

Abstract
Fibrous Dysplasia/McCune-Albright Syndrome (FD/MAS), arising from gain-of-function mutations in Gαs, and Cutaneous Skeletal Hypophosphatemia Syndrome (CSHS), arising from gain-of-function mutations in the Ras/MAPK pathway, are strikingly complex, mosaic diseases with overlapping phenotypes. Both disorders are defined by mosaic skin and bone involvement, and both are complicated by increased FGF23 production. These similarities have frequently led to mis-diagnoses, primarily in patients with CSHS who are often assumed to have FD/MAS. The intriguing similarities in skeletal involvement in these genetically distinct disorders has led to novel insights into FGF23 physiology, making an understanding of FD/MAS and CSHS relevant to both clinicians and researchers interested in bone and endocrine disorders. This review will give an overview of FD/MAS and CSHS, focusing on the roles of mosaicism and FGF23 in the pathogenesis and clinical presentation of these disorders.
Keywords: metabolic bone disease, phosphate metabolism, mosaicism, GNAS, RAS
INTRODUCTION
Mosaicism is defined as the presence of 2 or more cell populations with distinct genotypes within an organism, typically arising from post-zygotic mutational events. Mosaicism is increasingly recognized as an important contributor to human health and disease, in part due to genomic technologies that have enabled detection and characterization of an ever-growing number of disorders. Because mosaic diseases often involve genetic changes that would be lethal in their germline states, they provide unique opportunities to investigate physiologic processes and gain biological insights. However, these disorders also present unique challenges to diagnosis and management. The variable involvement of multiple tissue types can lead to dramatic phenotypic variations. Diagnoses in mosaic disorders are most often made clinically; however, because they are rare and non-inheritable, clinicians have limited opportunities to gain direct clinical experience.
The purpose of this review is to provide an overview of 2 mosaic disorders that have striking phenotypic overlap. Fibrous Dysplasia/McCune-Albright Syndrome (FD/MAS) and the more recently described Cutaneous Skeletal Hypophosphatemia Syndrome (CSHS) are both defined by mosaic skin and bone involvement, complicated by increased FGF23 production. These similarities have frequently led to mis-diagnoses, primarily in patients with CSHS who are often assumed to have FD/MAS. The intriguing similarities in skeletal involvement in these genetically distinct disorders has led to novel insights into FGF23 physiology, making an understanding of FD/MAS and CSHS relevant to both clinicians and researchers interested in bone and endocrine disorders.
GENETIC ETIOLOGY
FD/MAS and CSHS arise from de novo mutations in non-germline cells during embryogenesis. The etiologies of somatic mutations are complex, and may be related to errors in DNA replication, environmental insults, and other factors (1). The developmental timing and location of these mutational events determines the location and extension of affected tissues. When the mutation occurs at earlier stages of development, extensive affected postnatal tissue can be expected; when it occurs later in embryogenesis or during fetal development, fewer and smaller areas are affected. If the mutation occurs prior to gastrulation, more than one germ layer can be affected, leading to the involvement of tissues and organs of different embryonic origins.
FD/MAS results from gain-of-function mutations in GNAS, which encodes the α-subunit of the Gs G protein -coupled receptor (GPCR). In most cases mutations affect codon R201, replacing an arginine residue with histidine or cysteine (2). Mutations in GNAS codon G227 have also been reported, representing less than 5% of cases (3). GNAS mutations inactivate the regulatory GTPase motif of Gαs and its isoforms, leading to constitutive receptor signaling and dysregulated production of cyclic AMP (cAMP), protein kinase A (PKA) activation, and downstream signaling.
CSHS arises from gain-of-function mutations in a RAS gene. Only NRAS Q61R and HRAS G13R mutations have been associated with CSHS to date (4, 5), however NRAS G12, G13, P34; HRAS G12, Q61 and KRAS G12 mutations have been found in other cutaneous mosaic RASopathies (6, 7), and future studies may associate these mutations with CSHS. Like GNAS mutations in FD/MAS, RAS mutations in CSHS lie within a regulatory GTP-binding region, locking the protein into a permanently active state. This results in activation of the protein kinase activity of RAF kinase, leading to phosphorylation of MEK and constitutive activation of the MAPK signaling pathway.
The diverse clinical manifestations of FD/MAS and CSHS (described below) underscore the ubiquitous nature of GCPR/Gαs/PKA and tyrosine Kinase/Ras/MAPK pathways. The study and characterization of these diseases therefore has the potential to expand our understanding of the roles of these pathways in human physiology.
PREVALENCE
FD/MAS and CSHS are both rare, non-hereditable conditions, and there are no high-quality epidemiologic data to accurately determine prevalence. FD has been estimated to account for approximately 7% of benign bone tumors, however MAS (involving extraskeletal features) is much rarer, with an estimated prevalence of 1:1,000,000 to 1:100,000 (8). CSHS has only recently been characterized, and one metanalysis identified 56 cases with a phenotype consistent with CSHS in the published literature (4). It is therefore likely that CSHS is comparatively much rarer than FD/MAS.
TAXONOMY
Historically, most mosaic conditions have been grouped phenotypically into clinically-defined syndromes. Only recently have improvements in genetic testing techniques enabled the association of many of these syndromes with specific somatic mutations. This new body of evidence has enabled a more systematic approach to classifying mosaic diseases. However, disease taxonomy should not rely solely on genotype, because multiple additional factors, such as the pattern and time of expression and gene-environment interactions, strongly influence the phenotypes, comorbidities, and medical needs of individuals with somatic mutations. An illustrative example is an otherwise healthy patient with an acquired GNAS- or RAS-related neoplasm. This is essentially a form of mosaicism; however, this patient would bear little in common clinically with patients affected by FD/MAS or CSHS. Therefore, genotype, phenotype, and environment must all be taken into account when defining disease entities (9).
FD/MAS: Polyostotic FD as a skeletal disorder was initially described in 1938 by pathologist Louis Lichtenstein, who aptly speculated that FD results from “perverted activity of the specific bone-forming mesenchyme” (10). The association of FD with precocious puberty and skin hyperpigmentation was separately reported in 1936 by Donovan McCune and Fuller Albright, leading to the conception of MAS as a syndromic “triad” of these features (11). It was later recognized that other hyperfunctioning endocrinopathies and extraskeletal features (with or without FD) may occur in varying combination in patients with GNAS mutations, leading to decades of taxonomic confusion regarding the diagnostic criteria for FD/MAS. Recently, an international consortium of clinicians, researchers, and advocates have attempted to clarify FD/MAS taxonomy through published consensus guidelines (12). According to this proposed system, the term “FD” in isolation refers exclusively to skeletal disease. Extraskeletal features associated with MAS are 1) café-au-lait macules with characteristic features, 2) gonadotropin-independent sex steroid production, 3) characteristic thyroid lesions with or without non-autoimmune hyperthyroidism, 4) GH excess, 5) neonatal hypercortisolism. MAS is defined as the combination of FD and one or more extraskeletal features, OR the presence of two or more extraskeletal features. For inclusivity, the disease entity is commonly referred to as “FD/MAS” in order to encompass individuals with and without extraskeletal disease.
CSHS was defined in 2014 as a multisystemic disorder involving focal bone lesions, pigmented skin lesions, and elevated circulating levels of FGF23, arising from gain-of-function mutations in the Ras/MAPK pathway (4, 13). Historically, epidermal and congenital melanocytic nevi have been rarely reported in association with extracutaneous findings, including skeletal malformations, precocious puberty, neurologic abnormalities, and hypophosphatemia (14, 15). Depending on the pattern of involvement, a wide range of taxonomic terms have been applied, including epidermal nevus syndrome, Becker’s nevus syndrome, Schimmelpenning–Feuerstein–Mims syndrome, and others (16–18). The genetic etiologies of many of these syndromes are unknown, however mutations in RAS gene family members have been identified in isolated and syndromic keratinocytic, sebaceous, and melanocytic nevi, typically in patients with widespread involvement of skin and other organ systems (19–21). A cohort study identified RAS mutations in 5 patients with phenotypes involving focal bone lesions, pigmented skin lesions, and FGF23-mediated hypophosphatemia, establishing a shared genetic etiology for this constellation of features (22). Therefore, the divergence of this phenotype and its associated clinical complications, as well as the identification of Ras activation as the common causative etiology, prompted the definition of CSHS as a distinct entity.
FD/MAS and CSHS thus illustrate two approaches to disease taxonomy. FD/MAS aims to more broadly group the various phenotypic features resulting from Gαs activation into a single taxonomic entity. In contrast, CSHS groups rare clinical variants of several previously-defined dermatologic syndromes of Ras/MAPK pathway activation into a unique taxonomic entity, based on common phenotypic features that dictate clinical presentation and management.
PATHOGENESIS AND HISTOPATHOLOGY
FD is characterized by replacement of the hematopoietic and adipose bone marrow with fibrous stroma. This tissue is a mixture of mutant and wild-type bone marrow stromal cells (BMSCs) that acquire an abnormal fibroblastic phenotype under the influence of Gαs activation. Proliferation of these abnormal BMSCs leads to the formation and expansion of FD lesions.
The fibrous stroma in FD is low to moderately cellular, highly proliferative and variably dense and collagenized (Fig 1). FD BMSCs may give raise to abnormal osteoblasts, which produce woven bone matrix forming unconnected, curvilinear trabeculae. Trabeculae are sometimes absent in FD and vary in quantity and pattern among and within lesions. FD bone is poorly mineralized and often hypercellular, which may present with expanded osteocytic lacunae. FD osteoblasts are characterized by a flat-stellate rather than cuboidal morphology. This morphological change arises due to cAMP dysregulation, which leads to cytoskeleton reorganization and retraction of the cell body (23). FD osteoblasts deposit collagen fibers perpendicularly to the bone matrix, forming structures termed Sharpey fibers. Osteoclastogenesis is highly prevalent, featuring osteoclasts located either on the bone surface or at ectopic locations. As lesions age, red and yellow marrow can reappear in FD marrow spaces, and lesions trend to form fluid-filled cysts (24).
Figure 1.

Histopathologic images. Within each section, left and right panels are bright field (Goldner trichrome staining) and polarized light microscopy, respectively, shown at low and high power. FD tissue is overly fibrotic with woven undermineralized bone deposition. Note the absence of adipocytes and hematopoietic marrow. CSHS skeletal lesions demonstrate normal marrow cellularity and lamellar bone matrix, with focal patches of fibrous stroma and woven bone. Severe osteomalacia with prominent visible osteoid is apparent in both tissue types. Of note, the patient with FD had chronic longstanding hypophosphatemia and was on treatment at the time the specimen was obtained. The patient with CSHS did not have clinically significant hypophosphatemia. Ad=adipocyte, FS=fibrous stroma, Hp=hematopoiesis, LB=lamellar bone, Od=osteoid, Oc=osteoclasts, Os=osteoblasts, ShF=Sharpey fibers, V=vascularization, WB=woven bone.
CSHS: Due to the limited number of cases, the histopathological features of CSHS are poorly defined and still subject to further characterization. In this section, we collect observations from three published references (4, 13, 25) and bone samples from 2 patients seen at the NIH.
In contrast to FD, CSHS bone lesions do not seem to have altered hematopoiesis and adipogenesis (Fig 1). In fact, many marrow areas appear normal except for focal regions of fibrous stroma. Stromal dysplastic cells do not appear to replace or alter bone marrow cells. Like FD, skeletal CSHS lesions demonstrate marked osteomalacia, with up to 80% of the bone surfaces lined with osteoid and deep unmineralized regions. However, most of the bone matrix has a lamellar structure, with focal areas in which the parallel structure is interrupted by small patches of woven bone. These areas are irregularly mineralized, hypercellular, and associated with dysplastic stroma. CSHS trabeculae and cortical bone have an altered architecture, with unconnected trabeculae and porotic cortical bone. Although osteoclasts are prevalent in CSHS skeletal lesions, this appears to occur to a lesser degree than in FD, and unlike FD is never present in ectopic sites. CSHS skeletal lesions also feature numerous bone formation rims, in which osteoblasts have a normal cuboidal appearance.
Lesional osteomalacia is a prominent feature of FD and CSHS, however in both disorders it has been observed even in the absence of frank hypophosphatemia (for example, the CSHS patient in Fig 1 was normophosphatemic). It is therefore likely that osteoblast dysfunction is a key contributor, potentially due to deposition of abnormal matrix with impaired ability to support calcium phosphate precipitation. Further investigation is needed to determine the etiologies and mechanisms of osteomalacia in these disorders.
GNAS AND RAS IN FGF23 REGULATION
The degree and severity of hypophosphatemia is variable in both FD/MAS and CSHS. While elevated FGF23 levels have been well-documented in patients with both disorders (4, 26, 27), the prevalence of clinically significant hypophosphatemia has not been well-established in either disease. However based on current evidence it appears to be higher in CSHS: one large metanalysis reported 50% of published CSHS cases were hypophosphatemic (4), while a large FD/MAS cohort study has observed a 16% prevalence (personal observation, AMB). The particularities of cAMP vs MAPK signaling pathways in FGF23 transcription, translation, and posttranslational processing likely account in part for this variability.
FD/MAS: Murine and in vitro studies have shown that upregulation of cAMP signaling in osteogenic cells leads to increased FGF23 transcription (28, 29). Moreover, transfection of mutant Gαs into HEKF cells leads to increased FGF23 production (27). However, mice and human studies show conflicting data regarding the effects of PTH infusion (which activates Gαs) on serum FGF23 levels (30–32). This may be in part due to overlapping regulatory effects of PTH and FGF23 on both phosphate and vitamin D metabolism. Interestingly, cAMP signaling in bone cells stimulates FGF23 transcription but also its inactivation through cleavage (27, 28), and this mechanism appears to play an important role in determining the net amount of intact FGF23 secreted in the bloodstream. This has been further evidenced in patients with FD/MAS, who have an increased ratio of C-terminal (fragmented and inactive) to intact (and biologically active) FGF23 in their serum (27, 28). This suggests that increased FGF23 degradation due to altered processing might partially mitigate the hypophosphatemic effects of FGF23 overproduction in patients with FD/MAS.
CSHS: In contrast to Gαs, Ras/MAPK pathway signaling appears to have more “clean” and direct effects on FGF23 production. Several observations support that fibroblast growth factor receptor 1 (FGFR1) activation leads to FGF23 production through Ras/MAPK signaling. For example, pathogenic gain-of-function FGFR1 mutations have been detected in patients with osteoglophonic dysplasia, a rare form of achondroplasia associated with FGF23-mediated hypophosphatemia (33). Mouse models have consistently demonstrated that stimulation and inhibition of Fgfr1 in bone cells leads to increased and decreased biologically active FGF23 in the blood, respectively (34–36). The identification of a fibronectin and FGFR1 fusion protein as the molecular driver of ectopic FGF23 production in a subset of phosphaturic mesenchymal tumors further highlights the strong influence of MAPK signaling on FGF23 production (37). These data suggest that the Fgfr1/Ras/MAPK pathway is a stronger and more direct regulator of FGF23 production in comparison to GPCR/Gαs/PKA pathway and may explain the relatively higher prevalence of clinically significant hypophosphatemia in CSHS in comparison to FD/MAS.
CLINICAL DESCRIPTION
Mosaicism is inherent in the clinical features of both FD/MAS and CSHS. Skin lesions occur in a distinctive distribution following developmental lines of Blaschko, reflecting patterns of embryonic cell migration. Bone lesions likewise occur in a patchy, irregular distribution, and may range from trivial disease affecting only a few areas, to severe disease involving most of the skeleton. Endocrine and other extraskeletal features are variably involved, contributing to the broad clinical spectrum that varies greatly between individuals. Specific disease features are included in Table 1 and discussed in further detail below.
Table 1.
Representative clinical features
| Features | Fibrous dysplasia/McCune-Albright syndrome | Cutaneous Skeletal Hypophosphatemia Syndrome |
|---|---|---|
| Dermatologic | Café-au-lait macules | Café-au-lait macules |
| Hyperpigmented melanocytic nevi | ||
| Phakomatosis pigmentokeratotica | ||
| Giant congenital melanocytic nevi | ||
| Keratinocytic epidermal nevi | ||
| Nevus sebaceous | ||
| Skeletal | Focal dysplastic lesions | Focal dysplastic lesions |
| Hypoplastic cranial bones | ||
| Endocrine | FGF23-mediated hypophosphatemia | FGF23-mediated hypophosphatemia |
| Precocious puberty | Precocious puberty | |
| Hyperthyroidism | ||
| Growth hormone excess | ||
| Neonatal hypercortisolism | ||
| Neurologic | Neurodevelopmental abnormalities | Neurodevelopmental abnormalities |
| Ventriculomegaly | ||
| Macro- and micro-cephaly | ||
| Neurocutaneous melanosis | ||
| Malignancies | Thyroid cancer | Thyroid cancer |
| Skeletal sarcomas | Malignant melanoma | |
| Pancreatic cysts | Pheochromocytoma | |
| Breast cancer | Brain tumors | |
| Others | Others | |
| Others | Hematologic abnormalities | Hematologic abnormalities |
| Tachycardia, hypertension | Ophthalmologic abnormalities |
Dermatologic features
FD/MAS: Café-au-lait macules are a classically described feature of MAS (11), affecting approximately 2/3 of patients (38). Hyperpigmentation occurs due to constitutive signaling through the melanocyte stimulating hormone receptor in GNAS mutation-bearing melanocytic cells. Typical of mosaic lesions, these macules have jagged borders and are located along the midline of the body (Fig 2A). There does not appear to be a geographic relationship between the location of skin lesions and dysplastic bone lesions (39).
Figure 2.
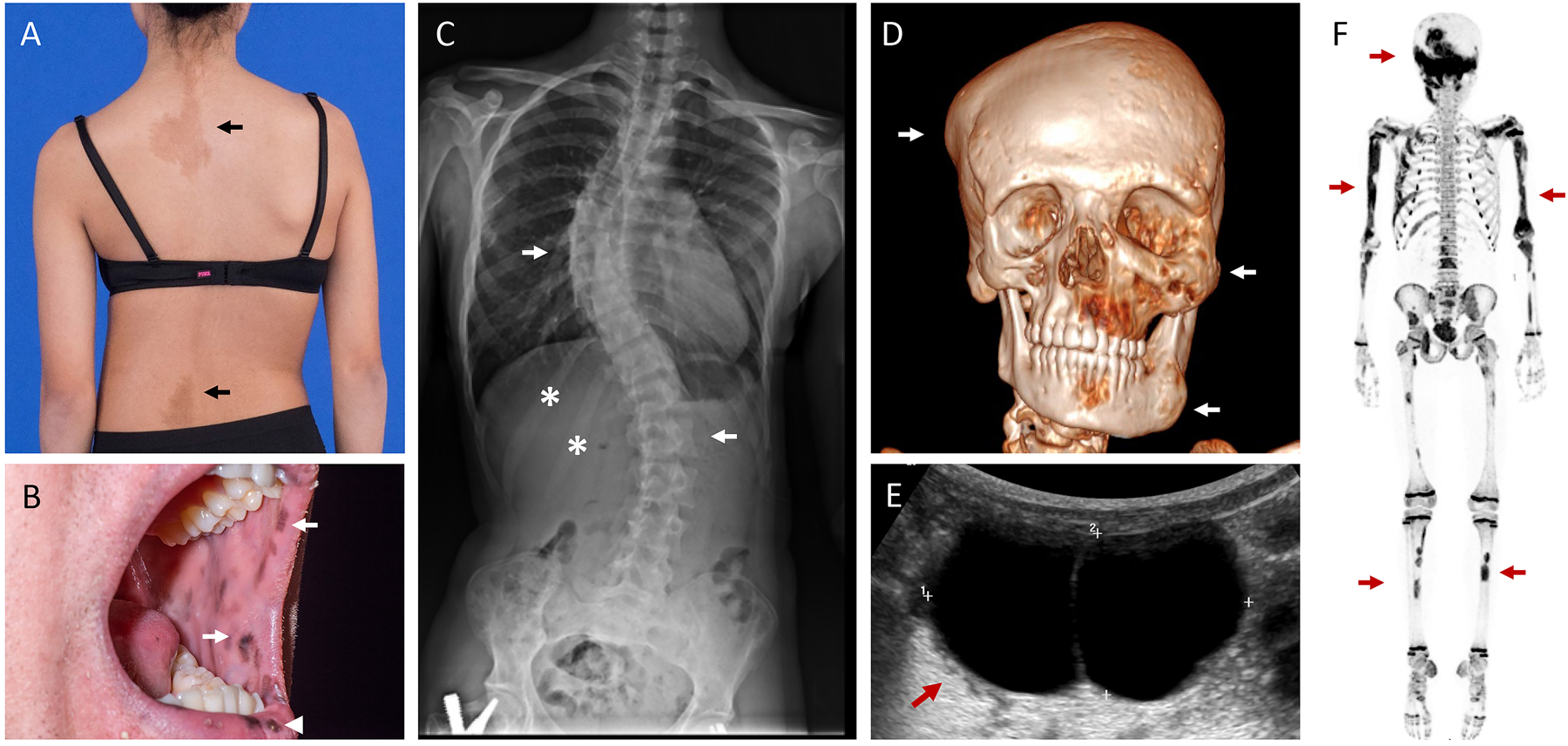
Clinical images in Fibrous Dysplasia/McCune-Albright Syndrome. A. Café-au-lait macules reflect along the spine of an adolescent girl (arrows), demonstrating typical jagged borders and distribution along the midline of the body. Note the obliquity in this patient’s hips and shoulders, reflective of underlying scoliosis. B. Lentigines involving the oral mucosa (arrows) and outer lips (arrowhead) in an adult patient. C. Radiograph from the patient in panel A show severe scoliosis involving the thoracic and lumbar spine (arrows). Expansile FD lesions are apparent in the right 11th and 12th ribs (asterisks). The patient has a surgical implant visible in the proximal right femur related to a previous fracture repair. D. Three-dimensional computed tomography scan demonstrates multiple expansile FD lesions involving the calvarium, maxilla, and mandible (arrows). E. Pelvic ultrasound in a 3-year-old girl with pubertal symptoms and elevated estrogen levels reveals a large ovarian cyst (arrow). F. 18F-NaF PET/CT scan shows multiple areas of tracer uptake in the skull, ribs, spine, and long bones, consistent with polyostotic FD (arrows). Note the expected physiologic, symmetric uptake in the metaphyses throughout in this growing child.
Pigmentation involving the lips and oral mucosa has been recently described as an additional cutaneous feature of FD/MAS (40)(Fig 2B). In contrast to café-au-lait macules, which are present at or shortly after birth, these lesions develop in adulthood and may progress with age. Interestingly, these resemble the oral lentigines that occur in patients with Carney Complex, a disorder arising from PRKAR1A mutations resulting in activation of the cAMP-dependent PKA, suggesting a degree of clinical and biochemical overlap between these disorders.
CSHS: Dermatologic features of CSHS share some commonalities with MAS but are more striking in their diversity (4, 41)(Fig 3A, C, E). This likely reflects the involvement of Ras mutation-bearing cells from multiple embryonic lineages (22). Epidermal nevi affect approximately 60% of patients and derive from cells of ectodermal origin. Keratinocytic epidermal nevi commonly occur on the trunk and extremities, appearing as either linear or whorled plaques that may vary from skin-colored to hyperpigmented. Nevus sebaceous, another subset of epidermal nevi typically found on the head and neck, present as smooth, waxy plaques and may be associated with alopecic patches and eczematous reactions. Mutations in neural crest-derived cells give rise to hyperpigmented melanocytic nevi, affecting approximately 40% of patients. Café-au-lait macules have a similar appearance and distribution to those found in MAS. Phakomatosis pigmentokeratotica refers to the association of nevus sebaceous and speckled lentiginous nevi, and likewise occurs in a patchy, segmental distribution. Giant congenital melanocytic nevi are a dramatic feature of CSHS that may involve large portions of the body. Hair follicles may be infiltrated and enlarged by mutation-bearing melanocytes, producing a hairy appearance. Patients often present with multiple superimposed cutaneous features, leading to strikingly complex skin findings.
Figure 3.

Clinical images in Cutaneous Skeletal Hypophosphatemia Syndrome (CSHS). A. Phakomatosis pigmentokeratotica is a subtype of melanocytic nevi characterized by the combination of epidermal nevi and speckled lentiginous nevi (black arrowheads). Nevus sebaceous is a subtype of epidermal nevus presenting as smooth, waxy plaques, and may be associated with alopecic patches (white asterisks). Café-au-lait macules are frequently superimposed (black arrows). B. The patient in Panel A has a hypoplastic, lytic-appearing calvarial lesion adjacent to the area of phakomatosis pigmentokeratotica on her scalp visible on a three-dimensional computed tomography scan (black arrow). C. A child with severe rickets leading to “windswept” deformities of the lower extremities. Note the presence of epidermal nevi on the chest and abdomen (black arrows). D. Radiographs of the lower extremities from the child in Panel C again show “windswept” deformities and evidence of rachitic metaphyseal changes (white arrows). Note the heterogeneity and cortical irregularity in the dysplastic left femur (black arrow). E. Photograph of a child’s hand shows widening at the wrist consistent with rickets (arrow), and expansile deformities of the 3rd and 4th digits (asterisks). Cutaneous melanocytic nevi are apparent throughout. F. Hand radiograph from the child in Panel E shows diffuse demineralization due to longstanding severe hypophosphatemia. Areas of dysplastic bone are apparent in the metacarpals and expanded 3rd and 4th digits (asterisks). G. Magnetic resonance imaging of the brain shows melanin deposition (arrow) consistent with neurocutaneous melanosis.
Skeletal features
FD/MAS: The clinical presentation in Fibrous Dysplasia (FD) depends on the location and extent of skeletal involvement. In the craniofacial region, complications arise due to FD lesion expansion, leading to facial asymmetry and rarely functional impairment such as vision and hearing loss (42, 43)(Fig 2D). In the appendicular skeleton, affected areas tend to fracture and deform under weight-bearing forces, such as the classic “shepherd’s crook” coxa vara deformity of the proximal femur (44)(Fig 4B). Spinal FD frequently leads to scoliosis (Fig 2A), which in rare cases may be progressive and potentially lethal (45). Metabolic abnormalities such as MAS endocrinopathies and FGF23-mediated hypophosphatemia (discussed below) can contribute to skeletal complications; screening and treatment of extraskeletal features is therefore a critical component of management. Bone pain is also a common feature. Its etiology is poorly understood; however, pain appears to increase with age, and is not associated with overall skeletal disease burden (46, 47).
Figure 4.

Radiographic skeletal features in Fibrous Dysplasia/McCune-Albright Syndrome (FD/MAS) and Cutaneous Skeletal Hypophosphatemia Syndrome (CSHS). A. Computed tomography images of the skull. The left panel depicts an expansile lesion in the infra-orbital region with “ground glass” homogeneity, typical of FD (arrow). The right panel shows a hypoplastic, lytic-appearing lesion in the same region in a patient with CSHS (arrow). B. Radiographs of the proximal femora. The left panel shows diffuse involvement with a homogeneous “ground glass” FD lesion (white arrows). Note the characteristically thin cortices. There is an expansile deformity of the proximal medial portion of the femur (black arrowhead). There is a mild coxa vara deformity of the femoral shaft. The right panel shows a femur from a patient with CSHS demonstrating a similar degree of involvement (white arrows). In comparison to the FD lesion in the left panel, this radiograph shows a more heterogeneous, “streaky” appearance with mixed sclerotic and lytic elements. The lesion appears to involve the cortices, but in contrast to FD, their thickness is relatively preserved. C. Radiograph of the humeri. The left panel shows diffuse involvement with FD, again demonstrating homogeneous radiolucency and cortical thinning (arrows). The right panel shows a lesion in a patient with CSHS, demonstrating a heterogeneous “streaky” appearance involving the cortices (arrows). D. Hand radiographs. The left panel shows expansile, homogeneous FD lesions affecting multiple metacarpals and proximal phalanges (asterisks). A patient with CSHS (right panel) demonstrates heterogeneous areas of involvement (asterisks). Note the diffuse demineralization and metaphyseal irregularity consistent with longstanding hypophosphatemia (white arrow).
FD has distinctive radiographic features apparent on multiple imaging modalities (48). Nuclear medicine scans such as technetium-99 scintigraphy or 18F-NaF PET/CT scans show increased tracer uptake in affected areas with high sensitivity (49, 50)(Fig 2F). Craniofacial lesions are best evaluated with computed tomography, where they demonstrate a homogeneous “ground glass” appearance that may develop heterogeneous areas of radiolucency with age (42)(Fig 4A). Appendicular lesions also show a distinctive “ground glass” character on X-rays, along with cortical thinning that may produce an appearance of endosteal scalloping (Fig 4B–D, right panels).
The mainstay of management in FD is surgical. Orthopedic procedures are focused on repairing fractures and correcting and preventing deformities. Techniques commonly used in other skeletal disorders, such as bone grafting, are frequently ineffective in FD, and many surgeons prefer use of intramedullary rods (51, 52). Outcomes from craniofacial procedures are often suboptimal due to post-operative FD regrowth in approximately 50–85% of patients (53), and should generally be limited to correction of functional deficits and severe deformities. Somewhat surprisingly, scoliosis appears to respond well to traditional spinal fusion techniques (45).
Antiresorptive therapies have been advocated as a potential treatment strategy due to the high levels of osteoclastogenesis in FD tissue. Retrospective studies have failed to detect any effect of bisphosphonates on FD histopathology (54), lesion size, or rate of new lesion formation (55), but have suggested potential pain relief with intravenous bisphosphonates (46, 56). A randomized, placebo-controlled trial of the oral bisphosphonate alendronate detected increased FD lesion density in children (but not adults) with FD, and did not improve pain, fracture, or other clinically-relevant outcome measures (57). Concerningly, osteonecrosis of the jaw was reported in 5% of bisphosphonate-treated patients in one large retrospective series, suggesting FD may potentially carry an increased risk for this complication (58). The current evidence therefore supports that bisphosphonate use in FD should be limited to treatment of bone pain, using the lowest effective dose and interval.
Recent evidence suggests that inhibition of receptor activator of nuclear kappa-B ligand (RANKL), a protein that promotes osteoclast differentiation, may be a potentially effective treatment. FD BMSCs express high levels of RANKL, and RANKL levels are increased in FD patient serum (59). In a mouse model of FD, treatment with an anti-RANKL antibody led to increased bone density and decreased marrow fibrosis (60–63). In patients with FD/MAS, treatment with the monoclonal anti-RANKL antibody denosumab has been associated with decreased pain and lesion expansion, however denosumab discontinuation has led to rebound increased bone turnover and life-threatening hypercalcemia (60, 64). At present, denosumab treatment in patients with FD should be limited to clinical trials, and on a compassionate basis with extreme caution for patients with severe morbidity.
CSHS: In comparison to FD, the skeletal dysplasia associated with CSHS has been described in considerably less detail. All skeletal compartments have been reported to be affected, supporting a wide range of potential involvement. Like in FD, patients frequently present with fractures and deformities of weight-bearing bones (Fig 3C&E). Hemibody asymmetry is reported in up to 20% of patients with CSHS (4), which may be due to skeletal deformities and/or growth plate abnormalities in limbs affected by dysplastic bone (41)(Fig 2E&F). Scoliosis appears to be a common feature, reported in up to 40% of patients (4). In contrast to craniofacial FD which typically causes morbidity due to lesion expansion, craniofacial involvement in CSHS often leads to hypoplastic bone (Fig 3B). Reports range from mild asymmetric calvarial thinning and maxillary hypoplasia to severe osteolysis with large calvarial defects (4, 25, 65–67). Of note, in reported cases epidermal nevi often overlay the affected regions of craniofacial bone, leading to speculation that post-zygotic paracrine effects from skin lesions may contribute to the development of skull disease (41, 68). Further studies are needed to investigate this potential association. In the extracranial skeleton there does not appear to be a geographic relationship between affected skin and the location of dysplastic bone lesions, indicating that lesions in these areas likely arise from primary dysplastic processes (4). The radiographic appearance of bone lesions has not been fully characterized, and lesions are often confused with FD on X-rays, contributing to frequent mis-diagnosis. In comparison to FD, CSHS skeletal lesions are more heterogeneous, with poorly defined cortical-medullary junctions and mixed lytic/sclerotic elements (Fig4B–D, right panels). The bony cortex is frequently involved, and may display some degree of endosteal scalloping, but is typically less thin in comparison to FD.
Clinical management in CSHS is comparatively less well-defined than FD, but similarly consists primarily of surgical repair and prevention of fractures and deformities. There are no medical therapies known to alter the disease course, and no reports of use of anti-resorptive medications. Unlike FD, osteoclastogenesis is not highly prominent in dysplastic tissue in CSHS, therefore anti-resorptives are a less intuitive potential treatment strategy.
FGF23-mediated Hypophosphatemia
Overproduction of FGF23 is a distinctive feature of both FD/MAS and CSHS, and an important contributor to skeletal morbidity. In both disorders, rickets results in impaired growth, metaphyseal abnormalities, and pseudofractures in both dysplastic and unaffected areas of the skeleton. However, dysplastic bone may be particularly vulnerable to the effects of hypophosphatemia. In patients with FD, hypophosphatemia is a demonstrated risk factor for long bone fractures (69), progressive scoliosis (45), and cranial base deformities (70). The association between hypophosphatemia and skeletal complications in CSHS has not been evaluated systematically, however there are case reports of improved ambulation and decreased pain after starting treatment with phosphorus and vitamin D analogs (4).
Clinical investigation has confirmed in FD and strongly suggests in CSHS that dysplastic bone is the source of FGF23 overproduction (22, 26). This is unsurprising, given that bone is the tissue source of FGF23 under normal physiologic conditions (71). This has important therapeutic implications in CSHS, because patients have frequently undergone painful and disfiguring skin resections under the mistaken assumption that dysplastic skin lesions produce FGF23 (4). Because dysplastic bone lesions appear to produce FGF23, the severity of phosphate wasting parallels the degree of skeletal involvement, leading to more frequent and severe hypophosphatemia in patients with higher skeletal disease burdens. Interestingly, data suggests that FGF23 production may decrease over time in both disorders. An age-related decrease in serum FGF23 and bone turnover markers has been well-established in FD/MAS (55); improved hypophosphatemia in adults has also been reported in CSHS, although some patients persist (4). This may reflect age-related apoptosis or senescence of mutation-bearing skeletal cells (72).
Treatment of hypophosphatemia in both FD/MAS and CSHS consists of oral phosphorus and vitamin D analogs (73). The recent approval of the anti-FGF23 antibody burosumab for treatment of X-linked hypophosphatemia raises the possibility that this medication may also be helpful for other forms of FGF23 excess, including FD/MAS and CSHS. Burosumab offers several advantages over conventional therapy, including greater ease of administration, greater gastrointestinal tolerability, and decreased hypercalciuria (74, 75). Unmineralized osteoid is a prominent feature of dysplastic lesions in both FD and CSHS, even in patients who are normophosphatemic on conventional therapy (Fig 1), and it is unknown whether burosumab treatment can alter this tissue-level osteomalacia. Another important unanswered question is whether systemic burosumab has potential effects on mineralization and cell proliferation in dysplastic lesions. Investigation into these tissue-specific effects is needed before burosumab can be used routinely in patients with FD/MAS and CSHS.
Other Endocrinopathies
FD/MAS: Hyperfunctioning endocrinopathies are a key feature of FD/MAS. Gs-mediated G-coupled protein receptors play a central role in multiple hormone signaling cascades, making this a frequent manifestation of Gαs activation.
The most common endocrinopathy is gonadotropin-independent sex steroid production, which affects ~85% of girls and women with FD/MAS (38). Recurrent estrogen-producing ovarian cysts leads to a relapsing-remitting course, presenting with acute onset breast development and linear growth acceleration (Fig 2E). Cyst resolution leads to withdrawal bleeding, which is often the event that prompts patients to seek care. Management is aimed at preventing premature skeletal maturation, preserving adult height, and preventing psychosocial distress from early development. The aromatase inhibitor letrozole is considered first-line treatment (76). Patients with significant bone age advancement may develop secondary central puberty as a complication of peripheral puberty and may benefit from adjuvant GnRH agonist therapy (8, 76). Recurrent ovarian cysts persist into adulthood and are frequently associated with dysfunctional uterine bleeding (77). While pregnancy is typically possible and safe in women with FD/MAS, fertility may be impaired (77).
Testicular lesions, comprised of Leydig and Sertoli cell hyperplasia, are found in ~85% of boys and men with FD/MAS (78). Lesions may present with diffuse macro-orchidism, and on ultrasonography appear as discrete hypo- and hyperechoic lesions with or without microlithiasis. Precocious puberty develops in only 10–15% of affected boys, and can typically be managed with a combination of testosterone receptor blockers and aromatase inhibitors (78).
Thyroid disease occurs in ~2/3 of patients, manifesting with thyromegaly and ultrasonographic findings such as discrete nodules and diffuse heterogeneity (38, 79). About half of affected patients have frank hyperthyroidism, which typically responds well to anti-thyroidal medications. Surgery is often preferred for long-term management. Clinicians may also consider radioablation, however due to mosaic involvement within the gland, there is a hypothetical risk of exposing non-affected tissue to a sub-therapeutic dose of radioiodine (12).
Growth hormone excess affects approximately 15% of patients with FD/MAS (38), and has been identified as a risk factor for craniofacial morbidity, including vision loss (80), hearing loss (43), and craniofacial FD expansion (53). Patients typically respond well to somatostatin analogs and growth hormone receptor blockers, used alone or in combination (81).
Neonatal hypercortisolism is a rare and potentially serious complication of FD/MAS, affecting <5% of patients (38, 82). Disease presents exclusively in the first year of life and can lead to severe illness and death if not recognized early. Most patients are treated with adrenalectomy, however spontaneous resolution of hypercortisolism has been reported in up to 1/3 of patients, therefore it may be appropriate to observe patients who are stable on medical therapy (82).
CSHS: Unlike Gαs, Ras does not play a central role in multiple hormone signaling pathways. Other than the notable exception of FGF23 excess, endocrinopathies do not appear to be a common feature of CSHS. Central precocious puberty has been reported in several patients (83, 84), however most had neurologic abnormalities, calling into question whether puberty was a primary manifestation of CSHS or a consequence of underlying brain anomalies. Pheochromocytoma has been reported in one adult patient (85), and there are case reports of benign thyroid nodules and medullary thyroid carcinoma in pediatric patients (4, 66).
Neuropsychiatric features
FD/MAS: Gαs and cAMP have long been recognized as potential contributors to synaptic plasticity and neuropsychiatric disease (86, 87). Neuropsychiatric abnormalities, including psychotic, speech, and language disorders, have been reported in patients with FD/MAS, but have not yet been systematically described (82, 88). Interestingly, transgenic mice expressing the activating Q227L mutation (reported in <5% of patients with FD/MAS) demonstrate neurobehavioral abnormalities (89). Defining the neuropsychiatric phenotype in FD/MAS is therefore a high priority for research and may inform understanding of more common disorders, including major depression and schizophrenia. However, like other features of FD/MAS, neuropsychiatric involvement is likely to be highly variable depending on the relative amount and distribution of mutation-bearing tissue in the central nervous system. At present, children with FD/MAS should be monitored for developmental abnormalities, with a low threshold for further evaluation and intervention if issues arise.
CSHS: The Ras pathway plays numerous roles in neurodevelopment and neuronal signaling (90); it is therefore not surprising that neurological abnormalities are frequently encountered in patients with CSHS and other RASopathies (91, 92). A large metanalysis reported neurologic disorders in 45% of patients with CSHS, the most frequent being neurodevelopmental delays and intellectual disability, followed by ventriculomegaly, and seizures (4). Other manifestations included macro- and microcephaly, spastic hemiplegia, hyperreflexia, and neurocutaneous melanosis (4). Subjects with giant congenital melanocytic nevus, particularly those with posterior and satellite nevi, are prone to develop neurocutaneous melanosis, characterized by melanocytic proliferation in the brain (93)(Fig 3G). The most likely etiology of these neurologic abnormalities is presence of mutation-bearing cells in the brain. Others have speculated that post-zygotic paracrine effects from skin lesions may also contribute, noting that nevi on the face and scalp appears to increase the likelihood of central nervous system involvement in patients with epidermal nevus syndromes (94). However further research is needed to investigate this potential association.
All patients with CSHS should be considered at risk for neurologic sequelae, and young children should be referred for early interventional services and neurodevelopmental evaluation. Brain imaging to evaluate for structural abnormalities should be strongly considered, especially in patients with giant melanocytic nevi.
Malignancies
FD/MAS: G-protein dysregulation commonly contributes to tumorigenesis, and studies have reported that up to 4.2% of all tumors in the general population harbor gain-of-function GNAS mutations (95). The Gαs R201 mutations associated with FD/MAS are commonly found in sporadic, hyperfunctioning pituitary tumors (28%), thyroid adenomas (5%), and pancreatic intraductal pancreatic mucinous neoplasms (IPMNs)(96–98). Pancreatic IPMNs are found in up to 50% in patients with FD/MAS, however malignant transformation to pancreatic adenocarcinoma appears to be a rare development, with only 1 reported case (99).
Women with FD/MAS were reported to have a 3.5–4-fold increased risk of breast cancer compared to the general population in one large series (100). Disease presented at a relatively young age (36–46 years), however outcomes were highly favorable, with 100% survival and no recurrence or metastases. Women with FD/MAS are therefore recommended to initiate breast cancer screening at an earlier age compared to the general population, i.e. age 50 years based on current guidelines (100).
The potential for transformation of FD into bone cancers has been well-established, including osteosarcoma, chondrosarcoma, and others (101–104). Patients typically present with acute expansion and/or focal pain, and imaging may demonstrate extension of the lesion through the bony cortex. Due to the absence of rigorous epidemiologic studies, the prevalence of malignant transformation in FD has not been established. In the NIH cohort of approximately 200 patients, 2 cases of osteosarcoma have been identified, suggesting a prevalence around 1% (personal observation, AMB). Risk factors for malignant transformation include therapeutic external beam radiation, a harmful practice in FD which has been largely abandoned (102, 105–107). Additional rarely reported tumors include thyroid cancer (108, 109), testicular cancer (78), ovarian tumors (110, 111), and hepatoblastoma (112).
CSHS: Activating RAS mutations, particularly those altering RAS’s GTPase domain, are well known tumor drivers (113). A variety of benign extracutaneous neoplasms have been reported in patients with CSHS, including thymoma, giant cell granuloma, bladder leiomyoma, ocular dermoids, benign vascular tumors, and others (4). Malignant transformation appears to be relatively uncommon, however cases of congenital rhabdomyosarcoma (84), medullary thyroid cancer (66), and a malignant brainstem tumor (114) have been reported. Although the vast majority of tumors identified in patients with CSHS have not been sequenced, given their early onset and known association with sporadic RAS mutations (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), they are likely directly disease-related.
The various skin lesions in patients with CSHS also harbor potential for malignant transformation. Giant cutaneous melanocytic nevi likely have the highest risk for transformation to malignant melanoma, reported in <1–5% of cases (115). Nevus sebacous commonly transform into secondary benign tumors such as trichoblastoma or syringocystoadenonma papilliferum, however the risk for malignant transformation (most frequently into basal cell carcinoma) is very low (<1%) (116). The risk of malignant transformation of keratinocytic epidermal nevi is extremely rare (117). It is not currently recommended to remove epidermal nevi for cancer prevention. However, skin lesions in patients with CSHS should be monitored regularly by a dermatologist, and patients should be educated about concerning signs and symptoms.
Interestingly, there are no reports of osteosarcoma or other skeletal malignancies in CSHS. This observation is consistent with the low prevalence of RAS mutations in sporadic bone tumors (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). However, the rarity of CSHS and lack of rigorous epidemiologic studies makes the skeletal malignancy risk difficult to ascertain, and this should be considered in the differential if patients present with concerning features such as rapid lesion expansion.
Other features
A wide range of tissue may harbor GNAS and RAS mutations, however numerous factors affect the likelihood of manifesting clinical disease. Depending on the physiologic role of the Gαs or Ras pathways, some tissues may display subtle or no detectable phenotype. In contrast, mutations may have strong deleterious effects in other cell types, selecting against cell survival and prohibiting disease in certain tissues. More detailed phenotyping studies are likely to uncover the presence (or absence) of additional manifestations of FD/MAS and CSHS, which will inform understanding of Gαs and Ras physiology.
FD/MAS: Although disruption of bone marrow architecture and hematopoiesis are key histologic features of FD (Fig 1A), somewhat surprisingly, patients do not typically develop clinically apparent hematologic disease. However, rare reports of bone marrow failure and extramedullary hematopoiesis associated with extensive skeletal disease have emerged, suggesting that FD may impact overall marrow function to some extent (118, 119). Platelet dysfunction has also been reported in patients with FD/MAS, although its clinical significance is unclear (120). Tachycardia and hypertension have been reported in patients with extensive bone disease; it is unknown whether this represents a primary effect of Gαs activation in cardiovascular tissue, or a secondary physiologic response to increased cardiac demand (38, 121).
CSHS: Ophthalmologic disorders have been reported in approximately ¼ of patients with CSHS, most commonly colobomas, limbal dermoids, strabismus and corneal opacities (4). Interestingly, like the brain and skin, the eyes are ectodermal in origin, suggesting that tissues derived from this germ layer feature prominently in the clinical presentation of CSHS.
Marrow function in CSHS has not been systematically evaluated. Hematopoiesis appears to be preserved in dysplastic skeletal tissue (Fig 1), however there are 2 known cases of chronic anemia and thrombocytopenia (122)(personal observation, DO & AMB), suggesting potential deleterious effects.
FUTURE DIRECTIONS
Critical knowledge gaps in both FD/MAS and CSHS have limited the development of novel diagnostics and treatments. Given the rarity of these diseases, collaboration among international investigators and patient advocacy organizations is a high priority to improve health outcomes. The FD/MAS International Consortium has made important contributions in promoting such collaborations, supporting patient registries, and developing consensus guidelines for evaluation and management (12, 123). The development of similar collaborative organizations for CSHS is needed.
CONCLUSION
FD/MAS and CSHS represent 2 strikingly complex disorders with distinct and overlapping phenotypes. The unique presentation with skeletal mosaicism and FGF23 overproduction in these genetically divergent diseases has led to increased understanding into the complexity of FGF23 physiology. Further investigation in these disorders is likely to lead to additional novel biological insights, and ultimately to improved diagnostics and treatments for patients.
Funding:
This work was supported by the Intramural Research Program of the NIDCR, NIH.
Footnotes
Declaration of interest:
AMB is the Principal Investigator of an investigator-sponsored study of denosumab treatment for fibrous dysplasia supported by Amgen, Inc through NIDCR. AMB is an investigator in an investigator-sponsored of burosumab treatment for CSHS supported by Ultragenyx through Children’s National Health System. DOC has participated as a member of an advisory board for Kyowa Kirin.
REFERENCES
- 1.Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355(6331):1330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lumbroso S, Paris F, Sultan C, European Collaborative S. Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome--a European Collaborative Study. J Clin Endocrinol Metab. 2004;89(5):2107–13. [DOI] [PubMed] [Google Scholar]
- 3.Idowu BD, Al-Adnani M, O’Donnell P, Yu L, Odell E, Diss T, Gale RE, Flanagan AM. A sensitive mutation-specific screening technique for GNAS1 mutations in cases of fibrous dysplasia: the first report of a codon 227 mutation in bone. Histopathology. 2007;50(6):691–704. [DOI] [PubMed] [Google Scholar]
- 4.Ovejero D, Lim YH, Boyce AM, Gafni RI, McCarthy E, Nguyen TA, Eichenfield LF, DeKlotz CM, Guthrie LC, Tosi LL, et al. Cutaneous skeletal hypophosphatemia syndrome: clinical spectrum, natural history, and treatment. Osteoporos Int. 2016;27(12):3615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park PG, Park E, Hyun HS, Kang HG, Ha IS, Cho TJ, Ko JM, Cheong HI. Cutaneous Skeletal Hypophosphatemia Syndrome in Association with a Mosaic HRAS Mutation. Ann Clin Lab Sci. 2018;48(5):665–9. [PubMed] [Google Scholar]
- 6.Hafner C, Groesser L. Mosaic RASopathies. Cell Cycle. 2013;12(1):43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134(10):2493–6. [DOI] [PubMed] [Google Scholar]
- 8.Boyce AM, Florenzano P, de Castro LF, Collins MT. Fibrous Dysplasia/McCune-Albright Syndrome In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. , editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993. [Google Scholar]
- 9.Robin NH, Biesecker LG. Considerations for a multiaxis nomenclature system for medical genetics. Genet Med. 2001;3(4):290–3. [DOI] [PubMed] [Google Scholar]
- 10.Lichtenstein L Polyostotic fibrous dysplasia. Arch Surg. 1938;36:874–98. [Google Scholar]
- 11.McCune D Osteitis fibrosa cystica; the case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child. 1936;52 743–4. [Google Scholar]
- 12.Javaid MK, Boyce A, Appelman-Dijkstra N, Ong J, Defabianis P, Offiah A, Arunde P, Shaw N, Pos VD, Underhil A, et al. Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: a consensus statement from the FD/MAS international consortium. Orphanet J Rare Dis. 2019;14(1):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim YH, Ovejero D, Sugarman JS, Deklotz CM, Maruri A, Eichenfield LF, Kelley PK, Juppner H, Gottschalk M, Tifft CJ, et al. Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Hum Mol Genet. 2014;23(2):397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Happle R The group of epidermal nevus syndromes Part I. Well defined phenotypes. J Am Acad Dermatol. 2010;63(1):1–22; quiz 3–4. [DOI] [PubMed] [Google Scholar]
- 15.Gathwala G, Dalal P, Dalal JS, Dayal S, Singh G. Giant Congenital Melanocytic Nevi: A Rare Association with Hypophosphatemic Rickets. The Indian Journal of Pediatrics. 2013;80(5):430–1. [DOI] [PubMed] [Google Scholar]
- 16.Feuerstein RC, Mims LC. Linear nevus sebaceus with convulsions and mental retardation. Am J Dis Child. 1962;104:675–9. [DOI] [PubMed] [Google Scholar]
- 17.Schimmelpenning GW. [Clinical contribution to symptomatology of phacomatosis]. Fortschr Geb Rontgenstr Nuklearmed. 1957;87(6):716–20. [PubMed] [Google Scholar]
- 18.Becker SW. Concurrent melanosis and hypertrichosis in distribution of nevus unius lateris. Arch Derm Syphilol. 1949;60(2):155–60. [DOI] [PubMed] [Google Scholar]
- 19.Aschinberg LC, Solomon LM, Zeis PM, Justice P, Rosenthal IM. Vitamin D-resistant rickets associated with epidermal nevus syndrome: demonstration of a phosphaturic substance in the dermal lesions. J Pediatr. 1977;91(1):56–60. [DOI] [PubMed] [Google Scholar]
- 20.Sugarman GI, Reed WB. Two unusual neurocutaneous disorders with facial cutaneous signs. Arch Neurol. 1969;21(3):242–7. [DOI] [PubMed] [Google Scholar]
- 21.Gathwala G, Dalal P, Dalal JS, Dayal S, Singh G. Giant congenital melanocytic nevi: a rare association with hypophosphatemic rickets. Indian J Pediatr. 2013;80(5):430–1. [DOI] [PubMed] [Google Scholar]
- 22.Lim YH, Ovejero D, Derrick KM, Collins MT, Choate KA. Cutaneous skeletal hypophosphatemia syndrome (CSHS) is a multilineage somatic mosaic RASopathy. J Am Acad Dermatol. 2016;75(2):420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riminucci M, Liu B, Corsi A, Shenker A, Spiegel AM, Robey PG, Bianco P. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol. 1999;187(2):249–58. [DOI] [PubMed] [Google Scholar]
- 24.Riddle ND, Bui MM. Fibrous Dysplasia. Archives of Pathology & Laboratory Medicine. 2013;137(1):134–8. [DOI] [PubMed] [Google Scholar]
- 25.Heike CL, Cunningham ML, Steiner RD, Wenkert D, Hornung RL, Gruss JS, Gannon FH, McAlister WH, Mumm S, Whyte MP. Skeletal changes in epidermal nevus syndrome: does focal bone disease harbor clues concerning pathogenesis? Am J Med Genet A. 2005;139a(2):67–77. [DOI] [PubMed] [Google Scholar]
- 26.Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112(5):683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhattacharyya N, Wiench M, Dumitrescu C, Connolly BM, Bugge TH, Patel HV, Gafni RI, Cherman N, Cho M, Hager GL, et al. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res. 2012;27(5):1132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knab VM, Corbin B, Andrukhova O, Hum JM, Ni P, Rabadi S, Maeda A, White KE, Erben RG, Juppner H, et al. Acute Parathyroid Hormone Injection Increases C-Terminal but Not Intact Fibroblast Growth Factor 23 Levels. Endocrinology. 2017;158(5):1130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010;299(4):F882–9. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17(5):1305–15. [DOI] [PubMed] [Google Scholar]
- 31.Singh RJ, Kumar R. Fibroblast growth factor 23 concentrations in humoral hypercalcemia of malignancy and hyperparathyroidism. Mayo Clin Proc. 2003;78(7):826–9. [DOI] [PubMed] [Google Scholar]
- 32.Tebben PJ, Singh RJ, Clarke BL, Kumar R. Fibroblast growth factor 23, parathyroid hormone, and 1alpha,25-dihydroxyvitamin D in surgically treated primary hyperparathyroidism. Mayo Clin Proc. 2004;79(12):1508–13. [DOI] [PubMed] [Google Scholar]
- 33.White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005;76(2):361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao L, Du E, Homer-Bouthiette C, Hurley MM. Inhibition of FGFR Signaling Partially Rescues Hypophosphatemic Rickets in HMWFGF2 Tg Male Mice. Endocrinology. 2017;158(10):3629–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao L, Naganawa T, Lorenzo J, Carpenter TO, Coffin JD, Hurley MM. Nuclear isoforms of fibroblast growth factor 2 are novel inducers of hypophosphatemia via modulation of FGF23 and KLOTHO. J Biol Chem. 2010;285(4):2834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS One. 2014;9(8):e104154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JC, Jeng YM, Su SY, Wu CT, Tsai KS, Lee CH, Lin CY, Carter JM, Huang JW, Chen SH, et al. Identification of a novel FN1-FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour. J Pathol. 2015;235(4):539–45. [DOI] [PubMed] [Google Scholar]
- 38.Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis. 2012;7 Suppl 1:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis. 2008;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pichard DC, Boyce AM, Collins MT, Cowen EW. Oral pigmentation in McCune-Albright syndrome. JAMA Dermatol. 2014;150(7):760–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asch S, Sugarman JL. Epidermal nevus syndromes: New insights into whorls and swirls. Pediatr Dermatol. 2018;35(1):21–9. [DOI] [PubMed] [Google Scholar]
- 42.Burke AB, Collins MT, Boyce AM. Fibrous dysplasia of bone: craniofacial and dental implications. Oral Dis. 2017;23(6):697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyce AM, Brewer C, DeKlotz TR, Zalewski CK, King KA, Collins MT, Kim HJ. Association of Hearing Loss and Otologic Outcomes With Fibrous Dysplasia. JAMA Otolaryngol Head Neck Surg. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ippolito E, Farsetti P, Boyce AM, Corsi A, De Maio F, Collins MT. Radiographic classification of coronal plane femoral deformities in polyostotic fibrous dysplasia. Clin Orthop Relat Res. 2014;472(5):1558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berglund JA, Tella SH, Tuthill KF, Kim L, Guthrie LC, Paul SM, Stanton R, Collins MT, Boyce AM. Scoliosis in Fibrous Dysplasia/McCune-Albright Syndrome: Factors Associated with Curve Progression and Effects of Bisphosphonates. J Bone Miner Res. 2018. [DOI] [PubMed] [Google Scholar]
- 46.Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int. 2008;19(1):57–63. [DOI] [PubMed] [Google Scholar]
- 47.Majoor BCJ, Traunmueller E, Maurer-Ertl W, Appelman-Dijkstra NM, Fink A, Liegl B, Hamdy NAT, Sander Dijkstra PD, Leithner A. Pain in fibrous dysplasia: relationship with anatomical and clinical features. Acta Orthop. 2019;90(4):401–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kushchayeva YS, Kushchayev SV, Glushko TY, Tella SH, Teytelboym OM, Collins MT, Boyce AM. Fibrous dysplasia for radiologists: beyond ground glass bone matrix. Insights Imaging. 2018;9(6):1035–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Collins MT, Kushner H, Reynolds JC, Chebli C, Kelly MH, Gupta A, Brillante B, Leet AI, Riminucci M, Robey PG, et al. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res. 2005;20(2):219–26. [DOI] [PubMed] [Google Scholar]
- 50.Papadakis G, Manikis G, Karantanas A, Florenzano P, Bagci U, Marias K, Collins M, Boyce A. (18) F-N aF PET/CT IMAGING IN FIBROUS DYSPLASIA OF BONE. J Bone Miner Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leet AI, Boyce AM, Ibrahim KA, Wientroub S, Kushner H, Collins MT. Bone-Grafting in Polyostotic Fibrous Dysplasia. J Bone Joint Surg Am. 2016;98(3):211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanton RP IE, Springfield D, Lindaman L, Wientroub S, Leet A. The surgical management of fibrous dysplasia of bone. Orphanet J Rare Dis. 2012;24 (7):ppl 1:S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyce AM, Burke A, Cutler Peck C, DuFresne CR, Lee JS, Collins MT. Surgical Management of Polyostotic Craniofacial Fibrous Dysplasia: Long-Term Outcomes and Predictors for Postoperative Regrowth. Plast Reconstr Surg. 2016;137(6):1833–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab. 2003;88(10):4569–75. [DOI] [PubMed] [Google Scholar]
- 55.Florenzano P, Pan KS, Brown SM, Paul SM, Kushner H, Guthrie LC, de Castro LF, Collins MT, Boyce AM. Age-Related Changes and Effects of Bisphosphonates on Bone Turnover and Disease Progression in Fibrous Dysplasia of Bone. J Bone Miner Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chapurlat RD, Gensburger D, Jimenez-Andrade JM, Ghilardi JR, Kelly M, Mantyh P. Pathophysiology and medical treatment of pain in fibrous dysplasia of bone. Orphanet J Rare Dis. 2012;7 Suppl 1:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boyce AM, Kelly MH, Brillante BA, Kushner H, Wientroub S, Riminucci M, Bianco P, Robey PG, Collins MT. A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab. 2014;99(11):4133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Metwally T, Burke A, Tsai JY, Collins MT, Boyce AM. Fibrous Dysplasia and Medication-Related Osteonecrosis of the Jaw. J Oral Maxillofac Surg. 2016;74(10):1983–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Castro LF, Burke AB, Wang HD, Tsai J, Florenzano P, Pan KS, Bhattacharyya N, Boyce AM, Gafni RI, Molinolo AA, et al. Activation of RANK/RANKL/OPG Pathway Is Involved in the Pathophysiology of Fibrous Dysplasia and Associated With Disease Burden. J Bone Miner Res. 2019;34(2):290–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boyce AM, Chong WH, Yao J, Gafni RI, Kelly MH, Chamberlain CE, Bassim C, Cherman N, Ellsworth M, Kasa-Vubu JZ, et al. Denosumab treatment for fibrous dysplasia. J Bone Miner Res. 2012;27(7):1462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ganda K, Seibel MJ. Rapid biochemical response to denosumab in fibrous dysplasia of bone: report of two cases. Osteoporos Int. 2014;25(2):777–82. [DOI] [PubMed] [Google Scholar]
- 62.Majoor BCJ, Papapoulos SE, Dijkstra PDS, Fiocco M, Hamdy NAT, Appelman-Dijkstra NM. Denosumab in Patients With Fibrous Dysplasia Previously Treated With Bisphosphonates. J Clin Endocrinol Metab. 2019;104(12):6069–78. [DOI] [PubMed] [Google Scholar]
- 63.Palmisano B, Spica E, Remoli C, Labella R, Di Filippo A, Donsante S, Bini F, Raimondo D, Marinozzi F, Boyde A, et al. RANKL Inhibition in Fibrous Dysplasia of Bone: A Preclinical Study in a Mouse Model of the Human Disease. J Bone Miner Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benhamou J, Gensburger D, Chapurlat R. Transient improvement of severe pain from fibrous dysplasia of bone with denosumab treatment. Joint Bone Spine. 2014;81(6):549–50. [DOI] [PubMed] [Google Scholar]
- 65.Camacho Martinez F, Moreno Gimenez JC. [Epidermal nevus syndrome (of Solomon, Fretzin and Dewald)]. Ann Dermatol Venereol. 1985;112(2):143–7. [PubMed] [Google Scholar]
- 66.Rustin MH, Bunker CB, Gilkes JJ, Robinson TW, Dowd PM. Polyostotic fibrous dysplasia associated with extensive linear epidermal naevi. Clin Exp Dermatol. 1989;14(5):371–5. [DOI] [PubMed] [Google Scholar]
- 67.Fritzsch C, Konig R, Jacobi G. [Schimmelpenning-Feuerstein-Mims syndrome and its neurologic manifestations. 6 personal cases and review of the literature]. Klin Padiatr. 1995;207(5):288–97. [DOI] [PubMed] [Google Scholar]
- 68.Ho N, Roig C, Diadori P. Epidermal nevi and localized cranial defects. Am J Med Genet. 1999;83(3):187–90. [PubMed] [Google Scholar]
- 69.Leet AI, Chebli C, Kushner H, Chen CC, Kelly MH, Brillante BA, Robey PG, Bianco P, Wientroub S, Collins MT. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res. 2004;19(4):571–7. [DOI] [PubMed] [Google Scholar]
- 70.Pan KS, Heiss JD, Brown SM, Collins MT, Boyce AM. Chiari I Malformation and Basilar Invagination in Fibrous Dysplasia: Prevalence, Mechanisms, and Clinical Implications. J Bone Miner Res. 2018;33(11):1990–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Edmonston D, Wolf M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat Rev Nephrol. 2019. [DOI] [PubMed] [Google Scholar]
- 72.Kuznetsov SA, Cherman N, Riminucci M, Collins MT, Robey PG, Bianco P. Age-dependent demise of GNAS-mutated skeletal stem cells and “normalization” of fibrous dysplasia of bone. J Bone Miner Res. 2008;23(11):1731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26(7):1381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, Padidela R, Van’t Hoff W, Mao M, Chen CY, et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N Engl J Med. 2018;378(21):1987–98. [DOI] [PubMed] [Google Scholar]
- 75.Insogna KL, Briot K, Imel EA, Kamenicky P, Ruppe MD, Portale AA, Weber T, Pitukcheewanont P, Cheong HI, Jan de Beur S, et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis. J Bone Miner Res. 2018;33(8):1383–93. [DOI] [PubMed] [Google Scholar]
- 76.Estrada A, Boyce AM, Brillante BA, Guthrie LC, Gafni RI, Collins MT. Long-term outcomes of letrozole treatment for precocious puberty in girls with McCune-Albright syndrome. Eur J Endocrinol. 2016;175(5):477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boyce AM, Casey RK, Ovejero Crespo D, Murdock CM, Estrada A, Guthrie LC, Brillante BA, Gomez-Lobo V, Nieman LK, Collins MT. Gynecologic and reproductive outcomes in fibrous dysplasia/McCune-Albright syndrome. Orphanet J Rare Dis. 2019;14(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boyce AM, Chong WH, Shawker TH, Pinto PA, Linehan WM, Bhattacharryya N, Merino MJ, Singer FR, Collins MT. Characterization and management of testicular pathology in McCune-Albright syndrome. J Clin Endocrinol Metab. 2012;97(9):E1782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tessaris D, Corrias A, Matarazzo P, De Sanctis L, Wasniewska M, Messina MF, Vigone MC, Lala R. Thyroid abnormalities in children and adolescents with McCune-Albright syndrome. Horm Res Paediatr. 2012;78(3):151–7. [DOI] [PubMed] [Google Scholar]
- 80.Akintoye SO, Chebli C, Booher S, Feuillan P, Kushner H, Leroith D, Cherman N, Bianco P, Wientroub S, Robey PG, et al. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J Clin Endocrinol Metab. 2002;87(11):5104–12. [DOI] [PubMed] [Google Scholar]
- 81.Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab. 2014:jc20133826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brown RJ, Kelly MH, Collins MT. Cushing syndrome in the McCune-Albright syndrome. J Clin Endocrinol Metab. 2010;95(4):1508–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ivker R, Resnick SD, Skidmore RA. Hypophosphatemic vitamin D-resistant rickets, precocious puberty, and the epidermal nevus syndrome. Arch Dermatol. 1997;133(12):1557–61. [PubMed] [Google Scholar]
- 84.Shahgholi E, Mollaian M, Haghshenas Z, Honarmand M. Congenital rhabdomyosarcoma, central precocious puberty, hemihypertrophy and hypophosphatemic rickets associated with epidermal nevus syndrome. J Pediatr Endocrinol Metab. 2011;24(11–12):1063–6. [DOI] [PubMed] [Google Scholar]
- 85.Bouthors J, Vantyghem MC, Manouvrier-Hanu S, Soudan B, Proust E, Happle R, Piette F. Phacomatosis pigmentokeratotica associated with hypophosphataemic rickets, pheochromocytoma and multiple basal cell carcinomas. Br J Dermatol. 2006;155(1):225–6. [DOI] [PubMed] [Google Scholar]
- 86.Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fujita M, Richards EM, Niciu MJ, Ionescu DF, Zoghbi SS, Hong J, Telu S, Hines CS, Pike VW, Zarate CA, et al. cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Mol Psychiatry. 2017;22(5):754–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Padma SSP. Rare association of schizophrenia and unilateral Graves’ disease with contralateral thyroid hemiagenesis in two cases of McCune-Albright syndrome. Iranian J Nucl Med 2015;23(2):124–7. [Google Scholar]
- 89.Kelly MP, Stein JM, Vecsey CG, Favilla C, Yang X, Bizily SF, Esposito MF, Wand G, Kanes SJ, Abel T. Developmental etiology for neuroanatomical and cognitive deficits in mice overexpressing Galphas, a G-protein subunit genetically linked to schizophrenia. Mol Psychiatry. 2009;14(4):398–415, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhong J RAS and downstream RAF-MEK and PI3K-AKT signaling in neuronal development, function and dysfunction. Biol Chem. 2016;397(3):215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim YE, Baek ST. Neurodevelopmental Aspects of RASopathies. Mol Cells. 2019;42(6):441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Asch S, Sugarman JL. Epidermal nevus syndromes. Handb Clin Neurol. 2015;132:291–316. [DOI] [PubMed] [Google Scholar]
- 93.Jakchairoongruang K, Khakoo Y, Beckwith M, Barkovich AJ. New insights into neurocutaneous melanosis. Pediatric radiology. 2018;48(12):1786–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grebe TA, Rimsza ME, Richter SF, Hansen RC, Hoyme HE. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. American journal of medical genetics. 1993;47(1):24–30. [DOI] [PubMed] [Google Scholar]
- 95.O’Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer. 2013;13(6):412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340(6236):692–6. [DOI] [PubMed] [Google Scholar]
- 97.Omori Y, Ono Y, Tanino M, Karasaki H, Yamaguchi H, Furukawa T, Enomoto K, Ueda J, Sumi A, Katayama J, et al. Pathways of Progression From Intraductal Papillary Mucinous Neoplasm to Pancreatic Ductal Adenocarcinoma Based on Molecular Features. Gastroenterology. 2018. [DOI] [PubMed] [Google Scholar]
- 98.Hollstein PE, Shaw RJ. GNAS shifts metabolism in pancreatic cancer. Nat Cell Biol. 2018;20(7):740–1. [DOI] [PubMed] [Google Scholar]
- 99.Parvanescu A, Cros J, Ronot M, Hentic O, Grybek V, Couvelard A, Levy P, Chanson P, Ruszniewski P, Sauvanet A, et al. Lessons from McCune-Albright syndrome-associated intraductal papillary mucinous neoplasms: : GNAS-activating mutations in pancreatic carcinogenesis. JAMA Surg. 2014;149(8):858–62. [DOI] [PubMed] [Google Scholar]
- 100.Majoor BC, Boyce AM, Bovee JV, Smit VT, Collins MT, Cleton-Jansen AM, Dekkers OM, Hamdy NA, Dijkstra PS, Appelman-Dijkstra NM. Increased Risk of Breast Cancer at a Young Age in Women with Fibrous Dysplasia. J Bone Miner Res. 2018;33(1):84–90. [DOI] [PubMed] [Google Scholar]
- 101.Beuerlein ME, Schuller DE, DeYoung BR. Maxillary malignant mesenchymoma and massive fibrous dysplasia. Arch Otolaryngol Head Neck Surg. 1997;123(1):106–9. [DOI] [PubMed] [Google Scholar]
- 102.Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer. 1994;73(5):1411–24. [DOI] [PubMed] [Google Scholar]
- 103.Yalniz E, Er T, Ozyilmaz F. Fibrous dysplasia of the spine with sarcomatous transformation: a case report and review of the literature. Eur Spine J. 1995;4(6):372–4. [DOI] [PubMed] [Google Scholar]
- 104.Kaushik S, Smoker WR, Frable WJ. Malignant transformation of fibrous dysplasia into chondroblastic osteosarcoma. Skeletal Radiol. 2002;31(2):103–6. [DOI] [PubMed] [Google Scholar]
- 105.Liu F, Li W, Yao Y, Li G, Yang Y, Dou W, Zhong D, Wang L, Zhu X, Hu H, et al. A case of McCune-Albright syndrome associated with pituitary GH adenoma: therapeutic process and autopsy. J Pediatr Endocrinol Metab. 2011;24(5–6):283–7. [DOI] [PubMed] [Google Scholar]
- 106.Hall MB, Sclar AG, Gardner DF. Albright’s syndrome with reactivation of fibrous dysplasia secondary to pituitary adenoma and further complicated by osteogenic sarcoma. Report of a case. Oral Surg Oral Med Oral Pathol. 1984;57(6):616–9. [DOI] [PubMed] [Google Scholar]
- 107.Hansen MR, Moffat JC. Osteosarcoma of the Skull Base after Radiation Therapy in a Patient with McCune-Albright Syndrome: Case Report. Skull Base. 2003;13(2):79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Collins MT, Sarlis NJ, Merino MJ, Monroe J, Crawford SE, Krakoff JA, Guthrie LC, Bonat S, Robey PG, Shenker A. Thyroid carcinoma in the McCune-Albright syndrome: contributory role of activating Gs alpha mutations. J Clin Endocrinol Metab. 2003;88(9):4413–7. [DOI] [PubMed] [Google Scholar]
- 109.Yang GC, Yao JL, Feiner HD, Roses DF, Kumar A, Mulder JE. Lipid-rich follicular carcinoma of the thyroid in a patient with McCune-Albright syndrome. Mod Pathol. 1999;12(10):969–73. [PubMed] [Google Scholar]
- 110.Boussaid K, Meduri G, Maiza JC, Gennero I, Escourrou G, Bros A, Leguevaque P, Bennet A, Caron P. Virilizing sclerosing-stromal tumor of the ovary in a young woman with McCune Albright syndrome: clinical, pathological, and immunohistochemical studies. J Clin Endocrinol Metab. 2013;98(2):E314–20. [DOI] [PubMed] [Google Scholar]
- 111.Chevalier N, Paris F, Fontana S, Delotte J, Gaspari L, Ferrari P, Sultan C, Fenichel P. Postpubertal Persistent Hyperestrogenemia in McCune-Albright Syndrome: Unilateral Oophorectomy Improved Fertility but Detected an Unexpected Borderline Epithelial Ovarian Tumor. J Pediatr Adolesc Gynecol. 2015;28(6):e169–72. [DOI] [PubMed] [Google Scholar]
- 112.Johansen L, Haller W, Thyagarajan M, Kelly D, McKiernan P. Hepatic Lesions Associated With McCune Albright Syndrome. J Pediatr Gastroenterol Nutr. 2019;68(4):e54–e7. [DOI] [PubMed] [Google Scholar]
- 113.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–30. [DOI] [PubMed] [Google Scholar]
- 114.Shieh CC, Wang PJ. Giant nevocellular nevi with rickets and brainstem tumor. Pediatr Neurol. 1991;7(6):452–4. [DOI] [PubMed] [Google Scholar]
- 115.Kinsler VA, O’Hare P, Bulstrode N, Calonje JE, Chong WK, Hargrave D, Jacques T, Lomas D, Sebire NJ, Slater O. Melanoma in congenital melanocytic naevi. The British journal of dermatology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: A study of 596 cases. Journal of the American Academy of Dermatology. 2000;42(2 Pt 1):263–8. [DOI] [PubMed] [Google Scholar]
- 117.Viana AC, Gontijo B, Bittencourt FV. Giant congenital melanocytic nevus. An Bras Dermatol. 2013;88(6):863–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Robinson C, Boyce AM, Estrada A, Kleiner DE, Mathew R, Stanton R, Frangoul H, Collins MT. Bone marrow failure and extramedullary hematopoiesis in McCune-Albright syndrome. Osteoporos Int. 2018;29(1):237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mahdi AJ, Connor P, Thakur I. McCune-Albright syndrome-associated bone marrow failure and extramedullary haematopoeisis secondary to fibrous dysplasia. Br J Haematol. 2017;178(2):179. [DOI] [PubMed] [Google Scholar]
- 120.Bajpai A, Greenway A, Zacharin M. Platelet dysfunction and increased bleeding tendency in McCune-Albright syndrome. J Pediatr. 2008;153(2):287–9. [DOI] [PubMed] [Google Scholar]
- 121.Ohata Y, Yamamoto T, Mori I, Kikuchi T, Michigami T, Imanishi Y, Satomura K, Ida S, Ozono K. Severe arterial hypertension: a possible complication of McCune-Albright syndrome. Eur J Pediatr. 2009;168(7):871–6. [DOI] [PubMed] [Google Scholar]
- 122.Srinivas UM, Tourani KL. Epidermal nevus syndrome with hypophosphatemic renal rickets with hypercalciuria: a bonemarrow diagnosis. Int J Hematol. 2008;88(2):125–6. [DOI] [PubMed] [Google Scholar]
- 123.Boyce AM, Turner A, Watts L, Forestier-Zhang L, Underhill A, Pinedo-Villanueva R, Monsell F, Tessaris D, Burren C, Masi L, et al. Improving patient outcomes in fibrous dysplasia/McCune-Albright syndrome: an international multidisciplinary workshop to inform an international partnership. Arch Osteoporos. 2017;12(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]