

Abstract
Frontotemporal dementia (FTD) is a common neurogenerative disorder characterized by progressive degeneration in the frontal and temporal lobes. Heterozygous mutations in the gene encoding progranulin (PGRN) are a common genetic cause of FTD. Recently, PGRN has emerged as an important regulator of lysosomal function. Here, we examine the impact of PGRN mutations on the processing of full-length prosaposin to individual saposins, which are critical regulators of lysosomal sphingolipid metabolism. Using FTD-PGRN patient-derived cortical neurons differentiated from induced pluripotent stem cells, as well as post-mortem tissue from patients with FTLD-PGRN, we show that PGRN haploinsufficiency results in impaired processing of prosaposin to saposin C, a critical activator of the lysosomal enzyme glucocerebrosidase (GCase). Additionally, we found that PGRN mutant neurons had reduced lysosomal GCase activity, lipid accumulation and increased insoluble α-synuclein relative to isogenic controls. Importantly, reduced GCase activity in PGRN mutant neurons is rescued by treatment with saposin C. Together, these findings suggest that reduced GCase activity due to impaired processing of prosaposin may contribute to pathogenesis of FTD resulting from PGRN mutations.
Introduction
Frontotemporal dementia (FTD) encompasses a group of heterogeneous neurodegenerative disorders characterized by atrophy in the frontal and temporal lobes (1, 2). FTD is a major cause of early-onset dementia (<65 years) and is estimated to affect 20 000–30 000 patients each year (2–4). Approximately 40% of FTD patients have a family history of dementia, highlighting a strong genetic component to the development of the disease (5, 6). Heterozygous mutations in PGRN, which encodes progranulin (PGRN), are a common genetic cause of FTD (7–9). To date, over 70 PGRN mutations have been identified (10, 11). These mutations result in decreased production of PGRN in mutant cells and cause disease through haploinsufficiency (8, 12–15). PGRN is a highly conserved, multifunctional protein that is expressed in neurons and microglia within the CNS. Although PGRN has been implicated in a wide array of biological functions including inflammation (16–18), wound repair (19–21) and neurite outgrowth (22–24), its exact function and the mechanism by which PGRN haploinsufficiency leads to neurodegeneration remains unclear.
Several lines of evidence implicate PGRN in the regulation of general lysosomal function. This link was first established with the discovery that rare homozygous PGRN mutations, which result in no detectable PGRN production, cause the neurodegenerative lysosomal storage disorder neuronal ceroid lipofuscinosis (25, 26). Furthermore, previous studies demonstrate that PGRN localizes to the lysosomal compartment (27–29) and that partial or complete reduction of PGRN in mice cause an age-dependent, progressive up-regulation of lysosomal genes (30–32). Additionally, we and others have shown that FTD-linked PGRN mutations lead to significantly reduced lysosomal proteolysis and development of pathological phenotypes that resemble the characteristic hallmarks of neuronal ceroid lipofuscinosis (33–35).
PGRN has also been shown to interact with another key lysosomal regulator protein called prosaposin (36, 37). Prosaposin is a secreted glycoprotein that is delivered to the lysosome either directly from the trans-Golgi network or from the extracellular space via the endocytic pathway. Within the lysosome, prosaposin is cleaved into four lysosomal coactivators, saposins A–D (38). Each saposin has a distinct role as an activator of specific lysosomal enzymes involved in sphingolipid metabolism. Loss or dysfunction of individual saposins results in distinct lysosomal storage disorders (39–47). This includes saposin C, a critical activator of the enzyme glucocerebrosidase (GCase). Loss of saposin C clinically resembles Gaucher disease (GD), which is typically associated with homozygous loss-of-function mutations in GBA1, the gene that encodes GCase (45, 47). Additionally, heterozygous GBA1 mutations are a significant risk factor for Lewy body dementia, which is closely related to FTD. Finally, several patients with FTD resulting from PGRN mutations were recently found to have co-existing LBD, further highlighting the relationship between these disorders (48).
Previous studies have shown that PGRN facilitates the trafficking of prosaposin to the lysosome (36). Additionally, we previously demonstrated that cathepsin D activity is decreased in PGRN mutant neurons (33). Since cathepsin D has been shown to be predominately responsible for the proteolytic cleavage of full-length prosaposin into saposins (49, 50), reduced cathepsin D activity may result in a lower conversion of full-length prosaposin to saposins. Thus, we hypothesized that PGRN deficiency may result in reduced saposin levels due to either impaired trafficking of prosaposin to the lysosome or impaired cleavage of prosaposin. Specifically, we aimed to examine if levels of saposin C, and consequently GCase activity, are altered due to PGRN deficiency.
To examine this question, we utilized FTD-PGRN patient-derived cortical neurons obtained via induced pluripotent stem cells (iPSCs) as well as isogenic corrected iPSC-derived cortical neurons. Using this model, we examined if PGRN deficiency alters either trafficking of prosaposin to the lysosome or its cleavage into individual saposins. While we did not observe any defects in prosaposin trafficking, we observed a significant reduction in processing of prosaposin in PGRN mutant cortical neurons relative to isogenic controls. A similar impairment in prosaposin processing was observed in post-mortem cortex from FTLD patients with PGRN mutations. This impaired processing led to a significant reduction in saposin C in iPSC-derived cortical neurons and in post-mortem tissue with PGRN deficiency. Furthermore, we observed a significant reduction in lysosomal GCase activity associated with accumulation of lipids and insoluble α-synuclein.
Results
Prosaposin processing is impaired in iPSC-derived PGRN mutant neurons and FTLD-PGRN patient cortical tissue
To investigate the effect of PGRN mutations on trafficking and processing of prosaposin, we generated iPSCs from skin-biopsy derived fibroblasts of a FTD patient with a heterozygous PGRN mutation (c.26 C > A, p.A9D) (PGRN mutant) (15). To ensure the specificity of observed phenotypes, we used a CRISPR/Cas9 editing strategy to correct the point mutation and generate an isogenic iPSC line (PGRN WT) (33). Correction of the PGRN mutation in the iPSC line was previously confirmed by Sanger sequencing and resulted in increased PGRN expression in both PGRN WT iPSCs and iPSC-derived neurons (33). Additionally, previous characterization demonstrated presence of pluripotency markers in the PGRN WT and mutant iPSC and confirmed a normal karyotype in both lines (33). As cortical neurons are predominantly affected in FTD, we differentiated the iPSCs into human cortical neurons using the previously described Ngn2-overexpression protocol (51). These neurons have been previously described to contain >98% Map 2 and vGlut positive cells and shown to express cortical neuron markers (51).
A previous study found that PGRN facilitates neuronal uptake of prosaposin and its delivery to lysosomes (36). Thus, we began by examining the trafficking of prosaposin in PGRN WT and mutant neurons. To examine whether prosaposin trafficking to the lysosome was impaired, we fixed and stained PGRN WT and mutant cortical neurons for prosaposin, LAMP1 (a marker of lysosomes) and β-III-tubulin (a general neuronal marker) (Fig. 1A). Lysosomal localization of prosaposin was assessed by examining its colocalization with LAMP1 using Pearson’s coefficient. Using this approach, we found no significant difference in localization of prosaposin in PGRN mutant neurons relative to PGRN WT neurons (Fig. 1B). We then examined whether PGRN mutations alter the processing of prosaposin by using western blot analysis to determine levels of prosaposin, saposin C and NSE (loading control) in lysates from PGRN WT and mutant neurons (Fig. 1C). Prosaposin and saposin C intensity was determined by densitometric analysis and normalized to the respective intensity of NSE. This analysis revealed a significant increase in prosaposin (Fig. 1D) as well as a significant reduction in saposin C (Fig. 1E) in PGRN mutant neurons relative to WT. Using the same analysis, we calculated the processing of prosaposin, as defined by the ratio of saposin C/prosaposin and found a significant reduction in overall processing of prosaposin in PGRN mutant neurons as compared with PGRN WT neurons (Fig. 1F).
Figure 1.
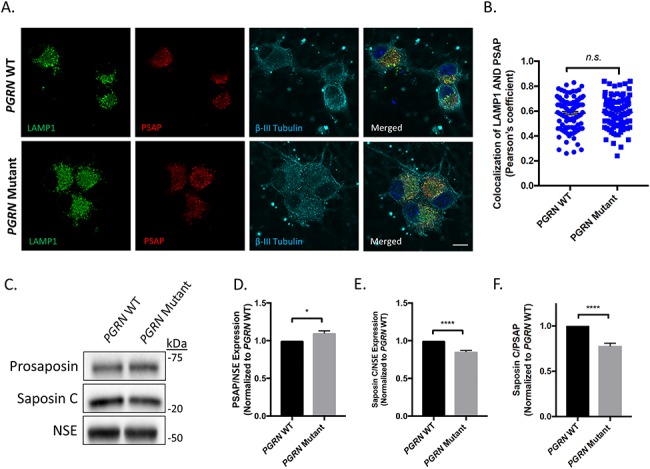
Prosaposin processing, but not localization, is affected in iPSC-derived PGRN mutant neurons. (A) PGRN WT and PGRN mutant cortical neurons were fixed (day 75 post-differentiation) and immunostained for LAMP1 (lysosomal marker), prosaposin (PSAP) and β-III tubulin (neuronal marker) and visualized using IF (n = 3, 27–36 cells/experiment) Scale bar = 10 μm. (B) Scatter plots showing LAMP1/prosaposin co-localization quantification as calculated by Pearson’s coefficient. Nuclei were visualized using DAPI. (C) Immunoblots for prosaposin, saposin C and NSE in PGRN WT and mutant neurons (day 125 post-differentiation) (n = 3). Quantification of (D) prosaposin and (E) saposin C determined by densitometric analysis of western blots. Prosaposin and saposin C expression levels were normalized to corresponding NSE values and divided by the mean value obtained from the control samples. (F) The ratio of saposin C to prosaposin was then used to determine degree of prosaposin processing in the neuronal samples. Data are presented as the mean ± SEM, n.s. (not significant), *P < 0.05, ****P < 0.0001 and two tailed Student’s t-test.
As we observed altered prosaposin processing in iPSC-derived PGRN mutant neurons, we aimed to determine if this effect would be seen in patients with different PGRN mutations. Thus, we examined prosaposin processing in post-mortem brain tissue of FTD patients with PGRN mutations. Homogenates were prepared from the frontal cortex of four patients with a confirmed neuropathological diagnosis of FTLD with PGRN mutations (FTLD-PGRN), four unaffected patients as population controls, and three FTLD patients without PGRN mutations (FTLD-PGRN WT) (Table 1). The patients with FTLD-PGRN were confirmed by sequencing to have heterozygous PGRN mutations (Table 1). These brain lysates were analyzed by western blot analysis for levels of prosaposin, saposin C, PGRN and GAPDH (loading control) (Fig. 2A–D). This analysis revealed a significant reduction in the total level of saposin C (Fig. 2C) as well as a significant impairment in processing of prosaposin (Fig. 2D) in patients with FTLD-PGRN that were not observed in either patient controls or FTLD patients without PGRN mutations. There was also a trend toward increased levels of prosaposin in FTLD-PGRN patients, but this did not reach significance in this data set (P = 0.084) (Fig. 2B).
Table 1.
Summary of demographic information for donors of cortical samples
| Case | Age | Sex | PGRN mutations | PMI (h) | Neuropathological diagnosis |
|---|---|---|---|---|---|
| CTRL-1 | 76 | F | n/a | 13 | Control |
| CTRL-2 | 81 | M | n/a | 17 | Control |
| CTRL-3 | 88 | M | n/a | 27 | Control |
| CTRL-4 | 95 | M | n/a | 13 | Control |
| FTLD-1 | 62 | F | c.102delC | 22 | FTLD-TDP type A |
| FTLD-2 | 65 | M | c.388_391delCAGT | 6 | FTLD-TDP type A |
| FTLD-3 | 56 | F | c.910_911dupTG | 11 | FTLD-TDP type A |
| FTLD-4 | 70 | M | c.−8 + 3A > G | 4 | FTLD-TDP type A |
| FTLD-1 | 73 | M | n/a | 23 | FTLD-TDP type A |
| FTLD-2 | 71 | M | n/a | 18 | FTLD-TDP type A |
| FTLD-3 | 82 | M | n/a | 20 | FTLD-TDP type A |
Figure 2.
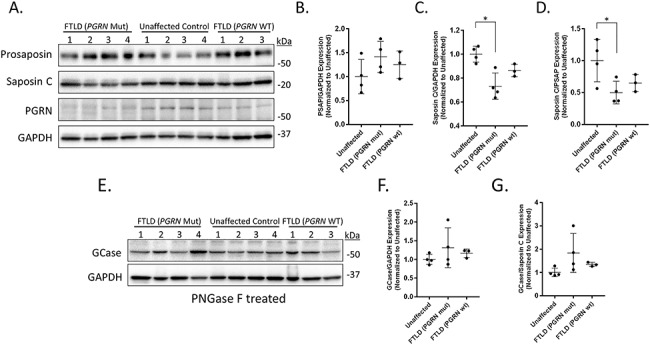
Prosaposin processing is impaired in the cortex of FTLD patients with PGRN mutations. Cortical tissue samples from patients with FTLD (PGRN mutation), unaffected control and FTLD (PGRN WT) were homogenized and examined via western blot analysis. (A) Immunoblots for prosaposin, saposin C, PGRN and GAPDH (loading control). Scatter plots showing quantification of (B) prosaposin and (C) saposin C for the individual cohorts. Relative prosaposin and saposin C expression levels were normalized to corresponding GAPDH values and divided by the mean value obtained from the unaffected samples. (D) The ratio of saposin C to prosaposin was then used for determination of the degree of prosaposin processing in the tissue. (E) Additional homogenate was pretreated with PNGase F prior to western blot analysis for GCase and GAPDH. Scatter plots showing quantification of (F) GCase expression and (G) the correlation of GCase and saposin C. Data are presented as the mean ± SD, n = 4 FTLD (PGRN Mut) and unaffected, n = 3 FTLD (PGRN WT), *P < 0.05, one-way ANOVA followed by Tukeys multiple comparisons post hoc test.
Since we observed decreased levels of saposin C in FTLD-PGRN patient samples, we then examined if PGRN mutations alter GCase protein levels within these samples. Prior to western blot analysis of GCase and GAPDH (loading control), the post-mortem tissue samples were pretreated with peptide-N-Glycosidase F (PNGase F) in order to remove glycosylation from GCase, which can lead to variable protein detection between different patient samples. This analysis found no significant difference in GCase protein levels (Fig. 2E and F) nor any correlation between GCase and saposin C levels among any of the groups (Fig. 2G), indicating that reduced saposin C levels observed in FTLD-PGRN patients did not significantly affect GCase levels.
Impaired processing of prosaposin leads to reduced lysosomal GCase activity in iPSC-derived PGRN mutant neurons
Recently, patients with loss-of-function mutations in saposin C were found to develop a disease similar to GD (47). Although GD is typically caused by loss-of-function mutations in the lysosomal hydrolase GCase, this loss of GCase function is also recapitulated by mutations in saposin C, highlighting the role of saposin C as an essential and potent activator of GCase enzymatic activity (52). Accordingly, we hypothesized that impaired processing of prosaposin to saposin C may lead to reduced lysosomal GCase activity in PGRN mutant neurons. To examine lysosomal GCase activity, we initially performed fractionation of the neuronal lysate using a lysosomal enrichment strategy (Fig. 3A). We then performed a GCase activity assay to measure levels of GCase activity in the lysosome-enriched fraction lysates of PGRN WT and mutant neurons and found that PGRN mutant neurons have significantly decreased levels of lysosomal GCase activity as compared with PGRN WT neurons (Fig. 3B).
Figure 3.
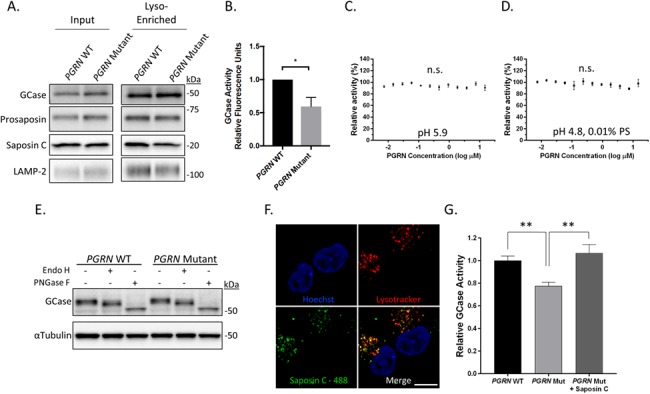
GCase activity is reduced in iPSC-derived PGRN mutant neurons and can be rescued by increasing saposin C. (A) Western blot of total lysate input and lysosome-enriched samples of PGRN WT and mutant neurons (day 65 post-differentiation) (n = 3). (B) Quantification of GCase activity assay using lysates of lysosome-enriched samples from PGRN WT and mutant neurons (n = 3). GCase activity assay using recombinant GCase incubated with recombinant PGRN at (C) pH 5.9 or (D) pH 4.8 with 0.01% phosphatidylserine (n = 3). (E) Western blot analysis of PGRN WT and mutant neuron lysates treated with Endo H, PNGase F or untreated control. (F) Confocal images of PGRN mutant neurons showing colocalization of Alexafluor-488 labeled saposin C with lysotracker positive compartments after 3 hour treatment with saposin C (200 ng/ml) (n = 3). (G) Quantification of lysosomal GCase activity in PGRN WT and mutant neurons with and without treatment with recombinant saposin C (200 ng/ml). Scale bar = 10 μm. Data are presented as mean ± SEM, n.s. (not significant), *P < 0.05, **P < 0.05, (A) two tailed Student’s t-test, (G) one-way ANOVA followed by Tukeys multiple comparisons post hoc test.
Recently, several studies have utilized co-immunoprecipitation assays to demonstrate that PGRN and GCase interact (53, 54). These observations highlight the possibility that PGRN can directly modulate GCase activity, which may explain the reduced lysosomal GCase activity we observed in PGRN mutant neurons. To test this hypothesis, we performed an in vitro GCase assay using recombinant GCase and increasing concentrations of recombinant PGRN at pH 5.9 (Fig. 3C). Additionally, we repeated this experiment using the lipid phosphatidylserine at pH 4.8 as previous studies have shown that phosphatidylserine is required for maximal activation of GCase by its known activator, saposin C (Fig. 3D) (40). In both experiments we found that PGRN had no effect, demonstrating that PGRN does not directly modulate GCase activity. These results suggest that the decreased lysosomal GCase activity observed in the patient-derived PGRN mutant neurons occurs through an alternate mechanism, such as decreased availability of its activator saposin C or impaired GCase trafficking.
We therefore examined if trafficking of GCase from the endoplasmic reticulum (ER) is impaired in PGRN mutant neurons. Since GCase is post-translationally modified as it travels along the secretory pathway, we utilized endoglycosidase H (Endo H) and PNGase F digestion to measure the extent of oligosaccharide processing that GCase has undergone along this pathway. Although PNGase F digestion removed all glycosylation, sensitivity to Endo H digestion indicates that a protein has been retained in the ER and would result in the appearance of an additional GCase immunopositive band at similar molecular weight to PNGase F treatment. iPSC-derived PGRN WT and mutant neurons treated with either Endo H or PNGase F were then analyzed using western blot analysis. These results show no difference between PGRN WT and mutant neurons upon Endo H digestion (Fig. 3E), suggesting that GCase trafficking is not impaired in patient-derived PGRN mutant neurons.
We then examined whether increasing levels of lysosomal saposin C can restore lysosomal GCase activity in PGRN mutant neurons. To accomplish this, we generated recombinant saposin C using previously established protocols (55) and added it to the cell culture media. Importantly, we began by examining if the recombinant saposin C would be taken up by the cells and properly targeted to the lysosome using live cell imaging. PGRN mutant neurons were plated on live-cell imaging dishes and treated with Alexa Fluor 488-labeled recombinant saposin C. After a 3 h incubation, lysotracker red was added to the iPSC-derived neurons and the cells were imaged by confocal microscopy. Examination of the confocal micrographs reveal significant colocalization of Alexa Fluor 488 with lysotracker red, indicating that saposin C was localized within lysosomes (Fig. 3F). To measure the effect of saposin C uptake on GCase activity, we cultured PGRN WT and mutant neurons in 96 well assay plates and examined lysosomal GCase activity upon application of recombinant saposin C (at a final concentration of 200 ng/ml) to the cell culture media. Following a 3 h incubation, the neurons were assayed for lysosomal GCase activity in real-time using the GCase substrate PFB-FDGlu as previous described (56). Utilizing this assay, we confirmed our previous findings that PGRN mutant neurons have significantly decreased levels of lysosomal GCase activity relative to WT neurons (Fig. 3G). Additionally, treatment of PGRN mutant neurons with recombinant saposin C significantly increased lysosomal GCase activity to levels comparable with those observed in PGRN WT neurons (Fig. 3G). A similar effect was observed for patient-derived fibroblasts with a heterozygous PGRN mutation (c.26 C > A, p.A9D), which were used to generate the iPSC lines in this study (Fig. S1A and B). Together these results demonstrate that recombinant saposin C can be used to increase lysosomal GCase activity levels in PGRN mutant neurons.
PGRN mutations result in accumulation of neutral lipids and insoluble α-synuclein
The effects of reduced GCase activity has been studied extensively, as GBA1 mutations are the most common genetic risk factor for synucleinopathies, including Lewy Body Dementia, and Parkinson’s disease (57, 58). Thus, we were interested in investigating if PGRN mutant neurons exhibit phenotypes typically associated with decreased GCase activity. We previously demonstrated that long-term cultures are required to detect pathological phenotypes in iPSC-derived patient neurons. As we observed decreased GCase activity in PGRN mutant neurons 65 days post-differentiation, we examined PGRN WT and mutant neurons 125 days post-differentiation for pathological phenotypes. We began by examining whether PGRN mutant neurons display lipid accumulation using BODIPY 493/503, a fluorophore which stains neutral lipids, since loss of GCase activity was previously shown to be associated with an accumulation of lipids (59, 60). BODIPY was added the media of PGRN WT and mutant neurons. After a 30 min incubation, PGRN WT and mutant neurons were fixed, stained with DAPI, and then imaged using confocal microscopy. Examination of the confocal micrographs revealed a significant increase in number of BODIPY puncta in PGRN mutant neurons as compared with WT (Fig. 4A). Quantification of the images demonstrated that the number of puncta per cell, average particle size and the overall BODIPY fluorescence (Fig. 4B–D) were significantly increased in PGRN mutant neurons relative to WT, indicating a significant accumulation of lipids in PGRN mutant neurons.
Figure 4.
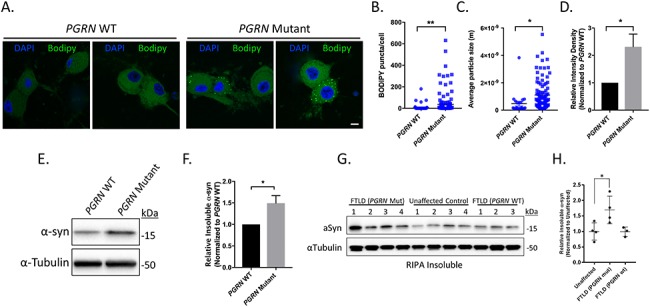
Lipid and insoluble α-synuclein accumulation in iPSC-derived PGRN mutant neurons and cortical tissue lysates from FTLD patients with PGRN mutations. (A) Neutral lipids in PGRN WT and mutant neurons were visualized using BODIPY 493/503 via IF (day 125 post-differentiation) (n = 3, 18–65 cells/experiment) Scale bar = 10 μm. Quantification of (B) BODIPY puncta/cell, (C) average BODIPY particle size, and (D) relative BODIPY intensity density in PGRN WT and mutant neurons. PGRN mutant neurons intensity samples were divided by the mean value obtained from PGRN WT samples. (E) Western blots of the insoluble protein fraction of PGRN WT and mutant neurons samples immunoblotted for α-synuclein and α-tubulin (loading control) (day 125 post-differentiation). (F) Quantification of insoluble α-synuclein in PGRN WT and mutant neuron lysates (n = 4). (G) Western blots of the insoluble protein fraction from cortical tissue samples from FTLD (PGRN mutant), unaffected control and FTLD (PGRN WT) immunoblotted for α-synuclein and α-tubulin (loading control). (H) Quantification of insoluble α-synuclein in cortical tissue samples. α-synuclein expression was normalized to the corresponding α-tubulin and divided by the mean value obtained from the unaffected samples. The data are presented as the mean ± SEM, *P < 0.05, **P < 0.01, (B,C,F) two tailed Student’s t-test, (H) one-way ANOVA followed by Tukeys multiple comparisons post hoc test.
Loss of GCase activity has also been shown to lead to increased α-synuclein aggregation (61, 62). To examine whether α-synuclein aggregation is occurring in PGRN mutant neurons, we further extracted the insoluble pellet by heating the sample in PBS containing 2% SDS. Western blot analysis revealed a significant increase of insoluble α-synuclein in PGRN mutant neurons as compared with PGRN WT neurons (Fig. 4E and F). We then examined if a similar effect could be observed in post-mortem tissue of FTLD-PGRN patients. The insoluble fraction from the previous experiment (Fig. 2) was further extracted in PBS containing 2% SDS and analyzed using western blot analysis (Fig. 4G). Quantification revealed a significant increase in insoluble α-synuclein levels in patients with FTLD-PGRN that were not observed in either patient controls or FTLD patients without PGRN mutations (Fig. 4H). Together these results highlight consequences of impaired prosaposin processing in PGRN mutant neurons.
Discussion
Using iPSC-derived patient PGRN mutant neurons and post-mortem brain samples of FTLD patients with PGRN mutations, we have shown that heterozygous PGRN mutations result in impaired processing of prosaposin and reduced cellular levels of saposin C, a critical activator of the lysosomal enzyme GCase. Furthermore, we found that PGRN mutant neurons have significantly decreased levels of lysosomal GCase activity as compared with isogenic controls, which can be restored by application of recombinant saposin C. Finally, we identified pathological phenotypes typically associated with decreased GCase activity present in PGRN mutant samples, including lipid accumulation in patient-derived PGRN mutant neurons and accumulation of insoluble α-synuclein in PGRN mutant neurons and post-mortem samples of FTLD-PGRN patients. Taken together, these data provide insight into the link between FTLD-PGRN, Parkinson’s disease and lysosomal storage disorders.
An important question that arises from these data is how prosaposin processing is affected in PGRN mutations. We and others have previously published that both full-length PGRN and granulin E interact with and increase the proteolytic activity of the lysosomal enzyme cathepsin D (33, 34). As cathepsin D was previously shown to be predominantly responsible for the proteolytic cleavage of full-length prosaposin into saposins (49, 50), there is a strong possibility that the reduced processing of prosaposin we observed in PGRN mutant neurons is caused by reduced cathepsin D activity. However, PGRN is also partially involved in prosaposin trafficking to the lysosome, so it is also possible that impaired trafficking of prosaposin to the lysosome in PGRN mutant neurons is responsible for the reduction in prosaposin processing. Our results however, demonstrated that trafficking of prosaposin to the lysosome is not affected in iPSC-derived PGRN mutant cortical neurons. Therefore, it seems more likely that impaired processing of prosaposin is a result of the decreased cathepsin D activity in PGRN mutant neurons.
A key observation in this work is that we observed reduced lysosomal GCase activity in PGRN mutant neurons. Heterozygous mutations in GBA1, which lead to reduced GCase activity, are a well-established genetic risk factor for Parkinson disease and Lewy body dementia (58, 63). Interestingly, mild parkinsonism is seen with high frequency (approximately 40–60%) in FTD patients with PGRN mutations during the late stages of the disease (64). Therefore, it is possible that reduced GCase activity contributes to the parkinsonism observed in FTLD-PGRN patients. Additionally, recent work has specifically shown that FTLD-PGRN patients also have coexisting Lewy body disease (48), highlighting the potential pathophysiological connection between these disorders. This connection is also strengthened by a recent unbiased lipidomic analysis, which suggested that PGRN is involved in lipid homeostasis (32). The study demonstrated that FTLD patients with PGRN mutations had distinct changes in lipid composition as compared with AD patients, FTD patients without PGRN mutation, and population controls. Further lipidomic analysis using PGRN+/+, PGRN+/− and PGRN−/− mice showed that the lipid composition alterations observed in the PGRN+/− and PGRN−/− mice were found to be PGRN dose-dependent (32). Although our study specifically examined saposin C and its effects on GCase activity, it is likely that impaired prosaposin processing would also affect other saposins, which are also critical co-factors in sphingolipid metabolism. Although further studies are required, we speculate that the observed lipid phenotypes resulting from PGRN mutations may be due to the reductions in saposins, including the effect of saposin C on GCase.
Although the reduction in GCase is likely one of the many contributing factors in disease pathogenesis, it highlights the possibility that therapies targeted at increasing GCase activity may be a viable therapeutic strategy for treating FTD patients with PGRN mutations. It would be interesting to examine whether GCase activating therapies that are currently being evaluated for Parkinson’s disease would be effective for FTD patients with PGRN mutations.
Materials and Methods
Generation of induced pluripotent stem cells and neuronal differentiation
The generation and characterization of PGRN WT and mutant iPSC lines were previously described (33). It was previously shown that expression of the transcription factor neurogenin-2 (Ngn2) induces rapid differentiation of iPSCs into glutamatergic neurons (51). PGRN WT and mutant iPSCs were co-transfected with TetO-Ngn2-Puro and reverse tetracycline-controlled transactivator (rtTA) lentiviruses with 8 μg/ml polybrene (Sigma, H9268) and 10 μM rock inhibitor Y27632 (Millipore, 688000) (day 1). On day 2, Ngn2 expression was induced with 2 μM doxycycline hyclate (Sigma, D9891) in KSR media alone with 10 μM SB431442 (R&D, 1614), 2 μM XAV939 (Stemgent, 04–0046) and 100 nM LDN-193189 (Stemgent, 04-0074) (doxycycline hyclate is maintained in all medias going forward). On day 3, cells were fed with a 1:1 ratio of KSR media + SB/XAV/LDN and N2-supplmented neural induction media with 2 μg/ml puromycin (Life Technologies, A1113803). On day 4, cells were fed with N2-supplemented neural induction media. On day 5, cells were dissociated with accutase (Sigma, A6964) and plated onto PDL-(Sigma, P1149) and laminin-(Life Technologies, 23 017-015) coated tissue culture plates. Cells were subsequently maintained with neurobasal media (Life Technologies, 21103049) supplemented with NeuroCult SM1 (StemCell Technologies, 05711) and 10 ng/ml brain-derived neurotrophic factor (R&D, 248-BD-005/CF) until collected. It was previously demonstrated that cortical neurons produced using this protocol form functional synapses at day 21 (51). For our analysis, cells were collected beginning on day 35.
Virus generation
Lentiviruses were produced in HEK293T cells by co-transfection of either TetO-Ngn2-Puro or rtTA plasmids with two helper plasmids (psPAX2 and pLP3) (3 μg of lentiviral vector and 2.25 and 0.75 μg of helper plasmids, respectively) using X-treme Gene HP DNA transfection reagent (Roche, 06366236001). Media was changed 12 h after transfection and media containing lentiviruses were collected and filtered (0.45 μM filter) 48 h after initial transfection. Lentiviruses were then concentrated 100X using Lenti-X Concentration (Takara, 631 232) and resuspended in mTeSR1 media. Viral titers were determined using ZeptoMetrix RETROtek HIV-1 p24 Antigen ELISA kit (FisherScientific, 22-156-700).
Antibodies
The antibodies used in this study were: Vinculin (Abcam, ab18058), LAMP1 (DSHB Hybridoma, H4A3-s) (for western blots), LAMP1 (Santa Cruz, sc-20 011) [for immunofluorescence (IF)], LAMP2 (DSHB Hybridoma, H4B4-s), PGRN (Abcam, ab108608), saposin C (Santa Cruz, sc-374 119), Prosaposin (Sigma, HPA004426), NSE (ThermoFisher, PA5-12374), LIMP2 (gift from Michael Schwake), GAPDH (Millipore, MAB374), Progranulin (Abcam, ab108608), α-synuclein (Santa Cruz, sc-7011), α-tubulin (Sigma, T5168) and B-III tubulin (Biolegend, 801201). All antibodies were used at a 1:1000 dilution for western blot analysis (in 5% Bovine Serum Albumin, 0.5% Sodium Azide) and a 1:200 dilution for IF analysis (in 5% normal goat serum, 1% BSA).
Western blot analysis of iPSC-derived cortical neurons
Western blot analysis was performed using cellular lysates from PGRN WT and mutant cortical neurons. PGRN mutant and WT neuron samples were collected at specific time points (days 55 and 125 post-differentiation) in Triton-X Buffer (1% Triton X-100, 20 mM HEPES, 150 mM NaCl, 10% glycerol, 1 mM EDTA and 1.5 mM MgCl2) containing cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Roche, 11836170001). The samples were homogenized on ice and subsequently centrifuged at 100 000×g for 30 min to obtain the triton soluble fraction. The Triton-insoluble pellet was washed with PBS three times and subsequently resuspended in SDS buffer (2% SDS, 50 mM Tris pH 7.4), boiled, sonicated and then centrifuged at 20 000×g for 20 min to obtain Triton-insoluble fraction. Fractions were then analyzed using western blot analysis.
Colocalization of prosaposin and LAMP1
PGRN WT and mutant neurons were plated onto nitric acid treated glass coverslips and fixed with 4% formaldehyde/PBS for 10 min. The coverslips were then washed with PBS for 5 min three times. Neurons were then blocked and permeabilized using blocking buffer (5% normal goat serum, 1% BSA and 0.1% saponin) for 30 min at room temperature. Neurons were incubated at 4°C with primary antibody overnight and secondary antibody for 2 h at room temperature in blocking buffer. The coverslips were mounted using VECTASHIELD Hard Set Antifade Mounting Medium with DAPI (Vector Laboratories, H-1500). Images were subsequently taken using a Leica confocal microscope at 63X magnification and analyzed using Coloc2 analysis in Fiji software. Colocalization between prosaposin and LAMP1 was quantified using Pearson’s coefficient.
Analysis of patient tissue
Research involving coded brain samples was granted an exemption from requiring ethics approval by the Northwestern University Institutional Review Board. Samples were obtained from the Northwestern Alzheimer’s Disease Center. Samples of human cortex were resuspended in RIPA buffer (50 mM Tris HCl, pH 7.4, 150 nM NaCl, 1% triton X-100, 0.1% SDS and 0.5% sodium deoxycholate) with HALT protease and phosphatase inhibitor cocktail (ThermoFisher). The tissue was homogenized using a Tissuemiser Homogenizer (Fisher Scientific). The homogenate was cleared by centrifugation (20 000×g) and the supernatant was analyzed by western blot analysis. Band intensities were determined via densitometric analysis using ImageJ software. To obtain the insoluble fraction, the pellet from the RIPA extraction was first washed twice by resuspending the pellet in RIPA followed by centrifugation and removal of the supernatant. The remaining pellet was resuspended in PBS pH 7.4 containing 2% SDS. The resuspended pellet was heated at 95°C for 5 min vortexed forcefully then heated at 95°C for an additional 5 min. The resulting suspension was cleared by centrifugation (100 000×g). The resulting supernatant was subjected to western blot analysis.
Endo H and PNGase F assay
Endo H and PNGase F glycan cleavage sensitivity experiments were performed according to the manufacturers protocol (New England Biolabs, P0702S, P0704S). A total of 10 μg protein from lysates derived from patient cortical lysates or from PGRN WT and PGRN mutant iPSC-derived cortical neurons were denatured and treated with 1 μL PNGase or 2 μL Endo H (iPSC-derived cortical neurons) in the supplied assay buffer for 1 h at 37°C. Digested lysates were subjected to western blot analysis. GCase that is retained in the ER is sensitive to Endo H treatment and results in a band at equivalent molecular weight to PNGase F treated samples, which removes all glycosylation.
BODIPY analysis
Levels of intracellular neutral lipids were quantified through incorporation of BODIPY 493/503 (4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene), a fluorescent dye that detects neutral lipids (ThermoFisher, D3922). BODIPY 493/503 was added to the media of PGRN WT and mutant neurons (final concentration of 10 μg/ml) and incubated for 30 min at 37°C, 5% CO2. Cells were subsequently washed with warm PBS and fixed with 4% formaldehyde/PBS for 10 min. The coverslips were mounted using VECTASHIELD Hard Set Antifade Mounting Medium with DAPI (Vector Laboratories, H-1500). Images were taken using a Leica confocal microscope at 63X magnification and analyzed using Fiji software.
Lysosomal enrichment of iPSC-derived cortical neurons
Lysosome-enriched samples were generated from PGRN WT and mutant neurons cells using Lysosome Enrichment Kit for Tissues and Cultured Cells (ThermoFisher, 89 839). Neurons were grown on 10 cm plates and collected 65–75 days post-differentiation. The samples were processed according to the provided protocol and the lysosome-enriched pellet was resuspended in Triton-X Buffer (1% Triton X-100, 20 mM HEPES, 150 mM NaCl, 10% glycerol, 1 mM EDTA and 1.5 mM MgCl2).
In vitro GCase activity assay with PGRN
In vitro GCase activity was measured as previously described (56, 65). Recombinant WT GCase enzyme (Cerezyme, Genzyme) was diluted (7.5 nM final concentration) in buffer containing 50 mM citric acid, 176 mM K2HPO4 and 0.01% Tween-20 at pH 5.9. Additional samples were prepared at pH 4.8 with the addition of 0.01% Phosphatidylserine (Sigma). Recombinant PGRN was added at the indicated concentrations. The reaction was initiated by addition of Methylumbelliferyl β-glucophyranoside (4-MU, Sigma-Aldrich, M3633) (final concentration of 1.5 mM). The reaction was terminated after 30 min by addition of 1 M glycine solution (pH 10). The fluorescence of the hydrolyzed substrate was measured using a Synergy H1 multimode plate reader (Biotek) at excitation and emission of 365 and 440 nm, respectively.
Glucocerebrosidase activity assay
The GCase activity assay was performed as previously described (59, 66) using lysosome-enriched samples from PGRN WT and mutant neurons. The GCase activity assay was performed in a black, flat bottom 96 well plate (ThermoFisher, 475 515) using Methylumbelliferyl β-glucophyranoside (4-MU, Sigma-Aldrich, M3633) (final concentration of 2 mM) and 0.5 μg of sample lysates in a final volume of 100 μL of activity assay buffer (0.25% (v/v) Triton X-100, 0.25% (w/v) Taurocholic acid (Sigma-Aldrich, T9034), 1% BSA, 1 mM EDTA, in citrate/phosphate buffer, pH 5.4). An additional set of each sample was prepared in the presence of the conduritol-b-epoxide (CBE) (FisherScientific, 21 103 049), an inhibitor of lysosomal GCase activity (GBA1). The samples were then incubated at 37°C for 20 min and the reaction was then stopped by addition of 100 μL of 1 M glycine, pH 12.5. Fluorescence was then measured at excitation wavelength of 355 nM and emission wavelength of 460 nM. The CBE sensitive signal was considered lysosomal specific hydrolysis of the substrate.
Purification and labeling of recombinant saposin C
The plasmid encoding saposin C was a gift from Dr Nico Tjandra (55). Recombinant saposin C was purified from BL21 (DE3) cells as previously described (55). Briefly, saposin C was expressed as a His-tagged protein using the pET-30b vector (Novagen). Protein expression was induced by adding IPTG (1 mM final concentration) for 4 h. The cells were harvested into buffer containing 20 mM Tris, pH 8.0, 150 mM NaCl, 20 mM imidazole, lysozyme, and PMSF. Cells were lysed by sonication then cleared by centrifugation. Protein was purified using cobalt conjugated agarose (Fisher, PI89964). His-tag was removed using biotinylated thrombin (Novagen). Protein purity was confirmed by gel filtration and SDS-PAGE. For fluorescent labeling, recombinant saposin C was buffer exchanged into PBS pH 8.0 and adjusted to 150 μM concentration. Alexa Fluor 488 NHS Ester (Fisher, A20000) was added at 3:1 mol:mol and allowed to incubate at room temperature for 1 h. Unreacted dye was removed using a P10 column (GE healthcare). The resulting labeled protein was concentrated and stored at −80°C prior to use.
Saposin C treatment of PGRN mutant iPSC-derived cortical neurons and fibroblasts
For analysis of saposin C uptake, iPSC-derived cortical neurons were plated onto a glass bottom dish (Cellvis). Neurons were treated with AlexaFluor 488-labeled recombinant saposin C (200 ng/ml final concentration). After 3 h treatment, Lysotracker Red (50 nM) (ThermoFisher) was added to the wells for an additional 30 min incubation. Cells were then washed with Phenol-Red free media and imaged using a Nikon A1R laser scanning confocal microscope with 100× oil immersion objective using NIS-Elements (Nikon). For analysis of the effect of saposin C in PGRN mutant fibroblasts, the experiment was performed identically except fibroblasts were cultured in media consisting of DMEM, 10% (v/v) FBS, penicillin (100 U/ml) and streptomycin 100 μg/ml.
Live-cell GCase activity assay in saposin C treated fibroblasts and iPSC-derived cortical neurons
Lysosomal GCase activity was performed as previously described (56). iPSC-derived cortical neurons or patient-derived fibroblasts were plated on a black 96-well plate at a density of 30 000 cells/well for neurons and 8000 cells/well for patient fibroblasts. Prior to measurement of GCase activity, cells were treated with recombinant saposin C (200 ng/ml final concentration) in phenol red free media and incubated at 37°C for 3 h. After 3 h, the media was replaced with 100 μL of phenol red free media containing 50 μg/ml PFB-FDGlu (Invitrogen). Additional wells with, or without saposin C were incubated with 100 μL of phenol red free media containing 50 μg/ml PFB-FDGlu and bafilomycin A1 (400 nm). After a 75 min pre-incubation to allow for loading of the substrate, substrate hydrolysis was monitored every 20 min using a plate reader measuring fluorescence at 485/525 nm (excitation/emission). The bafilomycin A1 sensitive signal was considered lysosomal specific hydrolysis of the substrate. Relative GCase activity was determined by normalizing the slope of untreated to saposin C treated fibroblasts.
Statistical analysis
Student’s t-test, unpaired, two-tailed statistical analysis and ANOVA were performed. P-values less than 0.05 were considered significant. Statistical analysis was performed using GraphPad Prism Software, Version 7.0b.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health [R01 NS076054 and R37 NS096241 to D.K., F99 NS105182 to C.V., and T32NS041234 to D.Y.]. This study was also supported in part by an Alzheimer’s Disease Core Center grant (P30 AG013854) from the National Institute on Aging to Northwestern University, Chicago Illinois. We gratefully acknowledge the assistance of the Northwestern Alzheimer’s Disease Center and its participants. Live Cell imaging analysis was performed at the Northwestern University Center for Advanced Microscopy generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center.
Conflicts of Interest statement. D.K. is the Founder and Scientific Advisory Board Chair of Lysosomal Therapeutics Inc. D.K. serves on the scientific advisory boards of The Silverstein Foundation, Intellia Therapeutics, and Prevail Therapeutics and is a Venture Partner at OrbiMed.
References
- 1. Onyike C.U. and Diehl-Schmid J. (2013) The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry, 25, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vieira R.T., Caixeta L., Machado S., Silva A.C., Nardi A.E., Arias-Carrion O. and Carta M.G. (2013) Epidemiology of early-onset dementia: a review of the literature. Clin. Pract. Epidemiol. Ment. Health, 9, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karageorgiou E. and Miller B.L. (2014) Frontotemporal lobar degeneration: a clinical approach. Semin. Neurol., 34, 189–201. [DOI] [PubMed] [Google Scholar]
- 4. Knopman D.S. and Roberts R.O. (2011) Estimating the number of persons with frontotemporal lobar degeneration in the US population. J. Mol. Neurosci., 45, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rohrer J.D., Guerreiro R., Vandrovcova J., Uphill J., Reiman D., Beck J., Isaacs A.M., Authier A., Ferrari R., Fox N.C. et al. (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology, 73, 1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Po K., Leslie F.V., Gracia N., Bartley L., Kwok J.B., Halliday G.M., Hodges J.R. and Burrell J.R. (2014) Heritability in frontotemporal dementia: more missing pieces? J. Neurol., 261, 2170–2177. [DOI] [PubMed] [Google Scholar]
- 7. Petkau T.L. and Leavitt B.R. (2014) Progranulin in neurodegenerative disease. Trends Neurosci., 37, 388–398. [DOI] [PubMed] [Google Scholar]
- 8. Baker M., Mackenzie I.R., Pickering-Brown S.M., Gass J., Rademakers R., Lindholm C., Snowden J., Adamson J., Sadovnick A.D., Rollinson S. et al. (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature, 442, 916–919. [DOI] [PubMed] [Google Scholar]
- 9. Gass J., Cannon A., Mackenzie I.R., Boeve B., Baker M., Adamson J., Crook R., Melquist S., Kuntz K., Petersen R. et al. (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet., 15, 2988–3001. [DOI] [PubMed] [Google Scholar]
- 10. Gass J., Prudencio M., Stetler C. and Petrucelli L. (2012) Progranulin: an emerging target for FTLD therapies. Brain Res., 1462, 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gijselinck I., Van Broeckhoven C. and Cruts M. (2008) Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum. Mutat., 29, 1373–1386. [DOI] [PubMed] [Google Scholar]
- 12. Finch N., Baker M., Crook R., Swanson K., Kuntz K., Surtees R., Bisceglio G., Rovelet-Lecrux A., Boeve B., Petersen R.C. et al. (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain, 132, 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chiang H.H., Rosvall L., Brohede J., Axelman K., Bjork B.F., Nennesmo I., Robins T. and Graff C. (2008) Progranulin mutation causes frontotemporal dementia in the Swedish Karolinska family. Alzheimers Dement., 4, 414–420. [DOI] [PubMed] [Google Scholar]
- 14. Mukherjee O., Wang J., Gitcho M., Chakraverty S., Taylor-Reinwald L., Shears S., Kauwe J.S., Norton J., Levitch D., Bigio E.H. et al. (2008) Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum. Mutat., 29, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wider C., Uitti R.J., Wszolek Z.K., Fang J.Y., Josephs K.A., Baker M.C., Rademakers R., Hutton M.L. and Dickson D.W. (2008) Progranulin gene mutation with an unusual clinical and neuropathologic presentation. Mov. Disord., 23, 1168–1173. [DOI] [PubMed] [Google Scholar]
- 16. Yin F., Banerjee R., Thomas B., Zhou P., Qian L., Jia T., Ma X., Ma Y., Iadecola C., Beal M.F. et al. (2010) Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med., 207, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tang W., Lu Y., Tian Q.Y., Zhang Y., Guo F.J., Liu G.Y., Syed N.M., Lai Y., Lin E.A., Kong L. et al. (2011) The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science, 332, 478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mundra J.J., Jian J., Bhagat P. and Liu C.J. (2016) Progranulin inhibits expression and release of chemokines CXCL9 and CXCL10 in a TNFR1 dependent manner. Sci. Rep., 6, 21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He Z., Ismail A., Kriazhev L., Sadvakassova G. and Bateman A. (2002) Progranulin (PC-cell-derived growth factor/acrogranin) regulates invasion and cell survival. Cancer Res., 62, 5590–5596. [PubMed] [Google Scholar]
- 20. He Z., Ong C.H., Halper J. and Bateman A. (2003) Progranulin is a mediator of the wound response. Nat. Med., 9, 225–229. [DOI] [PubMed] [Google Scholar]
- 21. Zhu J., Nathan C., Jin W., Sim D., Ashcroft G.S., Wahl S.M., Lacomis L., Erdjument-Bromage H., Tempst P., Wright C.D. et al. (2002) Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell, 111, 867–878. [DOI] [PubMed] [Google Scholar]
- 22. Van Damme P., Van Hoecke A., Lambrechts D., Vanacker P., Bogaert E., van Swieten J., Carmeliet P., Van Den Bosch L. and Robberecht W. (2008) Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol., 181, 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao X., Joselin A.P., Wang L., Kar A., Ray P., Bateman A., Goate A.M. and Wu J.Y. (2010) Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3beta. Protein Cell, 1, 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petkau T.L., Neal S.J., Milnerwood A., Mew A., Hill A.M., Orban P., Gregg J., Lu G., Feldman H.H., Mackenzie I.R. et al. (2012) Synaptic dysfunction in progranulin-deficient mice. Neurobiol. Dis., 45, 711–722. [DOI] [PubMed] [Google Scholar]
- 25. Almeida M.R., Macario M.C., Ramos L., Baldeiras I., Ribeiro M.H. and Santana I. (2016) Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol. Aging, 41(200), e201–e205. [DOI] [PubMed] [Google Scholar]
- 26. Smith K.R., Damiano J., Franceschetti S., Carpenter S., Canafoglia L., Morbin M., Rossi G., Pareyson D., Mole S.E., Staropoli J.F. et al. (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet., 90, 1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Almeida S., Zhou L. and Gao F.B. (2011) Progranulin, a glycoprotein deficient in frontotemporal dementia, is a novel substrate of several protein disulfide isomerase family proteins. PLoS One, 6, e26454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu F., Padukkavidana T., Vaegter C.B., Brady O.A., Zheng Y., Mackenzie I.R., Feldman H.H., Nykjaer A. and Strittmatter S.M. (2010) Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron, 68, 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanaka Y., Matsuwaki T., Yamanouchi K. and Nishihara M. (2013) Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience, 250, 8–19. [DOI] [PubMed] [Google Scholar]
- 30. Tanaka Y., Chambers J.K., Matsuwaki T., Yamanouchi K. and Nishihara M. (2014) Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol. Commun., 2, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lui H., Zhang J., Makinson S.R., Cahill M.K., Kelley K.W., Huang H.Y., Shang Y., Oldham M.C., Martens L.H., Gao F. et al. (2016) Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell, 165, 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Evers B.M., Rodriguez-Navas C., Tesla R.J., Prange-Kiel J., Wasser C.R., Yoo K.S., McDonald J., Cenik B., Ravenscroft T.A., Plattner F. et al. (2017) Lipidomic and transcriptomic basis of lysosomal dysfunction in progranulin deficiency. Cell Rep., 20, 2565–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Valdez C., Wong Y.C., Schwake M., Bu G., Wszolek Z.K. and Krainc D. (2017) Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum. Mol. Genet., 26, 4861–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beel S., Moisse M., Damme M., De Muynck L., Robberecht W., Van Den Bosch L., Saftig P. and Van Damme P. (2017) Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum. Mol. Genet., 26, 2850–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou X., Paushter D.H., Feng T., Pardon C.M., Mendoza C.S. and Hu F. (2017) Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol., 134, 151–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou X., Sun L., Bracko O., Choi J.W., Jia Y., Nana A.L., Brady O.A., Hernandez J.C.C., Nishimura N., Seeley W.W. et al. (2017) Impaired prosaposin lysosomal trafficking in frontotemporal lobar degeneration due to progranulin mutations. Nat. Commun., 8, 15277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou X., Sun L., Bastos de Oliveira F., Qi X., Brown W.J., Smolka M.B., Sun Y. and Hu F. (2015) Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J. Cell Biol., 210, 991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kishimoto Y., Hiraiwa M. and O'Brien J.S. (1992) Saposins: structure, function, distribution, and molecular genetics. J. Lipid Res., 33, 1255–1267. [PubMed] [Google Scholar]
- 39. Harzer K., Paton B.C., Christomanou H., Chatelut M., Levade T., Hiraiwa M. and O'Brien J.S. (1997) Saposins (sap) A and C activate the degradation of galactosylceramide in living cells. FEBS Lett., 417, 270–274. [DOI] [PubMed] [Google Scholar]
- 40. Qi X. and Grabowski G.A. (1998) Acid beta-glucosidase: intrinsic fluorescence and conformational changes induced by phospholipids and saposin C. Biochemistry, 37, 11544–11554. [DOI] [PubMed] [Google Scholar]
- 41. Sun Y., Qi X. and Grabowski G.A. (2003) Saposin C is required for normal resistance of acid beta-glucosidase to proteolytic degradation. J. Biol. Chem., 278, 31918–31923. [DOI] [PubMed] [Google Scholar]
- 42. Fischer G. and Jatzkewitz H. (1975) The activator of cerebroside sulphatase. Purification from human liver and identification as a protein. Hoppe-Seylers Z. Physiol. Chem., 356, 605–613. [DOI] [PubMed] [Google Scholar]
- 43. Morimoto S., Martin B.M., Kishimoto Y. and O'Brien J.S. (1988) Saposin D: a sphingomyelinase activator. Biochem. Biophys. Res. Commun., 156, 403–410. [DOI] [PubMed] [Google Scholar]
- 44. Diaz-Font A., Cormand B., Santamaria R., Vilageliu L., Grinberg D. and Chabas A. (2005) A mutation within the saposin D domain in a Gaucher disease patient with normal glucocerebrosidase activity. Hum. Genet., 117, 275–277. [DOI] [PubMed] [Google Scholar]
- 45. Schnabel D., Schroder M. and Sandhoff K. (1991) Mutation in the sphingolipid activator protein 2 in a patient with a variant of Gaucher disease. FEBS Lett., 284, 57–59. [DOI] [PubMed] [Google Scholar]
- 46. Spiegel R., Bach G., Sury V., Mengistu G., Meidan B., Shalev S., Shneor Y., Mandel H. and Zeigler M. (2005) A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans. Mol. Genet. Metab., 84, 160–166. [DOI] [PubMed] [Google Scholar]
- 47. Vaccaro A.M., Motta M., Tatti M., Scarpa S., Masuelli L., Bhat M., Vanier M.T., Tylki-Szymanska A. and Salvioli R. (2010) Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum. Mol. Genet., 19, 2987–2997. [DOI] [PubMed] [Google Scholar]
- 48. Forrest S.L., Crockford D.R., Sizemova A., McCann H., Shepherd C.E., McGeachie A.B., Affleck A.J., Carew-Jones F., Bartley L., Kwok J.B. et al. (2019) Coexisting Lewy body disease and clinical parkinsonism in frontotemporal lobar degeneration. Neurology, 92, e2472–e2483. [DOI] [PubMed] [Google Scholar]
- 49. Hiraiwa M., O'Brien J.S., Kishimoto Y., Galdzicka M., Fluharty A.L., Ginns E.I. and Martin B.M. (1993) Isolation, characterization, and proteolysis of human prosaposin, the precursor of saposins (sphingolipid activator proteins). Arch. Biochem. Biophys., 304, 110–116. [DOI] [PubMed] [Google Scholar]
- 50. Hiraiwa M., Martin B.M., Kishimoto Y., Conner G.E., Tsuji S. and O'Brien J.S. (1997) Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): its mechanism and inhibition by ganglioside. Arch. Biochem. Biophys., 341, 17–24. [DOI] [PubMed] [Google Scholar]
- 51. Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J. et al. (2013) Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron, 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tamargo R.J., Velayati A., Goldin E. and Sidransky E. (2012) The role of saposin C in Gaucher disease. Mol. Genet. Metab., 106, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jian J., Tian Q.Y., Hettinghouse A., Zhao S., Liu H., Wei J., Grunig G., Zhang W., Setchell K.D.R., Sun Y. et al. (2016) Progranulin recruits HSP70 to beta-Glucocerebrosidase and is therapeutic against Gaucher disease. EBioMedicine, 13, 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou X., Paushter D.H., Pagan M.D., Kim D., Nunez Santos M., Lieberman R.L., Overkleeft H.S., Sun Y., Smolka M.B. and Hu F. (2019) Progranulin deficiency leads to reduced glucocerebrosidase activity. PLoS One, 14, e0212382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de Alba E., Weiler S. and Tjandra N. (2003) Solution structure of human saposin C: pH-dependent interaction with phospholipid vesicles. Biochemistry, 42, 14729–14740. [DOI] [PubMed] [Google Scholar]
- 56. Zheng J., Jeon S., Jiang W., Burbulla L.F., Ysselstein D., Oevel K., Krainc D. and Silverman R.B. (2019) Conversion of quinazoline modulators from inhibitors to activators of beta-Glucocerebrosidase. J. Med. Chem., 62, 1218–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sidransky E. and Lopez G. (2012) The link between the GBA gene and parkinsonism. Lancet Neurol., 11, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nalls M.A., Duran R., Lopez G., Kurzawa-Akanbi M., McKeith I.G., Chinnery P.F., Morris C.M., Theuns J., Crosiers D., Cras P. et al. (2013) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol., 70, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mazzulli J.R., Xu Y.H., Sun Y., Knight A.L., McLean P.J., Caldwell G.A., Sidransky E., Grabowski G.A. and Krainc D. (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell, 146, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rothaug M., Zunke F., Mazzulli J.R., Schweizer M., Altmeppen H., Lullmann-Rauch R., Kallemeijn W.W., Gaspar P., Aerts J.M., Glatzel M. et al. (2014) LIMP-2 expression is critical for beta-glucocerebrosidase activity and alpha-synuclein clearance. Proc. Natl. Acad. Sci. U. S. A., 111, 15573–15578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yun S.P., Kim D., Kim S., Kim S., Karuppagounder S.S., Kwon S.H., Lee S., Kam T.I., Lee S., Ham S. et al. (2018) Alpha-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener., 13, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schondorf D.C., Aureli M., McAllister F.E., Hindley C.J., Mayer F., Schmid B., Sardi S.P., Valsecchi M., Hoffmann S., Schwarz L.K. et al. (2014) iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun., 5, 4028. [DOI] [PubMed] [Google Scholar]
- 63. Sidransky E., Nalls M.A., Aasly J.O., Aharon-Peretz J., Annesi G., Barbosa E.R., Bar-Shira A., Berg D., Bras J., Brice A. et al. (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med., 361, 1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kelley B.J., Haidar W., Boeve B.F., Baker M., Graff-Radford N.R., Krefft T., Frank A.R., Jack C.R. Jr., Shiung M., Knopman D.S. et al. (2009) Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol. Aging, 30, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zheng J., Chen L., Skinner O.S., Ysselstein D., Remis J., Lansbury P., Skerlj R., Mrosek M., Heunisch U., Krapp S. et al. (2018) Beta-glucocerebrosidase modulators promote dimerization of beta-glucocerebrosidase and reveal an allosteric binding site. J. Am. Chem. Soc., 140, 5914–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marshall J., McEachern K.A., Kyros J.A., Nietupski J.B., Budzinski T., Ziegler R.J., Yew N.S., Sullivan J., Scaria A., van Rooijen N. et al. (2002) Demonstration of feasibility of in vivo gene therapy for Gaucher disease using a chemically induced mouse model. Mol. Ther.: J. Am. Soc. Gene Ther., 6, 179–189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.