

Abstract
Duchenne muscular dystrophy is a severe pediatric neuromuscular disorder caused by the lack of dystrophin. Identification of biomarkers is needed to support and accelerate drug development. Alterations of metabolites levels in muscle and plasma have been reported in pre-clinical and clinical cross-sectional comparisons. We present here a 7-month longitudinal study comparing plasma metabolomic data in wild-type and mdx mice. A mass spectrometry approach was used to study metabolites in up to five time points per mouse at 6, 12, 18, 24 and 30 weeks of age, providing an unprecedented in depth view of disease trajectories. A total of 106 metabolites were studied. We report a signature of 31 metabolites able to discriminate between healthy and disease at various stages of the disease, covering the acute phase of muscle degeneration and regeneration up to the deteriorating phase. We show how metabolites related to energy production and chachexia (e.g. glutamine) are affected in mdx mice plasma over time. We further show how the signature is connected to molecular targets of nutraceuticals and pharmaceutical compounds currently in development as well as to the nitric oxide synthase pathway (e.g. arginine and citrulline). Finally, we evaluate the signature in a second longitudinal study in three independent mouse models carrying 0, 1 or 2 functional copies of the dystrophin paralog utrophin. In conclusion, we report an in-depth metabolomic signature covering previously identified associations and new associations, which enables drug developers to peripherally assess the effect of drugs on the metabolic status of dystrophic mice.
Introduction
Duchenne muscular dystrophy (DMD) is a rare neuromuscular disorder affecting 1 in 5000 male births (1). Patients with DMD become wheelchair dependent by the age of 10–12 years of age and die prematurely due to cardio-pulmonary complications in the 2nd–4th decade. The disease is caused by mutations in the dystrophin encoding DMD gene (2). There is currently no cure for the disease; however, the implementation of standards of care and chronic use of corticosteroids has significantly improved patients’ quality of life. In recent years, a number of therapeutic compounds have been tested in clinical trials in DMD (3), and two compounds received conditional and accelerated approval by the EU (4) and USA (5) regulatory authorities, respectively.
Therapies currently in development for DMD aim to slow down disease progression, which poses challenges on evaluating drug efficacy in clinical trials. While DMD is a progressive disease leading to the irreversible loss of function in patients, the functional decline during the duration of a clinical trial (6–24 months) as picked up with functional outcome measures is generally limited and can vary greatly between patients. In fact, many clinical trials failed to pick up a therapeutic effect (i.e. slower disease progression). This could be due to poor drug potency, but also to non-optimal trial design, insensitive and/or non-optimal outcome measures and to inter-individual variability in patients’ performance. Retrospective analysis showed that the power of clinical trials has been overestimated, due to assumptions that later turned out to be incorrect. For example, a recent phase 3 trial failed with an observed power of 0.53 in contrast to a pre-specified power of 0.9 (6).
These failures led to a number of research initiatives aimed at identifying biomarkers in blood and urine to be used as surrogate or secondary endpoints in clinical trials. Recent research has enabled the identification of multiple proteins (7–14) and miRNAs (15–19) able to separate healthy and DMD cases in cross-sectional studies. These studies have shown the diagnostic potential of these biomarkers without assessing their value in monitoring disease progression. So far, only a few studies have explored whether metabolites in the blood stream could serve as biomarkers in DMD patients (20–22) and DMD animal models (23,24). The focus of those studies has mainly been on the identification of a metabolic signature between healthy and disease in a cross-sectional manner. However, it is also important to know the long-term trajectories of these biomarkers, and how they correlate to or are predictive of pathology and function. Therefore, we here focus on the identification of longitudinal changes representative of disease progression in mice derived plasma samples. We present a prospective 7-months longitudinal study in which we obtained plasma samples from wild-type (WT) and mdx mice (the most used murine model of DMD carrying a nonsense mutation in the Dmd gene) at five different time points. We identified a signature of 31 metabolites in plasma able to discriminate between mdx and WT at different stages of the disease. We further compared the identified 31 metabolites in dystrophin negative mice carrying 2, 1 or no functional copies of the Utrn gene, encoding utrophin, which is a dystrophin paralog able to compensate to some extent for lack of dystrophin in mice. In fact, utrophin upregulation is one of the therapeutic approaches in development for DMD (25).
The presented study is the first of its kind with multiple phased longitudinal samples spanning a period of 7 months. The collected data show that metabolomic signatures are able to detect a disease progression signature beyond the known degeneration/regeneration phase known to occur in the first 10–12 weeks of the disease. The identified signature further shows how pathways targeted by drugs in development such as metformin could be monitored by studying the metabolomic signature in peripheral blood.
Results
WT and mdx mice were included in the experiment starting from 4 weeks of age. Plasma samples were obtained at five time points at 6, 12, 18, 24 and 30 weeks of age as indicated in Figure 1A. Skeletal muscles were isolated after sacrifice at 30 weeks of age. Mouse weight was recorded before and after fasting. Mdx mice were on average heavier compared with WT mice (Fig. 1B). H&E staining on tibialis anterior muscles obtained at 30 weeks of age showed an increase percentage of fibrotic/inflammatory/necrotic tissue compared with healthy mice, confirming the expected alterations due to the lack of dystrophin (Fig. 1C). A metabolomic approach was taken to identify non-invasive biomarkers during mdx mice disease progression. This analysis provided a list of 106 metabolites, most of which were amino acids, peptides and analogs (Fig. 1D). We performed unsupervised clustering on all samples as initial data exploration to assess whether clustering of WT and mdx mice could indicate the presence of a signature able to separate the two strains. Indeed, a reasonable clustering was observed between the two genotypes with a good but not complete separation of WT and mdx mice (Fig. 1E). This initial exploration showed that a potential signature is present in the data. To visualize the strength of correlations in the data set, we clustered the correlation matrix across metabolites showing that some correlation exist in the data set (Fig. 1F).
Figure 1.
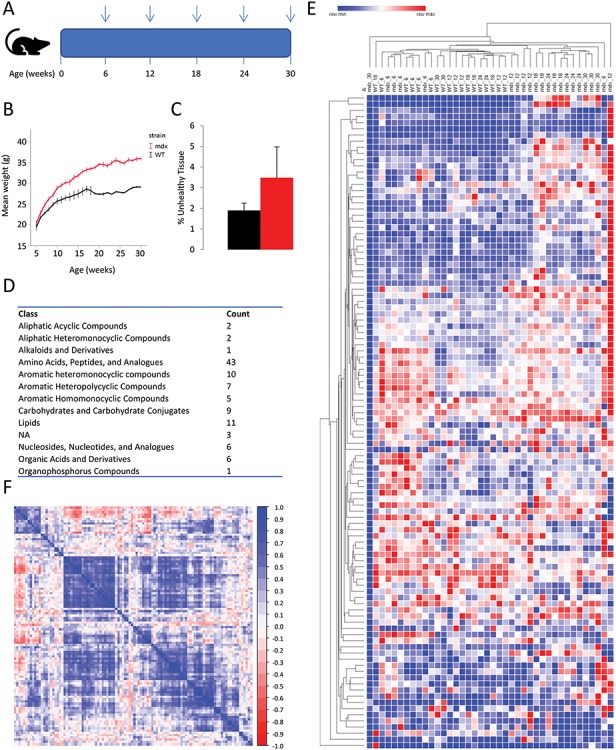
Overview of the study and obtained data. (A) Scheme showing the study design. Vertical arrows indicate at what age blood samples were obtained. (B) Line graph showing the weight progression in mdx and WT mice during the study. Weight before fasting is plotted on the y-axis, while age in weeks is plotted on the x-axis. (C) The percentage of unhealthy tissue composed of fibrotic tissue inflammatory infiltrate, and necrotic fibers was quantified after H&E staining of sections obtained from the tibialis anterior muscle of 30-weeks-old mdx (n = 4) and WT (n = 5) mice. (D) Table showing the counts of metabolites for each class identified by the analytical platform. (E) Heat map with all samples showing hierarchical clustering on both columns and rows based on Euclidean mean distance. Genotype and sampling time (in weeks) is presented for each column. (F) Heatmap of the Pearson correlation across metabolites in all samples shows moderate correlation across the identified metabolites.
Given the availability of up to five repeated measurements per mouse, we analyzed all metabolites with linear mixed models that account for the longitudinal nature of the data. We tested for differences in mean profiles at any time point between mdx and WT mice by testing whether both the main effect of group and its interaction terms with time are null. A comparison between the obtained and expected P-values (in case of no difference between groups) highlighted the presence of many metabolites differentially represented in mdx mice (Fig. 2A). A total of 31 metabolites showed significant differences between mdx and WT mice (FDR <5%). The majority of differences between mdx and WT mice were observed between weeks 12 and 24, with only minor differences observed at 6 weeks of age. Seven metabolites showed overall reduced relative levels in mdx compared with WT, while the remaining 24 were increased in mdx. Dipeptides normally highly represented in muscle tissue such as carnosine and anserine (Fig. 2B and C) showed a persistent increase in mdx compared with WT, while amino acids such as ornithine and glutamine (Fig. 2D and E) were reduced in mdx mice in accordance with previous reports in patients (20,21). A list of the significant differences between mdx and WT for each time point is presented in Supplementary Material, Table S1, while trajectory plots for metabolites are presented in Figure 2 and Supplementary Material, Figure S1. Only citrulline and tryptophan showed significant differences at all time points, with an inversion in directional changes at 6 weeks of age compared with the later sampling times (Supplementary Material, Figure S1). Metabolites in the guanidinoacetic acid, creatine and creatinine axis were elevated in mdx mice (Fig. 2F–H). While the elevation of creatine in mdx mice is in line with the previously reported elevation in patients (21,22), guanidinoacetic acid was previously reported to be reduced in patients (21). Creatinine showed a non-significant trend towards an increase in mdx mice, again showing a difference compared to patients (21). Supplementary Material, Figure S2 shows the individual mice trajectories for the significant associations presented in Figure 2. A number of nucleosides derivatives (methyladenosine, methylguanine and cytidine) were elevated in mdx mice as well multiple metabolites involved in amino acid conversions were significantly affected (Supplementary Material, Figure S1).
Figure 2.
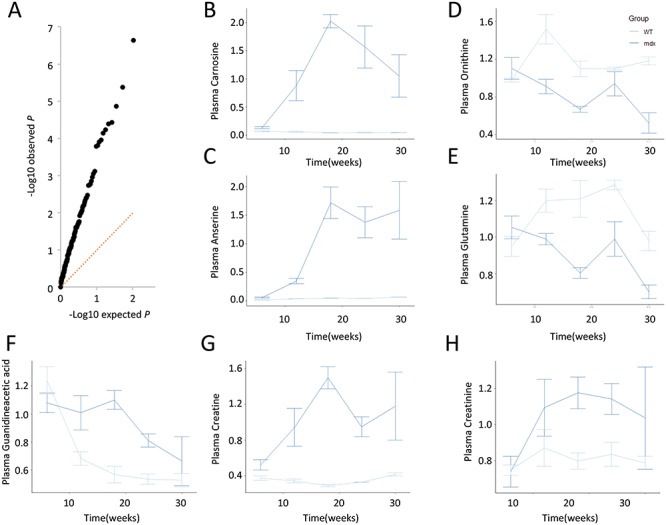
(A) Q-Q plot showing the enrichment in high −log10 observed P-global (black dots) for each metabolite compared with the expected distribution. (B–E) Line plots of scaled data showing examples for elevated levels of carnosine and anserine in mdx mice compared with WT mice, and decreased levels of ornithine and citrulline in mdx plasma. (F–H). Line plots of scaled data showing the plasma levels of guanidinoacetic acid, creatine and creatinine in mdx mice compared with WT mice.
Pathway analysis of the data was performed with the global test. A total of 25 pathways were found to be significant (Supplementary Material, Table S2). Pathways affected showed that some of the metabolites belonging to the same pathways provided a significant and opposite contribution to the pathway significance (Fig. 3A–D). Interestingly, network analysis showed how several metabolites with reduced plasma levels in mdx mice were amino acids downstream the NO precursor arginine, which was found to be elevated in mdx mice (Fig. 3E).
Figure 3.
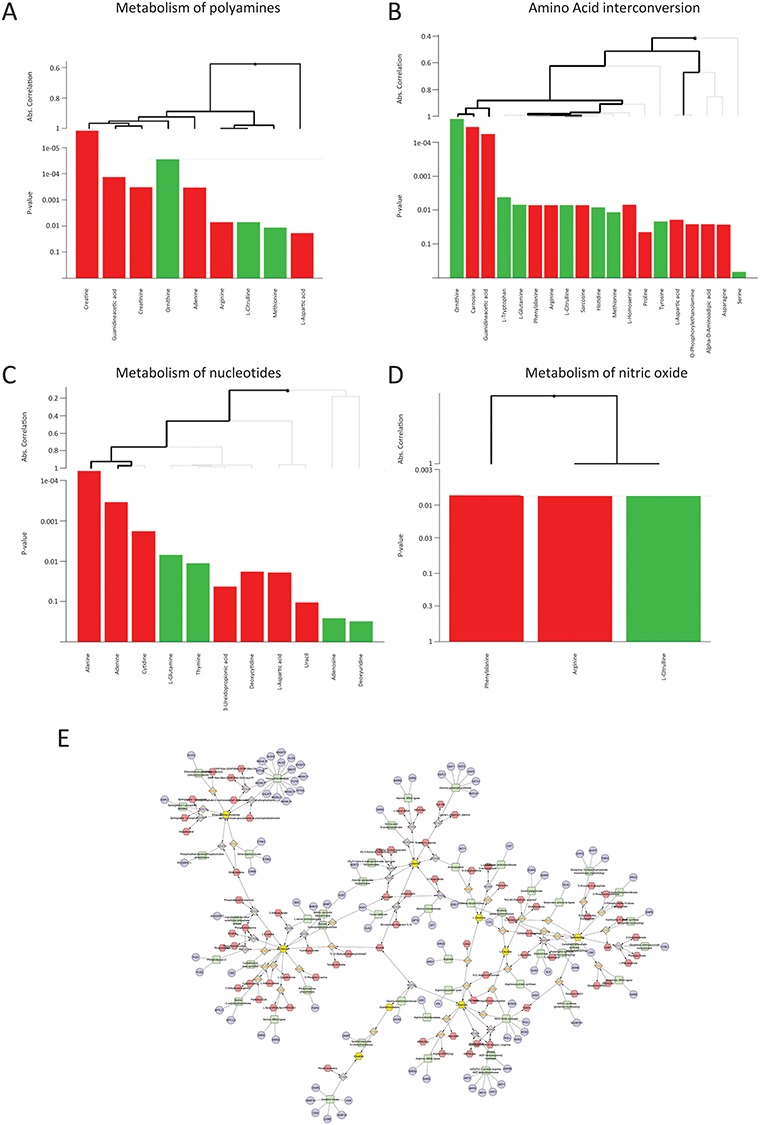
(A–D) Examples of pathways affected. Each panel shows a different pathway. Metabolites contributing to the pathway are graphed as bars at the bottom of each panel. The P-values displayed on the y-axis show how significant is the contribution of each metabolite to the pathway score. Bars in green are associated with the WT group (higher in WT mice), while bars in red are associated with the mdx group (higher in mdx mice). Hierarchical clustering is based on the absolute correlation distance between metabolites. Thick lines indicate a metabolite of a cluster of metabolites significantly contributing to the overall global test score. (E) Example of network involving significant amino acids identified in this study. Metabolites significantly affected are represented in yellow. The network was build using the Metscape app in Ctytoscape.
The data obtained in mdx mice revealed the presence of a metabolic endo-phenotype in these mice. The observed changes were consistent for the whole study duration supporting a role of these metabolites in disease progression. To further test whether the 31 metabolites identified in the comparison between mdx and WT mice were associated with disease trajectories and disease severity, we studied mdx mice carrying 0, 1 or 2 functional copies of the dystrophin paralog utrophin, as utrophin gene dosage has been linked to disease progression in mdx mice (26). Plasma samples were obtained at 6, 12, 18, 24 and 30 weeks of age for mice carrying either 1 or 2 functional Utrn alleles, while for double knock-out mice samples were only obtained at 6 weeks of age given the severe phenotype of these mice. The levels of propionylcarnitine and methylimidazoleacetic acid appear to relate to the number of functional Utrn copies (P < 0.05); however, the association was not significant after multiple testing correction (FDR > 5%). Line plots for propionylcarnitine and methylimidazoleacetic acid are presented in Figure 4, while line plots for the other 29 metabolites are presented in Supplementary Material, Figure S3. Double knock-out mice showed elevated levels of both metabolites at baseline compared with mice carrying at least one Utrn functional allele. Furthermore, mice carrying one functional Utrn gene showed higher levels of propionylcarnitine and methylimidazoleacetic acid compared with the mice with two functional alleles. Specifically, propionylcarnitine showed elevation at 12 (P = 0.02) and 24 weeks (P = 0.07) of age, while methylimidazoleacetic acid showed a significant increase at 24 weeks (P = 0.007). Interestingly, changes between mdx and WT mice were also identified for the same time points with propionylcarnitine being elevated at 12 weeks of age in mdx mice and methylimidazoleacetic acid being elevated at 18, 24 and 30 (Fig. 4C and D).
Figure 4.
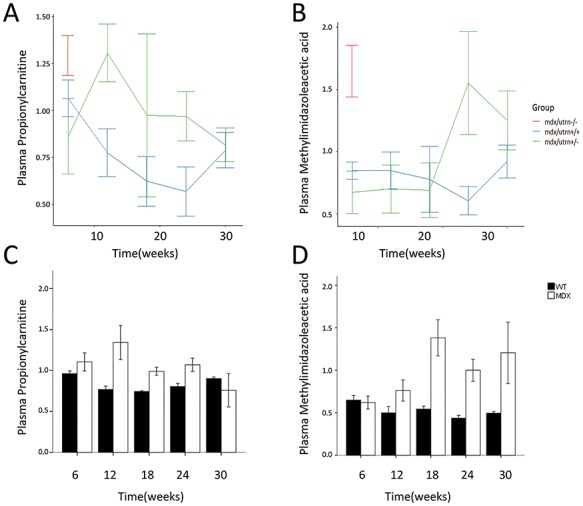
(A and B). Line graphs of normalized data showing the effect of utrophin dosage on the plasma levels of propionylcarnitine and methylimidazoleacetic acid. (C and D). Corresponding bar graphs showing the change in propionylcarnitine and methylimidazoleacetic acid in mdx mice compared with WT mice.
Discussion
DMD is rare genetic condition caused by mutations in the DMD locus resulting in the lack of dystrophin protein. A number of omics studies have been performed especially in skeletal muscle tissue obtained from patients and animal models to understand the biology of the disease, to identify drug targets and more recently to understand what molecules could be used as biomarkers. While the analysis of muscle biopsies provides direct information about the muscle, less invasive readouts are needed in order to provide objective biological information during disease progression and drug testing. The focus of biomarker research is the identification of pharmacodynamic biomarkers able to show dose-response to medicinal drugs, as well as biomarkers associated with disease progression in order to anticipate disease milestones and eventually substitute clinical endpoints in clinical trials. Most of the omics research focused on the identification of genetic modifiers (27), gene expression by microarray (28–32) and RNAseq (19), protein abundance by mass spectrometry (8,33–35) and affinity assays (7,9–11,36,37). Studies of metabolites involving muscle tissues have been somewhat less covered with one study showing analysis of muscle biopsies obtained from multiple forms of muscular dystrophy (38) and a few studies reporting changes in dystrophic mice (24) and golden retriever muscular dystrophy (GRMD) dogs (39). These studies clarified the pathophysiology of the disease including the energy deficit in DMD.
Clear associations included the impaired energy production in DMD muscle such as the reduction in glutamine levels in muscle tissue (38). Glutamine is mostly produced by muscle mass in the body (40), and it is one of the major energy sources for muscle cells (41). Glutamate/glutamine levels have been found to be increased in mdx muscle consistently at 3, 6 and 12 months of age (42,43) but reduced in DMD patients muscle (38). The described increase of glutamine levels in mdx muscle is opposed to our observation in plasma, where a persistent reduction is present starting from week 12 up to 30 weeks of age. It is possible that mice may compensate for the lack of energy in muscle fibers, caused by the known reduction in creatine concentration (43), with increased glutamine consumption; the augmented glutamine use by muscle would then be mirrored by reduced glutamine levels in circulation, such as in cachectic conditions (44,45). In DMD patients, glutamine levels are reduced in muscle (38) but unaffected in plasma (21), consistent with the inability of patients’ muscle to increase glutamine metabolism and compensate for the known reduction of creatine (46–48) and its energy buffering capacity. Glutamine synthesis depends on intermediates of the tricarboxylic acid (TCA) cycle such as fumarate, which has been shown to be reduced in DMD muscle biopsies (38), and alpha-keto glutarate. Metabolites of the TCA cycle have been shown to be affected in dystrophic mice (49) and dystrophic dogs (39). Further evidence from gene expression studies in GRMD dogs (50) but also from proteomic profiling in patients’ blood (10) show that enzymes of the TCA cycle are affected in DMD. Glutamine reduction can also be linked to the pathway leading to the synthesis of nitric oxide synthase (nNOS), a key player in DMD pathophysiology (51). Indeed, glutamine can be converted into citrulline, which is the only precursor for arginine synthesis (52). Our data show that both glutamine and citrulline are reduced in mdx mice, while arginine levels are elevated. Arginine is then converted in nitric oxide by nNOS, a direct dystrophin binder, which is typically displaced by the lack of dystrophin (53). Interestingly, supplementation of arginine and metformin showed improved muscle function in an open-label proof of concepts study (54). A follow-up double blind placebo controlled study was then planned substituting arginine with citrulline with the same intent (55). These metabolites could therefore be explored as pharmacodynamic readouts in such trials. Reduced glutamine levels could also be due to the reduced glutamine synthetase activity, which has been connected to the extrahepatic (and in particular muscular) ammonia detoxification (56), a pathway particularly significant in our data set.
Reduced energy capacity is also evident by the previously published evidence that the guanidinoacetic acid—creatine—creatinine axis is affected in DMD (22,57) and that the ratio between creatine and creatinine is associated with patients performance (21). Interestingly, mdx mice show increased plasma levels of all three metabolites, while patients show increased creatine and reduced guanidinoacetic acid and creatinine, underlining again the differences in metabolic capacity of mdx muscles compared with DMD muscles.
We further show that histidine, one of the glucogenic amino acids, is reduced in mdx mice plasma. Reduction of histidine levels could be related to the synthesis of carnosine, which is synthesized by carnosine synthase starting from histidine and beta-alanine. Carnosine levels were shown to be reduced in mdx muscles (43) and we report here an increase of carnosine levels in blood leading to postulate a release/leakage of carnosine to the blood stream in mdx mice. Carnosine is a dipeptide highly present in muscle, which is linked to muscle buffering capacity in muscle fibers. Elevated synthesis of carnosine via carnosine synthase could deplete histidine levels in an attempt to improve muscle buffering. This theory could be supported by the increased anserine levels in the plasma of mdx mice, which could also be produced by carnosine synthase starting from 3-methylhistidine.
By comparing mice carrying different functional utrophin copies, we did not identify metabolites able to separate mice with different functional Utrn copy number. However, propionylcarnitine and methylimidazoleacetic acid showed elevated profiles in more severely affected mice. The elevation of propionylcarnitine could be linked to the mitochondrial dysfunction. In type 2 diabetes, propionylcarnitine has been shown to be the predictive of mitochondrial dysfunction in muscle (58); DMD patients show some molecular characteristics of metabolic syndrome such as elevated serum leptin levels (7,59), and propionylcarnitine could indicate the known mitochondrial dysfunction associated with the lack of dystrophin. On the other hand, increased levels of methylimidazoleacetic acid could be related to performance via the histamine metabolism. Indeed, methylimidazoleacetic is the end product of the histamine catabolism, which is related to blood vessel dilation and permeability. Treatment with histamine and serotonin showed a beneficial effect on dystrophic mice (60). The net effect of histamine is however unclear as the improvement could also be due to the serotonin, which is in fact synthesized starting from tryptophan, which we describe here to be reduced in mdx compared with WT mice.
Our data partly overlap with recently published data (23,24,57) in dystrophic mice, such as alterations in metabolites mapping to the TCA cycle and glutamine identified in 4 to 6 months old Dmdmdx-4Cv mice (23) or the creatine increase identified in mdx BL10 mice (24). However, the differences across analytical platforms (such as in the study by Lee-McMullen et al. where NMR is used), study design (our study is longitudinal while previous studies are cross-sectional), genetic background (we use BL10 mdx, while Joseph et al. report on Dmdmdx-4Cv) and sample numbers are likely responsible for the different observations.
A strong point in our study is the availability of repeated measurements; published studies so far showed only single measurements per individual mouse, therefore limiting the possibility to model disease progression at the individual level. A weak point is the number of animals involved, which is limited to five per group but in line with previous publications; however, the availability of repeated measurement allowed us to increase the power to reliably identify the metabolic signature (61).
In this study, we have provided an in depth characterization of the circulating metabolites in four mouse models of DMD. The longitudinal follow-up of mice over a period of 30 weeks allowed us to model individual trajectories over the different phases of the disease, encompassing the highly regenerative phase up to the deteriorating phase. This enabled us to show that peripheral metabolic changes are less evident in young mice where performance is less affected but histological findings are more pronounced; in contrast, metabolites show a stronger signature at later stages when histology is improved but performance is worsening. We have shown how different metabolites are connected to the pathophysiology and to the mechanism of action of nutraceuticals and pharmaceuticals in development in the DMD space. The collected data will be helpful to evaluate the effects of drugs targeting dysregulated metabolic pathways such as the application of citrulline and metformin or serotonin and histamine. Furthermore, detailed reconstruction of metabolic flux over time could enable to propose therapeutic agents with potential beneficial effect in dystrophinopathies.
Materials and Methods
Mice
WT and mdx mice (five per group) were included in the experiment starting from 4 weeks of age. Only male mice were included in the experiment. Mice were kept in individually ventilated cages and were fed ad libitum with chow and had free access to water. Blood samples were obtained via the tail vein when mice were 6, 12, 18 and 24 weeks of age and from the eye at week 30. Mice were fasted for 4–6 h before sampling; during this time, they had free access to water. Mice were anesthetised with isoflurane before sampling, and a solution of lidocaine and adrenaline was applied on the tail cut before they were allowed to wake up from anaesthesia. Blood samples were obtained in heparin lithium tubes. Mice were sacrificed by cervical dislocation after the last sample was collected at 30 weeks of age. Muscles were then collected as well, and H&E staining was performed to quantify the proportion of unhealthy tissue (fibrosis, necrosis and inflammation) over the total as previously described (62). To test whether differences in disease severity existed, we included mice with different copy number of functional utrophin alleles. Five mice per group were included. Plasma samples were obtained at 6, 12, 18, 24 and 30 weeks of age for mice carrying 1 or 2 functional copies of utrophin, while for double knock-out mice, only samples at 6 weeks of age were collected. Double knock-out mice were sacrificed at 6 weeks of age as they were too severely affected to be kept. The experiment was evaluated and approved by the local animal welfare committee under DEC number 13154.
Sample preparation and data acquisition
Plasma samples were kept on ice and centrifuged at 18 000g for 5 min at 5°C. After centrifugation, the supernatant was aliquoted and frozen at −80°C pending use. The sample order of the sample preparation and the analytical batch was randomized to avoid bias. The procedure for data acquisition has been previously described for the analysis of human plasma (21). Briefly, plasma samples were introduced into a Transcend 1250 LC (Thermo Fisher Scientific) fitted with a Sequant ZICpHILIC 5 μm, 2.1 × 150 mm column (Merck). This was then coupled to a Q-Exactive mass spectrometer (Thermo Scientific) in both positive and negative ionization modes, alternatively.
Data analysis (peak picking and features annotation) was performed using TraceFinder 3.1 (Thermo Fisher Scientific). Annotation was based on the exact m/z ratio of the pseudo-molecular [M + H] + or [M − H] − ions in positive and negative mode, respectively (±5 ppm mass tolerance); retention time and isotopic pattern were also used to align to an in-house database of authentic standard compounds. The obtained data set was cleaned based on several parameters as described by Dunn et al. (63). The coefficients of variation of the areas of chromatographic peaks of features in QC samples (pool of each sample analyzed every five samples) should be below 30%, the coefficient of correlation between QC dilution factors (series of dilution of the QC sample) and areas of chromatographic peaks should be above 0.7 and the ratio of chromatographic peak areas of biological to blank samples above a value of 3.
After LC-MS analysis of samples and annotation of features, QC samples were re-injected for higher energy collisional dissociation MS/MS experiments in positive and negative ion modes on the same instrument set in targeted mode using inclusion lists. Only features that match with the MS/MS spectrum of the corresponding chemical standard were kept. These annotations correspond to the level 1 according to the Metabolomics Standards Initiative (64). Relative quantification was finally performed by comparing raw areas of identified compounds. For metabolites detected and identified in both negative and positive modes, only the data obtained in negative mode were included in the data analysis given that most metabolites were detected in that mode. Data are available as Supplementary Material with Supplementary Material, Table S3 providing the data used to compare WT and mdx mice and Supplementary Material, Table S4 reporting the data used to compare mdx mice with different functional copies of utrophin.
Statistics
A preliminary analysis of the raw metabolomics data highlighted considerable differences in the order of magnitude of metabolite concentrations; to correct for this, auto-scaling was employed to normalize the raw data, dividing the concentration levels of each metabolite at each time point by its standard deviation (65).
Visual exploration of the normalized data was performed by generating a heatmap where both samples and metabolites were clustered using the average linkage method in combination with the Euclidean distance. We employed linear mixed models (66) to study the dynamic evolution of each metabolite over time and to identify differences between WT and mdx mice at different time points. We considered a linear mixed model where the concentration of a metabolite in sample j from individual i, 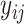 , depends on time (categorical), strain (WT and mdx) and their interactions as fixed effects, as well as on mice-specific random intercepts
, depends on time (categorical), strain (WT and mdx) and their interactions as fixed effects, as well as on mice-specific random intercepts 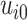 and slopes
and slopes 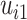 :
:
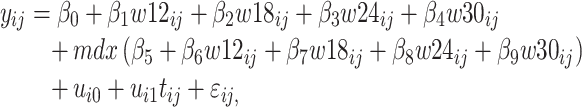 |
where 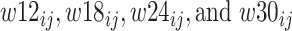 are dummy variables indicating whether sample j was obtained at week 12, 18, 24 or 30,
are dummy variables indicating whether sample j was obtained at week 12, 18, 24 or 30, 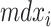 is a dummy whether mouse i is mdx (1) or WT (0),
is a dummy whether mouse i is mdx (1) or WT (0), 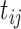 denotes time (in weeks),
denotes time (in weeks), 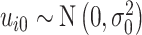 and
and 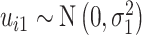 are Gaussian random effects and
are Gaussian random effects and 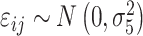 is a Gaussian error term. The statistical significance of differences between WT and mdx mice at any time point was assessed by testing the null hypothesis
is a Gaussian error term. The statistical significance of differences between WT and mdx mice at any time point was assessed by testing the null hypothesis  with the likelihood ratio test statistic; the P-values obtained from such test (‘p-global’) were then adjusted with the Benjamini–Hochberg method (FDR) (67) to account for multiple testing. Metabolites with statistically significant profiles between the two groups (FDR < 5%) were further investigated testing differences between the two groups at each time point using the Wald test.
with the likelihood ratio test statistic; the P-values obtained from such test (‘p-global’) were then adjusted with the Benjamini–Hochberg method (FDR) (67) to account for multiple testing. Metabolites with statistically significant profiles between the two groups (FDR < 5%) were further investigated testing differences between the two groups at each time point using the Wald test.
For the 31 metabolites that displayed significant differences between WT and mdx comparison, we further checked whether differences existed between mdx mice with different utrophin copy numbers, focusing our attention on mice with 1 and 2 copy numbers (due to the fact that all mice with 0 copy numbers were sacrificed after week 6 and no samples were thus available for the subsequent time points). We considered a linear mixed model where the concentration of each metabolite depends on time (categorical), strain (mdx mice with 1 or 2 copy numbers) and their interactions as fixed effects, as well as on mice-specific random intercepts and slopes. The statistical significance of differences between the two mice groups at any time point was tested with the likelihood ratio test, and P-values were adjusted with the Benjamini–Hochberg method (FDR) (67).
Pathway analysis was performed with the global test (68). As pathway analyses pipeline are developed for cross-sectional but not for longitudinal data, for each mouse, we derived a summary of the trajectories described by the metabolites computing the area under the profile of each metabolite. WikiPathways were used as source for the metabolic pathways (69). Pathway information from WikiPathways was mined using an internal workflow that interacts with the application programming interface services of WikiPathways (70). Workflows created with the open source software Taverna Workbench (71) can be found at http://www.myexperiment.org/packs/689. All pathways and corresponding metabolites were downloaded. P-values from the global test were adjusted using the max T test correction (72).
Cluster analysis of the metabolites was performed using Morpheus (73). The statistical analyses described in this paper were performed using R. We used the R package nlme (74) to estimate the linear mixed models and the R package globaltest (68) to compute the global test. Network visualization was performed using the MetScape App (75) in Cytoscape (76).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors would like to acknowledge Laura van Vliet for performing the H&E staining of muscle sections.
Conflict of Interest statement. The authors declare no competing interests.
Funding
European Commission through the projects Neuromics (No. 305121); RD-Connect (No. 305444); Duchenne Parent Project NL.
References
- 1. Mercuri E. and Muntoni F. (2013) Muscular dystrophies. Lancet (London, England), 381, 845–860. [DOI] [PubMed] [Google Scholar]
- 2. Tuffery-Giraud S., Saquet C., Thorel D., Disset A., Rivier F., Malcolm S. and Claustres M. (2005) Mutation spectrum leading to an attenuated phenotype in dystrophinopathies. Eur. J. Hum. Genet., 13, 1254–1260. [DOI] [PubMed] [Google Scholar]
- 3. Mercuri E. and Muntoni F. (2013) Muscular dystrophy: new challenges and review of the current clinical trials. Curr. Opin. Pediatr., 25, 701–707. [DOI] [PubMed] [Google Scholar]
- 4. Haas M., Vlcek V., Balabanov P., Salmonson T., Bakchine S., Markey G., Weise M., Schlosser-Weber G., Brohmann H., Yerro C.P. et al. (2015) European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul. Disord., 25, 5–13. [DOI] [PubMed] [Google Scholar]
- 5. Mendell J.R., Rodino-Klapac L.R., Sahenk Z., Roush K., Bird L., Lowes L.P., Alfano L., Gomez A.M., Lewis S., Kota J. et al. (2013) Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol., 74, 637–647. [DOI] [PubMed] [Google Scholar]
- 6. Goemans N., Mercuri E., Belousova E., Komaki H., Dubrovsky A., McDonald C.M., Kraus J.E., Lourbakos A., Lin Z., Campion G. et al. (2018) A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul. Disord., 28, 4–15. [DOI] [PubMed] [Google Scholar]
- 7. Spitali P., Hettne K., Tsonaka R., Charrout M., van den Bergen J., Koeks Z., Kan H.E., Hooijmans M.T., Roos A., Straub V. et al. (2018) Tracking disease progression non-invasively in Duchenne and Becker muscular dystrophies. J. Cachexia. Sarcopenia Muscle, 9, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oonk S., Spitali P., Hiller M., Switzar L., Dalebout H., Calissano M., Lochmüller H., Aartsma-Rus A.A., ’t Hoen P.E., van der B.Y. et al. (2015) Comparative mass spectrometric and immunoassay-based proteome analysis in serum of Duchenne muscular dystrophy patients. Proteomics. Clin Appl., 10. [DOI] [PubMed] [Google Scholar]
- 9. Hathout Y., Brody E., Clemens P.R., Cripe L., DeLisle R.K., Furlong P., Gordish-Dressman H., Hache L., Henricson E., Hoffman E.P. et al. (2015) Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci., 112, 7153–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ayoglu B., Chaouch A., Lochmüller H., Politano L., Bertini E., Spitali P., Hiller M., Niks E., Gualandi F., Pontén F. et al. (2014) Affinity proteomics within rare diseases: a BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol. Med., 6, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burch P.M., Pogoryelova O., Palandra J., Goldstein R., Bennett D., Fitz L., Guglieri M., Bettolo C.M., Straub V., Evangelista T. et al. (2017) Reduced serum myostatin concentrations associated with genetic muscle disease progression. J. Neurol., 264, 541–553. [DOI] [PubMed] [Google Scholar]
- 12. Burch P.M., Pogoryelova O., Goldstein R., Bennett D., Guglieri M., Straub V., Bushby K., Lochmüller H. and Morris C. et al. (2015) Muscle-derived proteins as serum biomarkers for monitoring disease progression in three forms of muscular dystrophy. J. Neuromuscul. Dis., 2, 241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coenen-Stass A.M.L., McClorey G., Manzano R., Betts C.A., Blain A., Saleh A.F., Gait M.J., Lochmüller H., Wood M.J.A. and Roberts T.C. et al. (2015) Identification of novel, therapy-responsive protein biomarkers in a mouse model of Duchenne muscular dystrophy by aptamer-based serum proteomics. Sci. Rep., 5, 17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guiraud S., Edwards B., Squire S.E., Babbs A., Shah N., Berg A., Chen H. and Davies K.E. (2017) Identification of serum protein biomarkers for utrophin based DMD therapy. Sci. Rep., 7, 43697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zaharieva I.T., Calissano M., Scoto M., Preston M., Cirak S., Feng L., Collins J., Kole R., Guglieri M., Straub V. et al. (2013) Dystromirs as serum biomarkers for monitoring the disease severity in Duchenne muscular dystrophy. PLoS One, 8, e80263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cacchiarelli D., Legnini I., Martone J., Cazzella V., D’Amico A., Bertini E. and Bozzoni I. (2011) miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med., 3, 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeanson-Leh L., Lameth J., Krimi S., Buisset J., Amor F., Le Guiner C., Barthélémy I., Servais L., Blot S., Voit T. and Israeli D. (2014) Serum profiling identifies novel muscle miRNA and cardiomyopathy-related miRNA biomarkers in golden retriever muscular dystrophy dogs and Duchenne muscular dystrophy patients. Am. J. Pathol., 184, 2885–98. [DOI] [PubMed] [Google Scholar]
- 18. Coenen-Stass A.M.L., Betts C.A., Lee Y.F., Mäger I., Turunen M.P., El Andaloussi S., Morgan J.E., Wood M.J.A. and Roberts T.C. (2016) Selective release of muscle-specific, extracellular microRNAs during myogenic differentiation.. Hum. Mol. Genet., 25, 3960–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coenen-Stass A.M.L., Sork H., Gatto S., Godfrey C., Bhomra A., Krjutškov K., Hart J.R., Westholm J.O., O’Donovan L., Roos A. et al. (2018) Comprehensive RNA-Sequencing Analysis in Serum and Muscle Reveals Novel Small RNA Signatures with Biomarker Potential for DMD. Mol. Ther. – Nucleic Acids, 13, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Srivastava N.K., Annarao S. and Sinha N. (2016) Metabolic status of patients with muscular dystrophy in early phase of the disease: in vitro, high resolution NMR spectroscopy based metabolomics analysis of serum. Life Sci., 151, 122–129. [DOI] [PubMed] [Google Scholar]
- 21. Spitali P., Hettne K., Tsonaka R., Sabir E., Seyer A., Hemerik J.B., Goeman J.J., Picillo E., Ergoli M., Politano L. and Aartsma-Rus A. (2018) Cross-sectional serum metabolomic study of multiple forms of muscular dystrophy. J. Cell. Mol. Med., 22, 2442–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boca S.M., Nishida M., Harris M., Rao S., Cheema A.K., Gill K., Wang D., An L., Gauba R., Seol H. et al. (2016) Correction: Discovery of Metabolic Biomarkers for Duchenne Muscular Dystrophy within a Natural History Study. PLoS One, 11, e0159895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Joseph J., Cho D. and Doles J. (2018) Metabolomic analyses reveal extensive progenitor cell deficiencies in a mouse model of Duchenne muscular dystrophy. Metabolites, 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee-McMullen B., Chrzanowski S.M., Vohra R., Forbes S.C., Vandenborne K., Edison A.S. and Walter G.A. (2019) Age-dependent changes in metabolite profile and lipid saturation in dystrophic mice. NMR Biomed., 32, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guiraud S., Squire S.E., Edwards B., Chen H., Burns D.T., Shah N., Babbs A., Davies S.G., Wynne G.M., Russell A.J. et al. (2015) Second-generation compound for the modulation of utrophin in the therapy of DMD. Hum. Mol. Genet., 24, 4212–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Putten M., Kumar D., Hulsker M., Hoogaars W.M.H., Plomp J.J., van Opstal A., van Iterson M., Admiraal P., van Ommen G.J.B., ’t Hoen P.aC and Aartsma-Rus A. (2012) Comparison of skeletal muscle pathology and motor function of dystrophin and utrophin deficient mouse strains. Neuromuscul. Disord., 22, 406–417. [DOI] [PubMed] [Google Scholar]
- 27. Bello L. and Pegoraro E. (2019) The “usual suspects”: genes for inflammation, fibrosis, regeneration, and muscle strength modify Duchenne muscular dystrophy. J. Clin. Med., 8, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pescatori M., Broccolini A., Minetti C., Bertini E., Bruno C., D’amico A., Bernardini C., Mirabella M., Silvestri G., Giglio V. et al. (2007) Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J., 21, 1210–1226. [DOI] [PubMed] [Google Scholar]
- 29. Chen Y.W., Zhao P., Borup R. and Hoffman E.P. et al. (2000) Expression profiling in the muscular dystrophies: identification of novel aspects of molecular pathophysiology. J. Cell Biol., 151, 1321–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roberts T.C., Blomberg K.E.M., McClorey G., Andaloussi S.EL, Godfrey C., Betts C., Coursindel T., Gait M.J., Edvard Smith C. and Wood M.J. (2012) Expression analysis in multiple muscle groups and serum reveals complexity in the MicroRNA Transcriptome of the mdx mouse with implications for therapy. Mol. Ther. – Nucleic Acids, 1, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bakay M., Zhao P., Chen J. and Hoffman E.P. (2002) A web-accessible complete transcriptome of normal human and DMD muscle. Neuromuscul. Disord., 12Suppl 1, S125–S141. [DOI] [PubMed] [Google Scholar]
- 32. Marotta M., Ruiz-Roig C., Sarria Y., Peiro J.L., Nuñez F., Ceron J., Munell F. and Roig-Quilis M. (2009) Muscle genome-wide expression profiling during disease evolution in mdx mice. Physiol. Genomics, 37, 119–132. [DOI] [PubMed] [Google Scholar]
- 33. Hathout Y., Marathi R.L., Rayavarapu S. et al. (2014) Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne muscular dystrophy patients. Hum. Mol. Genet., 23, 6458–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cynthia Martin F., Hiller M., Spitali P., Oonk S., Dalebout H., Palmblad M., Chaouch A., Guglieri M., Straub V., Lochmüller H. et al. (2014) Fibronectin is a serum biomarker for Duchenne muscular dystrophy. Proteomics – Clin. Appl., 8, 269–278. [DOI] [PubMed] [Google Scholar]
- 35. Doran P., Wilton S.D., Fletcher S. and Ohlendieck K. (2009) Proteomic profiling of antisense-induced exon skipping reveals reversal of pathobiochemical abnormalities in dystrophic mdx diaphragm. Proteomics, 9, 671–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mariot V., Joubert R., Hourdé C., Féasson L., Hanna M., Muntoni F., Maisonobe T., Servais L., Bogni C., Le Panse R. et al. (2017) Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun., 8, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lourbakos A., Yau N., De Bruijn P., Hiller M., Kozaczynska K., Jean-Baptiste R., Reza M., Wolterbeek R., Koeks Z., Ayoglu B. et al. (2017) Evaluation of serum MMP-9 as predictive biomarker for antisense therapy in Duchenne. Sci. Rep., 7, 17888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Srivastava N.K., Yadav R., Mukherjee S. et al. (2018) Perturbation of muscle metabolism in patients with muscular dystrophy in early or acute phase of disease: in vitro, high resolution NMR spectroscopy based analysis. Clin. Chim. Acta, 478, 171–181. [DOI] [PubMed] [Google Scholar]
- 39. Abdullah M., Kornegay J.N., Honcoop A., Parry T.L., Balog-Alvarez C.J., O’Neal S.K., Bain J.R., Muehlbauer M.J., Newgard C.B., Patterson C. and Willis M.S. (2017) Non-targeted metabolomics analysis of golden retriever muscular dystrophy-affected muscles reveals alterations in arginine and proline metabolism, and elevations in glutamic and oleic acid in vivo. Metabolites, 7, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Newsholme P., Lima M.M.R., Procopio J., Pithon-Curi T.C., Doi S.Q., Bazotte R.B. and Curi R. (2003) Glutamine and glutamate as vital metabolites. Brazilian J. Med. Biol. Res. = Rev. Bras. Pesqui. medicas e Biol., 36, 153–63. [DOI] [PubMed] [Google Scholar]
- 41. Zielke H.R., Zielke C.L. and Ozand P.T. (1984) Glutamine: a major energy source for cultured mammalian cells. Fed. Proc., 43, 121–125. [PubMed] [Google Scholar]
- 42. Griffin J.L., Williams H.J., Sang E., Clarke K., Rae C. and Nicholson J.K. (2001) Metabolic profiling of genetic disorders: a multitissue 1H nuclear magnetic resonance spectroscopic and pattern recognition study into dystrophic tissue. Anal. Biochem., 293, 16–21. [DOI] [PubMed] [Google Scholar]
- 43. Martins-Bach A.B., Bloise A.C., Vainzof M., Rahnamaye Rabbani S. (2012) Metabolic profile of dystrophic mdx mouse muscles analyzed with in vitro magnetic resonance spectroscopy (MRS). Magn. Reson. Imaging, 30, 1167–1176. [DOI] [PubMed] [Google Scholar]
- 44. Kinscherf R., Hack V., Fischbach T., Friedmann B., Weiss C., Edler L., Bärtsch P. and Dröge W. (1996) Low plasma glutamine in combination with high glutamate levels indicate risk for loss of body cell mass in healthy individuals: the effect of N-acetyl-cysteine. J. Mol. Med., 74, 393–400. [DOI] [PubMed] [Google Scholar]
- 45. Kuhn K.S., Muscaritoli M., Wischmeyer P. and Stehle P. (2010) Glutamine as indispensable nutrient in oncology: experimental and clinical evidence.. Eur. J. Nutr., 49, 197–210. [DOI] [PubMed] [Google Scholar]
- 46. Younkin D.P., Berman P., Sladky J., Chee C., Bank W. and Chance B. (1987) 31P NMR studies in Duchenne muscular dystrophy: age-related metabolic changes. Neurology., 37, 165–9. [DOI] [PubMed] [Google Scholar]
- 47. Newman R.J., Bore P.J., Chan L., Gadian D.G., Styles P., Taylor D. and Radda G.K. (1982) Nuclear magnetic resonance studies of forearm muscle in Duchenne dystrophy. Br. Med. J. (Clin. Res. Ed)., 284, 1072–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barbiroli B., Funicello R., Iotti S., Montagna P., Ferlini A. and Zaniol P. (1992) 31P-NMR spectroscopy of skeletal muscle in Becker dystrophy and DMD/BMD carriers. J. Neurol. Sci., 109, 188–195. [DOI] [PubMed] [Google Scholar]
- 49. Lindsay A., Chamberlain C.M., Witthuhn B.A., Lowe D.A., Ervasti J.M. (2019) Dystrophinopathy-associated dysfunction of Krebs cycle metabolism. Hum. Mol. Genet., 28, 942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brinkmeyer-Langford C., Chu C., Balog-Alvarez C., Yu X., Cai J.J., Nabity M. and Kornegay J.N. (2018) Expression profiling of disease progression in canine model of Duchenne muscular dystrophy. PLoS One, 13, e0194485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cacchiarelli D., Martone J., Girardi E., Cesana M., Incitti T., Morlando M., Nicoletti C., Santini T., Sthandier O., Barberi L. et al. (2010) MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the Dystrophin/nNOS pathway. Cell Metab., 12, 341–351. [DOI] [PubMed] [Google Scholar]
- 52. Ligthart-Melis G.C. and Deutz N.E.P. (2011) Is glutamine still an important precursor of citrulline? Am. J. Physiol. Endocrinol. Metab., 301, E264–E266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Molza A.-E., Mangat K., Le Rumeur E., Hubert J.-F., Menhart N. and Delalande O. (2015) Structural basis of neuronal nitric-oxide synthase interaction with Dystrophin repeats 16 and 17. J. Biol. Chem., 290, 29531–29541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hafner P., Bonati U., Erne B., Schmid M., Rubino D., Pohlman U., Peters T., Rutz E., Frank S., Neuhaus C. et al. (2016) Improved Muscle Function in Duchenne Muscular Dystrophy through L-Arginine and Metformin: An Investigator-Initiated, Open-Label, Single-Center, Proof-Of-Concept-Study. PLoS One, 11, e0147634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hafner P., Bonati U., Rubino D., Gocheva V., Zumbrunn T., Gueven N. and Fischer D. (2016) Treatment with l-citrulline and metformin in Duchenne muscular dystrophy: study protocol for a single-centre, randomised, placebo-controlled trial. Trials, 17, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. He Y., Hakvoort T.B.M., Köhler S.E., Vermeulen J.L.M., de Waart D.R., de Theije C., ten Have G.A.M., van Eijk H.M.H., Kunne C., Labruyere W.T., et al. (2010) Glutamine Synthetase in muscle is required for glutamine production during fasting and Extrahepatic ammonia detoxification. J. Biol. Chem., 285, 9516–9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thangarajh M., Zhang A., Gill K., Ressom H.W., Li Z., Varghese R.S., Hoffman E.P., Nagaraju K., Hathout Y. and Boca S.M. (2019) Discovery of potential urine-accessible metabolite biomarkers associated with muscle disease and corticosteroid response in the mdx mouse model for Duchenne. PLoS One, 14, e0219507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Abu Bakar M.H. and Sarmidi M.R. (2017) Association of cultured myotubes and fasting plasma metabolite profiles with mitochondrial dysfunction in type 2 diabetes subjects. Mol. Biosyst., 13, 1838–1853. [DOI] [PubMed] [Google Scholar]
- 59. Rodríguez-Cruz M., Cruz-Guzmán O.R., Escobar R.E., López-Alarcón M. (2016) Leptin and metabolic syndrome in patients with Duchenne/Becker muscular dystrophy. Acta Neurol. Scand., 133, 253–260. [DOI] [PubMed] [Google Scholar]
- 60. Mynatt R.L., Noland R.C., Elks C.M., Vandanmagsar B., Bayless D.S., Stone A.C., Ghosh S., Ravussin E., Warfel J.D. (2019) The RNA binding protein HuR influences skeletal muscle metabolic flexibility in rodents and humans. Metabolism, 97, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fitzmaurice G.M., Laird N.M. and Ware J.H. (2011) Applied longitudinal analysis. Applied longitudinal analysis; Wiley, Hoboken, New Jersey, 2011. [Google Scholar]
- 62. van M., de Winter C., van Roon-Mom W., van Ommen G.-J., ’t Hoen P.aC. and Aartsma-Rus A. (2010) A 3 months mild functional test regime does not affect disease parameters in young mdx mice. Neuromuscul. Disord., 20, 273–80. [DOI] [PubMed] [Google Scholar]
- 63. Dunn W.B., Broadhurst D., Begley P., Zelena E., Francis-McIntyre S., Anderson N., Brown M., Knowles J.D., Halsall A., Haselden J.N. et al. (2011) Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc., 6, 1060–1083. [DOI] [PubMed] [Google Scholar]
- 64. Sumner L.W., Amberg A., Barrett D., Beale M.H., Beger R., Daykin C.A., Fan T.W.M., Fiehn O., Goodacre R., Griffin J.L. et al. (2007) Proposed minimum reporting standards for chemical analysis. Metabolomics, 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van den Berg R.A., Hoefsloot H.C.J., Westerhuis J.A., Smilde A.K. and van der Werf M.J. (2006) Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics, 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McCulloch C.E., Searle S.R. and Neuhaus J.M. (2008) Generalized, Linear, and Mixed Models, 2nd Edition; Wiley, Hoboken, New Jersey, (2008). [Google Scholar]
- 67. Benjamini Y. and Hochberg Y. (1995) Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Source J. R. Stat. Soc. Ser. B, 57, 289–300. [Google Scholar]
- 68. Goeman J.J., Van de S., De Kort F. and van Houwellingen H.C. (2004) A global test for groups fo genes: testing association with a clinical outcome. Bioinformatics, 20, 93–99. [DOI] [PubMed] [Google Scholar]
- 69. Slenter D.N., Kutmon M., Hanspers K., Riutta A., Windsor J., Nunes N., Mélius J., Cirillo E., Coort S.L., DIgles D. et al. (2018) WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res., 46, D661–D667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kelder T., Pico A.R., Hanspers K., van Iersel M.P., Evelo C. and Conklin B.R. (2009) Mining biological pathways using WikiPathways web services. PLoS One, 4, e6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wolstencroft K., Haines R., Fellows D., Williams A., Withers D., Owen S., Soiland-Reyes S., Dunlop I., Nenadic A., Fisher P. et al. (2013) The Taverna workflow suite: designing and executing workflows of web services on the desktop, web or in the cloud. Nucleic Acids Res, 41, W557–W561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Westfall P.H. and Troendle J.F. (2008) Multiple testing with minimal assumptions. Biometrical J., 50, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. https://software.broadinstitute.org/morpheus .
- 74. Pinheiro J., Bates D., DebRoy S., Sarkar D. and R Core Team (2018) Linear and Nonlinear Mixed Effects Models. R package nlme version 3.1-137. Comprehensive R Archive Network (CRAN), (2018). [Google Scholar]
- 75. Karnovsky A., Weymouth T., Hull T., Tarcea V.G., Scardoni G., Laudanna C., Sartor M.A., Stringer K.A., Jagadish H.V., Burant C. et al. (2012) Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics, 28, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shannon P. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res., 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.