

Abstract
Abstract—The review summarizes literature data on the development of drugs based on natural and synthetic high-polymeric double-stranded RNA (dsRNA), their antiviral, immunoadjuvant, and antitumor properties. Special attention is paid to cell receptors responding to exogenous dsRNA, pathways of dsRNA-dependent antiviral reaction, ability of dsRNA to inhibit growth and induce apoptosis of malignant cells. It has been shown that enhancing the innate immune response with dsRNA can be an effective component in improving methods for treating and preventing infectious and cancer diseases. The further use of dsRNA for the correction of pathological processes of different origin is discussed.
Keywords: double-stranded RNA, interferon inducers, immunomodulators, antitumor and antiviral properties
INTRODUCTION
Recognition of viral nucleic acids and induction of interferon synthesis are the main components of innate antiviral immunity [1, 2]. It is generally accepted that double-stranded RNA (dsRNA) are one of the most important mediators of interferon (IFN) induction in response to viral infection. Viral infection is associated with the appearance in the cell of double-stranded forms of RNA, which either represent the actual viral genome of the virus or appear during the viral reproductive cycle [3]. The primary cascade of the body’s response to viral infection and administration of exogenous dsRNA (recognition, initiation of intracellular signaling pathways) share many similarities, and therefore dsRNA may be considered as regulators of antiviral reactions [1]. However, subsequent studies of mechanisms of dsRNA action have shown that their functions are more diverse . In this regard, a constant interest in dsRNA as a substance for creation of new drugs for prevention and treatment of infectious and non-infectious diseases has not been waning for many years. This review summarizes current trends in the pharmaceutical development of drugs based on high-polymeric dsRNA.
It should be noted that, unlike the situation in the field in 1960s−1970s, when the first dsRNA preparations have obtained and characterized, now much is known about the interaction of dsRNA with the cell and intracellular signal transduction. It has been found that the recognition of exogenous/extracellular dsRNA in the intracellular space can be performed using two main sensor systems [4–7]. One sensor system includes receptors of the well-studied family of Toll-like receptors of type 3 (Toll-like receptor 3, TLR3) [8, 9]. The other sensor system includes the cytoplasmic sensors RIG-I/MDA-5/LGP2 of the RIG-like receptor family (RLR) [10, 11]. Both types of these systems are able to induce production of type I interferons and pro-inflammatory cytokines, but the pathways for such activation differ.
1 TLR3-DEPENDENT MOLECULAR MECHANISMS FOR dsRNA RECOGNITION AND INDUCTION OF SIGNALING PATHWAYS
Human TLR3 is a type I transmembrane receptor with a molecular mass of 125 kDa, which has several structural domains: the extracellular or ectodomain (ECD), the transmembrane domain and the cytoplasmic signal domain [12]. The structural basis for TLR3 interaction with dsRNA is the extracellular variable domain. The TLR3 ectodomain has a horseshoe shape; it contains N- and C-terminal leucine-rich repeat domains (LRR). The transmembrane domain consists of a single alpha helix. The cytoplasmic signaling domain contains a highly conserved sequence of approximately 200 residues homologous to the intracellular domain of the IL-1β receptor, and therefore called the Toll-interleukin-1 receptor (TIR) domain. This signaling domain is involved in the interaction of an activated receptor with adapter components of the intracellular signaling pathways.
In the inactive state, TLR3 is a monomer, its activation occurs as a result of dimerization in response to dsRNA binding [13]. Crystal structure analysis of the complex consisting of two TLR3 ECD and dsRNA has shown that each ectodomain binds dsRNA molecules at two non-glycosylated sites located at opposite ends of the TLR3 horseshoe [12]. Despite the fact that receptor ectodomain binding to dsRNA is rather weak, the dimeric complex is highly stable due to intermolecular contacts in the TLR3 ECD C-terminal domains.
It has been found that TLR3 dimerization and corresponding signal can be induced by dsRNA of at least 45−50 base pairs (bp); this corresponds to the minimum spatially possible distance between the sites of dsRNA binding with receptor ectodomain monomers [12, 13]. This feature provides receptor recognition of only long dsRNAs appearing in the cell as a result of viral infection; shorter polyribonucleotides (tRNA, rRNA, microRNA) are not able to form stable complexes with TLR3 ECD. The second characteristic feature of TLR3 consists in the receptor binding of the ligand that occurs via surface interactions (hydrogen bonds, electrostatic interaction) with the sugar-phosphate backbone of RNA, and not with individual nucleotide bases, and depends only on the secondary structure of RNA [12]. This explains the universality of the ligand-receptor interaction: the ability of the same receptor to bind dsRNA of different primary structure. On the other hand, there is evidence that TLR3 recognizes such specific ligand characteristics as the presence or absence of hydroxyl group at the 2'-position of β-D-ribose, which discriminates dsRNA and DNA molecules [3, 12].
TLR3 is expressed mainly by specialized cells of the immune system, such as macrophages and dendritic cells (DC) [14]; this receptor is also found on somatic cells [15, 16]. In most cell types, TLR3 is located inside the cell compartments—on the membranes of endosomes and lysosomes, although some cells, such as epithelial cells and fibroblasts, also express it on the plasma membrane outer surface [17, 18]. During endocytosis of dsRNA-containing material, intracellular TLR3 is able to move (by means of the UNC-93B protein) from the endoplasmic reticulum to endosomes, where they acquire functional ability for ligand recognition [19].
Regardless of receptor localization, TLR3 binding to dsRNA is accompanied by receptor phosphorylation and its interaction with the TRIF adapter protein, which provides further signal transduction, forming intermediate complexes with the TRAF-3 regulatory proteins (first signaling pathway) or TRAF-6/RIP1 (second signaling way) (Fig. 1) [20–22].
Fig. 1.
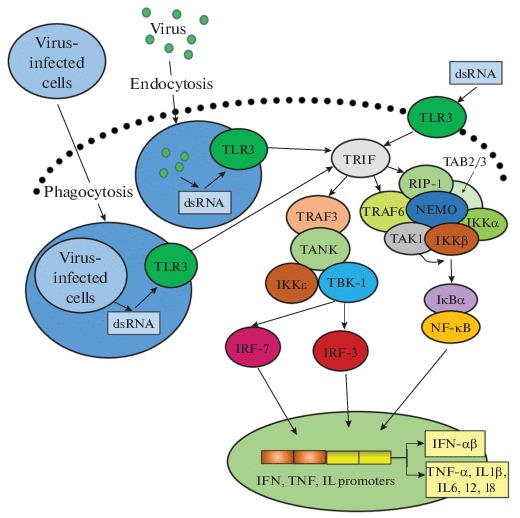
Induction of TLR3-dependent cell signaling pathways in response to exposure to dsRNA. Binding of extracellular dsRNA or dsRNA absorbed during endocytosis or phagocytosis to TLR3 leads to receptor phosphorylation and binding to the adapter molecule TRIF, which serves as a platform for assembly of proteins involved in triggering signaling pathways leading to activation of IRF-3 (7) or NF-κB. In the case of the NF-κB signaling pathway, activated TRIF interacts with TRAF6, after oligomerization the latter acquires ubiquitin-E3-ligase activity, causing ubiquitination of both TRAF6 and RIP1 protein. The polyubiquitin sequence is recognized by the TAK1-binding proteins TAB2 and TAB3, which promote TAK1 kinase binding to the complex. RIP1 with attached ubiquitin molecules is recognized by NEMO, resulted in IKK complex binding to the TRIF–RIP1–TRAF6–TAB–TAK1 complex. TAK1 phosphorylates the IKKb subunit of the IKK complex, promoting its activation. Phosphorylated IkB binds to ubiquitin undergoes proteasomal degradation with release of NF-κB, which migrates into the nucleus, where it binds to the IFN-β promoter. For activation of the IRF-3 (7) pathway TRAF3 binds to TANK or NAP1 and SINTBAD peptides, which, in turn, interact with TBK-1 and/or IKKε; their activated forms are able to phosphorylate IRF-3. Activation of the factor IRF-7 occurs similarly. In the nucleus, activated IRFs bind in the region of the IFN-β promoter with NF-κB and ATF-2/c-jun, resulting in the involvement of cofactors and RNA polymerase II and initiation of transcription. Abbreviations: TRIF—Toll—interleukin (IL)-1-resistance (TIR) domain-containing adaptor inducing IFN-β ; IRF—interferon regulatory factor; NF-κB—nuclear factor κB (nuclear factor kappa-light-chain-enhancer of activated B cells); TRAF6—TNF receptor-associated factor 6; RIP1—receptor-interacting protein 1; NEMO—NF-κB essential modifier; IKK—inhibitor of nuclear factor κB kinase; TAK1—transforming growth factor β-activated kinase 1. Adapted from [20].
In the case of the first signaling pathway, TRAF3 binds to TANK (or related peptides NAP1 and SINT-BAD), which, in turn, interact with TBK-1 and/or IKKε kinases; the activated kinases phosphorylate IFN-regulating factors 3 and 7 (IRF-3, IRF-7). In the nucleus, phosphorylated IRFs bind in the region of the IFN promoters with NF-κB and ATF-2/c-jun factors; this results in the attraction of cofactors, RNA polymerase II and initiation of interferon genes transcription.
Induction (due to dsRNA interaction with TLR3) of the second signaling pathway via regulatory molecules TRAF-6/RIP1 leads to increased activity of transcription factors NF-kB and AP-1 (Fig. 1). This causes production of IFN and a number of pro-inflammatory cytokines (TNF-α, IL-1b, IL-6, IL-12, IL-18) [26–28], involved in induction and regulation of a wide range of immune responses.
Interferons, in turn, induce expression of gene encoding proteins involved in realization of the antiviral response. Among IFN-stimulated genes, there are genes encoding 2'-5'-oligoadenylate synthetase/RNase L (OAS/RNase L), these enzymes cause degradation of newly synthesized cell mRNA (including viral ones) [23], and also the gene encoding protein kinase R (PKR), which inhibits protein synthesis [24, 25]. Most cells synthesize 2'-5'-OAS and PKR constitutively, but interferons increase the transcriptional activity of their genes several-fold. The mechanisms of activation of OAS/RNase L and PKR, as well as their biological effects, have been studied in details [24, 25].
1.1 dsRNA-Dependent Protein Kinase
Protein kinase R is a dsRNA-dependent IFN-induced Ser/Thr protein kinase [26]. In a non-phosphorylated state PKR is associated with ribosomes; a small part of the PKR is present in the nucleus. A key step in PKR activation is dsRNA binding to two structurally similar N-terminal motifs of the enzyme molecule, dsRBM1 and dsRBM2, followed by subsequent PKR autophosphorylation. After activation PKR phosphorylates a number of proteins, including alpha subunit of the eukaryotic initiation factor 2 (eIF-2); this has important consequences for the cell cycle. Phosphorylated eIF-2 binds to eIF-2B, thus causing the inhibition of the eIF-2B catalyzed reaction and protein synthesis (Fig. 2) [27]. In addition, PKR, activated by dsRNA, inhibits proliferation of various cell types and induces apoptosis [24].
Fig. 2.

Biochemical mechanisms of dsRNA-induced inhibition of translation and protein synthesis. DsRNA activated, IFN-induced protein kinase R (PKR) inhibits mRNA translation via phosphorylation of the alpha subunit of the eukaryotic initiation factor 2 (eIF-2α) and complex formation with eIF2B. Initiation of 2′-5′-oligoadenylate synthesis from ATP by dsRNA-activated 2′-5′-oligoadenylate synthetase (2′-5′OAS) leads to the dimerization of RNase L and non-specific RNA degradation. Adapted from [28].
1. 2 The Cascade 2',5'-Oligoadenylate Synthetase/Ribonuclease L (2',5'-OAS/RNase L)
OAS, like PKR, is an IFN-inducible enzyme; its activity depends on the presence of dsRNA in the cell. Unlike PKR, OAS does not have a separate domain for binding an inducer. dsRNA binds in the region of the positively charged groove of the enzyme; after conformational changes of the groove the OAS active center becomes spatially accessible. The activated enzyme catalyzes ATP polymerization resulted in formation of short mono-, di-, tri-, and tetra-oligoadenylates (2′, 5′-oligoA), which interact with RNase L (RNase L) and transform it from an inactive monomeric form to the active dimer (Fig. 2). RNase L, a protein with molecular mass of 83 kDa, consists of three domains: N-terminal regulatory domain, which contains nine ankyrin-like regions, protein kinase-like domain, and C-terminal RNase domain. 2'-5'A oligomers interact with the ankyrin-like sites of the RNase L N-terminal domain. The complex formation is accompanied by conformational changes, leading to dimerization of the enzyme molecule and the C-terminal ribonuclease domain becomes catalytically active. Destroying mRNA into fragments, RNase L suppresses formation of complete viral RNA, which leads to inhibition of both protein synthesis and assembly of viral particles [28, 29]. Thus, both OAS and PKR are dsRNA-dependent enzymes and become active only in the presence of dsRNA. This fact indicates an important role of dsRNA as markers of viral infection and triggers of the intracellular antiviral reaction.
2 THE SYSTEM OF CYTOPLASMIC SENSOR FACTORS RIG-I/MDA-5/LGP2
As already mentioned above, in addition to TLR3 present in almost all cell types, there is another dsRNA sensor system, which belongs to the family of RIG-like receptors [10, 30, 31]. RLRs are complex proteins with well-organized signaling and functional domains, with characteristic autoinhibition mechanism.
Proteins RIG-1 (retinoic acid-inducible gene 1) and MDA-5 (melanoma differentiation-associated gene 5) contain the C-terminal RNA recognition domain, the central DExD/H helicoid domain, and two N-terminal CARD domains (caspase activation and recruitment domain), which are able to bind CARD-containing adapters, such as Cardif/VISA/MAVS/IPS-1. These adapter proteins, in turn, serve as a platform for assembly of signaling proteins that trigger IRF-3 or NF-κB-signaling pathways (Fig. 1) [31–34].
There is a specificity in dsRNA recognition by individual sensors of this family. For example, RIG-1 recognizes single-stranded RNA with a double-stranded component containing terminal 5'-triphosphate, as well as relatively short (from 21 to 27 nucleotides) dsRNA formed due to action of activated RNase L [6]. Since partially complementary sequences in the 5' and 3' domains, containing dsRNA with 5'-terminal triphosphates, are characteristic of the “panhandle” structure—a double-stranded fragment of the negative-strand RNA virus genome, this mechanism seems to provide recognition specificity of dsRNA of such viruses as influenza A, rabies, measles, by the given sensory molecule [35–37].
In contrast to RIG-1, MDA-5 recognizes relatively long (more than 2 kb) dsRNA, and therefore it is a sensor of other types of viruses, particularly, encephalomyocarditis virus [35]. It is suggested that MDA-5 is the dominant cytoplasmic receptor for a complex of polyinosinic and polycytidylic acids (PolyI:PolyC) [38].
Until recently, helicase LGP2 (laboratory of genetics and physiology 2) was considered as an exclusively inhibiting factor of this family, preventing binding and activation of RIG-1 and MDA-5. However, several research groups have shown that LGP2 can be a positive regulator of the RLR-induced signal [39], which increases the recognition efficiency of viral dsRNA by MDA-5 [40].
Recently, it has been found that, in addition to the described receptor systems, there are other systems of intracellular sensors and signaling molecules that interact with dsRNA in cells; the specific features of ligand binding and the biological role of these systems are not yet fully understood. These include:
—the family of NLRs (NOD-like receptor, nucleotide-binding oligomerization domain-like receptors). NLRs are localized intracellularly and are expressed by many cell types, from immunocytes to epithelial cells. Induction of apoptosis in influenza infection is associated with the NOD receptor [41];
—extracellular dsRNA receptors—SR-As (scavenger class A receptors), responsible for dsRNA penetration into the cell, interaction with intracellular sensors, and synthesis of type I IFN [42, 43].
Thus, realization of the dsRNA effect on the cell involves several receptor systems that are specific in terms of dsRNA recognition. The receptor diversity provides reliable identification of foreign double-stranded structures and their differentiation from endogenous cell RNA. Finally, induction of intracellular signaling pathways leads to expression of cytokine genes, primarily IFN, and activation of dsRNA-binding enzyme, the key elements of the antiviral response.
The ability of dsRNA to induce synthesis of IFN and other cytokines is a basis for diversity of their immunomodulatory properties. It has been shown that dsRNA causes an increase in absorption, metabolic and bactericidal activity of phagocytic cells (macrophages, neutrophils), activity of natural killers (NK), and maturation and activation of DC; this leads to activation of CD8+ and CD4+ T cells [44]. Thus, dsRNAs play a role of some functional linkers between innate and acquired immunity systems, providing a fast and productive defensive response to an infectious challenge.
Another important aspect of the dsRNA effect on the cell is regulation of cell proliferation and apoptosis. It is currently known that apoptosis of infected cells occurs when different dsRNA-dependent pathways are activated [45], while the main mechanism for its implementation is activation of caspases [46].
The described effects of dsRNA are realized not only in infected cells, but also in cells subjected to malignant transformation. As it has been shown in several mouse and human cell lines, TLR3 agonists inhibit growth of tumor cells through two main mechanisms: suppression of proliferation and induction of apoptotic death [45]. In addition, dsRNAs are able to influence immune system cells by inducing antitumor immune responses. Stimulation of antigen-presenting DC by dsRNA leads to activation of NK cells and cytotoxic CD8+ T-lymphocytes causing death of tumor cells [47, 48]. Phenotype transformation of myeloid suppressor cells and tumor-associated macrophages, caused by dsRNA, resulted in an increase in their antitumor activity [45, 49].
Thus, being agonists of several types of intracellular receptors and sensors responsible for the “emergency response” system, dsRNAs perform an important function of its activation. The clinical use of exogenous dsRNA is directed to the use of these evolutionarily formed mechanisms for induction and activation of cytokines, enzymes and other cellular proteins involved in the development of the antiviral and antitumor reaction for therapeutic purposes.
3 DsRNA AS DRUG SUBSTANCES
High polymeric dsRNAs that are attractive in terms of their therapeutic potential can be subdivided into two types: natural and synthetic.
Sources of natural dsRNA, in addition to RNA-containing viruses that cause diseases in humans and animals, can be insect viruses [50], plants and microorganisms viruses [51], some fungi contain dsRNA elements [52]. On the basis of this type of dsRNA, Ridostin and Larifan preparations are developed and approved for use in clinical practice.
Ridostin is a drug based on dsRNA isolated from virus-like particles of the Saccharomyces cerevisiae killer strain as two individual forms, L and M (4.6 kb and 1.8−1.6 kb, respectively) [52]. It has been developed at the SRC VB “Vector” (Russia). Ridostin is a highly effective antiviral agent, an IFN inducer and an immunomodulator, which is used to treat influenza, herpes, and Chlamydia infection. Certain evidence exists that Ridostin increases the effectiveness of the antitumor therapy [53].
Larifan (produced by Larifan Ltd., Latvia) is a drug containing the replicative form of bacteriophage f2 dsRNA, obtained from Escherichia coli cells infected with the phage. Based on phage dsRNA, several dosage forms have been developed: the injection form of Larifan, an antiviral and immunostimulatory drug with antitumor activity; Larifan ointment and suppositories [54].
Limited information exists on the development of drugs based on other natural dsRNAs. One such example is dsRNA of bacteriophage φ6 isolated from infected Pseudomonas phaseolicola cells and representing a mixture of dsRNA of different lengths: 6.3 kb; 4.0 kb and 2.9 kb (L-, M- and S-forms, respectively). In a series of experimental studies, it has been shown that phage dsRNA exhibits high IFN-inducing and antiviral activity against viruses causing Omsk hemorrhagic fever and influenza virus A/Chicken/Kurgan/05/2005 (H5N1) [55–57].
Synthetic dsRNA is another type of high polymeric dsRNA widely used in pharmaceutical development. The history of synthetic double-stranded polyribonucleotide complexes began in 1955, when Grunberg-Manago and Ochoa showed that the enzyme polynucleotide phosphorylase (PNPase; EC 2.7.7.8) from Azotobacter vinelandii in the presence of Mg2+ could catalyze the reaction of polynucleotide synthesis of a certain structure up to 2–2.7 kb from ribonucleoside diphosphates with the release of stoichiometric amounts of orthophosphate [58]. Subsequently, double-stranded complexes were obtained from complementary polyribonucleotides by annealing. The development of simple methods for synthesis of high-polymer dsRNA has opened the possibility of producing them in sufficient quantities to study and create drugs on their basis.
In a series of studies by Vilner et al. [59, 60] it was shown that the antiviral activity of synthetic polyribonucleotides significantly depended on the polymer length (now it is clear, that it is determined by the specificity of receptor binding). Based on the results of biological studies, PolyA:PolyU, PolyG:PolyC, and PolyI:PolyC exhibiting the most pronounced IFN-inducing and antiviral activity were chosen for further studies.
As was shown in in vitro and in vivo experiments, PolyA:PolyU suppresses reproduction of herpes viruses, viruses of reproductive respiratory syndrome , transmissible gastroenteritis, and African swine fever [61, 62]. It is an active IFN inducer, although its ability to enhance interferon synthesis and to increase the survival of mice in experimental viral infections, was a bit lower than of PolyG:PolyC and PolyI:PolyC [61].
Currently, on the basis of PolyA:PolyU, the drug Poludan (manufactured by Lance-Farm LLC, Russia) has been developed and registered. It is used in the form of eye and nasal drops in viral eye diseases (adenoviral and herpetic keratoconjunctivitis, keratitis, keratoiridocyclitis, etc.), in complex therapy of influenza and other acute respiratory infections.
There are data on the clinical trials of the foreign drug analogue Polyadenur (PolyA:PolyU with a molecular mass ranged from 250 kDa to 1500 kDa). It has been developed by Beaufour Ipsen (France) together with Hemispherx Biopharma (USA), for using as a component of complex therapy (in combination with IFN-α) of hepatitis B and C, and also for treatment of breast cancer [63–65].
According to the classification proposed by Yershov et al., the PolyG:PolyC and PolyI:PolyC based drugs are referred to the group of highly active IFN inducers (IFN titers at the peak of the response of more than 1000 units/mL) with pronounced antiviral activity [61].
Studies of the complex of double-stranded polyriboguanylic and polyribocytidylic acids (PolyG:PolyC, Polyguacyl) performed in the 1970s–1980s, showed that the drug had a broad spectrum of antiviral activity and could be effective in the acute phase of viral hepatitis and encephalitis, for the treatment of influenza and rabies [61, 66]. However, a number of significant drawbacks, such as the uncertainty of the chemical structure, the lack of homogeneity and, as a result, the unpredictable pharmacokinetics and toxicity of Polyguacyl became a serious obstacle to its clinical use [46, 67].
In 2013, researchers from Riboxx GmbH (Germany) developed a modified method for producing PolyG:PolyC to overcome these drawbacks. The new drug RGC100 was characterized by a strictly defined length (100 bp) and a chemical structure, good solubility and pronounced immunomodulatory activity, particularly, the ability to cause DC activation and T-cell proliferation [46].
Further improvement of RGC100 resulted in appearance of the drug RGIC100—Poly(GI):PolyC; one its chain was PolyC, and the complementary chain contained G and I random quantitative combinations from 1 to n − 1, where n is the total number of nucleotides in the complementary chains. The pharmaceutical composition contains the active component Poly(GI):PolyC of at least 45 bp in length in combination with pharmaceutically acceptable carriers, excipients and/or solvents. According to the authors of the patent [68], Poly(GI):PolyC can be used for activation of innate immunity, prevention and treatment of infectious and neoplastic diseases.
It is interesting to note that, despite the comparable level of antiviral activity and the less pronounced toxic properties of the PolyG:PolyC complex [61, 69], until recently, the creation of drugs based on the complex PolyI:PolyC was developed more intensively in world pharmaceutical practice. Most likely, one of the reasons for such preference consists in the technological problems of obtaining PolyG [58, 70], although other reasons are also possible (e.g. differences in the width and manifestation of the immunomodulating action of these complexes).
Studies of biological properties of PolyI: PolyC, started in the 1970s, showed that the drug was a powerful activator of innate immunity and an inducer of tumor cell apoptosis [44]. Promising results have been obtained in clinical studies of PolyI:PolyC as a means of treating seasonal influenza [71], as well as treating malignant tumors of different localizations in adults and children [72–74].
An improved form of PolyI:PolyC, the drug Ampligen, is currently at the stage of clinical trials. Ampligen (PolyI:PolyC12U) is a double-stranded complex of polyriboinosinic acid and a copolymer of cytidyl and uridylic acids (with a C : U ratio of 12 : 1) and a length from 500 bp to 2000 bp (Hemispherx Biopharma, Inc., USA) [75]. It is an IFN inducer with a markedly reduced incidence of adverse effects as compared to the parent compound. In the ClinicalTrials.gov Database, 16 types of clinical trials of this drug are registered. The effectiveness of the drug Ampligen has been demonstrated in the framework of combination therapy of chronic fatigue syndrome and acquired immunodeficiency syndrome (AIDS). Currently, the drug has been approved in Argentina for the treatment of severe cases of myalgic encephalomyelitis/chronic fatigue syndrome [76].
In 2014, an improved version of the drug Ampligen, PolyI:PolyC30U, was developed [75, 77]. In contrast to Ampligen, the new drug is characterized by a higher content of C versus U (30 : 1), and therefore has a reduced ability to form branched dsRNA and as a result, it binds to TLR3 more actively.
One of the current problems associated with the development and production of synthetic polyribonucleotides is standardization of their structure. In this context, in 2018, Nakano et al. developed a new method for production of industrial quantities of PolyI:PolyC of a strictly defined length [78]. In this method prevention of an uncontrolled increase in the length of a polyribonucleotide during annealing was achieved by mixing several short PolyI molecules (0.1 kb) with one long PolyC molecule (0.4 kb) (Fig. 3). Using this method it is possible to obtain the preparation uPIC100-400 of a given length, which was characterized by a higher storage stability as compared to the original PolyI:PolyC [78].
Fig. 3.
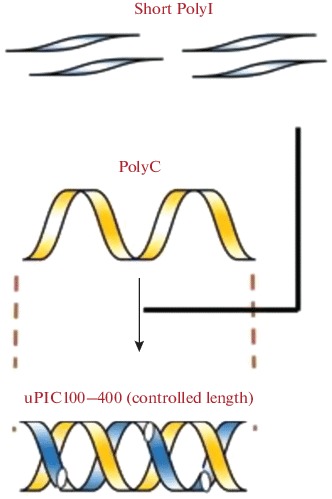
The scheme illustrating production of the double-stranded complex of PolyI:PolyC-uPIC100-400 with a controlled nucleotide length. Adapted from [78].
In other words, natural and synthetic dsRNA preparations have already been employed in clinical practice as antiviral and antitumor therapeutic agents. Among the modern areas of pharmaceutical development, two of the most actively developing trends are recognized: the improvement of dsRNA dosage forms to increase the effectiveness of the active substance and their wider use.
4 MODERN TRENDS IN THE DEVELOPMENT OF ANTI-VIRUS dsRNA BASED DRUGS
The widespread clinical use of dsRNA based preparations is often hampered by their instability in biological media, a high level of effective doses and side effects. Numerous efforts are undertaken to improve dsRNA efficiency; particular attention is paid to drug formulation that can enhance the action of the main substance or reduce side effects; preparations on polymeric carriers and the means of targeted delivery [79–81].
Nucleic acids penetration into the cell can be improved by frequently used cationic polymers. The negative charges of nucleic acids prevent their interaction with a negatively charged cell surface. After binding of nucleic acids to polycations their negative charge neutralizes and they reduce in size; this promotes nucleic acid transfection into cells. The most known preparation of dsRNA preparations stabilized by polymers is Hiltonol.
Hiltonol (PolyI:PolyC, stabilized by poly L-lysine and carboxymethylcellulose—Poly ICLC), was developed by Oncovir, Inc. (USA) [82]. Poly ICLC is characterized by increased resistance to hydrolysis. It induces IFN synthesis and thus exhibits a modulating effect on the immune system cells, including activation of T-cells, NK cells and DC, synthesis of cytokines (interleukins, TNF-α) and corticosteroids. Being less toxic than existing analogs it can be used in higher doses for the treatment of patients with advanced inoperable cancer [83]. The ClinicalTrials.gov database contains information about 97 clinical trials of Hiltonol for the treatment of oncological diseases of various localization, influenza, severe acute respiratory syndrome (SARS) and other viral diseases [76]. Hiltonol is an attractive potential drug for treatment for Ebola haemorrhagic fever [84].
Liposomes are another variant of the cationic delivery vehicle. Wong et al. showed that intranasal administration of PolyICLC, encapsulated in cationic liposomes, to mice prior to infection with a lethal dose of influenza A virus, provided a higher level of protection than the non-encapsulated drug [85]. In mice infected with Dengue virus, PolyICLC encapsulated in liposomes enhanced antiviral reactions, mainly by increasing expression of cytokines, particularly, IFN-γ [86]. Liposomes can be conjugated with antibodies, proteins and other ligands, providing their targeted delivery. For example, liposomes containing PolyI:PolyC, were prepared by incorporating Sendai virus fusogenic F protein into the lipid bilayer and targeted by a monoclonal antibody bound to the liposomes via Staphylococcus aureus protein A [87]. Immunoliposomes have been shown to provide a reasonable level of PolyI:PolyC internalization sufficient for antiviral response induction.
A composition containing yeast dsRNA (the active ingredient of the drug Ridostin) and the polymer polyvinylpyrrolidone exhibited prolonged activating effect on the system of nonspecific resistance and reduced toxicity than the initial drug [81].
In recent years, transport systems based on polyethyleneimine (PEI) have been actively developed for nucleic acid delivery. PEI polyplexes provide a higher level of transfection and protection of RNA from the action of nucleases compared to other polycations. This is associated with a high density of charges on PEI, contributing to condensation of nucleic acids into more compact complexes. To reduce the toxic properties of PEI polyplexes are modified with PEG. A study of such constructs containing PolyI:PolyC and epidermal growth factor showed that high concentrations of immobilized dsRNA were accumulated in the tumor tissue and caused regression of U87MGwetEGFR glioblastoma, A431 adenocarcinoma, and MDA-MB-468 breast cancer [88].
Polysaccharides are widely used in pharmacology, particularly, as encapsulation matrix for preparation of drug formulations [89]. Based on yeast dsRNA and polyglucin (dextran with molecular mass of 50−60 kDa), Ridostin Forte, an early-type IFN inducer characterized by high IFN-inducing and antiviral activity, was created [90]. Chitosan is another polysaccharide widely used in creation of drug dosage formulations due to its low toxicity, high sorption capacity, bactericidal activity, biocompatibility, and biodegradability [91].
The use of bisphosphonates, synthetic analogues of natural pyrophosphates is an interesting aspect in context of developing methods for drug delivery to bones. Bisphosphonates (BPs) are characterized by high affinity for apatite bone structures and also by antitumor activity. In order to provide an intracellular codelivery, PolyI:PolyC and the third-generation bisphosphonate zoledronic acid (ZOL), Chen et al. [92] obtained lipid coated calcium phosphate nanoparticles (LCP). It was shown that LCP loaded with PolyI:PolyC and ZOL exhibited significantly higher antitumor activity against B16BL6 melanoma cells than free PolyI:PolyC and ZOL preparations. Synergism of the antitumor action of PolyI:PolyC and ZOL in the LCP composition was also demonstrated in mice transplanted with B16BL6 melanoma.
dsRNA delivery systems are also developed on the basis of inorganic molecules. For example, iron oxide nanoparticles were used for PolyI:PolyC delivery to lymph nodes and this led to a significant increase in the immune response [93]. Immunization of mice with calcium phosphate particles containing PolyI:PolyC and CpG dinucleotide promoted their intensive uptake by DC and this was accompanied by the expansion of pathogen-specific CD4+ and CD8+ T-lymphocytes [94].
Another way to improve the therapeutic characteristics of dsRNA is the development of dosage forms indented for non-parenteral use. This is especially attractive in the case of treatment of infectious diseases, for which the mucous membranes are the “gateway of infection.” McNally et al. [95] demonstrated that a single intranasal administration of PolyI:PolyC to mice induced local production of Th1 chemokines in the lungs and upper respiratory tract, and generated a steady migration of T-lymphocytes into the respiratory tract. The observed effects depended on the length of dsRNA. The authors of another study [96] found that the intranasal administration of liposomes with PolyICLC before or shortly after infection of mice with H5N1 influenza virus inhibited viral replication. This resulted in a decrease in its lung titers, an increase in the level of specific humoral and cellular immune responses and survival of infected animals. In addition, liposomal PolyICLC prevented the development of pulmonary fibrosis, a serious complication of the infection.
5 dsRNA BASED DRUGS AS PERSPECTIVE ADJUVANTS OF ANTI-VIRUS VACCINES
Experimental and clinical studies have shown that dsRNA can be used as an adjuvant to modulate and optimize the antigen-specific immune response. Evaluation of the parameters of the innate immune response after administration of PolyI:PolyC stabilized by poly L-lysine and carboxymethylcellulose (PolyICLC) to volunteers showed that the administration of the drug caused formation of immune reactions similar to those observed after immunization with yellow fever vaccine, and therefore, the drug could be used to enhance the vaccine immunogenicity [97].
Martins et al. [98] studied the effect of PolyICLC on the efficacy of vaccination of mice, guinea pigs, and primates with virus-like particles (VLP) against filovirus. Administration of PolyICLC resulted in an increase of the level of TNF-α, IL-6 and chemokines, activation of DC, NK, and B-cells. The drug enhanced antigen-specific CD4+ and CD8+ T-cell response, increased production of antibodies and protection during viral infection.
Co-administration of PolyI:PolyC, lipoteichoic acid and CL097 (a TLR7/8 ligand , the imidazoquinoline compound with high antiviral activity) with an inactivated vaccine against the porcine reproductive and respiratory syndrome virus (PRRSV) conferred higher protection against infection [99]. An increased cellular immune response was found in guinea pigs immunized with VLP vaccine against the FMD virus in combination with PolyI:PolyC [100].
Reliable protection of mice against tick-borne encephalitis virus was observed after vaccination with a specific inactivated tick-borne encephalitis vaccine Encevir in combination with the drug Ridostin [101]. Ridostin also increased the protective properties of weakly immunogenic vaccines against tuberculosis and horse strangles disease [102].
The design of microcarriers for delivery of antigens in combination with immunoadjuvants to antigen-presenting cells (APC) is one of actively developing areas of vaccine construction. TLR3 ligands, particularly, PolyI:PolyC, are also used as candidate adjuvants in these systems. Such adjuvants activate not only “professional” APC (DC, macrophages), but also non-immune cells (epithelial cells, endothelial cells, fibroblasts); this can result in the development of autoimmune reactions or chronic inflammation. In order to avoid such undesirable reactions, Hafner et al. proposed an original PolyI:PolyC delivery system [103]. The microcarrier is a negatively charged polystyrene microsphere equipped with a “crown” of polyethylene glycol (PEG), which is formed by electrostatic coating with polycationic copolymers based on poly(L-lysine)-graft-poly(ethylene glycol). Experiments on primary culture of peripheral blood monocytes have shown that PolyI:PolyC adsorbed on the surface of the microspheres caused activation of monocyte-derived DC [103]. At the same time, the preparation drug did not affect expression of antigens of the major histocompatibility complex (MHC) class I or secretion of IL-6 by human skin fibroblasts and weakened their phagocytic activity; this indicates that the adsorbed PolyI:PolyC lacks the ability to induce adverse inflammatory reactions [104].
The use of non-parenteral routes of administration of dsRNA as an adjuvant is particular interesting. For example, mucosal treatment with PolyI: PolyC shortly after vaccination of mice with live attenuated influenza vaccines enhanced the functional activity of the respiratory tract DC, formation of CD8+ T-cells and production of neutralizing antibodies [105].
Kim et al. [106] found an increase in the production of antigen-specific serum IgG and mucosal IgA in response to vaccination of mice with trivalent hemagglutinin-subunits or split H1N1 vaccine antigen after administration of PolyI:PolyC as an eyedrop adjuvant. Importantly, there was no increase in mRNA expression of proinflammatory cytokines and mononuclear cell infiltration at the injection site; this indicates that the drug does not have a side effect in the form of a local inflammatory response.
Uchida et al. developed an original platform for improvement of mRNA vaccines [107]. It is based on the following. Eukaryotic mRNA with a polyA tail, formed as a result of mRNA processing, was hybridized with PolyU. The hybridization did not affect the efficiency of mRNA translation, and the double-stranded portion of the mRNA:PolyU had an immunostimulating effect. Activation of mouse and human dendritic cells by mRNA:PolyU was 10–100 times more efficient than in the case of single-stranded mRNA [107]. A study of immunostimulation mechanisms, performed on the HEK293 cell culture, characterized by a high level of TLR3 expression, and RAW264.7 characterized by MDA-5 and RIG-1 receptor knockout, showed that the immune response to mRNA hybridized with PolyU was mediated by TLR3 and RIG-I [107].
Thus, using dsRNA as vaccine adjuvants makes it possible to increase their protectivity. The adjuvant activity of dsRNA seems to be based on the ability to alter the balance of cytokines and modulate activity of cells mediating specific and nonspecific immune responses.
6 dsRNA-BASED DRUGS AS ANTI-TUMOR AGENTS
Although TLR3 has been first identified in cells of the immune system, more recent studies have shown that it is also expressed by malignant cells. The antitumor effects of the TLR3 agonist PolyI:PolyC have been demonstrated on a wide range of mouse and human tumor cell lines. For example, there is evidence that intracellular delivery of liposomal PolyI:PolyC to human AGS adenocarcinoma cells induced tumor cell apoptosis in vitro and significantly inhibited xenograft growth in nude mice [108].
Similar results were obtained by Chen et al. [109], who showed that transfection of PolyI:PolyC into HeLa cervical cancer cells induced IFN-β synthesis, formation of reactive oxygen species, causing DNA damage, activation of caspases 3 and 9, and finally apoptosis.
The antiproliferative, cytotoxic, and apoptogenic effects of PolyI:PolyC were observed in experiments on poorly differentiated human urinary bladder cancer cell lines MGH-U3 and RT4 [110]. In MGH-U3 cells the drug induced growth arrest at the G1-S transition; it also increased the immunogenicity of MGH-U3 and RT4 cells, inducing expression of MHC class I molecules on their surface and secretion of pro-inflammatory cytokines IL-8 and IL-6 [110]. The ability of PolyI:PolyC to induce apoptosis of prostate cancer cells was demonstrated on cell lines derived from bone and intracerebral metastases of a human tumor; the effect was more pronounced on the androgen-dependent LNCaP line [111]. The apoptogenic effects of PolyI:PolyC were also reported for tumors of other histotypes (e.g. tumors of the breast, head and neck, kidneys, colon, and lungs) [112].
Studies performed on cells of the human melanoma sectional material have shown that the apoptogenic effects of dsRNA can be enhanced during combined use with cytokines, such as IFN-α or IL-27 [113, 114].
Despite existence of intrinsic antitumor activity dsRNA preparations are more frequently studied as synergists and adjuvants to enhance the action of specific therapeutic agents and vaccines. For example, Ayari et al. have shown [110] that combination of PolyI:PolyC with the Bacillus Calmette-Guérin (BCG) vaccine enhanced the antitumor effect of BCG on bladder cancer transplants in MBT-2 mice. Similar results were obtained by Muthuswamy et al. [115]; these authors believe that the combined use of BCG with PolyI:PolyC and IFN-α may be a new method for increasing the effectiveness of immunotherapy for bladder cancer.
Alipour et al. [116] developed a PolyI:PolyC delivery system to enhance the antitumor immune response to the P5 peptide, containing multiple epitopes of the HER2/Neu glycopeptide characteristic of tumor cells. The P5 peptide was encapsulated into cationic nanoliposomes consisting of modified cholesterol (1,2-dioleoyl-3-trimethylammoniumpropane-cholesterol) that contained or did not contain PolyI:PolyC. In groups of mice immunized with liposomes carrying the P5 protein and PolyI:PolyC, as well as liposomes containing only PolyI:PolyC, a more significant level of IFN-γ secretion by splenocytes and an increase in the number of CD4+ and CD8+ T-cells compared with other groups were observed. The most pronounced inhibition of the growth of the experimental TUBO tumor with a high level of rHER2/neu expression during the whole experiment (90 days) was observed during treatment with liposomes containing P5 and PolyI:PolyC. This suggests that the combination of PolyI: PolyC and liposomes with a tumor antigen is able to induce an antitumor immune response even without the use of additional T-helper epitopes.
Intratumor administration of a plasmid carrying the apoptin gene with PolyI:PolyC not only significantly inhibited the growth of the 4T1 mammary tumor in Balb/c mice, but also induced a potent antitumor immune response, as evidenced by an increase in the number of CD4+ and CD8+ T-lymphocytes, increased secretion of cytokines into the circulation (IFN-γ, IL-2) and tumor tissue infiltration with immune cells [117, 118]. Similar data were obtained using PolyI:PolyC together with the canine parvovirus NS1 (CPV2.NS1) protein [117, 118].
More than 90% of common solid tumors and many hemoblastoses, such as lymphoma, leukemia and multiple myeloma, are known to express MUC1 glycopeptide (MUC1). However, fully synthetic glycopeptide tumor vaccines created on the basis of this glycopeptide are characterized by low immunogenicity. Glaffig et al. [119] found that the use of PolyI:PolyC as an adjuvant resulted in a significant increase, in response to the vaccination, of the production of specific IgG, which actively interacted with human breast tumor cells expressing MUC1, and the induction of a cellular immune response.
Kano et al. [120] investigated PolyI:PolyC and the recombinant protein encoded by the lymphocyte 3 activation gene (LAG-3) fused to human IgG Fc (LAG-3-Ig) as adjuvants to a peptide vaccine against the tumor antigen P1A. LAG-3 (CD223) is a protein expressed on the membrane of activated T-, B-cells, NK-сells, plasmacytoid DC and plays an important role in their functioning. In mice with grafted mastocytoma tumor P815, PolyI:PolyC in combination with LAG-3-Ig caused rejection of the grafted tumor and increased longevity of animals. The antitumor effect was due to increased proliferation and infiltration of tumor tissues by T-cells and their production of Th1-type cytokines. The combination of PolyI:PolyC with LAG-3-Ig suppressed expression of a number of proteins and receptors on T-cells, preventing their “depletion”; this is important for realization of a deep and long-lasting antitumor immune response [120].
DC dysfunction is a serious obstacle in the development of effective anti-tumor vaccines because these cells are often used for vaccine preparation. It has been found that tumor associated DC (TADC) are less mature than normal DC and respond poorly to stimulation by TLR agonists, associated possibly with hyperactivity of intracellular STAT3 .
In order to overcome DC dysfunction Luo et al. [121] developed the PMP/OVA/siRNA nanovaccine, which is a polypeptide micelle formed by poly(et-hyleneglycol)-b-poly(L-lysine)-b-poly(L-leucine) hybrid polypeptides (PEG-PLL-PLLeu) with encapsulated OVA antigen, STAT3-siRNA and PolyI:PolyC . Experiments have shown a significant (up to 200-fold) increase in the uptake of OVA antigen and siRNA by TADC after administration of the vaccine and a decrease (by more than 50%) in STAT3 expression. The PMP/OVA/siRNA vaccine increased expression of CD86 and CD40 markers and IL-12 production by DC, exerted an increase in the number of mature DCs and a decrease in the number of immunosuppressive cells in the lymph nodes, which ultimately contributed to the weakening of immunosuppression in the tumor microenvironment, development of antitumor immune responses and tumor regression . In other words, combination of an immunomodulator PolyI:PolyC and STAT3-siRNA with nanovaccines can be a promising strategy for increasing their therapeutic efficacy [121].
CONCLUSIONS
The considered literature data indicate that dsR-NAs are components of an evolutionarily formed system of nonspecific protection and play the role of functional linkers between innate and acquired immunity systems. Synthetic and natural dsRNA preparations are used in clinical practice as pharmaceutical substances for development of prophylactic antiviral and immunomodulatory agents, and there is a clear interest in expanding the sphere of their application and methods for improving therapeutic properties (Table 1).
Table 1. .
dsRNA based drugs: modern trends of pharmaceutical developments
| Name | Structure | Developer | Current status | Prescription | Reference |
|---|---|---|---|---|---|
| Natural dsRNAs | |||||
| Ridostin | Mixture of dsRNA and single stranded RNA from Saccharomyces cerevisiae | SRC VB “Vector” (Russia) | Approved for medical application | Influenza, herpes, chlamydia, tick-borne encephalitis | 53, 91, 101 |
| Ridostin Pro | Mixture of dsRNA and single stranded RNA from Saccharomyces cerevisiae, stabilized with polyvinylpyrrolidone | SRC VB “Vector” (Russia) | Registration stage | Influenza, SARS | 81 |
| Larifan | DsRNA of phage f2 | Larifan Ltd. (Latvia) | Approved for medical application | Arbovirus and rhabdovirus infections, herpetic diseases | 54, 56 |
| Rifastin | DsRNA of phage φ6 | SRC VB “Vector” (Russia) | Laboratory studies | Viral infections (Omsk hemorrhagic fever, influenza) | 55–57 |
| Double-stranded complexes of complementary homopolynucleotides | |||||
| PolyA:PolyU | |||||
| Poludan | Double-stranded complex polyriboadenylic and polyribouridylic acids | LANS-Pharm Ltd. (Russia) | Approved for medical application | Viral eye diseases (adenovirus and herpetic infections), influenza and SARS | 61, 62, 122, 123 |
| Polyadenur | Double-stranded complex polyriboadenylic and polyribouridylic acids | Beaufour Ipsen (France), Hemispherx Biopharma (USA) | Clinical trials | Hepatitis B and C (as a part of IFN-α based complex therapy), breast cancer | 63–65 |
| PolyG:PolyC | |||||
| Polyguacyl | Double-stranded complex of polyriboguanylic and polyribocytidylic acids | SRC VB “Vector” (Russia) | Laboratory studies | Viral infections (caused by viruses of encephalomyocarditis, tick-borne encephalitis, vesicular stomatitis) | 46, 61, 66, 67 |
| RGC100 | Double-stranded complex of polyriboguanylic and polyribocytidylic acids (100 bp) | Riboxx GmbH (Germany) | Laboratory studies | To increase efficiency of vaccination (activation of dendritic cells, T-cell proliferation) | 46 |
| PolyI:PolyC | |||||
| PolyI:PolyC | Double-stranded complex of polyriboinosinic and polyribocytidylic acids | University of Alabama at Birmingham, Hemispherx Biopharma, Inc. (USA); Medical University of South Carolina, Gibbs Cancer Center and Research Institute, Eli Lilly and Company (USA), etc. | Clinical trials | Hepatocellular carcinoma, hepatitis B, influenza | 71–74, 76 |
| Institute of Pharmaceutical Sciences, Institute of Molecular Biology and Biophysics (Switzerland); Nanjing Agricultural University (China); Icahn School of Medicine at Mount Sinai (USA); Yonsei University (South Korea); The University of Tokyo (Japan), etc. | Laboratory studies | For antitumor therapy of malignant neoplasms of different localization, enhancement of immunogenicity of antitumor vaccines | 88, 99, 100, 104, 105, 106, 109–112, 115, 117–121 | ||
| uPIC100-400 | Double-stranded complex of polyriboinosinic and polyribocytidylic acids (100–400 bp) |
Kyowa Hakko Bio Co., Ltd., Hofu (Japan); Hokkaido University, Sapporo (Japan) |
Laboratory studies | For prophylaxis and treatment of infectious and oncological diseases | 78 |
|
PolyI:PolyC, modified |
Double-stranded complex of polyriboinosinic and polyribocytidylic acids modified by polymers and/or encapsulated into particles | Research laboratories in Germany, China, Italy, the Netherlands, Canada, Belgium, Australia, USA | Laboratory studies | Oncological diseases of various localization (glioblastoma, adenocarcinoma, breast cancer, melanoma) | 74, 79–81, 92–96, 108, 116 |
|
Hitonol, PolyICLC |
Double-stranded complex of polyriboinosinic and polyribocytidylic acids with non-covalent adduct of poly-L-lysine and carboxymethylcellulose | Oncovir, Inc., Washington, DC (USA) | Clinical and laboratory trials | Oncological diseases of various localization, including inoperable cancer cases, viral infections, increased immunogenicity of vaccines | 74, 76, 82–87, 97, 98 |
| Double-stranded complexes of homopolyribonucleotide and polyribonucleotide copolymer | |||||
|
Ampligen Atvogen Rintatolimod Rintamod® PolyI:PolyC12U |
Double-stranded complex of polyriboinosinic acid and copolymer of polyribocytidylic and uridylic acids (at a ratio 12 : 1) | Hemispherx Biopharma, Inc., Philadelphia, PA (USA) | Clinical trials. Approved for medical application (Argentina) |
Complex therapy of syndromes of chronic fatigue and acquired immunodeficiency syndrome (AIDS), breast cancer, liver, prostate, ovarian cancer, colorectal cancer, mesothelioma Severe cases of myalgic encephalomyelitis/chronic fatigue syndrome |
75, 76 |
| PolyI:PolyC30U | Double-stranded complex of polyriboinosinic acid and copolymer of polyribocytidylic and uridylic acids (at a ratio 30 : 1) | Hemispherx Biopharma, Inc., Philadelphia, PA (USA) | Laboratory studies | For the prevention and treatment of infectious and oncological diseases | 75, 77 |
| RGIC100 PolyGI:PolyC | Double-stranded complex of polyribocytidylic acid and copolymer of polyguanylic and polyriboinosinic acids (100 bp) | Riboxx GmbH (Germany) | Laboratory studies | For the prevention and treatment of infectious and oncological diseases | 68 |
Results of preclinical and clinical trials, experimental studies reveal that dsRNAs exhibit inhibitory action against a wide range of viruses, as well as the adjuvant properties demonstrated in relation to vaccines against various viral infections. Modern trends of pharmaceutical development include improvement of dosage forms of dsRNA, the development of targeted delivery tools to improve antiviral efficacy and improve the therapeutic properties of such drugs.
A relatively new research direction includes the use of dsRNA as antitumor agents and antitumor vaccine adjuvants. It has been found hat these drugs inhibit growth of tumor cells through of proliferation suppression and induction of apoptotic cell death, enhance the antitumor immune response. Current clinical trials are aimed at investigating the antitumor properties of dsRNA as drugs for mono- and complex therapy in combination with antitumor agents, including vaccines. Further studies of the mechanisms of action, the role of dsRNA in the correction of pathological processes of various origins, will obviously help to optimize methods of drug usage to increase their effectiveness and safety.
FUNDING
The review has been prepared within the framework of the State Task of the SRC VB “Vector.”
CONFLICT OF INTEREST
All authors declare that they have no conflict of interest requiring disclosure in this article.
Footnotes
Translated by A. Medvedev
REFERENCES
- 1.Kawai T., Akira S. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Yoneyama M., Fujita T. Rev. Med. Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 3.de Faria I.J., Olmo R.P., Silva E.G., Marques J.T. J. Interferon Cytokine Res. 2013;33:239–253. doi: 10.1089/jir.2013.0026. [DOI] [PubMed] [Google Scholar]
- 4.Lee M.S., Kim Y.J. Annu. Rev. Biochem. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 5.Chow K.T., Gale M., Loo Y.M. Annu. Rev. Immunol. 2018;36:667–694. doi: 10.1146/annurev-immunol-042617-053309. [DOI] [PubMed] [Google Scholar]
- 6.Yu M., Levine S.J. Cytokine Growth Factor Rev. 2011;22:63–72. doi: 10.1016/j.cytogfr.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baum A., García-Sastre A. Amino Acids. 2010;38:1283–1299. doi: 10.1007/s00726-009-0374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sen G.C., Sarkar S.N. Cytokine Growth Factor Rev. 2005;16:1–14. doi: 10.1016/j.cytogfr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto M., Funami K., Oshiumi H., Seya T. Microbiol. Immunol. 2004;48:147–154. doi: 10.1111/j.1348-0421.2004.tb03500.x. [DOI] [PubMed] [Google Scholar]
- 10.Meylan E., Tschopp J., Karin M. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 11.Yoneyama M. Immunol. Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 12.Liu L., Botos I., Wang Y., Leonard J.N., Shiloach J., Segal D.M., Davies D.R. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y., Liu L., Davies D.R., Segal D.M. J. Biol. Chem. 2010;285:36836–36841. doi: 10.1074/jbc.M110.167973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwasaki A., Medzhitov R. Regulation of Adaptive Immunity by the Innate Immune System. Science. 2010;327(5963):291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shang T., Xiaoyan Z., Wang T. Endocrinology. 2011;152:2827–2836. doi: 10.1210/en.2011-0031. [DOI] [PubMed] [Google Scholar]
- 16.Agarwal, S., Wu, M., Livingston, C., Parks, D., Mayes, M., Arnett, F., and Tan, F., Arthritis Research & Therapy, 2011, http://arthritis-research.com/content/13/1/R3. [DOI] [PMC free article] [PubMed]
- 17.Zhang S., Herman M., Ciancanelli M., Perez de Diego R., Sancho-Shimizu V., Abel L., Casanova J.-L. Curr. Opin. Immunol. 2013;25:19–33. doi: 10.1016/j.coi.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lester S.N., Li K. J. Mol. Biol. 2014;426:1246–1264. doi: 10.1016/j.jmb.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee B.L., Moon J.E., Shu J.H., Yuan L., Newman Z.R., Schekman R., Barton G.M. Elife. 2013;2:e00291. doi: 10.7554/eLife.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Randall, R.E. and Goodbourn, S., J. Gen. Virol., 2008, vol. 89, pt 1 , pp. 1–47.
- 21.Yamamoto M., Sato S., Mori K., Hoshino K., Takeuchi O., Takeda K., Akira S. J. Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 22.Oshiumi H., Matsumoto M., Funami K., Akazawa T., Seya T. Nat. Immun. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 23.Silverman R.H. J. Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.García M.A., Gil J., Ventoso I., Guerra S., Domingo E., Rivas C., Esteban M. Microbiol. Molecular Biol. Revs. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang P., Samuel C.E. J. Virol. 2007;81:8192–8200. doi: 10.1128/JVI.00426-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole J.L. Trends Biochem. Sci. 2007;32:57–62. doi: 10.1016/j.tibs.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gantier M.P., Williams B.R. Cytokine Growth Factor Rev. 2007;18:363–371. doi: 10.1016/j.cytogfr.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koul, A., Deo, S., Booy, E.P., Orriss, G., Genung, M., and McKenna, S.A., Biochem. Cell. Biol., 2019, Apr 9. [Epub ahead of print].10.1139/bcb-2019-0060 [DOI] [PubMed]
- 29.Calderon B.M., Conn G.L. J. Biol. Chem. 2018;293:16115–16124. doi: 10.1074/jbc.RA118.004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato H., Takeuchi O., Sato S., Yoneyama M., Ya-mamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J., Yamaguchi O., Otsu K., Tsujimura T., Koh C.S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 31.Yoneyama M., Kikuchi M., Matsumoto K., Imaizumi T., Miyagishi M., Taira K., Foy E., Loo Y.M., Gale M., Akira S., Yonehara S., Kato A., Fujita T. J. Immunol. 2006;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 32.Yoneyama M., Fujita T. Uirusu. 2004;54:161–167. doi: 10.2222/jsv.54.161. [DOI] [PubMed] [Google Scholar]
- 33.Kato H., Sato S., Yoneyama M., Yamamoto M., Uematsu S., Matsui K., Tsujimura T., Takeda K., Fujita T., Takeuchi O., Akira S. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 34.Xu L.G., Wang Y.Y., Han K.J., Li L.Y., Zhai Z., Shu H.B. Mol. Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 35.Saito T., Gale M. J. Exp. Med. 2008;205:1523–1527. doi: 10.1084/jem.20081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui S., Eisenächer K., Kirchhofer A., Brzózka K., Lammens A., Lammens K., Fujita T., Conzelmann K.K., Krug A., Hopfner K.P. Mol. Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 37.Fujita T. Science. 2006;314:935–936. doi: 10.1126/science.1135756. [DOI] [PubMed] [Google Scholar]
- 38.DeWitte-Orr S.J., Mehta D.R., Collins S.E., Suthar M.S., Gale M., Mossman K.L. J. Immunol. 2009;183:6545–6553. doi: 10.4049/jimmunol.0900867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satoh T., Kato H., Kumagai Y., Yoneyama M., Sato S., Matsushita K., Tsujimura T., Fujita T., Akira S., Takeuchi O. Proc. Natl. Acad. Sci. USA. 2010;107:1512–1517. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruns A.M., Horvath C.M. Cytokine. 2015;74:198–206. doi: 10.1016/j.cyto.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martínez, I., Oliveros, J.C., Cuesta, I., de la Barrera, J., Ausina, V., Casals, C., de Lorenzo, A., García, E., García-Fojeda, B., Garmendia, J., González-Nicolau, M., Lacoma, A., Menéndez, M., Moranta, D., Nieto, A., Ortín, J., Pérez-González, A., Prat, C., Ramos-Sevillano, E., Regueiro, V., Rodriguez-Frandsen, A., Solís, D., Yuste, J., Bengoechea, J.A., and Melero, J.A., Front. Microbiol., 2017, vol. 8, 276. eCollection 201710.3389/fmicb.2017.00276 [DOI] [PMC free article] [PubMed]
- 42.Nellimarla S., Baid K., Loo Y.M., Gale M., Bowdish D.M., Mossman K.L. J. Immunol. 2015;195:3858–3865. doi: 10.4049/jimmunol.1501028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Semple S.L., Vo N.T.K., Poynter S.J., Li M., Heath D.D., DeWitte-Orr S.J., Dixon B. Dev. Comp. Immunol. 2018;89:93–101. doi: 10.1016/j.dci.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 44.Bianchi F., Pretto S., Tagliabue E., Balsari A., Sfondrini L. Cancer Biol. Ther. 2017;18:747–756. doi: 10.1080/15384047.2017.1373220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iordanov M.S., Kirsch J.D., Ryabinina O.P., Wong J., Spitz P.N., Korcheva V.B., Thorburn A., Magun B.E. Apoptosis. 2005;10:167–176. doi: 10.1007/s10495-005-6071-x. [DOI] [PubMed] [Google Scholar]
- 46.Naumann K., Wehner R., Schwarze A., Petzold C., Schmitz M., Rohayem J. Clin. Dev. Immunol. 2013;2013:283649. doi: 10.1155/2013/283649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salem M.L., Diaz-Montero C.M., El-Naggar S.A., Chen Y., Moussa O., Cole D.J. Vaccine. 2009;27:549–557. doi: 10.1016/j.vaccine.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shcheblyakov D.V., Logunov D.Yu., Tukhvatulin A.I., Shmarov M.M., Naroditzkiy B.S., Gintzburg A.L. Acta Naturae. 2010;2:28–37. doi: 10.32607/20758251-2010-2-3-21-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shime H., Matsumoto M., Oshiumi H., Tanaka S., Nakane A., Iwakura Y., Tahara H., Inoue N., Seya T. Proc. Natl. Acad. Sci. USA. 2012;109:2066–2071. doi: 10.1073/pnas.1113099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aliyari R., Wu Q., Li H.W., Wang X.H., Li F., Green L.D., Han C.S., Li W.X., Ding S.W. Cell. Host. Microbe. 2008;4:387–397. doi: 10.1016/j.chom.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tzanetakis I.E., Martin R.R. J. Virol. Methods. 2008;149:167–170. doi: 10.1016/j.jviromet.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 52.Wickner R.B. Microbiological Rev. 1996;60:250–265. doi: 10.1128/mr.60.1.250-265.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barinskiy I.F., Kosyakova N.P., Lavrukhina L.A., Posevaya T.A. Vopr. Virusol. 1994;39:179–182. [PubMed] [Google Scholar]
- 54.http://www.larifans.lv/index.php/ru/2016-03-23-14-48-05. Application date March 29, 2018 .
- 55.Nosik N.N., Pugatchev V.G., Yershov A.I. Antibiot. Khimioter. 1990;35:25–27. [PubMed] [Google Scholar]
- 56.Loginova S.Ya., Yefanova T.N., Kovaltchuk A.V., Faldina V.N., Androshchuk I.A., Pistzov M.N., Borisevich S.V., Kopylova N.K., Pashchenko Yu.I., Khamitov R.A., Maksimov V.A., Vasilyev N.T. Vopr. Virusol. 2002;47:27–30. [PubMed] [Google Scholar]
- 57.Bateneva A.V., Sysoeva G.M., Gamaley S.G., Skarnovitch M.O., Skarnovitch M.A., Shishkina L.N., Lebedev L.R., Danilenko E.D., Biofarmatzevt. Zhurnal. 2018;10:21–27. [Google Scholar]
- 58.Grunberg-Manago M., Ortiz P.J., Ochoa S. Science. 1955;122:907–910. doi: 10.1126/science.122.3176.907. [DOI] [PubMed] [Google Scholar]
- 59.Vilner L.M., Platonova G.A., Sidorova N.S., Timkovskiy A.L. Vopr. Virusol. 1985;3:337–339. [PubMed] [Google Scholar]
- 60.Vilner L.M., Kogan E.M., Platonova G.A. Antibiotiki. 1982;1:54–56. [PubMed] [Google Scholar]
- 61.Yershov F.I., Zhdanov V.M. Induktory interpherona. Moscow: Interferon Inducers; 1982. [Google Scholar]
- 62.Sharypova D.V., Zhukov I.Yu. Ali Mazlum, Vlasova, N.N., Puzankova, O.S., Gavrilova, V.L., and Igolkin, A.S. Veterinariya Segodnya. 2018;1:37–41. doi: 10.29326/2304-196X-2018-1-37-41. [DOI] [Google Scholar]
- 63.de Paillette, E., US Patent 6,509,154 B1, Appl. No. 09/485,086, Date of Patent January 21, 2003.
- 64.Fabrice, A. and Sabourin, J.-Ch., Int. Pub. No.: WO 2006/054129 A1, International Appl. No. PCT/IB2004/004093, Publication Date May 26, 2006.
- 65.https://www.thefreelibrary.com/Hemispherx+initiates+legal+action+against+Beaufour+Ipsen+for…-a063275880. Application date January 10, 2019.
- 66.Yershov F.I., Narovlyanskiy A.N. Vopr. Virusol. 2015;60:5–10. [PubMed] [Google Scholar]
- 67.Robinson R.A., DeVita V.T., Levy H.B., Baron S., Hubbard S.P., Levine A.S. J. Natl. Cancer. Inst. 1976;57:599–602. doi: 10.1093/jnci/57.3.599. [DOI] [PubMed] [Google Scholar]
- 68.Rohayem, J., US Patent No.10,023,871 B2, Appl. No. 15/105,467, Date of Patent July 17, 2018.
- 69.Masycheva V.I., Nadolinnaya I.G. Farmakol. Toksikol. 1981;44:353–357. [PubMed] [Google Scholar]
- 70.chem21.info/info/166822/Big Chemical Encyclopedia 21, p. 341. Application date April 10, 2019.
- 71.Overton E.T., Goepfert P.A., Cunningham P., Carter W.A., Horvath J., Young D., Strayer D.R. Vaccine. 2014;32:5490–5495. doi: 10.1016/j.vaccine.2014.07.078. [DOI] [PubMed] [Google Scholar]
- 72.Mehrotra S., Britten C.D., Chin S., Garrett-Mayer E., Cloud C.A., Li M., Scurti G., Salem M.L., Nelson M.H., Thomas M.B., Paulos C.M., Salazar A.M., Nishimura M.I., Rubinstein M.P., Li Z., Cole D.J. J. Hematol. Oncol. 2017;10:82. doi: 10.1186/s13045-017-0459-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Okada H., Weller M., Huang R., Finocchiaro G., Gilbert M.R., Wick W., Ellingson B.M., Hashimoto N., Pollack I.F., Brandes A.A., Franceschi E., Herold-Mende C., Nayak L., Panigrahy A., Pope W.B., Prins R., Sampson J.H., Wen P.Y., Reardon D.A. Lancet Oncol. 2015;16:e534–e542. doi: 10.1016/S1470-2045(15)00088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith Melody, García-Martínez Elena, Pitter Michael R., Fucikova Jitka, Spisek Radek, Zitvogel Laurence, Kroemer Guido, Galluzzi Lorenzo. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. OncoImmunology. 2018;7(12):e1526250. doi: 10.1080/2162402X.2018.1526250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carter, W.A. and Strayer, D.R., Pub. No.: US 2014/0335112 A1, Appl. No. 14/275,754, Publication Date November 13, 2014.
- 76.http://clinicaltrials.gov/. Application date April 9, 2019.
- 77.Carter, W.A. and Strayer, D.R., Int. Pub. No.: WO 2011/059505 A2, International Appl. No. PCT/US20 10/002970, International Publication Date May 19, 2011.
- 78.Nakano T., Yamamura E.T., Fujita H., Sone T., Asano K. Biosci. Biotechnol. Biochem. 2018;82:1889–1901. doi: 10.1080/09168451.2018.1501264. [DOI] [PubMed] [Google Scholar]
- 79.Torchilin V.P. Therapeutic Delivery. 2013;4:537–538. doi: 10.4155/tde.13.25. [DOI] [PubMed] [Google Scholar]
- 80.Ghosh S.K., Chowdhury R.R. Expert Opinion Drug Delivery. 2013;10:437–450. doi: 10.1517/17425247.2013.757591. [DOI] [PubMed] [Google Scholar]
- 81.Levagina, G.M., Alikin, Yu.S., Masycheva, V.I., Danilenko, E.D., Fadina, V.A., and Ignatyev, G.M., RUS Patent no. 2 172 631, Byull. Izobret., 2001, no. 24.
- 82.Levy, H.B., US Patent No.: US 4,349,538, Appl. No. 208,029, Publication Date September 14, 1982 .
- 83.Salazar, A.M., Oncovir, Inc. http://www.oncovir.com/id2.html. Application date January 11, 2018.
- 84.Mitchell W.M., Carter W.A. Emerg. Microbes Infect. 2014;3:e77. doi: 10.1038/emi.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wong J.P., Yang H., Nagata L., Kende M., Levy H., Schnell G., Blasetti K. Vaccine. 1999;17:1788–1795. doi: 10.1016/S0264-410X(98)00439-3. [DOI] [PubMed] [Google Scholar]
- 86.Hu Y., Hu Y., Sun L., Wong J., Wang M. Biochem. Biophys. Res. Commun. 2016;478:913–918. doi: 10.1016/j.bbrc.2016.08.050. [DOI] [PubMed] [Google Scholar]
- 87.Compagnon B., Milhaud P., Bienvenüe A., Philippot J.R. Exp. Cell Res. 1992;200:333–338. doi: 10.1016/0014-4827(92)90180-G. [DOI] [PubMed] [Google Scholar]
- 88.Shir A., Ogris M., Wagner E., Levitzki A. PLoS Med. 2006;3:e6. doi: 10.1371/journal.pmed.0030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Masycheva, V.I., Lebedev, L.R., Danilenko, E.D., Sysoeva, G.M., and Gamaley, S.G., RUS Patent no. 2 386 447, Byull. Izobret., 2010, no. 11.
- 90.Danilenko, E.D., Gamaley, S.G., Bateneva, A.V., Sysoeva, G.M., Levagina, G.M., and Masycheva, V.I., Zhurnal Mikrobiol., Epidemiol., Immunobiol., 2011, no. 3, pp. 57–61. [PubMed]
- 91.Howard K.A., Rahbek U.L., Liu X., Damgaard C.K., Glud S.Z., Andersen M.Ø., Hovgaard M.B., Schmitz A., Nyengaard J.R., Besenbacher F., Kjems J. Mol. Ther. 2006;14:476–484. doi: 10.1016/j.ymthe.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 92.Chen L., Ding Y., Wang Y., Liu X., Babu R., Ravis W., Yan W. Int. J. Nanomedicine. 2013;8:138–145. doi: 10.2147/IJN.S38928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cobaleda-Siles M., Henriksen-Lacey M., Ruiz de Angulo A., Bernecker A., Gómez Vallejo V., Szczupak B., Llop J., Pastor G., Plaza-Garcia S., Jauregui-Osoro M., Meszaros L.K., Mareque-Rivas J.C. Small. 2014;10:5054–5067. doi: 10.1002/smll.201401353. [DOI] [PubMed] [Google Scholar]
- 94.Knuschke T., Epple M., Westendorf A.M. Hum. Vaccin. Immunother. 2014;10:164–169. doi: 10.4161/hv.26203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McNally B., Willette M., Ye F., Partida-Sanchez S., Flaño E. PLoS One. 2012;7:e51351. doi: 10.1371/journal.pone.0051351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li Y., Hu Y., Jin Y., Zhang G., Wong J., Sun L.Q., Wang M. J. Gene Med. 2011;13:60–72. doi: 10.1002/jgm.1536. [DOI] [PubMed] [Google Scholar]
- 97.Caskey M., Lefebvre F., Filali-Mouhim A., Cameron M.J., Goulet J.P., Haddad E.K., Breton G., Trumpfheller C., Pollak S., Shimeliovich I., Duque-Alarcon A., Pan L., Nelkenbaum A., Salazar A.M., Schlesinger S.J., Steinman R.M., Sékaly R.P. J. Exp. Med. 2011;208:2357–2366. doi: 10.1084/jem.20111171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Martins Karen A. O., Steffens Jesse T., van Tongeren Sean A., Wells Jay B., Bergeron Alison A., Dickson Samuel P., Dye John M., Salazar Andres M., Bavari Sina. Toll-Like Receptor Agonist Augments Virus-Like Particle-Mediated Protection from Ebola Virus with Transient Immune Activation. PLoS ONE. 2014;9(2):e89735. doi: 10.1371/journal.pone.0089735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang L., Bai J., Liu J., Wang X., Li Y., Jiang P. Vet. Microbiol. 2013; 164:253–260. doi: 10.1016/j.vetmic.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 100.Terhuja M., Saravanan P., Tamilselvan R.P. Biologicals. 2015;43:437–443. doi: 10.1016/j.biologicals.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 101.Barinskiy I.F., Alimbarova L.M., Lazarenko A.A., Makhmudov F.R., Mordvintzeva E.Yu., Sergeev O.V. Immunologiya. 2015;36:95–98. [Google Scholar]
- 102.http://lib.fedpress.ru/person/donchenko-aleksandr-semenovich. Application date March 29, 2018 .
- 103.Hafner A.M., Corthésy B., Textor M., Merkle H.P. Biomaterials. 2011;32:2651–2661. doi: 10.1016/j.biomaterials.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 104.Hafner A.M., Burschowsky D., Corthésy B., Textor M., Merkle H.P. J. Control Release. 2012;59:204–214. doi: 10.1016/j.jconrel.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 105.Pérez-Girón J.V., Belicha-Villanueva A., Hassan E., Gómez-Medina S., Cruz J.L., Lüdtke A., Ruibal P., Albrecht R.A., García-Sastre A., Muñoz-Fontela C. J. Immunol. 2014;193:1324–1332. doi: 10.4049/jimmunol.1400222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim Eun-Do, Han Soo Jung, Byun Young-Ho, Yoon Sang Chul, Choi Kyoung Sub, Seong Baik Lin, Seo Kyoung Yul. Inactivated Eyedrop Influenza Vaccine Adjuvanted with Poly(I:C) Is Safe and Effective for Inducing Protective Systemic and Mucosal Immunity. PLOS ONE. 2015;10(9):e0137608. doi: 10.1371/journal.pone.0137608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Uchida S., Yoshinaga N., Yanagihara K., Yuba E., Kataoka K., Itaka K. Biomaterials. 2018;150:62–170. doi: 10.1016/j.biomaterials.2017.09.033. [DOI] [PubMed] [Google Scholar]
- 108.Qu J., Hou Z., Han Q., Zhang C., Tian Z., Zhang J. Int. Immunopharmacol. 2013;17:814–820. doi: 10.1016/j.intimp.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 109.Chen H., Wang D.L., Liu Y.L. Mol. Med. Rep. 2016;13:2689–2695. doi: 10.3892/mmr.2016.4848. [DOI] [PubMed] [Google Scholar]
- 110.Ayari C., Besançon M., Bergeron A., LaRue H., Bussières V., Fradet Y. Cancer Immunol. Immunother. 2016;65:223–234. doi: 10.1007/s00262-015-1789-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Palchetti S., Starace D., De Cesaris P., Filippini A., Ziparo E., Riccioli A. J. Biol. Chem. 2015;290:5470–5483. doi: 10.1074/jbc.M114.601625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Salaun, B., Lebecque, S., Matikainen, S., Rimoldi, D., and Romero, P., Clin. Cancer Res., 2007, vol. 13, no. 15 Pt1, pp. 4565–4574. 10.1158/1078-432.CCR-07-0274 [DOI] [PubMed]
- 113.Chiba Yukino, Mizoguchi Izuru, Mitobe Kana, Higuchi Kaname, Nagai Hiroshi, Nishigori Chikako, Mizuguchi Junichiro, Yoshimoto Takayuki. IL-27 Enhances the Expression of TRAIL and TLR3 in Human Melanomas and Inhibits Their Tumor Growth in Cooperation with a TLR3 Agonist Poly(I:C) Partly in a TRAIL-Dependent Manner. PLoS ONE. 2013;8(10):e76159. doi: 10.1371/journal.pone.0076159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Estornes Y., Micheau O., Renno T., Lebecque S. Oncogene and Cancer—from Bench to Clinic. Novi Sad: INTECH; 2013. [Google Scholar]
- 115.Muthuswamy, R., Wang, L., Pitteroff, J., Gingrich, J.R., and Kalinski, P., J. Immunother. Cancer, 2015, vol. 3, 6. eCollection 201510.1186/s40425-015-0050-8 [DOI] [PMC free article] [PubMed]
- 116.Alipour T.G., Ebrahimi Z., Badiee A., Mansourian M., Attar H., Arabi L., Jalali S.A., Jaafari M.R. Immunol. Lett. 2016;176:57–64. doi: 10.1016/j.imlet.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 117.Gupta S.K., Tiwari A.K., Gandham R.K., Sahoo A.P. Int. Immunopharmacol. 2016;35:163–173. doi: 10.1016/j.intimp.2016.03.034. [DOI] [PubMed] [Google Scholar]
- 118.Gupta S.K., Yadav P.K., Tiwari A.K., Gandham R.K., Sahoo A.P. Tumour. Biol. 2016;37:12089–12102. doi: 10.1007/s13277-016-5093-z. [DOI] [PubMed] [Google Scholar]
- 119.Glaffig M., Stergiou N., Schmitt E., Kunz H.I. Chem. Med. Chem. 2017;12:722–727. doi: 10.1002/cmdc.201700254. [DOI] [PubMed] [Google Scholar]
- 120.Kano Y., Iguchi T., Matsui H., Adachi K., Sakoda Y., Miyakawa T., Doi S., Hazama S., Nagano H., Ueyama Y., Tamada K. Cancer Sci. 2016;107:398–406. doi: 10.1111/cas.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Luo Z., Wang C., Yi H., Li P., Pan H., Liu L., Cai L., Ma Y. Biomaterials. 2015;38:50–60. doi: 10.1016/j.biomaterials.2014.10.050. [DOI] [PubMed] [Google Scholar]
- 122.https://products.veropharm.ru/products/cold/poludanum//. Application date May 26, 2019 .
- 123.Shumilov V.I., Ivannikov Yu.G., Ogarkov P.I., Lobastov S.P., Olontzev V.V. Voyenno-Med. Zhurnal. 2002;323:45–47. [PubMed] [Google Scholar]