35, 421–432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gencode , Human genome GRCh38.p7. ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_25/GRCh38.p7.genome.fa.gz. Accessed 11 December 2016.
- 66.Joung J., et al. , Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fairley S., Lowy-Gallego E., Perry E., Flicek P., The international genome sample resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 48, D941–D947 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abecasis G. R., et al. ; 1000 Genomes Project Consortium, An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Broad Institute , Genome Analysis Toolkit resource bundle. https://gatk.broadinstitute.org/hc/en-us/articles/360036212652-Resource-Bundle. Accessed 11 December 2016.
- 70.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bryant D. M., et al. , A molecular network for de novo generation of the apical surface and lumen. Nat. Cell Biol. 12, 1035–1045 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dull T., et al. , A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tange O., GNU Parallel 20150322 (“Hellwig”). 10.5281/zenodo.16303. Accessed 1 May 2017. [DOI]
- 74.McCarthy D. J., Chen Y., Smyth G. K., Differential expression analysis of multifactor RNA-seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.R Core Team , R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2014).
- 76.Wickham H., ggplot2: Elegant Graphics for Data Analysis (Springer, ed. 3, 2010), p. 213. [Google Scholar]
- 77.Wagih O., ggseqlogo: A versatile R package for drawing sequence logos. Bioinformatics 33, 3645–3647 (2017). [DOI] [PubMed] [Google Scholar]
- 78.FlowJoTM (Version 10, Becton, Dickinson and Company, Ashland, OR).
- 79.Roberts T. C., et al. , Quantification of nascent transcription by bromouridine immunocapture nuclear run-on RT-qPCR. Nat. Protoc. 10, 1198–1211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.