

Abstract
Traditional risk factors are incompletely predictive of cardiovascular disease development, a leading cause of death in the elderly. Recent epidemiological studies have shown that human aging is associated with an increased frequency of somatic mutations in the hematopoietic system, which provide a competitive advantage to a mutant cell, thus allowing for its clonal expansion, a phenomenon known as clonal hematopoiesis. Unexpectedly, these mutations have been associated with a higher incidence of cardiovascular disease, suggesting a previously unrecognized connection between somatic mutations in hematopoietic cells and cardiovascular disease. Here, we provide an up-to-date review of clonal hematopoiesis and its association with aging and cardiovascular disease. We also give a detailed report of the experimental studies that have been instrumental in understanding the relationship between clonal hematopoiesis and cardiovascular disease and have shed light on the mechanisms by which hematopoietic somatic mutations contribute to disease pathology.
Keywords: Somatic mutations, clonal hematopoiesis of indeterminate potential, CHIP, age-related clonal hematopoiesis, ARCH, TET2, DNMT3A, IL-1β
AGING AND CARDIOVASCULAR DISEASE: A MISSING PIECE OF THE PUZZLE
Cardiovascular disease is the most common cause of death worldwide, accounting for 31% of all deaths globally (1). In 2016, it was estimated that 17.9 million people died due to a cardiovascular cause, and of these deaths, 85% were attributed either to ischemic heart disease or stroke (1). In addition to death, cardiovascular disease poses an enormous financial burden and accounts for a substantial proportion of health-care spending and loss of productivity worldwide (2). Cardiovascular disease becomes much more prevalent with age, and, thus, with the world’s population aging, the incidence of chronic disease is expected to increase (3). Indeed, it has been predicted that by 2035 nearly one in four individuals will be 65 years or older (4). Despite strong epidemiological evidence linking cardiovascular disease with advancing age (3), we still have an incomplete understanding of how it drives disease progression. While, the effect of aging on many cardiovascular diseases reflects, to a great extent, long-term exposure to traditional risk factors, such as hypertension, diabetes mellitus, hyperlipidemia, smoking, physical inactivity, and excessive alcohol consumption, these are still incompletely predictive of disease development in the elderly (5). Overall, this suggests that there are other unidentified age-related causal risk factors that drive the development and progression of cardiovascular disease. Thus, identifying and understanding these risk factors may uncover new ways to reduce the burden of cardiovascular disease.
SOMATIC MUTATIONS IN THE HEMATOPOIETIC SYSTEM AND AGING
In an effort to better understand the etiology of cardiovascular disease, research during the past two decades has focused on identifying common germline mutations that contribute to disease pathogenesis (6–8). These are mutations that are inherited from one’s parents and are present in all tissues throughout the body. However, mutations may also arise spontaneously over the course of an individual’s lifetime. These mutations are referred to as somatic or postzygotic mutations, and, in most cases, they result in no or little phenotypic consequences to the individual (9). In a few instances, however, a somatic mutation confers a competitive advantage to a cell, leading to progressive expansion of the mutant clone (10). As a result, the individual becomes a mosaic of cells with different genotypes, a phenomenon known as somatic mosaicism (9–11). Like germline mutations, somatic mutations can give rise to disease, and a well-known example of this is cancer (10, 12). However, with advances in sampling techniques and high-throughput sequencing technology, we are becoming increasingly aware that somatic mutations are also associated with the development numerous other diseases, including cardiovascular disease (13–17). Given the burden that cardiovascular disease imposes worldwide, there is a need to better understand the role that somatic mutations play in disease pathogenesis.
With advancing age, we accumulate mutations in our somatic cells and, therefore, it is not surprising that somatic mosaicism is a hallmark of aging in many tissues (18). Somatic mosaicism is more frequently detected in highly proliferative tissues, such as the hematopoietic system, which is responsible for producing more than 100 billion blood cells per day. In particular, it has been calculated that human hematopoietic stem and progenitor cells (HSPCs), the precursors to all mature blood cells, develop 0.13 ± 0.02 exonic mutations per year of life, and thus, it can be estimated that by age 50, an individual will accumulate an average of 5 coding gene mutations within each HSPC (19). If, by chance, one of these mutations confers a competitive advantage to the HSPC—such as by promoting its proliferation, self-renewal, survival, or some combination of these—this may lead to expansion of the mutant clone at a disproportionate rate compared with other HSPCs(20). As a result, the HSPC-derived mutation will propagate through the hematopoietic system and into its immediate cellular progeny, giving rise to a genetically distinct population of mature blood cells.
Some of the earliest indications that clonal events occur within the aged hematopoietic system originated from studies examining the pattern of maternal-to-paternal X chromosome inactivation in the white blood cells of otherwise healthy females (21). The method used was originally devised to study the clonal origin of cancer, as early during female embryonic development each cell undergoes random inactivation of one of its X chromosomes, so a skewing of the predicted 1:1 ratio of the maternal:paternal X inactivation pattern can be suggestive of a clonal expansion. In these studies, it was observed that healthy females older than 60 years were more likely to exhibit significant skewing of one of their inactive X chromosomes when compared with younger females (21). A subsequent study undertaken more than a decade later identified that females who exhibited inactive X chromosome skewing in their blood were more likely to also possess a mutation in the epigenetic regulator and blood cancer driver gene ten eleven translocation 2 (TET2), suggesting that this particular genetic mutation was playing a role in driving the clonal expansion of blood cells within these women (22).
Recent, large exome-sequencing and chromosomal mosaicism studies have expanded this association, detecting the presence of commonly occurring aberrant clonal expansions in hematopoietic cells from elderly individuals (11, 13, 15, 23–25). Specifically, two studies published in 2012 detected the presence of mosaic chromosomal alterations in the blood of otherwise healthy individuals, and the frequency of these alterations increased sharply with age (23, 24). In 2014, three independent studies used whole-exome sequencing of peripheral blood to examine the frequency of mutations within specific genes recurrently mutated in hematological malignancies (13, 15, 25). Although mutations in more than 100 candidate driver genes were associated with these clonal events within the hematopoietic system, most mutations occurred in just three genes that encode epigenetic regulators, specifically DNA methyltransferase 3 alpha (DNMT3A), TET2, and additional sex combs-like 1 (ASXL1) (13, 15, 25). Further, these studies showed that the frequency of these mutations increased with age, with approximately 10% of individuals older than 70 possessing at least one mutation in their white blood cells (13, 15, 25). While it was identified that individuals who harbored these mutations had an increased risk of developing a subsequent blood cancer, epidemiological studies showed that most will never develop a malignancy, as these conditions are relatively rare and generally require the acquisition of multiple oncogenic mutations, in accordance with the multistep leukemogenesis theory (13, 15, 26). Thus, based on these observations, this condition has been referred to as clonal hematopoiesis of indeterminate potential, or CHIP(27). In the literature, CHIP is defined by the presence of an expanded blood cell clone carrying a mutation in a known driver gene of hematological malignancy at a variant allele frequency (VAF) of at least 2% without meeting the standard diagnostic criteria for malignancy (i.e., cytopenia or abnormal blood cell counts, or both) (27). This distinction, while convenient, is largely based on the limits of mutation detection by sequencing methodology, rather than being based on epidemiological data or biological principles. Therefore, some researchers have labeled this definition as provisional and have suggested that it may be subject to change as we gain better insight into the impact that CHIP has on health and disease (27, 28). It is also important to note that clonal hematopoiesis can occur without the presence of a known driver mutation, perhaps by genetic drift in the HSPC pool during normal aging, by a mutation in an unidentified driver gene, or by a heritable epigenetic trait (29–31). In fact, studies estimate that mutations in unknown driver genes account for a substantial proportion of clonal events within the hematopoietic system (15, 31). Therefore, the term age-related clonal hematopoiesis (ARCH) has also been used to describe the presence of a clonal event observed in the hematopoietic system that is associated with advancing age (29).
Since the above-described epidemiological studies were reported, there has been increased attention paid to examining the prevalence of clonal hematopoiesis in other cohorts of individuals across the life span (31). Specifically, a study by Zink et al. (31) used nonbiased whole-genome sequencing to identify the presence of clonal hematopoiesis in the peripheral blood of 11,262 Icelanders. Unlike some of the earlier studies that used whole-exome sequencing (13, 25), this study did not select for candidate driver gene mutations and, thus, was able to detect clonal hematopoiesis at an even greater prevalence within the study population (31). Indeed, it was found that the overall frequency of mutations increased from 0.5% in individuals younger than 35 years to >50% in individuals older than 85 years, which is much higher than previous estimates. Interestingly, when the authors examined the frequency of mutations in candidate driver genes, it was found that they accounted for only a small proportion of the total number of clonal events, suggesting that there are other unidentified mutations that drive ARCH (Figure 1).
Figure 1.
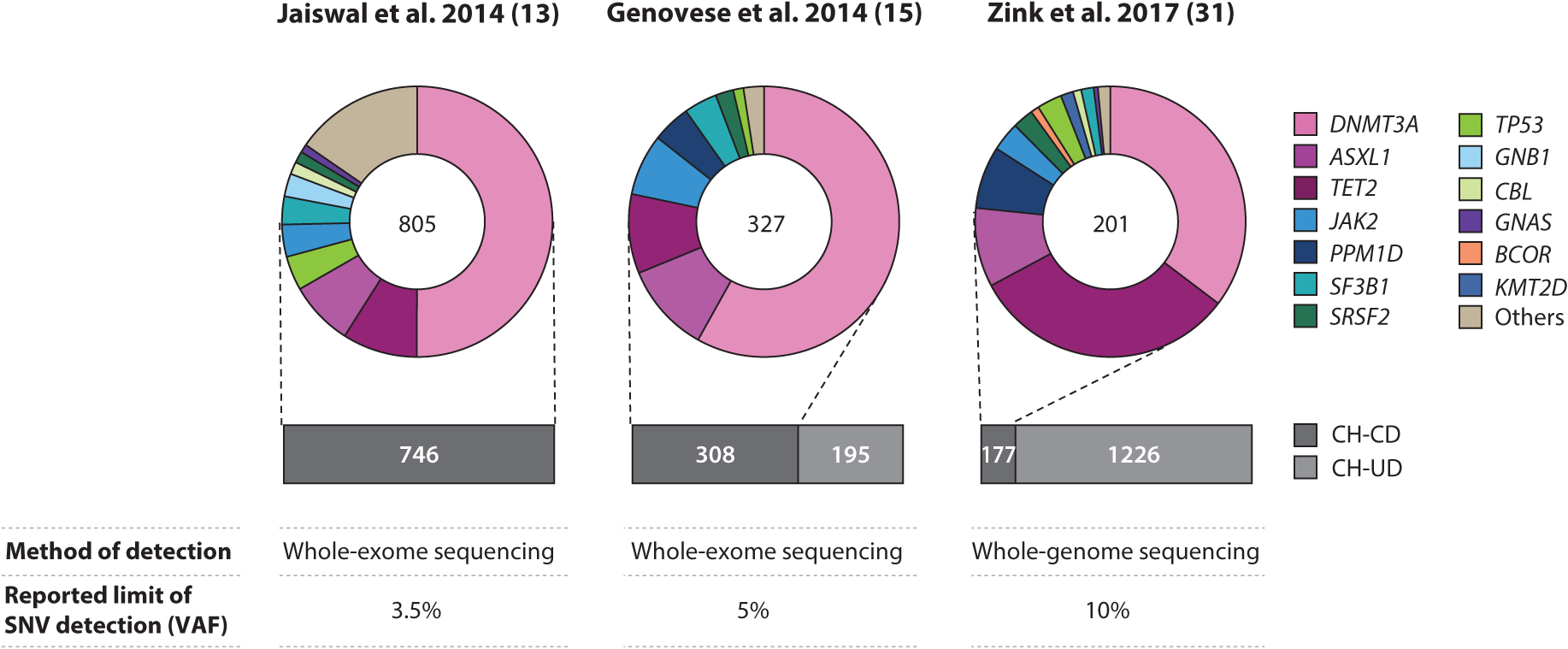
Summary of reported prevalence of clonal hematopoiesis. The bar graphs represent the total number of individuals with clonal hematopoiesis associated with (magenta) and without (gray) known driver gene mutations reported in three independent studies (13, 15, 31). The donuts summarize the frequencies of somatic mutations in candidate driver genes; the numbers within the donuts indicate the total number of driver gene mutations detected. Abbreviations: CH-CD, clonal hematopoiesis with candidate driver mutations; CH-UD, clonal hematopoiesis with unknown driver mutations; SNV, single nucleotide variant; VAF, variant allele frequency.
Related to this issue, advances in sequencing technology have allowed researchers to examine mutations at an even greater depth, and data arising from using this technology also suggest that clonal hematopoiesis is more prevalent than previously estimated, even when the analysis focuses only on candidate driver genes. For instance, ultradeep sequencing of mutational hot spots within select candidate driver genes has projected that the mean mutation frequency is >12% in individuals in their 60s, >19% in those in their 70s, >40% in those in their 80s, and >74% in those in their 90s (32). Similarly, a study using error-corrected sequencing, which allows for detection of clonal events at as low as 0.03% VAF, found that mutations in select candidate driver genes occurred in 95% of individuals aged 50–70 years, an age group for which previous studies estimated that only 5% of individuals had detectable clonal hematopoiesis (33). Overall, the findings from these studies reveal that clonal hematopoiesis is almost ubiquitous by middle age, and the expansion of these clones occurs in a portion of individuals later in life. However, exactly what drives the clonal expansion of these mutations in some individuals and not others remains speculative. It is likely that genetics and environmental factors play roles in this process, perhaps by modulating the kinetics of clonal expansion or by altering the bone marrow niche to favor survival of the fittest HSPC clone (34). Certainly, the aging process itself has effects on hematopoiesis in terms of the output and function of HSPCs (35). However, longitudinal data on individuals with clonal hematopoiesis are limited. Current data suggest that mutant clones are relatively stable for more than a 10-year period (33, 36, 37). These findings suggest that their expansion is episodic rather than linear, perhaps indicating that variations in exposure to environmental stressors may alter the overall hematopoietic demand or lead to changes in the bone marrow niche per se. Nevertheless, it is apparent that further studies are warranted to better understand and identify the factors that drive the clonal expansion of mutant cells within the hematopoietic system.
CLONAL HEMATOPOIESIS AND ALL-CAUSE MORTALITY: AN UNEXPECTED CONNECTION
To date, several independent reports have associated clonal hematopoiesis, either with or without candidate driver gene mutations, with an increase in all-cause mortality. The first and most prominent reports of this association originated from two large longitudinal studies published concurrently in the New England Journal of Medicine in 2014 (13, 15). These studies, which are also described above, used whole-exome sequencing of peripheral blood mononuclear cells from more than 29,000 individuals who were unselected for cancer or hematological malignancies (13,15). It was found that clonal hematopoiesis linked to mutations in known driver genes of hematological malignancy or CHIP was associated with a 40–50% increase in all-cause mortality (13,15). This association appeared to be correlated with age, as it was stronger in individuals who had their blood sequenced after the age of 70 (13). Further, the study by Genovese et al. (15) used nonbiased exome sequencing analysis, which allowed for the detection of clonal hematopoiesis with unknown driver mutations. It was documented that clonal hematopoiesis with mutations in unknown driver genes accounted for ~40% of all detectable clonal events; however, all-cause mortality was slightly lower for individuals bearing these types of mutations (15). Both of these studies observed that there was a marked increase in the frequency of hematological cancer in individuals with clonal hematopoiesis (13, 15), which is to be expected as mutations in driver genes represent an early step in the progression toward a hematological malignancy. However, hematological malignancies occur at a relatively low frequency in the general population, and, thus, could not fully explain the increase in all-cause mortality observed in these two studies (13, 15). In particular, the study by Jaiswal et al. (13) found that only one individual died due to a hematological neoplasm, and cause-specific analyses of 5,132 individuals confirmed a lack of association with cancer death. This lack of attributable cause in all-cause mortality led researchers to the question: What is the underlying cause of this increase in all-cause mortality in individuals who exhibit clonal hematopoiesis? In an unplanned secondary analysis, it was discovered that there was an association between clonal hematopoiesis and an increased incidence of coronary heart disease and ischemic stroke, suggesting that the increase in all-cause mortality could be explained by an increase in the incidence of these two conditions (13). Indeed, a cause-specific analysis confirmed the association between clonal hematopoiesis and death due to cardiovascular conditions (13). This surprising discovery has sparked increased interest in the relationship between somatic mutations in the hematopoietic system and cardiovascular disease, and the findings of the resultant studies are discussed in greater detail below.
Since these two studies were undertaken, additional epidemiological data have been published supporting an association between clonal hematopoiesis and an increase in all-cause mortality. The Icelandic study, also described above, which used barcoded, nonbiased whole-genome sequencing, reported an association between clonal hematopoiesis and all-cause mortality (31). In this particular study population, clonal hematopoiesis was detected at a high frequency, and it was found that the risk of mortality was similar for clonal hematopoiesis associated with and without candidate driver gene mutations, which differs somewhat from the findings of Genovese et al. (15). Most strikingly, however, it was found that individuals with detectable clonal hematopoiesis had a risk of all-cause mortality similar to those who had ever smoked (31), indicating that clonal hematopoiesis carries a similar risk to a well-established risk factor for all-cause mortality. Although this study did not examine cardiovascular outcomes, significant associations were detected between clonal hematopoiesis and smoking, smoking-related diseases, chronic pulmonary disease, and psychiatric disease (31). Expanding this association beyond specific gene mutations, a recent study by Loh et al.(30) reported an association between large clonal mosaic chromosomal alterations and all-cause mortality. Mutations involving large alterations of chromosomes have been an understudied area of clonal hematopoiesis, likely due to limitations in the methods of detection and the requirement for large study populations (11, 28). In the study by Loh et al. (30), 8,342 mosaic chromosomal alterations were detected within the blood of 151,202 participants in the UK Biobank using single nucleotide polymorphism–based computational techniques. Within the cohort, 71% of the detected alterations were classified as gains, deletions, or copy-number-neutral loss of heterozygosity, and the remaining 29% of events could not be inferred definitively (30). It was observed that the frequency of these types of mutations increased with age and was associated with a doubling in the risk of all-cause mortality (30). Similar to previous reports, this association could only partly be explained by cancer deaths after correcting for age, sex, and smoking status, and, thus, the authors speculated that some of these deaths might reflect the effects of these mutations on cardiovascular health (30). It should be noted that some of these aberrations likely affected genomic regions that include candidate driver genes, such as DNMT3A and TET2 (30), which could possibly account for some of the increase in all-cause mortality observed in this study. Nevertheless, this study provides evidence that multiple DNA mutations beyond those that target known candidate driver genes can give rise to clonal hematopoiesis and have consequences for mortality.
CLONAL HEMATOPOIESIS AND CARDIOVASCULAR DISEASE: EVIDENCE FROM THE CLINIC
The discovery that cardiovascular disease could account for a substantial amount of the increase in all-cause mortality associated with clonal hematopoiesis (13) has led to further clinical investigations into the putative connection between the two phenomena (14, 38). As briefly discussed above, the study by Jaiswal et al. (13) in which this association was first detected, found that individuals with CHIP had a significantly increased risk of coronary heart disease (hazard ratio: 2.0) and ischemic stroke (hazard ratio: 2.6) after adjusting for age, sex, type 2 diabetes, systolic blood pressure, and body mass index. It was also noted that individuals with a VAF >10% had a higher risk of cardiovascular disease, suggesting that clone size may be correlated with risk (13). Prominently, the risks of coronary heart disease and ischemic stroke were higher in carriers of CHIP than in individuals with traditional risk factors, such as hypertension and high body mass index (13). However, the main aim of this study was to evaluate the frequency of hematopoietic somatic mutations in the general population rather than determine their association with cardiovascular disease. Therefore, a follow-up investigation was conducted to directly test the hypothesis that CHIP is associated with an increase in the incidence of cardiovascular disease (14). For this analysis, whole-exome sequencing was performed on blood samples obtained from four case–control studies (two prospective and two retrospective) consisting of 4,726 individuals with coronary heart disease and 3,529 controls. In the prospective study cohorts, it was found that carriers of CHIP had an increased risk of coronary heart disease (combined hazard ratio: 1.9), which was comparable with what was found in the 2014 study (13, 14). Analogous to previous studies, most mutations were detected in just four genes, namely DNMT3A, TET2, ASXL1, and Janus kinase 2 (JAK2). To examine whether there were any gene mutation–specific differences in the risk of coronary heart disease, a meta-analysis was performed by combining the data from the prospective cohorts with the risks reported in previously published works (14). While there was no statistically significant difference in the risk of coronary heart disease for individuals carrying mutations in DNMT3A (hazard ratio: 1.7), TET2 (hazard ratio: 1.9), or ASXL1 (hazard ratio: 2.0), individuals who carried JAK2 mutations displayed a marked increase in disease risk (hazard ratio: 12.0) (14). However, it should be noted that the sample of individuals who carried JAK2 mutations was small, and this work should be viewed as preliminary. Further, an analysis of individuals within one of the prospective studies (known as BioImage) found that carriers of CHIP were more likely to have coronary artery calcification, and this appeared to be correlated with clone size (14). Analyses of the retrospective studies found there was also an association between carriers of CHIP and early-onset myocardial infarction (14). Overall, these findings add further weight to the initial report that CHIP is associated with an increased incidence of cardiovascular disease.
To assess the potential prognostic significance of CHIP in patients with cardiovascular disease, a more recent study by Dorsheimer et al. (38) examined the long-term prognosis of patients with CHIP and chronic heart failure (CHF) of ischemic origin. In a cohort of 200 CHF patients, the frequency of CHIP was assessed in bone marrow–derived mononuclear cells using deep, targeted amplicon sequencing of 56 genes previously associated with CHIP and myeloid malignancies (38). It was found that 18.5% of patients within this cohort were carriers of CHIP and had a VAF of at least 2%. Most mutations were detected in DNMT3A and TET2, which is similar to what has been reported in previous studies assessing clonal hematopoiesis in the general population. The authors also noted that there were smaller numbers of patients with mutations in lysine demethylase 6A (KDM6A) and BCL6 corepressor (BCOR). Consistent with earlier reports, carriers of CHIP were significantly older compared with noncarriers, although it was found that CHIP carriers did not differ with respect to clinical parameters of heart function and disease stage (38). However, in a subsequent analysis of patients harboring either DNMT3A or TET2 mutations, it was found that patients with these gene-specific mutations had overall a worse long-term clinical outcome in terms of both death and death combined with rehospitalization for CHF compared with noncarriers, even after adjusting for age (38). To determine whether this association was correlated with clone size, the threshold of mutation calling was lowered to a VAF of 0.5% (38). Using this new threshold, it was identified that there were an additional 66 and 53 patients with DNMT3A and TET2 mutations, respectively, with clone sizes between 0.5% and 2% VAF. When patients were grouped based on VAF (i.e., no mutation, VAF < 1%, VAF between ≥ 1% and < 2%, or VAF > 2%), it was found that there was a dose-dependent relationship between clone size and clinical outcome, including clone sizes between 1% and 2% VAF, which is lower than the proposed threshold for CHIP designation (38). Regardless, this small clinical study supports the overarching hypothesis that somatic mutations in the hematopoietic system, and particularly those in the candidate driver genes DNMT3A and TET2, are associated with progression and poorer prognosis in patients with CHF. More recently, Mas-Peiro et al. (39) found an association between mutations in DNMT3A and TET2 and poorer prognosis in 279 patients treated for aortic valve stenosis. However, further studies using larger cohorts will be required to validate and examine these associations more deeply.
In addition to the above-described studies, there has been a report that the presence of CHIP in patients undergoing autologous stem cell transplantation for lymphoma is associated with worse survival and death due to cardiovascular conditions (40). Interestingly, in this study, it was documented that there was a much higher frequency of mutations in protein phosphatase magnesium/manganese dependent 1D (PPM1D) and tumor protein 53 (TP53), both of which are classic tumor suppressor genes. In individuals undergoing cytotoxic therapy prior to stem cell transplantation, the hematopoietic system is likely under extreme stress, and it is thought that mutations in tumor suppressor genes, such as TP53 and PPM1D, confer the mutated HSPCs with a survival advantage against genotoxic stress induced by chemotherapy, a phenomenon known as therapy-associated clonal hematopoiesis (41–44). Indeed, it was found that 30% of individuals undergoing autologous stem cell transplantation for lymphoma were carriers of CHIP (40). This finding is similar to other studies, which have reported enrichment of PPM1D mutations within the blood of patients who have been previously treated for solid-organ tumors (45–47). Thus, in general, this study may suggest that somatic mutations in genes such as TP53 and PPM1D may also be associated with an increased incidence of cardiovascular disease, particularly in patients who have previously undergone chemotherapy (40). While of interest, further studies are required to directly assess this relationship, particularly as these genes are not as frequently mutated in the general population.
CLONAL HEMATOPOIESIS AND CARDIOVASCULAR DISEASE: EVIDENCE FROM THE LABORATORY
The descriptive nature of the aforementioned human studies does not allow us to determine whether a causal relationship exists between CHIP and an increased risk of cardiovascular disease, as the association between these two phenomena could be secondary to confounding factors or simply reflect a shared consequence of the aging process. Moreover, in clinical studies it is difficult to assess directionality and the study factors that may drive the clonal expansion of cells. This is important, as it has been suggested that chronic inflammation and other stresses associated with cardiovascular disease may promote somatic mutagenesis and the clonal expansion of mutant cells (48–50). Hence, animal models have recently proved to be powerful tools for gaining a better understanding of the relationship between clonal hematopoiesis and cardiovascular disease (14, 51–54) (Figure 2). The forthcoming sections describe recent experimental work examining the effects of clonal hematopoiesis associated with mutations in the key driver genes TET2, DNMT3A, and JAK2 on cardiovascular disease and the wider implications of these findings for human cardiovascular health.
Figure 2.
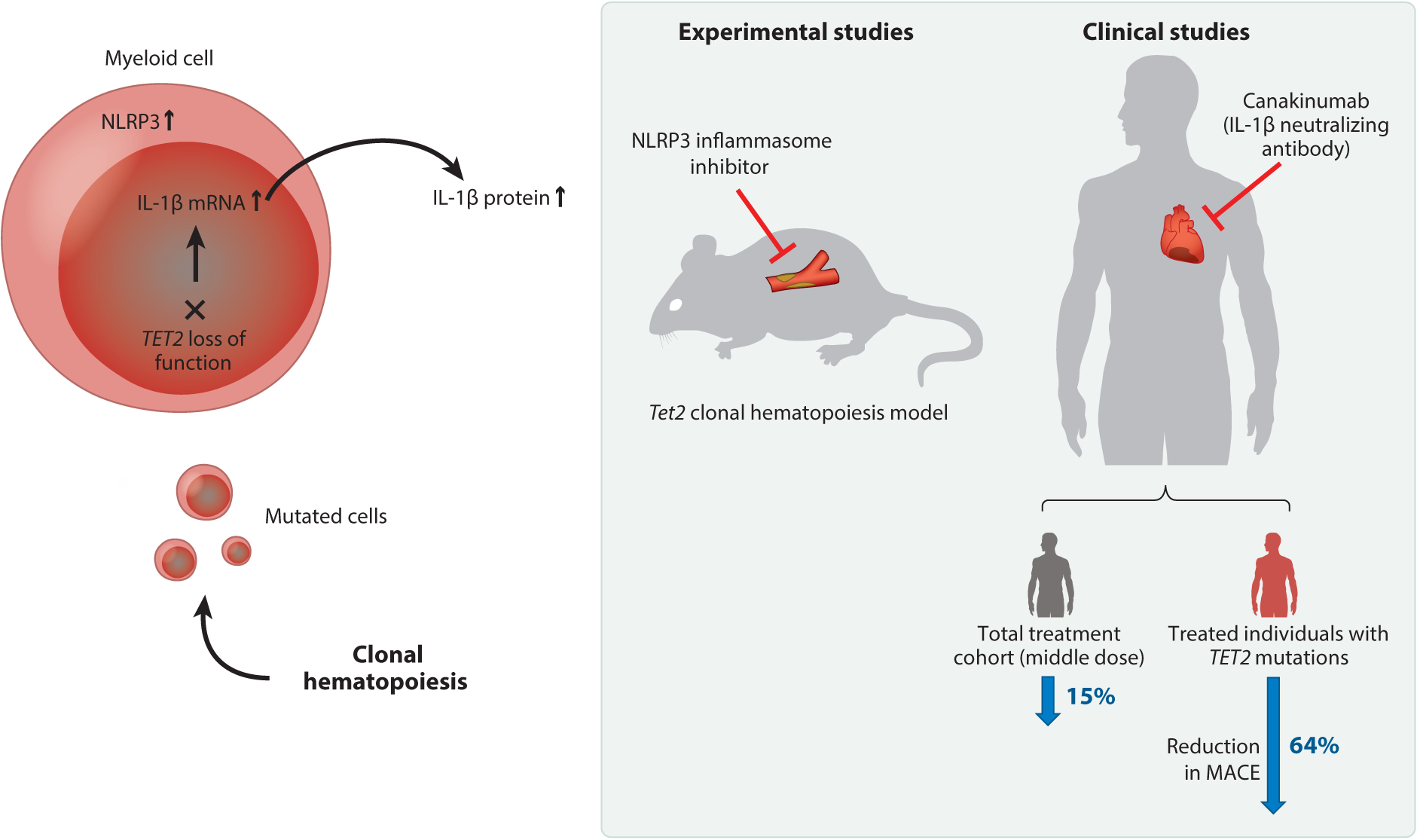
Central overview of age-related clonal hematopoiesis and how it drives cardiovascular disease progression. During the aging process, healthy hematopoietic stem cells (HSCs) may acquire a mutation in a driver gene. This mutation provides the mutant HSC with a competitive advantage, allowing for its expansion at a disproportionate rate compared with other HSCs within the bone marrow niche (clonal expansion). As a result, the HSC-derived mutation propagates through the hematopoietic system and into its immediate leukocyte progeny, giving rise to a genetically distinct population of mature white blood cells. During cardiovascular disease, inflammatory cells migrate to the injury site and are involved in promoting disease pathogenesis through a variety of mechanisms. In the setting of clonal hematopoiesis, HSC-derived mutations may result in changes to the inflammatory profile of the leukocyte progeny(i.e., elevated cytokine production). As a result, this altered inflammatory profile of leukocytes can exacerbate cardiovascular injury processes, driving disease progression.
TET2
As indicated above, one of the most commonly mutated genes associated with an increased incidence of and death due to cardiovascular conditions is TET2 (13, 14, 38). The protein encoded by TET2 is an α-ketoglutarate and an Fe2+-dependent enzyme that catalyzes DNA methylation by converting 5-methylcytosine into 5-hydroxymethylcytosine, thus promoting the transcriptional activation of genes (55, 56). Furthermore, depending on the cellular and molecular environment, TET2 may also induce the repression of genes by recruiting histone deacetylases to gene promotors (51, 57). TET2 was the first gene reported to exhibit acquired mutations in the blood cells of individuals without hematological malignancies (22), and, to date, more than 130 different TET2 mutations have been identified in the blood cells of humans (13, 15, 22, 25, 31, 56, 58). Most of these mutations are small insertions or deletions, or nonsense or missense point mutations, often resulting in changes to the catalytic site and, hence, a loss of protein function (13, 15, 56). It has also been reported that large alterations in chromosome 4q may lead to TET2 loss of function in some instances (59). Since the discovery of Tet2 in 2009, studies in rodents have shed light on its function within the hematopoietic system and have indicated that it plays an important role in regulating the self-renewal of HSPCs as well as myeloid cell differentiation (60–62). In particular, Tet2 deficiency in mice leads aberrant patterns of hematopoiesis, and Tet2-deficient HSPCs display increased self-renewal and proliferative capacity in competition assays compared with their wild-type counterparts (62, 63). Further, animals deficient in Tet2 exhibit myeloid lineage skewing as well as extramedullary hematopoiesis, characterized by an increase in monocytes and neutrophils within the spleen (62, 64). Notably, studies have observed that not all Tet2-deficient mice will develop a hematological malignancy, consistent with the stepwise concept of blood cancer development (49).
To determine whether a causal relationship existed between TET2 clonal hematopoiesis and cardiovascular disease, we recently assessed the impact of Tet2 deficiency in HSPCs in several murine models of cardiovascular disease (51–53). For our initial assessment, a mouse model of atherosclerosis was chosen, as atherosclerosis represents a root cause of both coronary heart disease and ischemic stroke, the two conditions in which an association was initially reported with clonal hematopoiesis (13). To model clonal hematopoiesis, competitive bone marrow transplantation (BMT) experiments were performed to introduce a small number of Tet2-deficient hematopoietic cells into atherosclerosis-prone Ldlr−/− mice (51). This experimental setting mimics the human scenario of individuals carrying a TET2 somatic mutation, since these mutations are initially carried by a small number of HSPCs, which will gradually expand over time. Consistent with this concept, it was found that following transplantation, Tet2-deficient cells expanded progressively in the bone marrow, spleen, and blood, and they exhibited a slight myeloid bias, with preferential expansion into the Ly6Chigh monocyte population (51). Importantly, there was no effect of HSPC Tet2 deficiency on the total number of white blood cells, which is analogous to what is observed in cancer-free individuals who exhibit clonal hematopoiesis associated with a TET2 mutation (13, 15, 25). Notably, the expansion of Tet2-deficient cells in this manner accelerated atherosclerosis, leading to a marked increase in plaque size (51). To more closely model the human scenario, whereby most individuals bear a mutation in only one allele, equivalent experiments were performed using cells that were heterozygous for Tet2. In these experiments, heterozygosity of Tet2 was sufficient to accelerate atherosclerosis, although the phenotype was milder than what was observed following the transplantation of homozygous cells (51). It was also noticed that the kinetics of clonal expansion were slower for heterozygous cells, indicating a gene dose–dependent effect. Further, myeloid-specific ablation of Tet2 was sufficient to promote atherosclerosis, indicating that cells of myeloid origin and likely macrophages as well, play major roles in accelerating atherosclerosis associated with the clonal expansion of Tet2-deficient HSPCs (51). Credence was given to these findings when an independent group also reported similar outcomes in a mouse model of atherosclerosis (14). In that study, a conventional BMT approach was taken, whereby all transplanted hematopoietic cells were deficient in Tet2 (14). In contrast to the approach taken by Fuster et al. (51), the approach in the study reporting similar outcomes led to the widespread presence of xanthomas, enlargement of the spleen, and macrophage infiltration into various tissues (14). Also, total hematopoietic deletion of Tet2 resulted in a substantial increase in plaque size in this model of atherosclerosis (14). This study also found that myeloid-specific deletion of Tet2 led to an augmented atherosclerotic lesion, providing additional evidence that the effects of hematopoietic Tet2 loss of function are mediated through myeloid cells (14).
To corroborate and extend these studies, we recently examined the effect of Tet2 clonal hematopoiesis on experimental heart failure (52, 53). In the initial studies, two distinct models of heart failure were employed, specifically the left anterior descending coronary artery ligation model of myocardial infarction and the transverse aortic constriction model of pressure overload–induced cardiac hypertrophy. It was found that hematopoietic Tet2 inactivation, achieved by competitive BMT, led to more pronounced pathological cardiac remodeling in these models, as indicated by poorer cardiac function and greater myocardial hypertrophy, fibrosis, and inflammation(53). Further, it was found that myeloid-specific deletion of Tet2 also led to accelerated heart failure, indicating that myeloid cell populations are likely involved in accelerating the pathology (53). An ensuing study strengthened these findings using an angiotensin II–induced pressure overload model of heart failure (52). In this study, Tet2 inactivation was achieved using a lentivirus-mediated CRISPR-Cas9 system to create Tet2 mutations in a small number of lineage-negative hematopoietic cells ex vivo. Following lentiviral transduction, BMT experiments were performed, similar to our earlier studies using competitive BMT (52). Importantly, the CRISPR-edited cells expanded in the bone marrow and blood, and the expansion characteristics were similar to what was observed in prior studies using cells from Tet2-deficient mice. Further, mice transplanted with CRISPR-edited cells displayed exacerbated pathological cardiac remodeling following angiotensin II infusion, characterized by worse cardiac function and greater levels of fibrosis and inflammation (52). Notably, the profile of cardiac remodeling following angiotensin II infusion was similar between our CRISPR-mediated and conventional competitive BMT approaches, indicating that the CRISPR-Cas9 system is an efficient method to study clonal hematopoiesis associated with candidate driver mutations (52). Overall, the findings from these two studies in experimental heart failure indicate that clonal hematopoiesis associated with Tet2 mutations accelerates adverse cardiac remodeling leading to heart failure.
Data suggested that macrophages were playing a key role in the mechanism by which Tet2 loss of function exacerbated cardiovascular pathology (51–53). However, Tet2 deficiency did not affect macrophage proliferation, apoptosis, oxidized low-density lipoprotein uptake, or the expression of regulators that control cholesterol uptake (51). Thus, we hypothesized that Tet2-deficient hematopoietic cells may accelerate atherosclerosis by generating a pool of macrophages with enhanced proinflammatory actions. In support of this notion, isolated Tet2-deficient macrophages were found to display augmented inflammatory gene expression profiles and to produce greater amounts of the proinflammatory cytokine interleukin 1β (IL-1β) when treated with lipopolysaccharide (LPS) (51). Increased transcript and protein levels of IL-1β were also detected in the plaques of atherosclerotic mice, possibly suggesting a causative role for IL-1β (Figure 3). To better understand the connection between Tet2 and IL-1β, additional experiments were performed in isolated macrophages to interrogate how Tet2 regulates IL-1β production. These experiments revealed that Tet2 plays a role in inhibiting the transcription of IL-1β via histone deacetylase–mediated histone deacetylation rather than by promoting DNA methylation (51). This finding is analogous to a previous report that Tet2 represses transcriptional activation of specific inflammatory genes in macrophages by recruiting histone deacetylase 2 to the gene promotor (57).
Figure 3.
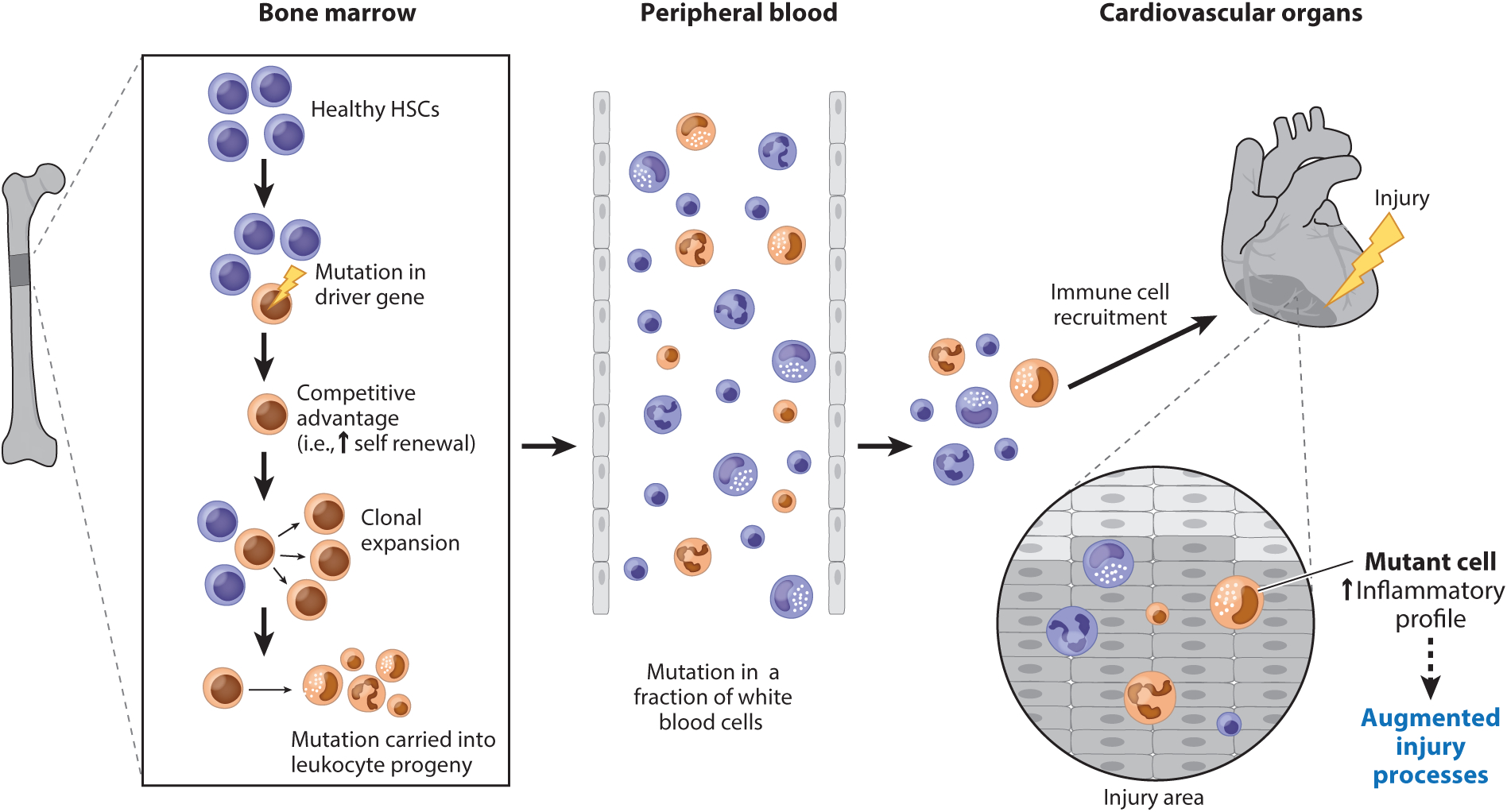
TET2-mediated clonal hematopoiesis accelerates cardiovascular disease through aberrant IL-1β signaling. Somatic mutations in TET2 within hematopoietic stem and progenitor cells lead to their clonal expansion, giving rise to myeloid cells that promote cardiovascular disease via aberrant activation of the NLRP3/IL-1β pathway (51–53). Blockade of the NLRP3 inflammasome complex has been shown to reduce disease pathology associated with Tet2 clonal hematopoiesis in mouse models of atherosclerosis and heart failure (51, 53). Moreover, a subanalysis of the CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcome Study) trial indicated that individuals with somatic mutations in TET2 responded more favorably to the IL-1β neutralizing antibody canakinumab than did individuals without TET2 mutations (66–68). Abbreviations: IL, interleukin; MACE, major adverse cardiovascular event.
It is well established that IL-1β is synthesized as a precursor protein (pro-IL-1β) and requires cleavage for activity, often by an immune complex called the NLRP3 inflammasome (65). Studies in the atherosclerosis model found that Tet2 regulates components of the NLRP3 inflammasome complex and that selective inhibition of this complex with a small molecule inhibitor reduced lesion burden specifically within animals transplanted with Tet2-deficient bone marrow cells, thus confirming a role for IL-1β and the NLRP3 inflammasome in the augmented pathology (51). In terms of the downstream actions of IL-1β, findings suggested that IL-1β released from Tet2-deficient plaque macrophages was perpetuating inflammation by promoting endothelial cell activation and P-selectin expression (51). In turn, this led to an increase in the recruitment of monocytes to the lesion site, regardless of the Tet2 genotype, thereby augmenting vascular inflammation. Therefore, it can be speculated that a few Tet2-mutant cells in the plaque act in a catalytic manner by amplifying and perpetuating vascular inflammation and, thus, drive atherosclerotic plaque progression. Similarly, Jaiswal et al. (14) also reported that macrophages from Tet2-deficient animals were more proinflammatory and exhibited increased levels of various cytokines and chemokines, including IL-1β, following stimulation with LPS. Moreover, in that study, atherosclerotic animals, which had transplants of Tet2-deficient bone marrow, appeared to have enhanced levels of circulating cytokines and chemokines compared with animals that had transplants of wild-type cells. The authors speculated that Tet2 loss of function was accelerating atherosclerosis by promoting monocyte recruitment to the lesion site via enhanced CXCR2 signaling, although this concept was not examined directly. In subsequent studies using mouse models of heart failure, it was found that IL-1β was involved in the augmented pathology associated with Tet2 clonal hematopoiesis (52, 53). Specifically, increased transcript and protein levels of IL-1β were observed in the hearts of animals transplanted with Tet2-deficient cells (52, 53). Similarly, inhibition of the NLRP3 inflammasome complex led to reduced cardiac remodeling and heart failure in the left anterior descending and transverse aortic constriction models (53). The mechanistic findings from these studies indicate that TET2 regulates IL-1β production at multiple levels, and, thus, loss of function of Tet2 results in aberrant IL-1β signaling, which, in turn, accelerates cardiovascular disease (51–53).
Implications for the CANTOS Trial
These findings on TET2-mediated clonal hematopoiesis may have implications for understanding the outcomes of the CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcome Study) trial. CANTOS was a large, placebo-controlled clinical trial that examined the effects of escalating doses of canakinumab, a neutralizing IL-1β antibody, in patients who were post-myocardial infarction and had sustained levels of high-sensitivity C-reactive protein (hsCRP), a measure of inflammation. In this high-risk group, targeting IL-1β at the middle dose (150 mg every 3 months) led to a significantly lower frequency of major adverse cardiovascular events (MACE) compared with the placebo group (66). In a prespecified secondary analysis of the reduction in CRP relative to events in CANTOS, it was found that MACE and all-cause mortality were highly dependent on the degree of CRP reduction (67). For canakinumab-treated patients who exhibited a reduction in hsCRP to <2 mg/L there were also reductions of 25%, 31%, and 31% in, respectively, MACE, cardiovascular mortality, and all-cause mortality (67). In contrast, the nonresponders (i.e., those who on treatment had hsCRP levels of ≥2 mg/L) displayed no reductions in these end points. It had previously been hypothesized that individuals with TET2-mediated clonal hematopoiesis would have a better response to therapies that target the IL-1β/NLRP3 inflammasome pathway (51). Support for this hypothesis was provided by a recent exploratory analysis of nearly 4,000 individuals enrolled in the CANTOS trial (68) that found patients with mutations in TET2 displayed a 64% reduction in MACE in response to canakinumab therapy (Figure 3).
DNMT3A
DNMT3A belongs to a family of cytosine methylases and plays a key role in epigenetic regulation by catalyzing the addition of methylation marks to genomic DNA (69). DNMT3A is the most frequently mutated candidate driver gene associated with clonal hematopoiesis in the elderly (13, 15, 25, 31), and, as indicated above, it is associated with an increased risk of cardiovascular disease (13, 14, 39). Experimental studies examining the role of Dnmt3a within the hematopoietic system indicate it is involved in regulating HSPC self-renewal, and the loss of function of this enzyme has been reported to impair HSPC differentiation (70, 71). Moreover, mice that have Dnmt3a selectively deleted from their hematopoietic cells exhibit hallmarks of primitive hematopoiesis, including an upregulation of fetal hematopoietic gene expression and preferential myelopoiesis, as well as expansion of myeloid cells within the liver (72). Thus, it has been suggested that Dnmt3a regulates genes involved in the early development of the hematopoietic system.
To better understand the connection between clonal hematopoiesis associated with DNMT3A mutations and cardiovascular disease, our laboratory recently assessed the effect of hematopoietic Dnmt3a deficiency on experimental heart failure induced by angiotensin II infusion (52). Dnmt3a loss of function was achieved by genetic editing of lineage-negative hematopoietic cells using the same CRISPR-Cas9 approach that was used to produce Tet2 loss of function, described above. Following lentiviral transduction, BMT experiments were performed to introduce a small number of Dnmt3a-mutated cells into mice. Unlike mutations in Tet2, Dnmt3a loss of function did not lead to increased chimerism of the edited cells during the 4-month course of this study (52). This finding is analogous to previous reports showing that Dnmt3a-deficient HSPCs expand only in aged animals or after sequential BMT (70, 73, 74), raising potential concerns regarding the use of murine models to study clonal hematopoiesis associated with mutations in DNMT3A. Nevertheless, in mice with Dnmt3a-mutated cell chimerism, between 5% and 10% exhibited worse cardiac function following angiotensin II infusion and greater cardiac remodeling, as indicated by increased cardiac fibrosis and myocyte cross-sectional area (52).
From a mechanistic point of view, DNMT3A is thought to play a role in regulating inflammatory pathways. For example, it has been reported that DNMT3A can restrain inflammatory pathways in mast cells (75) as well as regulate T helper cell polarization (76–78) and macrophage functions (78, 79). Consistent with these previous reports, our in vitro and in vivo data suggested that hematopoietic DNMT3A loss of function was potentially promoting heart failure by augmenting inflammatory pathways (52). It was observed that mice transplanted with Dnmt3a-mutated cells had greater macrophage accumulation in the heart and increased expression of immune cell markers following angiotensin II infusion. Moreover, a Dnmt3a-deficient macrophage cell line displayed increased expression of the proinflammatory molecules Il-6, Cxcl1, Cxcl2, and Ccl5 following stimulation with LPS. Interestingly, this inflammatory profile differed from that observed for Tet2-deficient macrophages, which showed increases in Il-1β, Il-6, and Ccl5, but not Cxcl1 or Cxcl2, following LPS stimulation (52). Although further experiments using primary cells from mice and humans are required, these findings highlight the concept that mutations in different driver genes may result in clones with divergent functions, which may ultimately drive disease progression through different mechanisms. This could conceivably have implications for the design of future therapies, particularly as it has been observed in a subanalysis of the CANTOS trial that individuals with hematopoietic TET2 mutations respond more favorably to canakinumab than do individuals without mutations in this gene (68). Overall, the findings from this study indicate that Dnmt3a loss of function within hematopoietic cells promotes inflammatory processes and heart failure (52). However, given the complex immunomodulatory properties of DNMT3A in multiple immune cell types (75–79), additional experimental studies are required to better understand how DNMT3A affects the pathogenesis of cardiovascular disease.
JAK2
Another relatively common gene that is mutated within the aged hematopoietic system is JAK2 (1–3, 80). JAK2 is a member of the Janus family of cytoplasmic non-receptor tyrosine kinases and plays an important role in immune signaling processes (81). In particular, it is involved in many cytokine signal transduction pathways, such as those of erythropoietin and thrombopoietin(81). The most frequent type of JAK2 mutation associated with both clonal hematopoiesis and myeloproliferative disorders, such as polycythemia vera, is the V617F mutation, which is often denoted as JAK2V617F (1–3, 80, 82). This mutation is a guanine-to-thymidine substitution, resulting in an amino acid substitution of valine for phenylalanine at codon 617 of JAK2 (83). The mutation is thought to cause changes to the autoinhibitory site of JAK2 kinase, resulting in a constitutively active protein; thus, this type of mutation is considered a gain-of-function mutation(84). It has been suggested that JAK2 mutations may occur in lineage-restricted stem cells, such as myeloid-restricted stem cells (85–87); however, studies have challenged this and have observed that individuals who harbor the JAK2V617F mutation possess it in all blood cell compartments (88,89). Moreover, it has been found that some common germline variants have a higher risk of developing mutations in JAK2 (80, 90); however, the mechanisms by which these germline variants predispose individuals to developing mutations in JAK2 remain unknown. A recent study examining the prevalence of the JAK2V617F mutation in the peripheral blood of 19,958 adult Danish citizens found that 3.1% of individuals harbor this mutation (91). Of these JAK2V617F-positive individuals, only 2.3% were diagnosed with a myeloproliferative neoplasm (15). While previous studies have documented individuals who possess the JAK2V617F allele in their blood cells and do not exhibit overt changes in the levels of erythrocytes, platelets, or leukocytes, this study found that 42% of mutation-positive individuals had a more hyperproliferative blood cell count (15), which challenges the current definition of CHIP.
Recently, our laboratory examined the relationship between clonal hematopoiesis associated with JAK2 gain of function and cardiovascular disease (54). Using a transgenic mouse strain that expressed human JAK2V617F, competitive BMT experiments were performed to characterize the expansion characteristics of JAK2 mutant cells. Interestingly, JAK2V617F mutants expanded exclusively within the myeloid cell population, and this expansion was accelerated following myocardial infarction (54). However, at 16 weeks following competitive BMT, all mice showed signs of myeloproliferative disease, with elevated numbers of platelets and red blood cells; thus, this model was not ideal for studying CHIP (54). To avoid such complications, a lentiviral approach was developed, whereby exogenous JAK2 expression was under the control of a myeloid-specific promotor and enhancer. Lineage-negative bone marrow cells were then transduced with the lentivirus encoding JAK2WT (control) or JAK2V617F (mutant) and transplanted into lethally irradiated mice. Importantly, mice transplanted with JAK2V617F cells did not display alterations in blood cells or platelets, suggesting this was a superior model for studying clonal hematopoiesis in the setting of cardiovascular disease (54). Using the left anterior descending artery (or LAD) ligation model of myocardial infarction and the transverse aortic constriction (or TAC) model of pressure overload, mice transplanted with a small number of JAK2V617F cells showed augmented indices of heart failure compared with animals transplanted with JAK2WT cells (54). Moreover, it was noted that there were elevated levels of proinflammatory mediators within the hearts of animals that received JAK2V617F mutant cells (54). Overall, this indicates that clonal hematopoiesis associated with the JAK2V617F mutation can accelerate heart failure, likely through the overactivation of inflammatory pathways. It has also been reported that total hematopoietic JAK2 gain of function accelerates experimental atherosclerosis (92). Specifically, it has been documented that the transplantation of JAK2VF bone marrow to Ldlr−/− atherosclerosis-prone mice results in larger and more complex atherosclerotic lesions following a high-fat, high-cholesterol diet (92). However, caution should be taken when interpreting these findings as the entire bone marrow was reconstituted with JAK2 mutant cells, and mice exhibited a myeloproliferative disease phenotype that is not representative of CHIP.
In terms of the potential underlying mechanism by which hematopoietic JAK2 gain-of-function mutations may promote cardiovascular disease, it has been documented that the mutation results in an increase in extracellular trap formation by neutrophils (NETs). More specifically it has been found that JAK2V617F human and mouse neutrophils have increased NET formation in response to ionomycin treatment in vitro (93). Moreover, it has been observed that JAK2V617F mutant cells from humans had increased expression of PAD4, an enzyme involved in the formation of NETs (93). Indeed, studies have reported that NETs can promote cardiac dysfunction in the setting of myocardial infarction (94, 95) and pressure overload (96); thus, increased NET formation may represent a potential mechanism by which hematopoietic JAK2 gain-of-function mutations promote cardiovascular disease. Furthermore, it is known that NET formation is involved in thrombosis, and it has been reported that mice heterozygous for the JAK2V617F allele in their hematopoietic cells show an increased propensity for developing spontaneous pulmonary thrombosis (93). Consistent with this, a case–control study has documented an association between JAKV617F-positive CHIP carriers and major venous thrombotic events (93). Thus, an increase in thrombosis may account for the increase in the incidence of of cardiovascular disease (i.e., myocardial infarction and stroke) observed in JAKV617F-positive carriers.
PERSPECTIVES AND CONCLUDING REMARKS
Clonal hematopoiesis is an inevitable consequence of normal human aging, and recent clinical studies indicate that this condition is associated with an increased incidence of mortality and cardiovascular conditions (13–15, 31, 38–40). Recent experimental work has provided evidence that this relationship is causal and has shed light on the mechanisms through which mutations in Tet2 and Dnmt3a, and JAK2V617F in hematopoietic cells contribute to pathology in models of atherosclerosis and heart failure (14, 51–54). Despite this progress, this area of research is very much in its infancy. To date, only a small number of candidate driver genes have been evaluated in models of cardiovascular disease. Furthermore, clonal hematopoiesis not attributed to known drivers has also been associated with an increase in all-cause mortality, and conceivably some of these deaths could be due to cardiovascular conditions (15, 30, 31). Thus, identifying these mutations and their relationship with cardiovascular disease will be an important step forward. Due to mechanistic differences, different genetic mutations will likely confer divergent functions on hematopoietic cells. Therefore, some mutations may be highly pathological, whereas others may be benign or perhaps even cardioprotective. It is also important to note that the same mutation may affect components of the cardiovascular system differently, and, thus, future experimental studies should examine the effects of clonal hematopoiesis on cardiovascular diseases beyond atherosclerosis and heart failure. It also remains to be determined how clonal hematopoiesis can affect other age-related diseases, particularly those that are driven by inflammation. Continued research into this area could provide a rationale for the prognostic evaluation of somatic mutations in the hematopoietic system, as it could add to the predictive capabilities of traditional risk factors for cardiovascular disease, which were deduced ~50 years ago. Moreover, understanding how different mutations affect pathology may ultimately uncover novel approaches to treat cardiovascular disease.
ACKNOWLEDGMENTS
This work was supported by US National Institutes of Health grants to K.W. (HL138014, HL1238014, HL126141, and HL132564) and grants to S.S. from the American Heart Association Postdoctoral Fellowship program (17POST33670076) and the Kanae Foundation for the Promotion of Medical Science.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.WHO (World Health Organ.). 2017. Cardiovascular diseases (CVDs). WHO https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)
- 2.Bloom D, Cafiero E, Jané-Llopis E, Abrahams-Gessel S, Bloom L, et al. 2011. The Global Economic Burden of Non-Communicable Diseases. Geneva: World Econ. Forum [Google Scholar]
- 3.Sniderman AD, Furberg CD. 2008. Age as a modifiable risk factor for cardiovascular disease. Lancet 371:1547–49 [DOI] [PubMed] [Google Scholar]
- 4.Steenman M, Lande G. 2017. Cardiac aging and heart disease in humans. Biophys. Rev 9:131–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, et al. 2012. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2224–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khera AV, Kathiresan S. 2017. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat. Rev. Genet 18:331–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Touzé E, Rothwell PM. 2007. Heritability of ischaemic stroke in women compared with men: a genetic epidemiological study. Lancet Neurol. 6:125–33 [DOI] [PubMed] [Google Scholar]
- 8.O’Donnell CJ, Nabel EG. 2011. Genomics of cardiovascular disease. N. Engl. J. Med 365:2098–109 [DOI] [PubMed] [Google Scholar]
- 9.Loewe L, Hill WG. 2010. The population genetics of mutations: good, bad and indifferent. Philos. Trans. R. Soc. B 365:1153–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martincorena I, Campbell PJ. 2015. Somatic mutation in cancer and normal cells. Science 349:1483–89 [DOI] [PubMed] [Google Scholar]
- 11.Forsberg LA, Gisselsson D, Dumanski JP. 2017. Mosaicism in health and disease—clones picking up speed. Nat. Rev. Genet 18:128–42 [DOI] [PubMed] [Google Scholar]
- 12.Luzzatto L 2011. Somatic mutations in cancer development. Environ. Health 10(Suppl. 1):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, et al. 2014. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371:2488–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, et al. 2017. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med 377:111–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, et al. 2014. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371:2477–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gollob MH, Jones DL, Krahn AD, Danis L, Gong X-Q, et al. 2006. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med 354:2677–88 [DOI] [PubMed] [Google Scholar]
- 17.Bonnefond A, Skrobek B, Lobbens S, Eury E, Thuillier D, et al. 2013. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat. Genet 45:1040–43 [DOI] [PubMed] [Google Scholar]
- 18.Vijg J 2014. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev 26:141–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, et al. 2012. The origin and evolution of mutations in acute myeloid leukemia. Cell 150:264–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowman RL, Busque L, Levine RL. 2018. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 22:157–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busque L, Mio R, Mattioli J, Brais E, Blais N, et al. 1996. Nonrandom X-inactivation patterns in normal females: Lyonization ratios vary with age. Blood 88:59–65 [PubMed] [Google Scholar]
- 22.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, et al. 2012. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet 44:1179–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, et al. 2012. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet 44:651–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, et al. 2012. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet 44:642–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, et al. 2014. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med 20:1472–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vineis P, Schatzkin A, Potter JD. 2010. Models of carcinogenesis: an overview. Carcinogenesis 31:1703–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, et al. 2015. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126:9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silver AJ, Jaiswal S. 2019. Clonal hematopoiesis: pre-cancer PLUS. Adv. Cancer Res 141:85–128 [DOI] [PubMed] [Google Scholar]
- 29.Shlush LI. 2018. Age-related clonal hematopoiesis. Blood 131:496–504 [DOI] [PubMed] [Google Scholar]
- 30.Loh PR, Genovese G, Handsaker RE, Finucane HK, Reshef YA, et al. 2018. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559:350–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, et al. 2017. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130:742–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, et al. 2015. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 10:1239–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young AL, Challen GA, Birmann BM, Druley TE. 2016. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun 7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henry CJ, Marusyk A, DeGregori J. 2011. Aging-associated changes in hematopoiesis and leukemogenesis: What’s the connection? Aging 3:643–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konieczny J, Arranz L. 2018. Updates on old and weary haematopoiesis. Int. J. Mol. Sci 19:2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, et al. 2018. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med 24:1015–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, et al. 2018. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559:400–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, et al. 2019. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 4:25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mas-Peiro S, Hoffmann J, Fichtlscherer S, Dorsheimer L, Rieger MA, et al. 2019. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J. 2019:ehz591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, et al. 2017. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J. Clin. Oncol 35:1598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takahashi K, Wang F, Kantarjian H, Doss D, Khanna K, et al. 2017. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 18:100–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sano S, Wang Y, Walsh K. 2018. Clonal hematopoiesis and its impact on cardiovascular disease. Circ. J 83:2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, et al. 2017. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21:374–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kahn JD, Miller PG, Silver AJ, Sellar RS, Bhatt S, et al. 2018. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 132:1095–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swisher EM, Harrell MI, Norquist BM, Walsh T, Brady M, et al. 2016. Somatic mosaic mutations in PPM1D and TP53 in the blood of women with ovarian carcinoma. JAMA Oncol. 2:370–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zajkowicz A, Butkiewicz D, Drosik A, Giglok M, Suwinski R, Rusin M. 2015. Truncating mutations of PPM1D are found in blood DNA samples of lung cancer patients. Br. J. Cancer 112:1114–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Artomov M, Rivas MA, Genovese G, Daly MJ. 2017. Mosaic mutations in blood DNA sequence are associated with solid tumor cancers. NPJ Genom. Med 2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abegunde SO, Buckstein R, Wells RA, Rauh MJ. 2018. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol 59:60–65 [DOI] [PubMed] [Google Scholar]
- 49.Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, et al. 2018. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557:580–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hasselbalch HC. 2012. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 119:3219–25 [DOI] [PubMed] [Google Scholar]
- 51.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, et al. 2017. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355:842–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. 2018. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ. Res 123:335–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, et al. 2018. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol 71:875–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sano S, Wang Y, Yura Y, Sano M, Oshima K, et al. 2019. JAK2V617F-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl. Sci In press 10.1016/j.jacbts.2019.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ito S, Shen L, Dai Q, Wu SC, Collins LB, et al. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333:1300–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fuster JJ, Walsh K. 2018. Somatic mutations and clonal hematopoiesis: unexpected potential new drivers of age-related cardiovascular disease. Circ. Res 122:523–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, et al. 2015. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525:389–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, et al. 2017. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 130:753–62 [DOI] [PubMed] [Google Scholar]
- 59.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, et al. 2009. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 113:6403–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, et al. 2009. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 23:905–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, et al. 2009. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet 41:838–42 [DOI] [PubMed] [Google Scholar]
- 62.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, et al. 2011. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20:11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, et al. 2011. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20:25–38 [DOI] [PubMed] [Google Scholar]
- 64.Li Z, Cai X, Cai CL, Wang J, Zhang W, et al. 2011. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118:4509–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He Y, Hara H, Nunez G. 2016. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci 41:1012–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, et al. 2017. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med 377:1119–31 [DOI] [PubMed] [Google Scholar]
- 67.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, et al. 2018. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 391:319–28 [DOI] [PubMed] [Google Scholar]
- 68.Svennson E, Madar A, Campbell C, He Y, Sultan M, et al. 2018. TET2-driven clonal hematopoiesis predicts response to canakinumab in the CANTOS trials: an exploratory analysis. Circulation 138(Suppl.1):15111 [Google Scholar]
- 69.Lyko F 2018. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet 19:81–92 [DOI] [PubMed] [Google Scholar]
- 70.Challen GA, Sun D, Jeong M, Luo M, Jelinek J, et al. 2011. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet 44:23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H. 2007. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med 204:715–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guryanova OA, Lieu YK, Garrett-Bakelman FE, Spitzer B, Glass JL, et al. 2016. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia 30:1133–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang X, Su J, Jeong M, Ko M, Huang Y, et al. 2016. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet 48:1014–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cole CB, Russler-Germain DA, Ketkar S, Verdoni AM, Smith AM, et al. 2017. Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J. Clin. Investig 127:3657–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, et al. 2017. Dnmt3a restrains mast cell inflammatory responses. PNAS 114:E1490–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gamper CJ, Agoston AT, Nelson WG, Powell JD. 2009. Identification of DNA methyltransferase 3a as a T cell receptor–induced regulator of Th1 and Th2 differentiation. J. Immunol 183:2267–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pham D, Yu Q, Walline CC, Muthukrishnan R, Blum JS, Kaplan MH. 2013. Opposing roles of STAT4 and Dnmt3a in Th1 gene regulation. J. Immunol 191:902–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu Q, Zhou B, Zhang Y, Nguyen ET, Du J, et al. 2012. DNA methyltransferase 3a limits the expression of interleukin-13 in T helper 2 cells and allergic airway inflammation. PNAS 109:541–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li X, Zhang Q, Ding Y, Liu Y, Zhao D, et al. 2016. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol 17:806–15 [DOI] [PubMed] [Google Scholar]
- 80.Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, et al. 2016. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 128:1121–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hammarén HM, Virtanen AT, Raivola J, Silvennoinen O. 2019. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 118:48–63 [DOI] [PubMed] [Google Scholar]
- 82.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, et al. 2005. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365:1054–61 [DOI] [PubMed] [Google Scholar]
- 83.Shammo JM, Stein BL. 2016. Mutations in MPNs: prognostic implications, window to biology, and impact on treatment decisions. Hematology 2016:552–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, et al. 2005. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7:387–97 [DOI] [PubMed] [Google Scholar]
- 85.Mead AJ, Mullally A. 2017. Myeloproliferative neoplasm stem cells. Blood 129:1607–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, et al. 2005. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434:1144–48 [DOI] [PubMed] [Google Scholar]
- 87.Lasho TL, Mesa R, Gilliland DG, Tefferi A. 2005. Mutation studies in CD3+, CD19+ and CD34+ cell fractions in myeloproliferative disorders with homozygous JAK2V617F in granulocytes. Br. J. Haematol 130:797–99 [DOI] [PubMed] [Google Scholar]
- 88.Larsen TS, Christensen JH, Hasselbalch HC, Pallisgaard N. 2007. The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders. Br. J. Haematol 136:745–51 [DOI] [PubMed] [Google Scholar]
- 89.Ishii T, Bruno E, Hoffman R, Xu M. 2006. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood 108:3128–34 [DOI] [PubMed] [Google Scholar]
- 90.Chiang Y-H, Chang Y-C, Lin H-C, Huang L, Cheng C-C, et al. 2017. Germline variations at JAK2, TERT, HBS1L-MYB and MECOM and the risk of myeloproliferative neoplasms in Taiwanese population. Oncotarget 8:76204–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cordua S, Kjaer L, Skov V, Pallisgaard N, Hasselbalch HC, Ellervik C. 2019. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 134:469–79 [DOI] [PubMed] [Google Scholar]
- 92.Wang W, Liu W, Fidler T, Wang Y, Tang Y, et al. 2018. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2V617F mice. Circ. Res 123:e35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, et al. 2018. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med 10:eaan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou Z, Zhang S, Ding S, Abudupataer M, Zhang Z, et al. 2019. Excessive neutrophil extracellular trap formation aggravates acute myocardial infarction injury in apolipoprotein E deficiency mice via the ROS-dependent pathway. Oxidative Med. Cell. Longev 2019:1209307–07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, et al. 2014. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 123:141–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, et al. 2017. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med 214:439–58 [DOI] [PMC free article] [PubMed] [Google Scholar]