

Abstract
Down syndrome (DS), caused by the triplication of human chromosome 21, leads to significant alterations in brain development and is a major genetic cause of intellectual disability. While much is known about changes to neurons in DS, the effects of trisomy 21 on non-neuronal cells such as astrocytes are poorly understood. Astrocytes are critical for brain development and function, and their alteration may contribute to DS pathophysiology. To better understand the impact of trisomy 21 on astrocytes, we performed RNA-sequencing on astrocytes from newly produced DS human induced pluripotent stem cells (hiPSCs). While chromosome 21 genes were upregulated in DS astrocytes, we found consistent up- and down-regulation of genes across the genome with a strong dysregulation of neurodevelopmental, cell adhesion and extracellular matrix molecules. ATAC (assay for transposase-accessible chromatin)-seq also revealed a global alteration in chromatin state in DS astrocytes, showing modified chromatin accessibility at promoters of cell adhesion and extracellular matrix genes. Along with these transcriptomic and epigenomic changes, DS astrocytes displayed perturbations in cell size and cell spreading as well as modifications to cell-cell and cell-substrate recognition/adhesion, and increases in cellular motility and dynamics. Thus, triplication of chromosome 21 is associated with genome-wide transcriptional, epigenomic and functional alterations in astrocytes that may contribute to altered brain development and function in DS.
Introduction
Down syndrome (DS) is a major genetic cause of disability and affects ~ 1 in 700 births worldwide (1). In DS, triplication of HSA21 (Homo sapiens chromosome 21) causes DS individuals to have pronounced intellectual disability that often coincides with brain disorders/diseases including epilepsy, autism, mental disability and Alzheimer’s disease (ad) (2–9). Analysis of DS brain and animal modeling studies has shown reduced brain volume and neuronal density (10, 11). This is attributed to reduced neurogenesis, increased neuronal apoptosis and over-representation of glial lineages (12–16). Abnormal dendritic arborization, spine morphology and spine density have also been reported, indicating altered formation and maintenance of neuronal circuits in DS (17–20).
While changes to neurons in DS have been reported, molecular and physiological disruptions to other brain cells are less understood. Among the cell types whose alteration may affect multiple aspects of brain development, function and response to injury are astrocytes, as they regulate many processes including synapse formation and plasticity, extracellular ion and neurotransmitter homeostasis and neurovascular coupling (21–25). Emerging evidence implicates astrocyte dysfunction in a variety of neurodevelopmental disorders and neurodegenerative diseases (26–29), raising the possibility of a direct contribution of astrocytes to DS pathophysiology. Consistent with this, astrocytes are more abundant in DS fetal cortex and appear more morphologically mature (16). Elevated levels of glial fibrillary acidic protein (GFAP) and S100ß, as well as altered cellular morphology also indicate increased astrocyte reactivity in the adult DS brain (30, 31). In DS mouse models, astrocytes decrease neuronal activity of cholinergic neurons (32) while fetal tissue-derived DS astrocytes have reduced secretion of the synaptogenic factor thrombospondin-1 and are believed to contribute to the alteration of dendritic spine structure and reduced dendrite arborization (33). DS astrocytes have also been shown to have disrupted mitochondrial morphology and function (34) but with potentially greater anti-oxidant capacity versus euploid astrocytes (35). Recent advances with human induced pluripotent stem cell (hiPSC) technologies have shown increased oxidative stress in iPSC-derived DS astrocytes which results in the release of factors that promote neuronal apoptosis, implicating astrocytes in neuronal death and overall reduction in neuronal number (36). DS astrocytes also contribute to elevations in mTOR signaling and synaptic marker expression in neurons (37). Thus, astrocytes may be primary effectors in DS pathophysiology. However, a detailed analysis of how trisomy 21 impacts the overall molecular profile and functional properties of astrocytes is still required.
To understand how trisomy 21 affects the molecular properties and function of astrocytes, we created astrocytes from newly produced DS hiPSCs. RNA-sequencing (RNA-seq) analysis showed genome-wide perturbations in gene expression in DS astrocytes with a strong dysregulation of molecules that function during nervous system development including those involved in axon guidance, cell adhesion/recognition and extracellular matrix (ECM) organization. Interestingly, the same gene sets were not disrupted in isogenic DS neural precursor cells (NPCs), demonstrating that the genome-wide changes in astrocytes are not transposed from an NPC state. Using ATAC-seq, we also observed global changes to the chromatin landscape in DS astrocytes and differential accessibility to the promoters of genes involved in cell adhesion and ECM organization. Consistent with these transcriptomic and epigenomic modifications, DS astrocytes have altered cell spreading, cell-cell and cell-substrate adhesion, motility and specific changes to protocadherin (PCDH)-mediated cellular adhesion. Overall, this study reveals that hiPSC-derived DS astrocytes have widespread transcriptomic and epigenomic changes that disrupt the adhesion profile of DS astrocytes, offering insight into non-neuronal changes that may occur in the developing and mature DS brain.
Results
Generation of DS and control hiPSCs, NPCs and astrocytes
We created six new hiPSC lines from three DS and three control (CTL) fibroblast cell lines using episomal reprogramming techniques (Fig. 1A). All DS and CTL fibroblast lines were obtained from the Coriell Institute for Medical Research. An additional DS hiPSC line was acquired from the American Type Culture Collection (ATCC). Fibroblast samples were matched for sex, age and ethnicity (Table S1). All generated hiPSC lines expressed the pluripotency markers OCT4, TRA-1-60 and SSEA4 (Figs 1C and S2A). All three CTL hiPSC lines showed a normal karyotype and three DS hiPSC lines were trisomic for chromosome 21 (Fig. 1B). One DS line (AG04823) showed a disomic karyotype and an insertion in the X chromosome and hence was not used for subsequent studies. CTL and DS lines will be made available to the scientific community.
Figure 1.
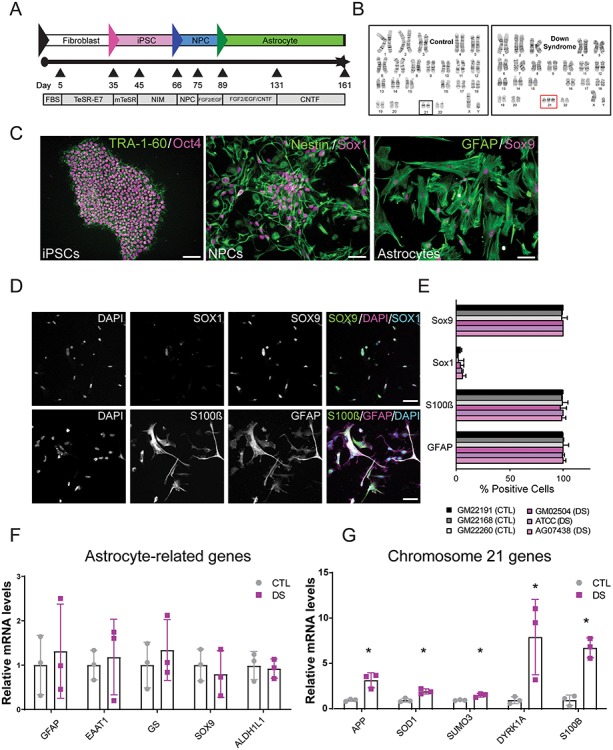
Generation and validation of CTL and DS astrocytes from fibroblasts and hiPSCs. (A) Production workflow of hiPSCs, NPCs and astrocytes. (B) Karyotypes of CTL and DS lines. (C) Expression of pluripotency markers in hiPSCs (TRA-1-60 and Oct4), and selective markers in neuronal precursor cell (NPC) markers (Sox1 and Nestin) and astrocyte cultures (GFAP and Sox9). Scale = 20 μm in TRA-1-60/Oct4 image and 50 μm in all others. (D) At 90 days, astrocytes express markers GFAP, S100B and Sox9 and low levels of the NPC marker Sox1. Scale = 50 μm. (E) Astrocyte differentiation causes a reduction of the NPC marker Sox1 and upregulation of astrocytic markers S100B, GFAP and Sox9. (F) CTL and DS astrocytes express equal levels of astrocyte-related genes GFAP, EAAT1, glutamine synthetase (GS), SOX9 and ALDH1L1 (shown by qPCR). (G) Chromosome 21 genes are upregulated in DS astrocytes (shown by qPCR). Data are represented as mean ± SEM. Two-tailed, unpaired t-tests were performed *P ≤ 0.05, n = 3 (three experiments each performed in the three DS and three CTL cell lines). FBS: fetal bovine serum; NIM: neural induction medium; NPC: neural precursor cells.
DS and CTL hiPSC lines were differentiated into NPCs using a monolayer, double-Smad inhibition protocol (38, 39) (Fig. 1A), after which all lines expressed NPC markers (Nestin, SOX1 and PAX6; Figs 1C and S2B). A monolayer protocol was then used to differentiate NPCs into astrocytes using differentiation protocols adapted from Roybon et al. and Shaltouki et al. (40, 41). Approximately 90 days after initiating astrocyte differentiation, the majority of cells expressed the astrocytic markers GFAP, S100ß and SOX9 and downregulated the NPC marker SOX1 (Figs 1D, E, S4A, and S5A). Cells also displayed more complex morphologies typical of astrocytes in vitro (Fig. 1D). Quantitative reverse transcriptase polymerase chain reaction (RT-PCR; now referred to as qPCR) analysis showed that CTL and DS astrocytes showed similar levels of astrocytic mRNAs including GFAP, EAAT1/GLAST, GS, SOX9 and ALDH1L1 (Fig. 1F). This was consistent with immunofluorescence labeling which showed that DS and CTL astrocytes expressed similar amounts of ALDH1L1 and EAAT1/GLAST protein (Figs S4B and S5B). CTL and DS astrocytes lacked expression of the neuronal marker Smi-312 and the oligodendrocyte marker CC1 (Fig. S3A). CTL and DS astrocytes also had comparable levels of GFAP and pro-inflammatory markers (data not shown), indicating that DS astrocytes do not upregulate markers of glial reactivity. qPCR analysis of DS astrocytes confirmed upregulation of chromosome 21 genes including APP, SOD1, SUMO3, DYRK1A and S100B (Fig. 1G). Thus, DS astrocytes express known markers of differentiated astrocytes and upregulate genes on chromosome 21.
DS astrocytes show normal spontaneous and ATP-evoked Ca2+ responses
A prominent feature of astrocytes is their ability to show spontaneous and evoked intracellular Ca2+ elevations which have been associated with numerous functional properties of astrocytes (42). To determine if DS astrocytes have altered Ca2+ elevations (43), we performed live cell imaging of Ca2+ dynamics in CTL and DS astrocytes. CTL and DS astrocytes showed similar spontaneous Ca2+ events with no significant differences in the percentage of active cells or changes in Ca2+ event amplitude (Fig. S3B–C). A recent study demonstrated an increase in spontaneous activity in DS astrocytes (44). However, this was not the case in this study. It should be noted that a different cell line was used in Mizuno et al. (44) with an isogenic control. It would be interesting to further examine the potential for line-specific differences in spontaneous Ca2+ dynamics in DS astrocytes. To determine if DS and CTL astrocytes differed in evoked Ca2+ responses, ATP pulses were delivered to the cells. DS and CTL astrocytes were similar with respect to the percentage of ATP-responsive cells and Ca2+ event amplitude (Fig. S3D–F). Thus, our results indicate that CTL and DS astrocytes display similar spontaneous and evoked Ca2+ dynamics.
Genome-wide transcriptional dysregulation in DS astrocytes
To determine how trisomy 21 affects the transcriptome of astrocytes, we performed RNA-seq on the three DS and three CTL astrocyte lines. Analysis of CTL astrocytes showed that they expressed many genes found in mature human astrocytes (45) with some residual expression of immature astrocyte genes (Table S6). This suggests that astrocytes in this study have a transcriptional profile intermediate to fetal and mature astrocytes. We performed differential gene expression analysis of DS and CTL astrocytes which revealed 725 protein-coding genes with altered mRNA levels in DS astrocytes (adjusted P-value < 0.05). Heatmaps of the top 50 differentially expressed genes, as well as PCA analysis and sample clustering were generated for differential analysis (Fig. S7). These analyses highlight the correct segregation of samples from each cell line as well as the variability between the cell lines. Inter-cell line variability has been reported in other studies (46, 47) and highlights the complexity of working with different human samples which can possess heterogeneous transcriptome profiles. Utilization of isogenic lines from each sample is one approach to reduce such variability.
More detailed analysis of RNA-seq datasets showed that approximately 20% of all chromosome 21 genes were upregulated. This is consistent with a previous microarray study of multiple regions of DS brain tissue (Fig. 2A and B) (48). A total of 51 of 52 differentially expressed chromosome 21 genes were upregulated, including known DS targets such as APP, DYRK1A, SOD1 and SUMO3, with a median absolute fold change of 1.56. Interestingly, we observed consistent genome-wide transcriptional changes in DS astrocytes. Of 725 differentially expressed genes, 673 genes (~93%) were located outside of chromosome 21 and were widely distributed across the genome (Fig. 2C). Furthermore, whereas chromosome 21 genes were predominantly upregulated (98.1%), differentially expressed mRNAs from other chromosomes were both up- and downregulated (51.5% upregulation and 48.5% downregulation) (Fig. 2 C,D). These results show that trisomy 21 leads to a robust, genome-wide perturbation of transcriptional activity in astrocytes.
Figure 2.
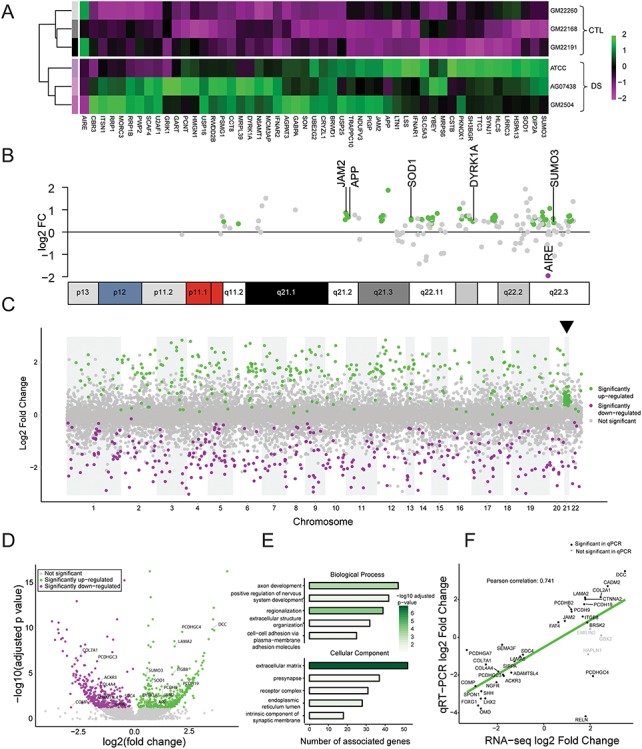
DS astrocytes show global transcriptome misregulation. (A) Heatmap of the differential expression of chromosome 21 genes in DS and CTL astrocytes. Heatmap displays relative expression level of each gene across samples (magenta, lower expression; green, higher expression). (B) Mapping of differentially expressed chromosome 21 genes onto the chromosome 21 ideogram. (C) Manhattan plot showing differentially expressed genes and illustrating global transcriptome dysregulation in DS astrocytes. Significantly upregulated genes are shown in green and downregulated genes are shown in magenta. Arrowhead indicates chromosome 21. (D) Volcano plot showing symmetrical changes in gene expression, with a similar proportion of up- and downregulation in DS astrocytes. (E) Top five biological processes and cellular enriched in differentially expressed genes according to GO analysis. Bars encode the number of genes associated with the ontology term and are colored by –log10 of adjusted P-value. (F) Correlation between RNA-seq and qPCR data (Pearson’s: 0.741, P ≤ 0.01).
DS astrocytes show strong alteration of genes involved in neuronal development, cell adhesion and ECM organization
Gene ontology (GO) analysis revealed that the 725 differentially expressed genes were primarily involved in nervous system development, axon guidance, regionalization, ECM organization and cell adhesion (Fig. 2E). Differentially expressed genes in DS astrocytes were also enriched in cellular components related to the ECM (Fig. 2E). Interestingly, pathway analysis also reported alterations to ECM-receptor interactions, ECM proteoglycans and Laminin interactions (Fig. S12A). To rule out potential false-positives of RNA-seq, we validated the change in gene expression by performing qPCR on the top 25 differentially expressed genes associated with GO terms, as well as a few additional genes of interest. BRSK2, CADM2, COL2A1, DCC, FAT4, ITGB8, JAM2, LAMA2, PCDH9, PCDH19 and PCDHB2 showed increases in mRNA expression (Fig. S8A) while ACKR3, ADAMTSL4, COL4A4, COL7A1, COMP, FOXG1, LAMA5, LHX2, NGFR, OMD, PCDHA7, PCDHB7, PCDHGC3, PCDHGC4, RELN, SDC4, SEMA3F, SHH, SIRPA and SPON1 showed reductions in mRNA levels in DS astrocytes (Fig. S8B). This revealed strong correspondence between the fold changes in gene expression derived from RNA-seq and qPCR (Pearson correlation coefficient: 0.741) (Fig. 2F). PCDHGC4 and RELN were the only mRNAs for which changes determined by the two experimental approaches did not directly correspond. Taken together, these findings demonstrate that trisomy 21 in astrocytes has a profound genome-wide impact on gene expression, with a particularly large effect on genes related to cell adhesion and ECM organization.
Altered chromatin state in DS astrocytes
Recent studies have shown that in addition to causing whole transcriptome dysregulation, trisomy 21 causes changes in epigenetic mechanisms which can alter gene expression. Indeed, changes in DNA methylation have been shown to be associated with DS, and more specifically, hypermethylation has been observed in DS brain (49–55). To investigate if chromatin accessibility in DS astrocytes is altered, we performed ATAC-seq (56) on three CTL and DS astrocyte lines. This revealed approximately fourteen thousand differentially accessible peaks. Differential chromatin accessibility was distributed across the genome, with an even split of increased and decreased accessibility (Fig. 3A). Approximately 6% of these sites were located in promoter regions (defined as within 3 kb of the transcription start site). Remarkably, we found that upregulated and downregulated genes identified by RNA-seq do not have corresponding differential accessibility at promoters in CTL and DS astrocytes (Figs 3B and S9). We performed GO analysis to identify the molecular function of genes with altered chromatin accessibility at their promoter regions. This analysis showed that the top five biological processes enriched for genes with differentially accessible chromatin include processes related to cell growth, cell adhesion and ECM such as: regulation of actin, cell substrate adhesion and extracellular matrix organization (Fig. 3C). Cellular component analysis also revealed that the products of those genes are largely related to cell adhesion and the ECM. These results reveal complex epigenomic changes in DS astrocytes, especially related to genes involved in cell adhesion and ECM organization. Furthermore, the results suggest that changes to the transcriptome of DS astrocytes are not simply due to direct epigenetic modifications to promoter regions of dysregulated mRNAs.
Figure 3.
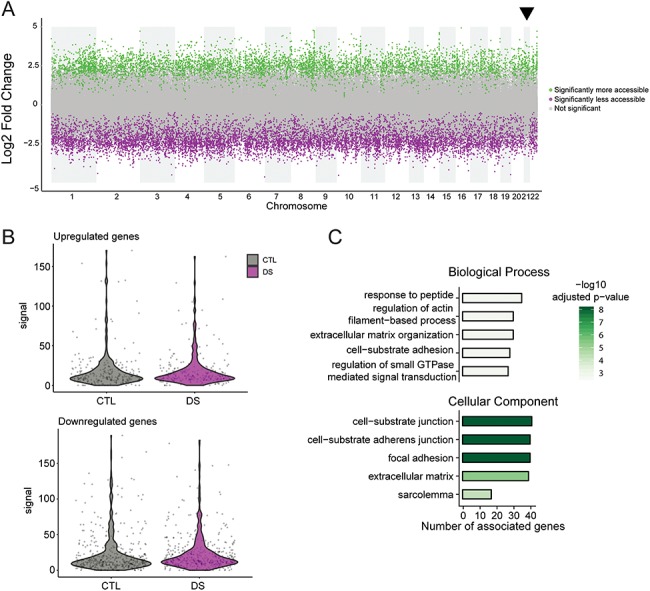
ATAC-seq reveals an altered chromatin state in DS astrocytes, with differential chromatin accessibility in the promoters of cell adhesion and ECM genes. (A) Plot displaying the differentially accessible peaks in DS astrocytes revealing both an increase and decrease in chromatin accessibility. (B) Violin plots of the correspondence between ATAC-seq and RNA-seq data sets show that upregulated and downregulated genes identified by RNA-seq do not have differential accessibility at promoters in CTL and DS astrocytes. (C) Top five biological processes and cellular components enriched in genes with differentially accessible promoters according to GO analysis. Bars encode number of genes associated with the ontology term and are colored by –log10 of adjusted P-value.
Differential changes in gene expression in DS NPCs and astrocytes
Since trisomy 21 is known to cause alterations to stem cells and precursor cells (57), it is possible that the differences in expression of ECM and adhesion genes in DS astrocytes are established at the NPC stage and prior to their differentiation into astrocytes. To determine if this is the case, we analyzed NPCs for the top 25 gene targets found in DS astrocytes and validated by qPCR. DS and CTL NPCs showed similar expression levels of PAX6, SOX1 and SOX2, and DS NPCs upregulated chromosome 21 genes DYRK1A, APP and SUMO3 (Fig. 4A), indicating that CTL and DS NPCs have comparable expression of NPC-enriched mRNAs and DS-associated genes, respectively. However, surprisingly, only 5 of 27 validated dysregulated ECM and cell adhesion mRNAs in DS astrocytes showed changes in DS NPCs (Fig. 4B and C). Of these, only two mRNAs were downregulated in both DS NPCs and DS astrocytes (COL7A1 and PCDHGC3) and three were regulated in the opposite direction in DS NPCs and DS astrocytes (ITGB8, PCDH9 and PCDH19) (Fig. 4D). These findings indicate that trisomy 21 has selective effects on the expression of ECM and cellular adhesion genes in astrocytes and that the transcriptional changes observed in DS astrocytes are not established in NPCs prior to their differentiation.
Figure 4.
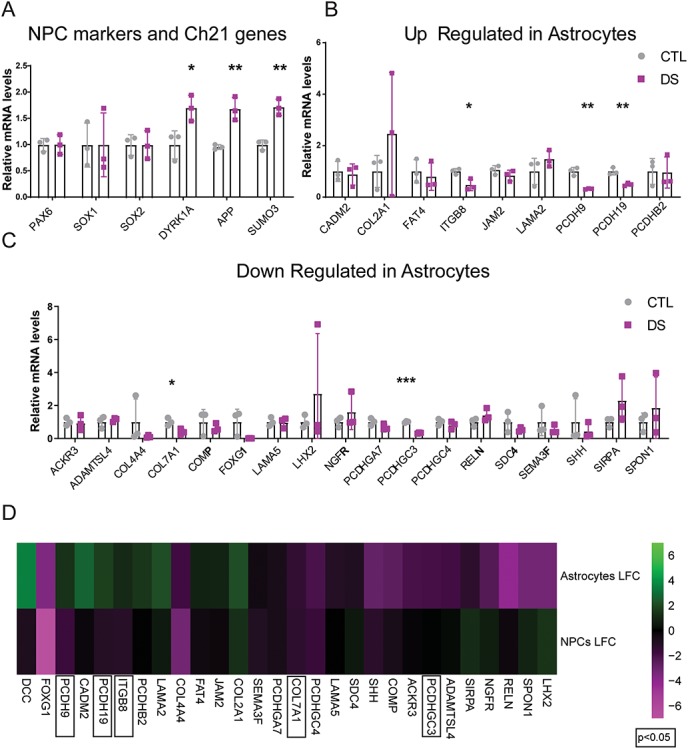
DS NPCs and astrocytes differ in their dysregulation of non-chromosome 21 genes. (A) qPCR demonstrates similar expression of Pax6, Sox1 and Sox2 in CTL and DS NPCs and upregulation of chromosome 21 genes: DYRK1A, APP and SUMO3 in DS NPCs. Data are represented as mean ± SEM. Two-tailed, unpaired t-tests were performed *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, n = 3 (three experiments each performed in the three DS and three CTL cell lines). (B) ITGB8, PCDH9 and PCDH19 are significantly downregulated in DS NPCs, whereas they were upregulated in DS astrocytes; CadM2, COL2A1, FAT4, JAM2, LAMA2 and PCDHB2 are unchanged in DS NPCs, whereas they were upregulated in DS astrocytes. Data are represented as mean ± SEM. Two-tailed, unpaired t-tests were performed *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, n = 3 (three experiments each performed in the three DS and three CTL cell lines). (C) COL7A1 and PCDHGC3 were downregulated in both DS NPCs and astrocytes, whereas ACKR3, ADAMTSL4, COL4A4, COMP, FOXG1, LAMA5, LHX2, NGFR, PCDHGA7, PCDHGC4, RELN, SDC4, SEMA3F, SHH, SIRPA and SPON1 are unchanged in DS NPCs and were downregulated in DS astrocytes. Data are represented as mean ± SEM. Two-tailed, unpaired t-tests were performed *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, n = 3 (three experiments each performed in the three DS and three CTL cell lines). (D) Heatmap of the expression of differentially expressed genes in DS astrocytes and NPCs (magenta: lower expression, green: higher expression).
Altered cell adhesion and recognition of DS astrocytes
Considering the transcriptomic and epigenomic changes in DS astrocytes related to genes involved in cell adhesion and ECM organization, we investigated if cell recognition and adhesion properties of DS astrocytes were perturbed. To determine if dysregulation of specific molecules identified in RNA-seq and qPCR analysis led to changes in cell adhesion/recognition, we performed assays involving protocadherins (PCDHs). PCDHs are a family of clustered genes that play critical roles in cell recognition and are important for processes during nervous system development including synaptogenesis and axonal and dendritic growth (58–60), which are processes known to be defective in DS (61). RNA-seq analysis showed many PCDHs to be dysregulated in DS astrocytes (Figs 2D, F and S8). This is consistent with a previous study in which DS fetal cortical lysates were also shown to have downregulated gamma PCDHs (PCDHG) (62). We first assessed if PCDHG protein levels were changed in DS astrocytes. We utilized a pan-PCDHG antibody to screen for overall changes in expression of this PCDH family using western blot analysis. We found that PCDHG protein was significantly reduced (3.85 fold) in DS astrocytes (Figs 5A, B, and S10). To investigate if the alteration of PCDHG protein in DS astrocytes caused specific functional changes to PCDHG-mediated homophilic cell recognition/adhesion, we performed cell binding assays on selective PCDH substrates. Interestingly, DS astrocytes showed a selective loss of binding to a PCDHGC3 substrate (Fig. 5C), consistent with the downregulation of PCDHGC3 mRNA (Fig. 2D and F). In contrast, DS astrocytes showed similar levels of binding to substrates of PCDH10 and PCDH12 as CTL astrocytes (Fig. 5C). Thus, DS astrocytes have selective impairments in PCDHG-mediated cellular recognition/adhesion.
Figure 5.
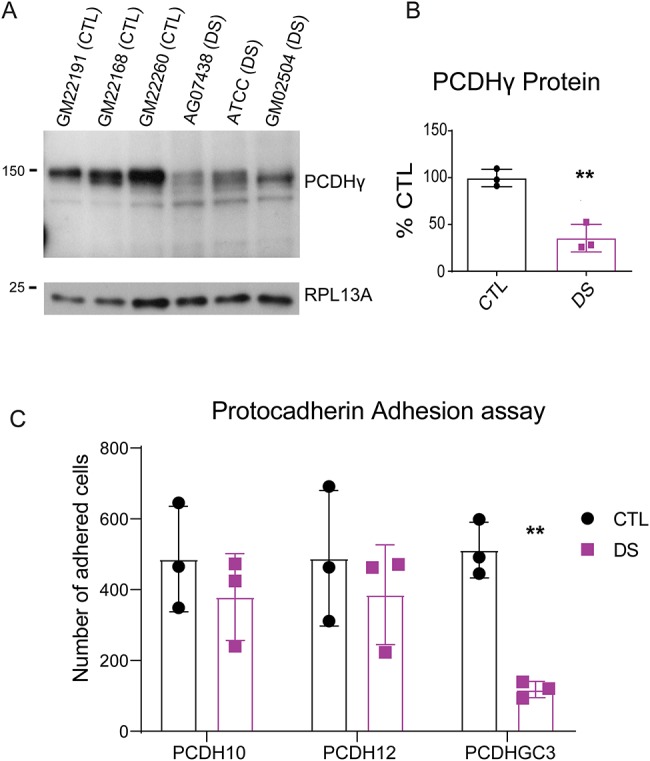
DS astrocytes display alterations in PCDH-mediated adhesion: (A, B) western blot analysis and quantification showing the downregulation of PCDHGs in DS astrocytes. Data are represented as mean ± SEM. Two-tailed, unpaired t-tests were performed *P ≤ 0.05 (n = 3), **P ≤ 0.01 (n = 3 experiments, each performed in three CTL and three DS cell lines). (C) CTL astrocytes adhere significantly more to PCDHGC3-coated coverslips than DS astrocytes. Data are represented as mean ± SEM. Two-tailed, unpaired t-tests performed *P ≤ 0.05 n = 3 (three experiments in which five images were taken in all three CTL cell lines and three DS cells lines).
During the course of this study, we noted that DS astrocytes appeared larger, more spread and adherent to the surface of the tissue culture vessel/dish than CTL astrocytes. To investigate this, we labeled astrocytes for the membrane bound glutamate transporter EAAT1 and measured the area covered by individual astrocytes. We found that DS astrocytes were significantly larger than CTL astrocytes (Fig. 6A and B). DAPI labeling showed that CTL and DS astrocytes have similar sized nuclei (Fig. 6A and B). Flow cytometry further showed that CTL and DS astrocytes have similar sizes when in suspension (Figs 6C, S11A, and B). The change in cell size of DS astrocytes did not appear to be caused by differences in cell proliferation or senescence, as the astrocyte cultures were passaged at similar frequencies, and no differences were observed in the expression of senescence markers (63) (data not shown). To assess if DS astrocytes have increased cell spreading ability which affects their size, we performed a cell spreading assay. Astrocytes were fixed and labeled for actin with phalloidin-488 after 30, 60, 90 and 120 minutes (Fig. 6D). Cell area at each time point was measured allowing us to obtain the rate of cell spreading. We found that DS astrocytes have a significantly increased rate of spreading (Fig. 6E). Thus, the observed increases in the size of DS astrocytes are likely related to their increased cell spreading ability.
Figure 6.
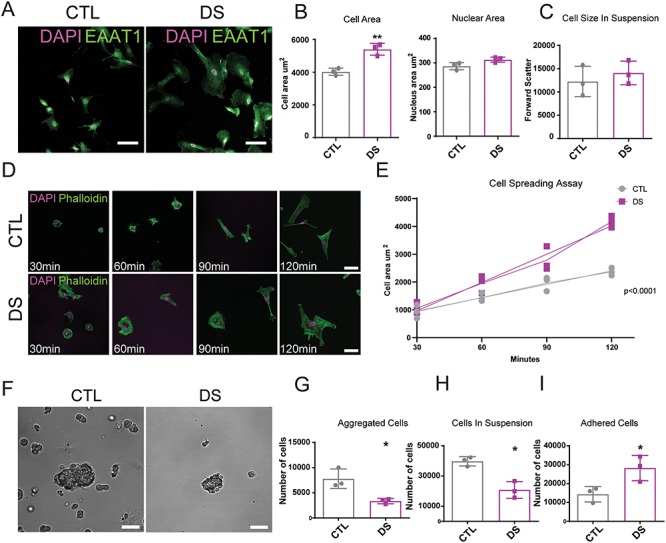
DS astrocytes have increased cell spreading and an altered adhesion profile: (A) DS astrocytes are significantly larger than CTL astrocytes. Composite images of EAAT1 and DAPI-positive astrocytes, scale = 100 μm. (B) Cell area and nuclear area quantifications (8 weeks post-differentiation). N > 900 cells from three separate experiments with three cell lines per conditions. (C) Cell size in suspension obtained by flow cytometry (forward scatter). Data are represented as mean ± SEM. Two tailed, unpaired t-test were performed *P ≤ 0.05 (D) DS astrocyte display increased cell spreading. Composite images of Phalloidin-488 and DAPI-positive astrocytes imaged at 30 min, 60 min, 90 min and 120 min of cell spreading, scale = 50 μm. (E) Mean cell area calculated for each time point, n = 3 (this experiment was performed three times in all three DS and all three CTL cell lines, five images were analyzed per cell line at each time point). Linear regressions were performed on the rate of spreading, and the slopes were determined to be significantly different from each other P < 0.0001. (F) DS astrocytes show differential cell-cell and cell-substrate adhesion profiles. After 2 hours in suspension DS astrocytes form smaller aggregates than CTL astrocytes, scale = 150 μm. (G) Aggregate comparison after two hours in suspension. n = 3 (This experiment was performed three times in all three DS and all three CTL cell lines, five images were analyzed per cell line at each time point). Data are represented as mean ± SEM. Two-tailed, unpaired t-test was performed *P ≤ 0.05. (H) The number of cells in suspension is lower for DS astrocytes. n = 3 (this experiment was performed three times in all three DS and all three CTL cell lines; the number of cells in suspension was counted with a cell counter). Data are represented as mean ± SEM. Two tailed, unpaired t-test was performed *P ≤ 0.05. (I) The number of cells having adhered is higher for DS astrocytes. n = 3 (this experiment was performed three times in all three DS and three CTL cell lines; the number of cells having adhered was counted with a cell counter following trypsinization). Data are represented as mean ± SEM. Two-tailed, unpaired t-test was performed *P ≤ 0.05.
To determine if DS astrocytes have alterations in cell adhesion, we performed a series of experiments to examine both cell-cell and cell-substrate adhesion. Cell aggregation assays revealed an altered cell-cell adhesion profile of DS astrocytes. DS astrocytes placed in suspension formed smaller aggregates than their CTL counterparts (Fig. 6F and G). Interestingly, this altered cell-cell adhesion property was accompanied by an increased number of astrocytes directly adherent to the uncoated petri dish and a reduced number of non-aggregated DS astrocytes remaining in suspension (Fig. 6H and I). Thus, DS astrocytes have an altered cell-cell adhesion profile, showing a preferential attachment to the tissue culture dish. Repeating the experiments using ultralow adhesion plates, in which cells were not capable of adhering to the bottom of the dish, showed no difference in the size of cell aggregates (Fig. S11C and D), suggesting that DS astrocytes do not have a cell-cell adhesion deficit but rather a preferential attachment to the tissue culture dish.
Finally, we performed cellular motility assays to investigate whether DS astrocytes have altered motility or dynamics under basal conditions. This was done using live cell imaging and automated cell tracking (64). These experiments revealed that over the course of 190 minutes, DS astrocytes plated at the same density as CTL astrocytes have increased cell motility (Fig. 7A), as indicated by increased net displacement and migration persistence (Fig. 7B–D). Interestingly, differences in autonomous cell velocity were not observed (Fig. 7E), suggesting that although DS astrocytes move at similar speeds to CTL astrocytes, DS astrocytes cover longer distances and have more persistent directed motility then their CTL counterparts. Altogether, these results suggest that DS astrocytes possess an altered functional adhesion state that impacts cell size, cell spreading, cell-cell and cell-substrate adhesion and cellular dynamics (Fig. 7F).
Figure 7.
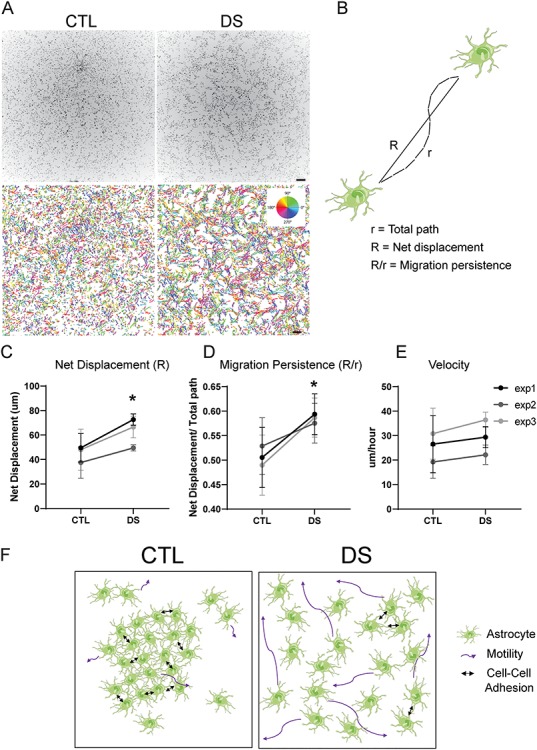
DS astrocytes have increased cell motility: (A) DS astrocytes have an increased motility at basal conditions, as determined by live imaging and nuclear tracking over 190 min. Automatically detected nuclear trajectories are colored based of the direction of movement (scale bars, 250 μm). (B) Schematic of the measurements used to determine migration persistence: migration persistence (R/r) is measured through the quotient of the net displacement (R) and the total distance travelled (r). (C–E) Over 190 min at basal conditions, DS astrocytes display an increase in net displacement and migration persistence, but not in velocity. (n = 3 independent experiments, in all three CTL and DS cell lines; data are represented as mean ± SEM. Two tailed, paired t-test was performed *P ≤ 0.05 **P ≤ 0.01). (F) Schematic summarizing the results of Figures 6 and 7. DS astrocytes show increased cell size through increased cell spreading, decreased cell-cell adhesion and increased motility.
Discussion
DS is a genetic disorder associated with neurodevelopmental alterations and aging-related brain pathologies, thus presenting complex neurological challenges for affected individuals throughout their lifespan (4). As astrocytes are implicated in a host of neurodevelopmental disorders and neurodegenerative diseases (26, 28, 65, 66), trisomy 21 in these cells may contribute to multiple aspects of DS etiology (3). Indeed, alterations in astrocyte structure and molecular composition have been observed in the developing and adult DS brain (16, 30, 31, 67). Here, we utilized newly created DS and CTL hiPSC lines to provide a comprehensive analysis of perturbations in gene expression and functional properties of DS astrocytes. Using RNA-seq, we observed a consistent genome-wide disruption in gene expression. Interestingly, while chromosome 21 genes show an expected upregulation (68), their increase represented only a small fraction (~7%) of the overall changes occurring within DS astrocytes. Gene ontology analysis further revealed extensive dysregulation of molecules that participate in cell adhesion and ECM organization. ATAC-seq revealed an alteration in chromatin accessibility in DS astrocytes, and notably showed a preferential dysregulation in chromatin accessibility of promoter regions of cell adhesion and ECM genes. Finally, we investigated the functional properties of DS astrocytes and uncovered changes to cell size, cell spreading, cell-cell adhesion, and cell motility, as well as alterations in PCDHG-mediated cellular adhesion/recognition. These collective results demonstrate a significant perturbation in the molecular and functional cell adhesion state of DS astrocytes.
As triplication of chromosome 21 is the cause of DS, most studies over the last several decades have focused on cellular pathways related to chromosome 21 genes. Recently, however, genome-wide disruptions in gene expression have been identified in various DS tissues and cell types (69–73). We now show that trisomy 21 causes a specific genome-wide transcriptome dysregulation in astrocytes, which are key cells that contribute to brain development, homeostasis and function. Our study also identified genome-wide changes in the chromatin state of DS astrocytes, which is in line with other studies suggesting that epigenomic changes may be a common feature in DS and linked to genome-wide transcriptome dysregulation. However, the lack of direct correspondence between mRNAs/genes identified by RNA-seq and ATAC-seq highlights the complex nature of the genetic and transcriptional perturbation found in DS astrocytes. This lack of correspondence has been previously reported in DS fibroblasts (54). Our results suggest a complex cascade of epigenomic and transcriptional alterations occurring in astrocytes with trisomy 21 that may include misregulation of transcription factors and non-coding RNAs, as well as alterations to epigenetic machinery. Our results, along with those from the above-mentioned studies, help to understand the global landscape of epigenomic and transcriptional modifications in DS and provide a better comprehension of this complex genetic condition with respect to non-neuronal cells of the brain.
Our transcriptomic analysis revealed extensive dysregulation of molecules that participate in cell adhesion and ECM organization. Remarkably, alterations in cell adhesion and ECM molecules have been reported in various non-nervous system cell types in DS. In DS fetal samples, ECM genes have been shown to be upregulated in cardiac tissues (74). Altered expression of ECM molecules (collagen VI and hyaluronan) has also been identified in skin fibroblasts and in the umbilical cord of DS individuals (75, 76). In addition, DS individuals have been shown to have an altered ECM composition known to be protective against solid tumors (77, 78), which is thought to be the cause of their decreased susceptibility to developing certain types of cancers (79). The altered expression of adhesion and ECM molecules observed in DS astrocytes may have important repercussions during brain development and also contribute to their response to CNS injury and disease (80). The altered adhesion profile of astrocytes could affect their migration, maturation and subsequent integration into brain microenvironments during development (81–83). Interestingly, recent RNA-seq studies performed on hiPSC-derived DS neurons (84, 85) have also shown gene expression changes in ECM and cell adhesion molecules, implying that such changes may occur globally in the DS brain, affecting both neurons and astrocytes. Such changes may have broad implications in early neural circuit wiring and synaptogenesis, which are known to be altered in the DS brain. Importantly, however, it should be noted that while similar results were found by GO analysis, only a small percentage of differentially expressed genes in astrocytes identified in our study are shared with the studies on neurons (Fig. S12B and C). Thus, while cell adhesion and ECM organization are generally altered in hiPSC-derived DS neurons and astrocytes, the specific genes responsible for such alterations appear to be largely different. Additional factors to be taken into consideration in future studies will be the influence of sex, age and ethnicity on transcriptional changes in both of these cell types. Understanding the underlying mechanisms for such selective defects in DS in neurons and astrocytes will help dissect the possible causes of intellectual disability and provide additional paths for more specific medical interventions during development.
Genome-wide changes in gene expression in astrocytes caused by trisomy 21 may also affect the ability of astrocytes to respond to CNS injury or disease. Astrocytes play a complex role following injury and in disease, forming glial scars and recruiting other cell types including immune cells (86, 87). Astrocytes become reactive and motile in order to respond to sites of injury such as in stroke (88), traumatic injury (89) or Aβ plaque deposition (90, 91) and participate in axonal regeneration and remyelination after injury (92–94). The altered cell adhesion and recognition properties of DS astrocytes identified in this study may be relevant for DS-associated ad (a pathology known to occur late in the life of DS individuals), where astrocytes become reactive around Aß plaques (95). Targeting the altered adhesive state and motility of DS astrocytes could be a novel target to mitigate injury-related and progressive AD pathology later in life.
Materials and Methods
Generation of hiPSCs
Six patient fibroblast cell lines were obtained from the Coriell Institute and one line from American Type Culture Collection (ATCC) (Table S1) and cultured in DMEM/F-12 supplemented with 20% heat inactivated FBS. Effort was made to match gender, age, ethnicity and origin of fibroblasts. The fibroblasts were electroporated with episomal vectors containing Oct4, Sox2, c-Myc and Klf4 as well as a puromycin selection cassette (Alstem). Puromycin selection was performed 48 hours post electroporation. The cells were then cultured for 30 days in TeSR-E7 reprogramming medium (Stem Cell Technologies) with daily medium changes, and hiPSC colonies were picked manually and plated on matrigel (Corning)-coated plates. Three clones were selected per cell line and grown and expanded in mTeSR maintenance medium (Stem Cell Technologies). Karyotyping was performed by the Center for Applied Genomics (The Hospital for Sick Children, Toronto).
Production of neural precursor cells and astrocytes
Neural precursor cells (NPCs) were generated from hiPSCs using a double-Smad inhibition protocol (38). The hiPSCs were dissociated and grown as a monolayer on matrigel-coated plates in DMEM/F-12 (Sigma) supplemented with N2, B27, BSA (1 mg/mL), SB431542 (10 μM) and noggin (200 ng/mL) for three weeks (neural induction medium; NIM) (39). The cells were then dissociated and grown in suspension in NPC maintenance medium (Stem Cell Technologies) for 7–9 days. Finally, the cells were plated on matrigel and cultured in NPC maintenance medium.
Astrocytes were derived from NPCs using a monolayer differentiation protocol (Fig. 1A) adapted from Roybon et al., Shaltouki et al. and Serio et al. (40, 41, 96). NPCs were passaged onto poly-D-lysine and matrigel-coated plates in DMEM/F-12 medium supplemented with FBS (10%; Gibco), EGF (20 ng/mL; Genscript) and FGF2 (20 ng/mL; Cell Signaling) for 2 weeks and were passaged as needed. The astrocytes were then cultured on tissue culture-treated culture dishes in DMEM/F-12 medium supplemented with FBS (10%; Gibco), EGF (20 ng/mL) (FGF2 (20 ng/mL) and CNTF (5 ng/mL; Genscript) (Protocol A) or cultured in DMEM/F-12 medium supplemented with FBS (10%; Gibco), EGF (20 ng/mL) and FGF2 (20 ng/mL) (Protocol B) for 6 weeks and were passaged as needed. Astrocytes were then matured in DMEM/F-12 medium supplemented with 10% FBS and CNTF (5 ng/mL) for an additional 4 weeks. Many studies have reported a gliogenic shift and an overall increase in the numbers of astrocytes in the DS brain (12–16). We observe no difference in the efficiency of astrocyte differentiation or astrocyte numbers between our DS and control cell lines. This is likely due to the fact that we are inducing astrocyte differentiation using a specific protocol.
Immunolabeling
Cells were fixed using 4% paraformaldehyde and permeabilized with 0.2% Triton-X100 (Sigma) in PBS for 15 minutes before blocking with normal horse serum (NHS; 2%) in PBS with 0.2% Triton-X100 for 1 hour. Primary antibodies (listed below) were applied for 18 hours in 2% NHS at 4°C. After washing three times, Alexa-conjugated secondary antibodies (Invitrogen, 1:500) were applied for 2 hours before washing three times. Coverslips were mounted using Prolong Gold with DAPI (Life Technologies) prior to imaging. The images were acquired using an Olympus FV-1000 laser scanning microscope and Fluoview FV10 software (Olympus). Primary antibodies were used at the following concentrations: guinea-pig anti-GFAP (Synaptic Systems) 1:500, goat anti-Sox1 1:250 (Thermo Fisher), rabbit anti-Sox2 1:250 (Thermo Fisher), rabbit anti-Sox9 1:500 (Millipore), rabbit anti-Pax6 1:250 (Thermo Fisher), mouse anti-Nestin 1:250 (Thermo Fisher), rabbit anti-Oct4 1:250 (Thermo Fisher), mouse anti-TRA-1-60 1:250 (Thermo Fisher), mouse anti-S100b 1:500 (Sigma), mouse anti-SMI-312 1:500 (Covance), mouse anti-CC1 1:200 (Calbiochem), mouse anti-MAP2 1:500 (Millipore), rabbit anti-EAAT1 1:500 (Abcam) and mouse anti-Aldh1l1 1:200 (Neuromab). Analysis of Sox9, Sox1, GFAP and S100b immunolabeling was performed with a DAPI co-label. Cells positive for DAPI and the above-mentioned markers were quantified using the ImageJ cell counter. A total of 10 images per condition were analyzed.
Ca2+ imaging
CTL and DS astrocytes (~90 days post-differentiation) were seeded onto coverslips and loaded with the single wavelength calcium indicator Fluo4-AM (Thermo Fisher) dissolved in DMSO. Immediately prior to loading, Fluo4-AM was diluted to 1 μM in culture medium. Cell culture medium was replaced entirely with this solution and the cells were incubated for 20–50 min at 37°C 5% CO2. Coverslips were transferred onto the stage of a customized Olympus FV1000 laser scanning microscope and continuously superfused with artificial cerebrospinal fluid (aCSF; 126 mM NaCl, 2.5 mM KCl, 1.3 mM MgCl2, 10 mM d-glucose, 2.4 mM CaCl2, 1.24 mM NaH2PO4, and 26 mM NaHCO3 saturated with 95% O2 and 5% CO2). Fluo4-AM was excited at 488 nm and images were recorded at a frame rate of 0.5 Hz. Spontaneous Ca2+ activity was acquired for 10 minutes. We counted all cells in which the F/F0 trace reached values higher than a threshold of 1.30 (30% signal increase over baseline) at any point during the 10 minute baseline recordings as spontaneously active cells. To obtain the fraction of spontaneously active cells, we divided this number by the total number of cells image-analyzed. To record ATP-evoked Ca2+ events, 2 minutes of additional baseline recordings were obtained, after which the cells were superfused with 100 μM ATP in aCSF for 1 minute. A total of three 1 minute 100 μM ATP pulses with 2 minutes of baseline in between were recorded. Image analysis was performed using the open source software FIJI. F/F0 was calculated separately for each pulse by dividing the fluorescence intensity of each image by the average intensity of 10 frames during the preceding baseline. The average F/F0 intensity over time was measured in manually defined ROIs corresponding to cells.
Quantitative reverse transcriptase polymerase chain reaction (qPCR)
At approximately 90 days post-differentiation, RNA of astrocytes was extracted using the RNeasy plus mini kit (Qiagen), and cDNA libraries were made using the Quantitect reverse transcription kit (Qiagen). qPCR was performed using Sybr Green Master Mix (Applied Biosystems) on a StepOne Plus thermocycler (Applied Biosystems). Relative levels of mRNA were calculated using the ΔΔCT method with RPL13A as the internal control.
RNA-sequencing and data analysis
At approximately 90 days post-differentiation, RNA was extracted from cells using the RNeasy Plus Mini Kit (Qiagen). Sample quality was assessed using a Bioanalyser (Agilent) and sequenced at 12 samples per lane on an Illumina HiSeq4000 for 100 bp paired-end reads at the McGill University and Genome Quebec Innovation Center. All libraries passed an initial quality control step using the FASTQC pipeline. The Fast-X toolkit was used to trim the first and last 10 bases of each read. TruSeq-specific paired-end adapters and low-quality stretches were removed using Trimmomatic (97), while poly-A tails were trimmed using PrinSeq (98). Surviving paired and orphaned reads were separately aligned using STAR to the GRCh37 human genome (99). HTseq was used to count pairs of reads mapping to specific genes (100). Using the DESeq2 package, we normalized read counts and applied a variance-stabilizing transformation to the data (101).
Differential expression analysis between CTL and DS astrocytes was performed using DESeq2 (101). Statistically significant genes (adjusted P-value < 0.05) were considered differentially expressed. Shrinkage of log-fold changes was applied using DESeq2, and shrunken estimates were used throughout the analysis. Genes were annotated using Ensembl BioMart and filtered to retain only protein-coding genes (102). Normalized and variance-stabilized transformed data was used for all heatmaps. Differentially expressed chromosome 21 genes were visualized on the chromosome 21 ideogram using the karyoploteR package (103).
Gene ontology analysis and functional annotation were performed with enrichGO from the R/Bicoonductor package clusterProfiler (version 3.10.0) (104), using all differentially expressed genes compared to a custom background gene list consisting of all expressed genes (defined as all genes with a baseMean greater than the first decile). FPKM values were calculated using exonic gene lengths with DESeq2, which estimates library size using the median of ratios method. Gene haploinsufficiency data were obtained from Lek et al., (105). Transcription factors were identified from the AnimalTFDB 3.0 resource (106). Targets in each gene ontology category were ranked by fold change, and genes with less than 30 base mean reads were omitted. The top 25 targets were then chosen to be validated by qPCR. Of those, 22 targets were validated (3 targets were too low in abundance) 19 of which were in concordance with RNAseq results.
ATAC-sequencing and data analysis
Astrocytes were cultured for 13 weeks as described above, with the exception that they were maintained on Matrigel-coated dishes throughout. ATAC-seq was performed as previously described using 50 000 nuclei (107, 108). Briefly, fresh cell pellets from 50 000 cells were exposed to a hypotonic cell lysis buffer (0.1% (wt/vol) sodium citrate tribasic dihydrate and 0.1% (v/v) Triton X-100) for 30 minutes at 4°C followed by treatment with normal cell lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% (v/v) IGEPAL CA-630) for 30 minutes at 4°C. To perform the transposition reaction, the nuclei were incubated with 25 μL of Transposase Master Mix (2.5 μL 10× TD buffer, 10 μL H2O and 12.5 μL Tn5 enzyme from an Illumina Nextera kit; FC-121–1031) for 30 minutes at 37°C. DNA was then purified and enriched by PCR using Nextera indexed primers, and the library was recovered with GeneRead Purification columns (Qiagen), eluted in 35ul EB buffer. The library was assessed using a bioanalyzer (Agilent) and sequenced on an Illumina HiSeq4000 instrument to produce 100 bp paired-end reads.
Enrichment of open chromatin was verified using qPCR primers amplifying open-chromatin positive (housekeeping genes GAPDH and B2M) versus open-chromatin negative (gene desert) regions. A 11 (B2M)-40 (GAPDH)-fold enrichment was observed (not shown). All libraries passed an initial quality control step using the FASTQC pipeline. Low quality and adapter sequences were trimmed using Trim Galore. Reads were aligned using to the GRCh37 human genome using bwa mem with default settings and duplicates removed using Picard MarkDuplicates. To obtain reads representing nucleosome-free DNA, BAM files were filtered to reads < 100 bp, following Buenrostro et al. (56). Peaks were called using MACS2 callpeak with the default FDR cutoff of 0.05 and used for downstream analysis (109).
Differentially accessible peaks between genotypes were identified using the R/Bioconductor package DiffBind (version 2.10.0) and annotated with the nearest gene and genomic compartment using the R/Bioconductor package ChIPseeker (version 1.18.0) (110). Bigwig tracks were generated using deepTools bamCoverage with RPKM normalization and a bin size of 1 (111). Peaks were defined to be within promoter regions if they were annotated as within 3 kb of a gene transcription start site by ChIPseeker. Average ATAC-seq signal in 5kb intervals centered at these peaks were then used to cluster these regions using k-means (k = 4) clustering implemented in deepTools. deepTools was used to generate average ATAC-seq signal profile over these peaks for each sample and to calculate tag density in regions upstream of gene transcription start sites. Gene ontology analysis was performed using enrichGO, applied to genes with differentially accessible peaks within promoter regions.
Code availability
All R codes to produce the RNA-seq and ATAC-seq analysis presented in this study will be made available at https://github.com/murailab/Ponroy2020_HMG.
Western blot analysis
Approximately 90 days post-differentiation, astrocytes were lysed in 400 μL RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 20 mM Tris pH 8.0, 150 mM NaCl and 1 mM EDTA) containing 1 μg/ml each of leupeptin, aprotinin, pepstatin, 10 mMNaF, 1 mM sodium ortho-vanadate and 1 mM PMSF. Lysates were diluted with 3X sample buffer and equal quantities of each lysate were run on a 10% polyacrylamide gel and transferred to PVDF membranes following standard protocols. Membranes were blocked for 40 minutes with 5% BSA/TBS-0.1% Tween, and incubated overnight at 4°C with an anti-mouse PCDHG antibody (1:1000, Neuromab) and an anti-rabbit RPL13A antibody (1:2000, Cell Signaling) as the loading control. The next day membranes were incubated for 1 hour at room temperature with secondary antibodies conjugated to horse radish peroxidase. Chemiluminescent signal was obtained using Amersham ECL Plus Western Blotting Detection Reagents (GE Healthcare) and captured on X-ray film. Densitometry was performed using the gel analysis function in ImageJ.
PCDH adhesion assay
Glass coverslips were coated with PCDHs for 2 hours at the following concentrations: Pcdhgc3 2 ug/mL (R&D Systems), Pcdh12 4 ug/mL (R&D Systems) and Pcdh10 4 ug/mL (RD systems). CTL and DS astrocytes at approximately 2.5 months of age were seeded onto glass coverslips with and without PCDH coating at 1 × 105 cells per well and were left to adhere for 30 minutes and subsequently agitated at 1500 rotations per minute for 30 minutes. The astrocytes were plated in astrocyte medium without serum, in order to avoid potential coating by the serum. Astrocytes were then fixed and stained with DAPI using the above-described immunohistochemistry protocols. Images were acquired using an Olympus FV-1000 laser scanning microscope and Fluoview FV10 software (Olympus). Images were analyzed and cells were counted using FIJI software.
Astrocyte cell size comparison
Approximately 8 weeks post-differentiation, astrocytes were stained with a rabbit anti-EAAT1 antibody (Abcam) with a DAPI co-label, and the images were acquired using an Olympus FV-1000 laser scanning microscope and Fluoview FV10 software (Olympus). The cell area was calculated using the freehand tool in ImageJ to draw the outline of the cells and the measure function. A total of 45 images per condition were analyzed from three independent experiments, in which five images were analyzed in three cell lines per condition.
Relative size and internal complexity (i.e. granularity) of the cells in suspension were extrapolated from forward scatter (FSC) and side scatter (SSC) parameters acquired using a Cyan ADP flow cytometer, and data were analyzed using the FlowJo software (Treestar).
Cell spreading assay
At approximately 8 weeks post-differentiation, 10 000 astrocytes were plated on PolyD-lysine-coated coverslips for 30, 60, 90 and 120 minutes in medium without FBS in order to avoid potential coating by the serum. Astrocytes were then fixed and stained with DAPI and Phalloidin-488 (Cell Signaling) using the above-described immunohistochemistry protocols. Five images per time point were acquired in all three CTL and 3DS cell lines using an Olympus FV-1000 laser scanning microscope and Fluoview FV10 software (Olympus). Images were analyzed and cell area was measured using FIJI software.
Cell aggregation assay
At approximately 8 weeks post-differentiation, 30 000 astrocytes were placed in suspension in non-coated petri-dishes for 2 hours, in low serum-containing medium (1% FBS). After two hours, the cell clumps were imaged on an Ultraview spinning disk confocal system (Perkin Elmer). The aggregate area was calculated using the freehand tool in ImageJ to draw the outline of the aggregate and the measure function. A total of 45 images per condition were analyzed from three independent experiments, in which five images were analyzed in three cell lines per condition. The medium containing the cells in suspension was then removed, spun down, trypsinized and resuspended, after which the cells were counted using a cell counter. The cells which had adhered to the petri dish were trypsinized and counted using a cell counter.
Cell motility assay
At approximately 8 weeks post-differentiation, astrocytes were seeded onto plastic tissue culture-treated 96 well plates (Corning 3904) at a density of 5000 cells per well and left to adhere for 24 hours. They were then incubated with Hoechst 33342 (0.2 μg/ml) to stain cell nuclei for approximately one hour after which the medium was switched for phenol-free DMEMF12 with the same previously described supplements, as well as 20 mM HEPES (pH 7.4). Time-lapse sequences were then acquired (20 frames, at 10 min intervals, 190 min total) at 37°C using an automated widefield fluorescence microscope (ImageXpress Micro XL, Molecular Devices), equipped with an Andor Zyla 5.5 sCMOS camera and using a 4x objective. Cell tracking was performed based on the trajectories of segmented cell nuclei using custom-written MATLAB routines, as previously described (64). Cell velocity was averaged over 20 frames and migration persistence computed as net displacement/total path length over 20 frames.
Experimental design and statistical analysis
Data are shown as the mean ± SEM. Statistical analysis between two groups was performed using unpaired two-tailed t-test, (using Graphpad software version 5.01). Standard symbols were used to report significance: n.s.—not significant (P ≥ 0.05), Unless stated otherwise, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Supplementary Material
Acknowledgements
We thank Marianna Bego for her help with the flow cytometry experiment. We thank the Molecular Imaging Platform of the RI-MUHC and the Advanced BioImaging Facility (ABIF) at McGill University for access to imaging equipment and services provided. We also thank A. Montpetit and the Genome Quebec staff for assistance with sequencing.
Contributions
BPB and KKM conceived the project and wrote the manuscript. BPB contributed to all experiments. WTF and SJ contributed to RNA-seq and ATAC-seq analysis and manuscript editing. EVJ contributed to biochemistry and immunolabeling experiments and preparation of cells for ATAC-seq and helped with hiPSC-derived astrocytes, as well as manuscript editing. AM helped with preparation and planning of the ATAC-seq experiment. AM and JD edited the manuscript. JL provided PCDH antibodies and edited the manuscript. BK performed Ca2+ imaging experiments and analysis. AH helped with the motility assays and performed their analysis and edited the manuscript. HP and CE provided support with generating hiPSCs, and CE edited the manuscript.
Funding
The Canadian Institutes of Health Research (OGB 111152, PJT148569, 156247 to K.K.M.); Natural Sciences and Engineering Research Council of Canada (408044-2011 and 69404 to K.K.M. and 05831-2018 to A.H.); Canada Research Chairs Program (K.K.M.; C.E.); Brain Canada/Weston Foundation (K.K.M.) and the Sandra and Alain Bouchard Intellectual Disability Research Program (K.K.M). Studentships from the McGill Faculty of Medicine and the Canada First Research Excellence Fund for the Healthy Brains for Healthy Lives initiative to B.P.B.
Conflict of Interest statement
None declared.
Data Availability
All data in this study is available through NCBI (PRJNA600245).
Abbreviations
DS, Down syndrome; hiPSCs, human induced pluripotent stem cells; ATAC, assay for transposase-accessible chromatin; HSA21, Homo sapiens chromosome 21; AD, Alzheimer’s disease; GFAP, glial fibrillary acidic protein; RNA-seq, RNA-sequencing; ECM, extracellular matrix; NPCs, neural precursor cells; PCDH, protocadherin; CTL, control; ATCC, American type culture collection; qPCR, uantitative reverse transcriptase polymerase chain reaction; GO, gene ontology
References
- 1. Parker S.E., Mai C.T., Canfield M.A., Rickard R., Wang Y., Meyer R.E., Anderson P., Mason C.A., Collins J.S., Kirby R.S. et al. (2010) Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res. A Clin. Mol. Teratol., 88, 1008–1016. [DOI] [PubMed] [Google Scholar]
- 2. Asim A., Kumar A., Muthuswamy S., Jain S. and Agarwal S. (2015) Down syndrome: an insight of the disease. J. Biomed. Sci., 22, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antonarakis S.E., Lyle R., Dermitzakis E.T., Reymond A. and Deutsch S. (2004) Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat. Rev. Genet., 5, 725–738. [DOI] [PubMed] [Google Scholar]
- 4. Lott I.T. (2012) Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res., 197, 101–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lott I.T. and Dierssen M. (2010) Cognitive deficits and associated neurological complications in individuals with Down's syndrome. Lancet Neurol., 9, 623–633. [DOI] [PubMed] [Google Scholar]
- 6. Antonarakis S.E. (2017) Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet., 18, 147–163. [DOI] [PubMed] [Google Scholar]
- 7. Arya R., Kabra M. and Gulati S. (2011) Epilepsy in children with Down syndrome. Epileptic Disord., 13, 1–7. [DOI] [PubMed] [Google Scholar]
- 8. Glasson E.J., Sullivan S.G., Hussain R., Petterson B.A., Montgomery P.D. and Bittles A.H. (2002) The changing survival profile of people with Down's syndrome: implications for genetic counselling. Clin. Genet., 62, 390–393. [DOI] [PubMed] [Google Scholar]
- 9. Roizen N.J. and Patterson D. (2003) Down's syndrome. Lancet, 361, 1281–1289. [DOI] [PubMed] [Google Scholar]
- 10. Pinter J.D., Brown W.E., Eliez S., Schmitt J.E., Capone G.T. and Reiss A.L. (2001) Amygdala and hippocampal volumes in children with Down syndrome: a high-resolution MRI study. Neurology, 56, 972–974. [DOI] [PubMed] [Google Scholar]
- 11. Teipel S.J., Schapiro M.B., Alexander G.E., Krasuski J.S., Horwitz B., Hoehne C., Moller H.J., Rapoport S.I. and Hampel H. (2003) Relation of corpus callosum and hippocampal size to age in nondemented adults with Down's syndrome. Am. J. Psychiatry, 160, 1870–1878. [DOI] [PubMed] [Google Scholar]
- 12. Busciglio J. and Yankner B.A. (1995) Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature, 378, 776–779. [DOI] [PubMed] [Google Scholar]
- 13. Guidi S., Bonasoni P., Ceccarelli C., Santini D., Gualtieri F., Ciani E. and Bartesaghi R. (2008) Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol., 18, 180–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu J., Esposito G., Scuderi C., Steardo L., Delli-Bovi L.C., Hecht J.L., Dickinson B.C., Chang C.J., Mori T. and Sheen V. (2011) S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors. PLoS One, 6, e22126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wisniewski K.E. (1990) Down syndrome children often have brain with maturation delay, retardation of growth, and cortical dysgenesis. Am. J. Med. Genet. Suppl., 7, 274–281. [DOI] [PubMed] [Google Scholar]
- 16. Zdaniuk G., Wierzba-Bobrowicz T., Szpak G.M. and Stepien T. (2011) Astroglia disturbances during development of the central nervous system in fetuses with Down's syndrome. Folia Neuropathol., 49, 109–114. [PubMed] [Google Scholar]
- 17. Garner C.C. and Wetmore D.Z. (2012) Synaptic pathology of Down syndrome. Adv. Exp. Med. Biol., 970, 451–468. [DOI] [PubMed] [Google Scholar]
- 18. Becker L.E., Armstrong D.L. and Chan F. (1986) Dendritic atrophy in children with Down's syndrome. Ann. Neurol., 20,520–526. [DOI] [PubMed] [Google Scholar]
- 19. Belichenko P.V., Kleschevnikov A.M., Salehi A., Epstein C.J. and Mobley W.C. (2007) Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. J. Comp. Neurol., 504, 329–345. [DOI] [PubMed] [Google Scholar]
- 20. Takashima S., Iida K., Mito T. and Arima M. (1994) Dendritic and histochemical development and ageing in patients with Down's syndrome. J. Intellect. Disabil. Res., 38(Pt 3), 265–273. [DOI] [PubMed] [Google Scholar]
- 21. Farmer W.T. and Murai K. (2017) Resolving astrocyte heterogeneity in the CNS. Front. Cell. Neurosci., 11, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Drejer J., Larsson O.M. and Schousboe A. (1982) Characterization of L-glutamate uptake into and release from astrocytes and neurons cultured from different brain regions. Exp. Brain Res., 47, 259–269. [DOI] [PubMed] [Google Scholar]
- 23. Parpura V., Basarsky T.A., Liu F., Jeftinija K., Jeftinija S. and Haydon P.G. (1994) Glutamate-mediated astrocyte-neuron signalling. Nature, 369, 744–747. [DOI] [PubMed] [Google Scholar]
- 24. Denis-Donini S., Glowinski J. and Prochiantz A. (1984) Glial heterogeneity may define the three-dimensional shape of mouse mesencephalic dopaminergic neurones. Nature, 307, 641–643. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y. and Barres B.A. (2010) Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr. Opin. Neurobiol., 20, 588–594. [DOI] [PubMed] [Google Scholar]
- 26. Sloan S.A. and Barres B.A. (2014) Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr. Opin. Neurobiol., 27, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y., Higashimori H. and Morel L. (2013) Developmental maturation of astrocytes and pathogenesis of neurodevelopmental disorders. J Neurodev Disord, 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Phatnani H. and Maniatis T. (2015) Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol., 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yates D. (2015) Neurodegenerative disease: factoring in astrocytes. Nat. Rev. Neurosci., 16, 67. [DOI] [PubMed] [Google Scholar]
- 30. Jorgensen O.S., Brooksbank B.W. and Balazs R. (1990) Neuronal plasticity and astrocytic reaction in Down syndrome and Alzheimer disease. J. Neurol. Sci., 98, 63–79. [DOI] [PubMed] [Google Scholar]
- 31. Mito T. and Becker L.E. (1993) Developmental changes of S-100 protein and glial fibrillary acidic protein in the brain in Down syndrome. Exp. Neurol., 120, 170–176. [DOI] [PubMed] [Google Scholar]
- 32. Nelson P.G., Fitzgerald S., Rapoport S.I., Neale E.A., Galdzicki Z., Dunlap V., Bowers L. and v Agoston D. (1997) Cerebral cortical astroglia from the trisomy 16 mouse, a model for down syndrome, produce neuronal cholinergic deficits in cell culture. Proc. Natl. Acad. Sci. U. S. A., 94, 12644–12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garcia O., Torres M., Helguera P., Coskun P. and Busciglio J. (2010) A role for thrombospondin-1 deficits in astrocyte-mediated spine and synaptic pathology in Down's syndrome. PLoS One, 5, e14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helguera P., Seiglie J., Rodriguez J., Hanna M., Helguera G. and Busciglio J. (2013) Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab., 17, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sebastia J., Cristofol R., Pertusa M., Vilchez D., Toran N., Barambio S., Rodriguez-Farre E. and Sanfeliu C. (2004) Down's syndrome astrocytes have greater antioxidant capacity than euploid astrocytes. Eur. J. Neurosci., 20, 2355–2366. [DOI] [PubMed] [Google Scholar]
- 36. Chen C., Jiang P., Xue H., Peterson S.E., Tran H.T., McCann A.E., Parast M.M., Li S., Pleasure D.E., Laurent L.C. et al. (2014) Role of astroglia in Down's syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun., 5, 4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Araujo B.H.S., Kaid C., De Souza J.S., Gomes da Silva S., Goulart E., Caires L.C.J., Musso C.M., Torres L.B., Ferrasa A., Herai R. et al. (2018) Down syndrome iPSC-derived astrocytes impair neuronal synaptogenesis and the mTOR pathway in vitro. Mol. Neurobiol., 55, 5962–5975. [DOI] [PubMed] [Google Scholar]
- 38. Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M. and Studer L. (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol., 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Scott Bell N.C.H., Silveira H., Peng H., Wu H., Jefri M., Antonyan L., Zhang Y., Zhang X. and Ernst C. (2019) Differentiation of human induced pluripotent stem cells (iPSCs) into an effective model of forebrain neural progenitor cells and mature neurons. Bioanalysis, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roybon L., Lamas N.J., Garcia A.D., Yang E.J., Sattler R., Lewis V.J., Kim Y.A., Kachel C.A., Rothstein J.D., Przedborski S. et al. (2013) Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep., 4, 1035–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shaltouki A., Peng J., Liu Q., Rao M.S. and Zeng X. (2013) Efficient generation of astrocytes from human pluripotent stem cells in defined conditions. Stem Cells, 31, 941–952. [DOI] [PubMed] [Google Scholar]
- 42. Perea G., Navarrete M. and Araque A. (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci., 32, 421–431. [DOI] [PubMed] [Google Scholar]
- 43. Scemes E. and Giaume C. (2006) Astrocyte calcium waves: what they are and what they do. Glia, 54, 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mizuno G.O., Wang Y., Shi G., Wang Y., Sun J., Papadopoulos S., Broussard G.J., Unger E.K., Deng W., Weick J. et al. (2018) Aberrant calcium signaling in astrocytes inhibits neuronal excitability in a human Down syndrome stem cell model. Cell Rep., 24, 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y., Sloan S.A., Clarke L.E., Caneda C., Plaza C.A., Blumenthal P.D., Vogel H., Steinberg G.K., Edwards M.S., Li G. et al. (2016) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron, 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lang C., Campbell K.R., Ryan B.J., Carling P., Attar M., Vowles J., Perestenko O.V., Bowden R., Baig F., Kasten M. et al. (2019) Single-cell sequencing of iPSC-dopamine neurons reconstructs disease progression and identifies HDAC4 as a regulator of Parkinson cell phenotypes. Cell Stem Cell, 24, e106, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Switonska K., Szlachcic W.J., Handschuh L., Wojciechowski P., Marczak L., Stelmaszczuk M., Figlerowicz M. and Figiel M. (2018) Identification of altered developmental pathways in human juvenile HD iPSC with 71Q and 109Q using transcriptome profiling. Front. Cell. Neurosci., 12, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olmos-Serrano J.L., Kang H.J., Tyler W.A., Silbereis J.C., Cheng F., Zhu Y., Pletikos M., Jankovic-Rapan L., Cramer N.P., Galdzicki Z. et al. (2016) Down syndrome developmental brain transcriptome reveals defective oligodendrocyte differentiation and myelination. Neuron, 89, 1208–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pogribna M., Melnyk S., Pogribny I., Chango A., Yi P. and James S.J. (2001) Homocysteine metabolism in children with Down syndrome: in vitro modulation. Am. J. Hum. Genet., 69, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chango A., Abdennebi-Najar L., Tessier F., Ferre S., Do S., Gueant J.L., Nicolas J.P. and Willequet F. (2006) Quantitative methylation-sensitive arbitrarily primed PCR method to determine differential genomic DNA methylation in Down syndrome. Biochem. Biophys. Res. Commun., 349, 492–496. [DOI] [PubMed] [Google Scholar]
- 51. Kerkel K., Schupf N., Hatta K., Pang D., Salas M., Kratz A., Minden M., Murty V., Zigman W.B., Mayeux R.P. et al. (2010) Altered DNA methylation in leukocytes with trisomy 21. PLoS Genet., 6, e1001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bacalini M.G., Gentilini D., Boattini A., Giampieri E., Pirazzini C., Giuliani C., Fontanesi E., Scurti M., Remondini D., Capri M. et al. (2015) Identification of a DNA methylation signature in blood cells from persons with Down syndrome. Aging (Albany NY), 7, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jones M.J., Farre P., McEwen L.M., Macisaac J.L., Watt K., Neumann S.M., Emberly E., Cynader M.S., Virji-Babul N. and Kobor M.S. (2013) Distinct DNA methylation patterns of cognitive impairment and trisomy 21 in Down syndrome. BMC Med. Genomics, 6, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Letourneau A., Santoni F.A., Bonilla X., Sailani M.R., Gonzalez D., Kind J., Chevalier C., Thurman R., Sandstrom R.S., Hibaoui Y. et al. (2014) Domains of genome-wide gene expression dysregulation in Down's syndrome. Nature, 508, 345–350. [DOI] [PubMed] [Google Scholar]
- 55. Lu J., McCarter M., Lian G., Esposito G., Capoccia E., Delli-Bovi L.C., Hecht J. and Sheen V. (2016) Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum. Mol. Genet., 25, 1714–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Buenrostro J.D., Wu B., Chang H.Y. and Greenleaf W.J. (2015) ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol, 109, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu B., Filippi S., Roy A. and Roberts I. (2015) Stem and progenitor cell dysfunction in human trisomies. EMBO Rep., 16, 44–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Garrett A.M. and Weiner J.A. (2009) Control of CNS synapse development by {gamma}-protocadherin-mediated astrocyte-neuron contact. J. Neurosci., 29, 11723–11731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Molumby M.J., Keeler A.B. and Weiner J.A. (2016) Homophilic protocadherin cell-cell interactions promote dendrite complexity. Cell Rep., 15, 1037–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Light S.E.W. and Jontes J.D. (2017) Delta-protocadherins: organizers of neural circuit assembly. Semin. Cell Dev. Biol., 69, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Becker L., Mito T., Takashima S. and Onodera K. (1991) Growth and development of the brain in Down syndrome. Prog. Clin. Biol. Res., 373, 133–152. [PubMed] [Google Scholar]
- 62. El Hajj N., Dittrich M., Bock J., Kraus T.F., Nanda I., Muller T., Seidmann L., Tralau T., Galetzka D., Schneider E. et al. (2016) Epigenetic dysregulation in the developing Down syndrome cortex. Epigenetics, 11, 563–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Althubiti M., Lezina L., Carrera S., Jukes-Jones R., Giblett S.M., Antonov A., Barlev N., Saldanha G.S., Pritchard C.A., Cain K. et al. (2014) Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis., 5, e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hayer A., Shao L., Chung M., Joubert L.M., Yang H.W., Tsai F.C., Bisaria A., Betzig E. and Meyer T. (2016) Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol., 18, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ricci G., Volpi L., Pasquali L., Petrozzi L. and Siciliano G. (2009) Astrocyte-neuron interactions in neurological disorders. J. Biol. Phys., 35, 317–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Verkhratsky A., Steardo L., Parpura V. and Montana V. (2016) Translational potential of astrocytes in brain disorders. Prog. Neurobiol., 144, 188–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Colombo J.A., Reisin H.D., Jones M. and Bentham C. (2005) Development of interlaminar astroglial processes in the cerebral cortex of control and Down's syndrome human cases. Exp. Neurol., 193, 207–217. [DOI] [PubMed] [Google Scholar]
- 68. Mao R., Zielke C.L., Zielke H.R. and Pevsner J. (2003) Global up-regulation of chromosome 21 gene expression in the developing Down syndrome brain. Genomics, 81, 457–467. [DOI] [PubMed] [Google Scholar]
- 69. Mao R., Wang X., Spitznagel E.L. Jr., Frelin L.P., Ting J.C., Ding H., Kim J.W., Ruczinski I., Downey T.J. and Pevsner J. (2005) Primary and secondary transcriptional effects in the developing human Down syndrome brain and heart. Genome Biol., 6, R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lockstone H.E., Harris L.W., Swatton J.E., Wayland M.T., Holland A.J. and Bahn S. (2007) Gene expression profiling in the adult Down syndrome brain. Genomics, 90, 647–660. [DOI] [PubMed] [Google Scholar]
- 71. FitzPatrick D.R., Ramsay J., McGill N.I., Shade M., Carothers A.D. and Hastie N.D. (2002) Transcriptome analysis of human autosomal trisomy. Hum. Mol. Genet., 11, 3249–3256. [DOI] [PubMed] [Google Scholar]
- 72. Saran N.G., Pletcher M.T., Natale J.E., Cheng Y. and Reeves R.H. (2003) Global disruption of the cerebellar transcriptome in a Down syndrome mouse model. Hum. Mol. Genet., 12, 2013–2019. [DOI] [PubMed] [Google Scholar]
- 73. Briggs J.A., Sun J., Shepherd J., Ovchinnikov D.A., Chung T.L., Nayler S.P., Kao L.P., Morrow C.A., Thakar N.Y., Soo S.Y. et al. (2013) Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells, 31, 467–478. [DOI] [PubMed] [Google Scholar]
- 74. Conti A., Fabbrini F., D'Agostino P., Negri R., Greco D., Genesio R., D'Armiento M., Olla C., Paladini D., Zannini M. et al. (2007) Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genomics, 8, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grossman T.R., Gamliel A., Wessells R.J., Taghli-Lamallem O., Jepsen K., Ocorr K., Korenberg J.R., Peterson K.L., Rosenfeld M.G., Bodmer R. et al. (2011) Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS Genet., 7, e1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Karousou E., Stachtea X., Moretto P., Viola M., Vigetti D., D'Angelo M.L., Raio L., Ghezzi F., Pallotti F., De Luca G. et al. (2013) New insights into the pathobiology of Down syndrome--hyaluronan synthase-2 overexpression is regulated by collagen VI alpha2 chain. FEBS J., 280, 2418–2430. [DOI] [PubMed] [Google Scholar]
- 77. Zorick T.S., Mustacchi Z., Bando S.Y., Zatz M., Moreira-Filho C.A., Olsen B. and Passos-Bueno M.R. (2001) High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur. J. Hum. Genet., 9, 811–814. [DOI] [PubMed] [Google Scholar]
- 78. Benard J., Beron-Gaillard N. and Satge D. (2005) Down's syndrome protects against breast cancer: is a constitutional cell microenvironment the key? Int. J. Cancer, 113, 168–170. [DOI] [PubMed] [Google Scholar]
- 79. Yang Q., Rasmussen S.A. and Friedman J.M. (2002) Mortality associated with Down's syndrome in the USA from 1983 to 1997: a population-based study. Lancet, 359, 1019–1025. [DOI] [PubMed] [Google Scholar]
- 80. Wiese S., Karus M. and Faissner A. (2012) Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharmacol., 3, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rakic P. (1972) Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol., 145, 61–83. [DOI] [PubMed] [Google Scholar]
- 82. Rakic P. (2007) The radial edifice of cortical architecture: from neuronal silhouettes to genetic engineering. Brain Res. Rev., 55, 204–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schmechel D.E. and Rakic P. (1979) A Golgi study of radial glial cells in developing monkey telencephalon: morphogenesis and transformation into astrocytes. Anat. Embryol., 156, 115–152. [DOI] [PubMed] [Google Scholar]
- 84. Gonzales P.K., Roberts C.M., Fonte V., Jacobsen C., Stein G.H. and Link C.D. (2018) Transcriptome analysis of genetically matched human induced pluripotent stem cells disomic or trisomic for chromosome 21. PLoS One, 13, e0194581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Huo H.Q., Qu Z.Y., Yuan F., Ma L., Yao L., Xu M., Hu Y., Ji J., Bhattacharyya A., Zhang S.C. et al. (2018) Modeling Down syndrome with patient iPSCs reveals cellular and migration deficits of GABAergic neurons. Stem Cell Reports, 10, 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tate C.C., Tate M.C. and LaPlaca M.C. (2007) Fibronectin and laminin increase in the mouse brain after controlled cortical impact injury. J. Neurotrauma, 24, 226–230. [DOI] [PubMed] [Google Scholar]
- 87. Sofroniew M.V. and Vinters H.V. (2010) Astrocytes: biology and pathology. Acta Neuropathol., 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wilhelmsson U., Bushong E.A., Price D.L., Smarr B.L., Phung V., Terada M., Ellisman M.H. and Pekny M. (2006) Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc. Natl. Acad. Sci. U. S. A., 103, 17513–17518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Burda J.E., Bernstein A.M. and Sofroniew M.V. (2016) Astrocyte roles in traumatic brain injury. Exp. Neurol., 275Pt 3, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Olabarria M., Noristani H.N., Verkhratsky A. and Rodriguez J.J. (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia, 58, 831–838. [DOI] [PubMed] [Google Scholar]
- 91. Hou L., Liu Y., Wang X., Ma H., He J., Zhang Y., Yu C., Guan W. and Ma Y. (2011) The effects of amyloid-beta42 oligomer on the proliferation and activation of astrocytes in vitro. In Vitro Cell. Dev. Biol. Anim., 47, 573–580. [DOI] [PubMed] [Google Scholar]
- 92. Properzi F., Carulli D., Asher R.A., Muir E., Camargo L.M., van Kuppevelt T.H., ten Dam G.B., Furukawa Y., Mikami T., Sugahara K. et al. (2005) Chondroitin 6-sulphate synthesis is up-regulated in injured CNS, induced by injury-related cytokines and enhanced in axon-growth inhibitory glia. Eur. J. Neurosci., 21, 378–390. [DOI] [PubMed] [Google Scholar]
- 93. Gotz B., Scholze A., Clement A., Joester A., Schutte K., Wigger F., Frank R., Spiess E., Ekblom P. and Faissner A. (1996) Tenascin-C contains distinct adhesive, anti-adhesive, and neurite outgrowth promoting sites for neurons. J. Cell Biol., 132, 681–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Clemente D., Ortega M.C., Melero-Jerez C. and de Castro F. (2013) The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front. Cell. Neurosci., 7, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Frost G.R. and Li Y.M. (2017) The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol., 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Serio A., Bilican B., Barmada S.J., Ando D.M., Zhao C., Siller R., Burr K., Haghi G., Story D., Nishimura A.L. et al. (2013) Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U. S. A., 110, 4697–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bolger A.M., Lohse M. and Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Schmieder R. and Edwards R. (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Anders S., Pyl P.T. and Huber W. (2015) HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Love M.I., Huber W. and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G. et al. (2018) Ensembl 2018. Nucleic Acids Res., 46, D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gel B. and Serra E. (2017) karyoploteR: an R/bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics, 33, 3088–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yu G., Wang L.G., Han Y. and He Q.Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hu H., Miao Y.R., Jia L.H., Yu Q.Y., Zhang Q. and Guo A.Y. (2019) AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res., 47, D33–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mayran A., Khetchoumian K., Hariri F., Pastinen T., Gauthier Y., Balsalobre A. and Drouin J. (2018) Pioneer factor Pax7 deploys a stable enhancer repertoire for specification of cell fate. Nat. Genet., 50, 259–269. [DOI] [PubMed] [Google Scholar]
- 108. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y. and Greenleaf W.J. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yu G., Wang L.G. and He Q.Y. (2015) ChIPseeker: an R/bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics, 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- 111. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F. and Manke T. (2016) deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res., 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data in this study is available through NCBI (PRJNA600245).