

Abstract
By using the Cre-mediated genetic switch technology, we were able to successfully generate a conditional knock-in mouse, bearing the KIF2A p.His321Asp missense point variant, identified in a subject with malformations of cortical development. These mice present with neuroanatomical anomalies and microcephaly associated with behavioral deficiencies and susceptibility to epilepsy, correlating with the described human phenotype. Using the flexibility of this model, we investigated RosaCre-, NestinCre- and NexCre-driven expression of the mutation to dissect the pathophysiological mechanisms underlying neurodevelopmental cortical abnormalities. We show that the expression of the p.His321Asp pathogenic variant increases apoptosis and causes abnormal multipolar to bipolar transition in newborn neurons, providing therefore insights to better understand cortical organization and brain growth defects that characterize KIF2A-related human disorders. We further demonstrate that the observed cellular phenotypes are likely to be linked to deficiency in the microtubule depolymerizing function of KIF2A.
Keywords: KIF2A, microtubule dynamics, knock-in, mouse model, malformations of cortical development, cortex, hippocampus, in utero electroporation, neuronal migration, apoptosis, videomicroscopy
Introduction
Proper development of the human cerebral cortex is based on three major, highly regulated processes: progenitor’s proliferation, followed by migration of post-mitotic neurons in the cortical plate (CP) and finally their differentiation. Alterations in one of these processes can lead to malformations of cortical development (MCD), often associated with intellectual disability and pharmaco-resistant epilepsy. Our team and others have previously identified mutations in the KIF2A gene in patients with MCD, diagnosed by magnetic resonance imaging sequences with posterior pachygyria and microcephaly (1–3). KIF2A encodes for a kinesin from the Kinesin-13 family, implicated in the regulation of microtubule (MT) plus-end dynamics (4,5) and individuals with missense mutations in this gene present cortical gyration abnormalities and microcephaly causing pharmaco-resistant epilepsy and intellectual disability (1–3). Using in utero electroporation and overexpression approaches, we have recently shown disease causing missense variants disrupt projection neurons positioning, interneuron migration and progenitor proliferation (6). Altered neuronal migration dynamics, together with defects in the coupling between cell cycle and ciliogenesis, were suggested to underlie the pathogenesis of the related cortical malformations.
In order to better understand the in vivo implications of KIF2A mutations in the pathophysiology of MCD, we have designed and engineered a conditional knock-in (cKI) mouse model, expressing the heterozygous human p.His321Asp mutation. This variant was identified by whole exome sequencing as causative mutation in a subject with MCD (1). Likewise, several other de novo heterozygous missense mutations in KIF2A were linked to MCD and have been shown to disrupt neurodevelopmental processes (6).
We have developed the cKI KIF2A mouse model by using an improved strategy of the Cre-dependent genetic switch (FLEx switch) previously reported by Schnütgen et al. (7). This system that is based on rearrangement events mediated by the Cre recombinase and two pairs of both wild-type and mutant LoxP sites allows shifting genetic expression from one sequence to another of interest. The FLEx technology was shown to be efficient and functional as long as the two sequences are sufficiently different. However, in case of high degree of homology between an endogenous exonic sequence and the sequence bearing a missense mutation of interest, the system leads to the formation of unstable RNA secondary structure followed by an excision of the endogenous exon and thereby to a model expressing a deleted transcript rather than a point mutation model. Thus, generating conditional knock-in mouse models bearing single, point mutations remains futile with the FLEx switch system. To overcome this limitation, we have developed an improved conditional FLEx switch system (FLExII) with the aim to avoid RNA recombination and subsequent exonic excision. The novelty in this FLExII switch system illustrated in Fig. 1 resides in the degeneration of the exonic sequence in antisense orientation to reduce identity and overcome the abovementioned constraints.
Figure 1.
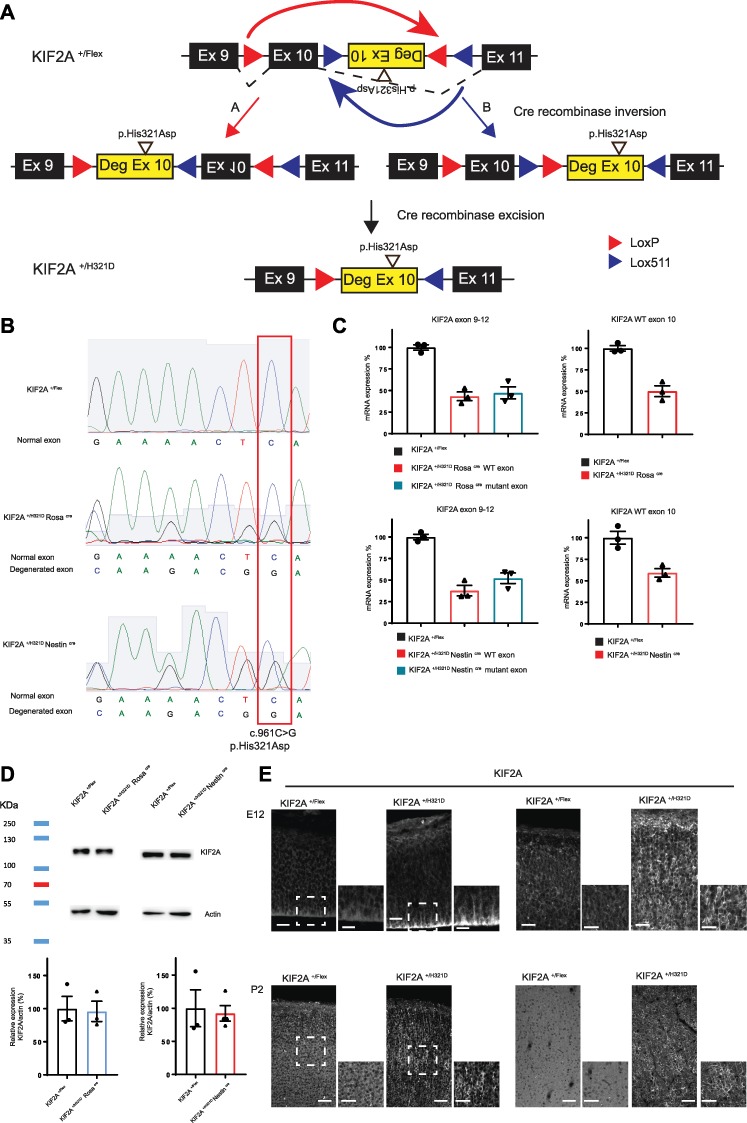
Generation of KIF2A Flex II mice. (A) Schematic representation of the plasmid construct used for the generation of the KIF2A Flex II mouse model. Mouse exons 9, 10 and 11 are represented as black rectangles. Human degenerated exon 10 carrying the His321Asp variant is presented as a yellow rectangle. LoxP and Lox511 sites are represented as red and purple triangles, respectively. Upper panel depicts the construct before action of the Cre recombinase. Middle panel shows the two constructs resulting from the Cre-induced inversion. Lower panel shows the final construct after excision. (B) Sequencing of cDNA obtained by RT-PCR on cortical extracts from KIF2A +/Flex, KIF2A+/H321DRosaCre and KIF2A+/H321DNestinCre mice showing the presence in KIF2A+/H321DRosaCre and KIF2A+/H321DNestinCre mice of the variant at a heterozygous state. (C) Quantitative PCR analysis of KIF2A levels in cortical extracts from KIF2A +/Flex, KIF2A+/H321DRosaCre and KIF2A+/H321DNestinCre mice, normalized to actin and represented as a percentage of control (n = 3 mice per group). (D) KIF2A protein levels in cortical extracts from KIF2A +/Flex, KIF2A+/H321DRosaCre and KIF2A+/H321DNestinCre mice detected by immunoblotting. Relative quantification of KIF2A protein expression normalized by actin and represented as percentage of control. Data are represented as mean ± SEM, n ≥ 3 mice per group. (E) Coronal sections of embryonic mouse cortices (E12, E15, E18), P2 pups and adult animals stained for KIF2A. Insets delimited with a dotted square show higher magnification of the cortex. Scale bar: E12: 20 μm and 10 μm for zoom, E18: 30 μm and 20 μm for zoom and P2 and adult: 60 μm and 40 μm for zoom.
In this study, we demonstrate the efficiency of the strategy through crosses with three different Cre-expressing mouse lines. Thus, Cre mediated cellular-specific expression of the p.His321Asp mutation allowed us to independently study effects on radial glial cells and post-mitotic neurons. We show that knock-in KIF2A+/H321D mice present microcephaly, hippocampal structure abnormalities and cortical layer disorganization resulting from abnormal neuronal migration and increased cell death. Furthermore, we show that the observed cellular phenotypes are likely to be linked to deficiency in the MT depolymerizing function of KIF2A.
Results
Design and validation of the strategy used to develop the conditional knock-in KIF2A mouse model
To study the pathophysiological mechanisms of MCDs resulting from KIF2A mutations, we generated a KIF2A+/H321D conditional knock-in mouse. Since all identified MCD-related KIF2A variants are heterozygous missense mutations and exert their pathogenicity through a dominant negative mechanism, conditional knock-in mouse model emerged as the most appropriate strategy to dissect neurodevelopmental processes underlying KIF2A-related MCD.
In order to develop the conditional knock-in Kif2a mouse model, we generated a plasmid construct (that was used for electroporation in ES cells) containing one pair of wild-type loxP sites and one pair of lox511 sites, with an alternate organization a head-to-head orientation within each pair of sites. The plasmid contained the DNA encoding the mouse exon 10 sequence and mouse intronic flanking sequences in the sense orientation and the modified ‘degenerated’ human sequence of exon 10 bearing the mutation (c.961C>G, p.His321Asp) and its flanking intronic sequences in the antisense orientation (Fig. 1A). Using this strategy, sequence comparison between WT and mutated exons showed that the homology was decreased from 96% homology (for the human sequence) and 100% homology (for the mouse sequence) to only 42% homology. Bioinformatics simulations predicted this ‘neo-exon’, with significantly reduced homology with the mouse sequence, flanked by human intronic sequences could be spliced at the expected canonical splicing sites.
As illustrated in Fig. 1A, both loxP and lox511 sites are recognized by Cre-recombinase; however, lox511 sites recombine efficiently with themselves but not with loxP sites. Thus, Cre-mediated recombination first induces inversion of the DNA at either loxP or lox511 sites generating a repeat of either two loxP or two lox511 sites. Further Cre-mediated excision then results in the elimination of the DNA sequence contained between the two loxP or lox511 sites. As a result, the allele construct contains single loxP and lox511 sites making further inversion impossible, and the promoter drives the stable expression of the mutant Kif2a instead of wild-type Kif2a.
Prior to the development of the mouse models, we first validated in ES cell clones the FLExII switch system through an assessment of the correct splicing and expression of the allele bearing the construct before and after the Cre-mediated inversion/excision of engineered cassette. Indeed, to verify the correct Cre-mediated rearrangement and RNA expression, we performed RT-PCR, qPCR and Sanger sequencing. Sequencing of cDNA showed the heterozygous presence of the c.961C<G (p.His321Asp) mutation and also the presence of the human degenerated sequence (Supplementary Material, Fig. S1A). Next, we performed, quantitative RT-PCR using two primer pairs: one between the exons 9 and 12 including the full KIF2A FLExII construction and one specific to the cDNA sequence of the WT KIF2A mouse exon 10 (Supplementary Material, Fig. S1B). Using the first couple of primers and before action of the Cre recombinase, the levels of KIF2A exon 9–12 comprising the KIF2A WT mRNA and the FLExII construct are comparable between WT and KIF2A+/Flex cells (Supplementary Material, Fig. S1C). However, after the action of the Cre, two distinct melting peaks are visible, due to the presence of mismatches between transcripts produced by the WT and the mutant alleles, allowing therefore to distinguish the two isoforms. The expression of each WT and mutant allele represented about 50% of total mRNA (Supplementary Material, Fig. S1C). Using the second pair of primers allowing to monitor the levels of the WT KIF2A allele, we observe similar levels between WT condition and KIF2A+/Flex before action of the Cre. After action of the Cre, in KIF2A+/H321D, we observe a decrease of about 50% in WT mRNA levels showing a switch in expression from the WT to the mutant allele (Supplementary Material, Fig. S1C). Thus, having confirmed correct functioning of the construct in ES cells, we proceeded to generating KIF2A+/Flex animals. Mice heterozygous for the FLExII allele, KIF2A+/Flex, were then crossed with transgenic animals expressing the Cre recombinase ubiquitously (RosaCre mice) (8), under the neuronal progenitor-specific promoter Nestin (NestinCre mice) (9) or under the control of the post-mitotic neuron-specific promoter Nex (NexCre mice) (10). We then verified the correct Cre-mediated inversion/excision, RNA expression and presence of the c.961C<G (p.His321Asp) mutation on cortical extracts (Fig. 1B). Additionally, using quantitative RT-PCR, we obtained results similar to those in ES cells (Fig. 1C). After breeding with RosaCre and NestinCre mice, we observe a decrease of about 50% in WT mRNA levels, demonstrating a switch from WT to mutant allele (Fig. 1C).
Altogether these observations show that in the absence of Cre recombinase the engineered allele bearing the FLExII cassette produces a normal non-rearranged stable transcripts at a level that is similar to the one produced by the WT allele. While after Cre-recombinase expression, the FLExII cassette undergoes inversion/excision events resulting in the expected switching from the mouse WT Kif2a exon 10 to the mutant Kif2a degenerated exon and an expression of a non-rearranged mutant transcript at a level that is equivalent to the one produced by the WT allele.
We further assessed KIF2A protein expression in mouse cortices by Western blot and confirmed a stable and non-truncated protein in both KIF2A+/H321D NestinCre and KIF2A+/H321D RosaCre conditions (Fig. 1D). Finally, we performed immunohistochemistry on mouse cortices in KIF2A+/Flex and KIF2A+/H321DRosaCre mice, at different embryonic (E12, E15, E18) and postnatal (P2, 10 weeks) stages. KIF2A is expressed throughout the cortex in both embryonic and adult cortices; however, abnormal subcellular localization is clearly visible for the mutant protein. In KIF2A+/Flex embryos, the protein presented with a homogenous cytoplasmic distribution (Fig. 1E, left panel), while in KIF2A+/H321D NestinCre embryos, the signal was particularly strong at the base of radial glial cells, marked by nestin staining, in the CP, stained by CTIP2 marker, and in neuronal processes, visible by TUJ1 staining (Supplementary Material, Fig. S2A and B). This abnormal localization was also visible in mutant ES cells in which KIF2A localized along MTs (Supplementary Material, Fig. S2C) was even more evident in adult cortices, where KIF2A staining was highly concentrated in axons and dendrites (Supplementary Material, Fig. S2D) and in mouse embryonic fibroblasts with a strong colocalization between KIF2A and α-tubulin (Supplementary Material, Fig. S2E). Together these observations highlight that the recombinant allele behaves exactly as predicted and that p.His321Asp variant strongly affects subcellular KIF2A localization, in agreement with our previous results in patient fibroblasts and in utero electroporated mouse cortices, showing an abnormal localization of mutant KIF2A at MTs (1,3,6).
KIF2A+/H321D mice present with global neuroanatomical defects resulting in behavioral and epileptic anomalies
We began our studies by crossing KIF2A+/Flex mice with RosaCre mice to achieve ubiquitous expression of the mutation and thereby physiological conditions, mimicking the ones in which the variant was identified. Obtained KIF2A+/H321D RosaCre animals were viable and born within the expected Mendelian ratios. However, when KIF2A+/H321D RosaCre mice were further crossed with C57Bl6/N WT mice to eliminate the presence of the Cre, litters with unexpected Mendelian ratios were obtained. When male KIF2A+/H321DRosaCre animals were bred with C57Bl6/N WT females, the litters were normally sized (8–10 pups), nonetheless contained only WT pups. When female KIF2A+/H321D RosaCre mice were bred with WT males, litters were abnormally reduced in size (4–5 pups) and pups’ mortality was elevated due to insufficient care from the mother. Due to these constraints, we carried on our investigations by systematically crossing KIF2A+/Flex with RosaCre mice.
KIF2A+/H321DRosaCre and KIF2A+/H321DNestinCre animals presented underdeveloped appearance with overall smaller size and lower weight compared to WT littermates (Fig 2A). Moreover, a reduction in brain size for both lines was already visible at early postnatal stages (P2) (Fig. 2B). We confirmed the presence of microcephaly in adult KIF2A+/H321DRosaCre mice by comprehensive neuroanatomical studies using a recently developed approach for the assessment of 166 brain parameters across 22 distinct brain regions (Supplementary Material, Table S1) (11). This consisted of a systematic quantification of the same sagittal brain region of male mice at exactly 16 weeks of age. Results revealed several major anomalies in KIF2A+/H321D RosaCre mice with an overall trend of reduced size of multiple areas when compared to WT (Fig. 2C and D). The cerebellum was the most severely affected brain structure exhibiting a decrease in size of 25.9% associated to a reduction of its internal granular layer (−30.1%) (Fig. 2E). The hippocampus showed a wide array of neuroanatomical defects: reduced area and length of the pyramidal layers (−31 and −19%, respectively), reduced height of the lacunosum molecular and radiatum layers (−25 and −26%, respectively) and abnormal CA1 layer organization (Fig. 2F).
Figure 2.
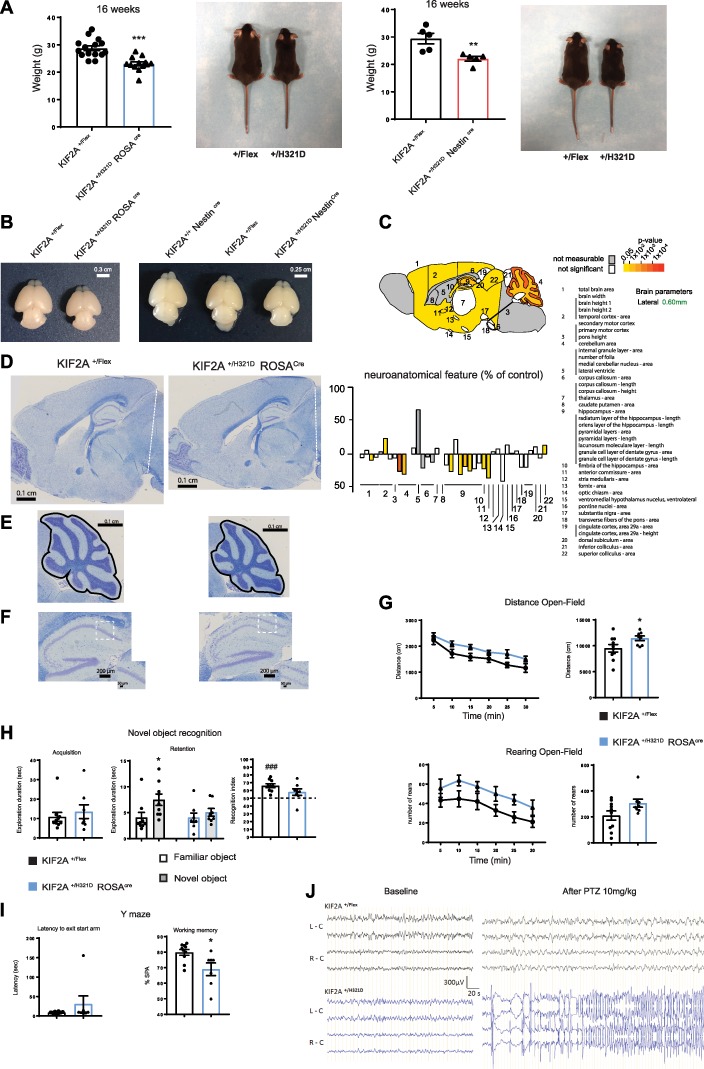
Adult KIF2A+/H321D mice present with neuroanatomical and behavioral anomalies. (A) Weight and appearance of KIF2A+/H321DRosaCre mice and corresponding controls (left panel) and KIF2A+/H321DRosaCre mice and corresponding controls (right panel). Histograms represent mean ± SEM, from at least five animals per group. (B–D) Sagittal sections from KIF2A+/H321DRosaCre and control mice stained with cresyl violet/luxol blue illustrating brain microcephaly (B, scale bar 0.1 cm); cerebellum atrophy (C, scale bar 0.1 cm) and abnormal organization of the hippocampal pyramidal layer (D, scale bar 200 μm) with insets showing the CA1 region (scale bar 50 μm). (E) Neuroanatomical features of KIF2A+/H321DRosaCre mice. (E) Upper panel: schematic representation of a brain section at lateral +0.60 mm colored according to P-values. Colored regions indicated the presence of at least one significant parameter within the brain region. White coloring indicates a P-value higher than 0.05 and gray shows not enough data to calculate a P-value. Lower panel: Histogram showing variation (decreased—minus scale or increased—positive scale) in areas and lengths expressed as percentage of WT together with a color map indicating the significance level. n = 4 mice per group, unpaired two-tailed t-test. Numbers indicate assessed brain regions listed on the right. (F) Brains from KIF2A+/H321DRosaCre (left panel) and KIF2A+/H321DRosaCre (right panel) P2 pups and corresponding controls. (G–I) Behavioral tests. (G) Open field assay. Locomotor activity is presented by the distance travelled (upper panel) and the rearing activity (lower panel). Graphical representations on the left show activity per 5-min interval. Histograms on the right show total activity during the 30-min session. (H) Novel object recognition test. Histograms represent the time spent exploring the objects during the acquisition (two identical objects) and the retention (one familiar and one novel object) phases. (I) Y maze test. Histograms show the latency to exit the start arm (sec) and the working memory index corresponding to the spontaneous alteration (SPA). Data are represented as mean ± SEM, n = 10 KIF2A+/H321DRosaCre and n = 9 control mice. (J) Representative EEG traces from superficial cortical recordings in KIF2A+/H321DRosaCre and WT mice. Left: EEG recordings before PTZ injection without discharge activity. Right: EEG recordings after 10 mg/kg of PTZ showing a generalized tonicoclonic seizure for KIF2A+/H321DRosaCre animals. L-C: left cortex; R-C: right cortex, n = 3 adult mice per group. ***A: p Value < 0,0001; **B: p Value = 0,0083; *G: p Value = 0,0491; *H: Retention: p Value = 0.0006; ###H: Recognition Index: p Value = 0.0003.
Next, we asked whether the aforedescribed neuroanatomical defects affect the behavior of mice. We tested for behavioral abnormalities by performing a battery of tests evaluating locomotor activity, learning and memory and motor coordination of KIF2A+/H321D RosaCre mice.
We began by investigating the global activity in the open field assay for 30 min. KIF2A+/H321D RosaCre mice travelled significantly longer distances in total and at all individual time points, compared to WT littermates (Fig. 2G). Additionally, mutant animals presented a trend (P = 0.0706) of increased number of rearings. Combined, these observations suggest that KIF2A+/H321D RosaCre mice are hyperactive. We continued our exploration by evaluating learning and memory, which are often affected in mouse models bearing mutations involved in human intellectual disability (12). In the object recognition task, we observed an impaired novel object recognition performance in KIF2A+/H321D RosaCre mice compared to WT littermates. When animals were allowed to explore an object during the acquisition phase, exploration duration was comparable between WT and knock-in mice (Fig. 2H). During the retention phase, knock-in mice explored equally the familiar and the new object and their recognition index was around 50%, while WT mice displayed recognition performance significantly above the chance level, presenting clear preference for exploring the new object than the familiar one. Additionally, in the Y-maze paradigm, mutant mice showed lower percentage of spontaneous alternations as compared to WT (Fig. 2I), suggesting deficient working memory and supporting results obtained in the object recognition task. We then assessed specific motor abilities in knock-in mice, which could be affected by the previously observed reduction in the size of the cerebellum, with the grip test, rotarod and notched-bar paradigms. The grip test, which measures muscular strength independently of coordination, revealed reduced absolute force in KIF2A+/H321D RosaCre mice (Supplementary Material, Fig. S3A). It should, however, be noted that these animals also presented a significantly decreased body weight correlating with the decrease in their muscular force. No alterations in motor coordination were observed with the notched-bar and rotarod test (Supplementary Material, Fig. S3B and C).
Finally, we analyzed epileptic activity in KIF2A+/H321D RosaCre animals, by recording superficial cortical electroencephalograms (EEG). No abnormal spontaneous activity was detected for either WT or mutant mice (Fig. 2J). However, when KIF2A+/H321D RosaCre animals were injected with a pro-convulsive dose (10 mg/kg) of the seizure provoking agent PTZ, an inhibitor of the γ-aminobutyric acid type 1 (GABAA) receptor widely used for inducing acute seizures (13), a generalized tonicoclonic seizure was recorded 1 min following the injection, while no seizure activity was recorded in WT mice. Altogether our behavioral analysis indicated that KIF2A+/H321D RosaCre mice exhibit memory deficits and hyperactivity, which are a common trait in mouse models of intellectual disability. These features, combined with their increased susceptibility to epilepsy, correlate with the phenotype described in human subjects with mutations in KIF2A (2).
KIF2A p.His321Asp mutation causes hippocampal CA1 heterotopia
We next sought to better characterize the defective organization of the hippocampal structure revealed by our neuroanatomical examination of KIF2A+/H321D RosaCre mouse brains. To this aim, we crossed KIF2A+/Flex with NestinCre mice, thereby limiting the expression of the mutation to the brain and compared results to those obtained in KIF2A+/H321D RosaCre animals. Immunohistological analysis revealed hippocampal heterotopia in the CA1 region with an additional layer of cells present in the stratum oriens of both KIF2A+/H321D NestinCre mice (Fig. 3A) and KIF2A+/H321D RosaCre mice (Supplementary Material, Fig. S3D). Moreover, staining with NeuN showed that heterotopic cells are differentiated neurons and staining with KIF2A confirmed the distinctive abnormal subcellular localization of the protein in both normally and abnormally positioned neurons (Supplementary Material, Fig. S3E and F). To define the time at which the heterotopia appears, and since layering in the hippocampus begins during late developmental stages and continues on at early postnatal stages (14), we chose two time-points for further analysis—E18.5 and P2. At E18.5, no visible difference could be detected between WT and KIF2A+/H321D NestinCre embryos (Fig. 3B). In P2 KIF2A+/H321D NestinCre pups, staining revealed an already split organization of the pyramidal layer in the CA1 region (Fig. 3C). To study specifically neuronal migration, independently of proliferation, we used Nexcre mice (10) to express the mutation exclusively in post-mitotic brain cells. First, we verified that the crossing with Nexcre works properly by verifying the KIF2A phenotype between E16.5 and adult stages in CP. At E16.5, we observed that the KIF2A phenotype are present only in TUJ1-positive cells in the CP and absent in intermediary progenitor TBR2 positive (Supplementary Material, Fig. S4B). At E18.5, the KIF2A phenotype is located only in the CP (Supplementary Material, Fig. S4B), and at adult stages, we can observed the same KIF2A mislocalization observed previously in KIF2A+/H321D NestinCre and KIF2A+/H321D RosaCre mice in the CP (Supplementary Material, Fig. S4C). Under these conditions, when the mutation is only expressed in post-mitotic neurons, P2 and adult KIF2A+/H321D NexCre mice still present hippocampal CA1 heterotopia (Fig. 3D), pointing to a defective neuronal migration. Together, our data suggest that the hippocampal heterotopia observed in adult mice appears to be predominantly linked to post-mitotic pathophysiological mechanisms.
Figure 3.

KIF2A+/H321D mice present with hippocampal heterotopia. (A) Coronal sections of the hippocampus in adult KIF2A+/H321DNestinCre and control mice, stained with CTIP2 (magenta), TBR1 (green) and counterstained with DAPI (blue), scale bar 0.2 cm. Insets delimited with a dotted square show higher magnification of the CA1 region, scale bar 0.1 mm. (B, C) Coronal sections of the hippocampus in E18 KIF2A+/H321DNestinCre and control embryos (B, scale bar 0.1 cm) and P2 pups (C, scale bar 0.2 cm), stained with CTIP2 (red) and counterstained with DAPI (blue). (D) Immunofluorescence staining of CTIP2 (green) and DAPI (blue) on hippocampal slices from adult KIF2A+/H321D Nexcre mice. Scale bar 0.2 cm. Insets delimited with a dotted square show higher magnification of the CA1 region, scale bar 0.1 cm.
KIF2A+/H321D mice show cortical layering and neuronal positioning abnormalities
We then asked whether abnormalities can be found in the developing cortex of the KIF2A mouse model. To investigate cortical layering, we performed immunohistochemistry analysis on E18.5 KIF2A+/H321DNestinCre and KIF2A+/H321D RosaCre embryos by using SATB2, CTIP2 and TRB1 antibodies, labeling respectively the upper II–IV cortical layers, layer V and layer VI. We observed no differences in the total thickness of the CP between KIF2A+/H321DNestinCre, KIF2A+/H321DRosaCre and WT E18.5 embryos. However, analysis of individual layers revealed significant anomalies in cortical organization. SATB2 staining showed the presence of cells destined to the II–IV cortical layers positioned at the interface between the intermediate zone and the CP in both KIF2A+/H321D NestinCre and KIF2A+/H321DRosaCre mice, which is absent in the WT mice. However, the total thickness of these upper cortical layers was only slightly increased in KIF2A+/H321D NestinCre, but not in KIF2A+/H321DRosaCre embryos compared to controls (Fig. 4A and Supplementary Material, Fig. S5A). Furthermore, CTIP2 staining revealed that layer V presents with disorganized structure, with an increase in thickness and a decrease in the cell density compared to the WT (Fig. 4A and B and Supplementary Material, Fig. S5A and B). Finally, TBR1 staining highlighted reduced thickness of layer VI in mutant KIF2A+/H321D NestinCre but not in KIF2A+/H321DRosaCre embryos. We further carried the same immunohistochemistry analysis in KIF2A+/H321DNestinCre P2 mice. Even though overall phenotype was milder for mutant pups, disorganization in layer V and heterotopic cells in the IZ destined for the II–IV cortical layers were still visible (Fig. 4C and D). To confirm alterations in positioning of pyramidal neurons in the neocortex, suggested by the abnormal layering, we injected pregnant mice with a pulse of EdU at E14.5, to stain specifically layer II–IV cells that are proliferating at this stage (Supplementary Material, Fig. S5C). Analysis at E18.5 showed an accumulation of cells in the intermediate zone of both KIF2A+/H321DNestinCre and KIF2A+/H321DRosaCre embryos, correlating with a decrease in the number of cells in the upper CP (Fig. 4E and Supplementary Material, Fig. S5D). To unravel the underlying causes, we examined neuronal migration by time-lapse recordings and verified the integrity of radial glial fibers. Our videomicroscopy studies showed no major differences in the migration velocity or pausing of neurons between KIF2A+/H321D NestinCre and WT embryos (Supplementary Material, Fig. S5E and F and Supplementary Material, Videos 1 and 2). However, there was a trend of decrease in the number of multipolar cells transiting to a bipolar morphology and increase in the number of multipolar cells persisting multipolar (Supplementary Material, Fig. S5G), suggesting a possible defect in the morphology transition step preceding locomotion. To further evaluate this anomaly, we performed in utero electroporation on E14.5 KIF2A+/H321D NestinCre embryos to label cells with a fluorescent reporter and then studied their morphology in the intermediate zone 3 days later. In control brains, the majority of newborn neurons displayed bipolar morphology (63% on average), around 25% were round and around 10% multipolar (Fig. 4F). In KIF2A+/H321D NestinCre embryos, the population of round and multipolar cells in the IZ were significantly increased while the number of bipolar cells was reduced, confirming our hypothesis of defective multipolar to bipolar transition.
Figure 4.
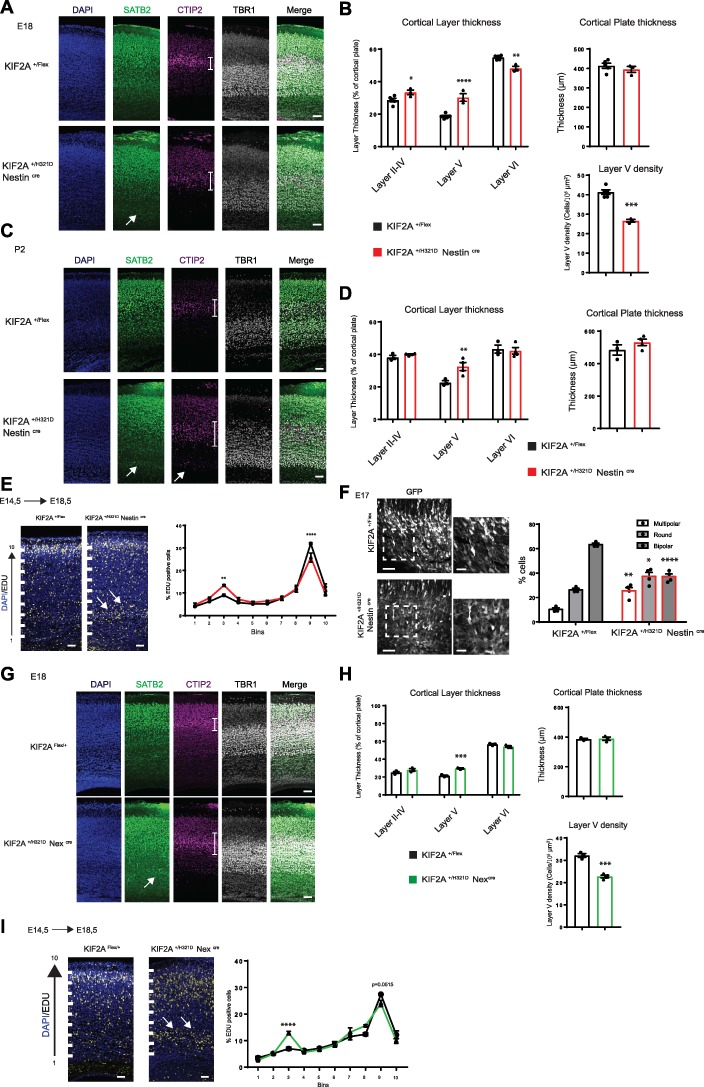
KIF2A+/H321D mice present cortical layering anomalies. (A) Coronal sections of E18 embryonic cortices from KIF2A+/H321DNestinCre and control mice, stained with SATB2 (green), CTIP2 (magenta), TBR1 (gray) and counterstained with DAPI (blue), scale bar 60 μm. White arrow shows SATB2+ cells in the intermediate zone, white lines delimit layer V. (B), Histograms showing relative thickness of layers II–IV, V and VI as percentage if the CP (left), total thickness of the CP (right) and cell density of layer V (lower panel). Data are represented as mean ± SEM, n ≥ 3 mice per group. (C) Coronal sections of P2 cortices from KIF2A+/H321DNestinCre and control mice, stained with SATB2 (green), CTIP2 (magenta), TBR1 (gray) and counterstained with DAPI (blue), scale bar 60 μm. White arrow shows SATB2+ or CTIP2+ cells in the intermediate zone, white lines delimit layer V. (D) Histograms showing relative thickness of cortical layers (left) and total thickness of the CP (right). Data are represented as mean ± SEM, n ≥ 3 mice per group. (E) Coronal sections of E18 embryonic cortices from KIF2A+/H321DNestinCre and control mice, after EdU staining (yellow) counterstained with DAPI (blue), scale bar 30 μm. The cortex was divided into 10 equal bins. White arrows show EdU+ cells accumulation in the intermediate zone. Graphical representation shows the percentage the EDU+ cells in each bin. Data are represented as mean ± SEM, n ≥ 3 mice per group. Scale bar 60 and 30 μm for insets. (F) Coronal sections of E17 embryonic cortices from KIF2A+/H321DNestinCre and control mice, electroporated at E14 with Venus-GFP. Histograms showing percentage of multipolar, round and bipolar cells. Data are represented as mean ± SEM, n ≥ 3 mice per group. (G) Coronal sections of E18 embryonic cortices from KIF2A+/H321DNexCre and control mice, stained with SATB2 (green), CTIP2 (magenta), TBR1 (gray) and counterstained with DAPI (blue), scale bar 60 μm. White arrow shows SATB2+ cells in the intermediate zone, white lines delimit layer V. (H) Histograms showing relative thickness of cortical layers, total thickness of the CP and cell density of layer V (respectively right, middle and left panels) in E18.5 KIF2A+/H321DNexCre embryos. Data are represented as mean ± SEM, n ≥ 3 mice per group. (I) Coronal sections of E18 embryonic cortices from KIF2A+/H321DNexCre and control mice, after EdU staining (yellow) counterstained with DAPI (blue), scale bar 30 μm. The cortex was divided into 10 equal bins. White arrows show EdU+ cells accumulation in the intermediate zone. Graphical representation shows the percentage of the EDU+ cells in each bin. Data are represented as mean ± SEM, n ≥ 3 mice per group. *B: Layer II–IV: p Value = 0.0356; ****B: Layer V: p Value > 0,0001; **B: Layer VI: p Value = 0,0026; ***B: Layer V Density p Value = 0.0001; **D: Layer V: p Value = 0,0054; **E: Bin 3: p Value = 0,0075; ****E: Bin 9: p Value > 0,0001; **F: Multipolar: p Value = 0,0013; *F: Round: p Value = 0,0155; ****F: Bipolar: p Value > 0,0001; ***H: Layer V: p Value = 0,0001; ***H: Layer V Density: p Value = 0,0002; ****I: Bin3: p Value > 0,0001.
Finally, we assessed the glial scaffold in the CP at E15.5 by in utero electroporating GFP (green fluorescent protein) under the control of a BLBP promoter (15). We observed no major anomalies in radial glial fibers (Supplementary Material, Fig. S5H). Given these results, it is possible to propose that anomalies in neuronal positioning in the neocortex are linked to a defective multipolar to bipolar morphology transition.
Next, again we took advantage of our mouse model performed the same analyses of layering and migration on KIF2A+/H321D Nexcre mice, focusing exclusively on post-mitotic effects of the mutation. At E18.5, anomalies were again visible in the organization of cortical layer V, with an increase in thickness and a decrease in cell density (Fig. 4G and H). Similarly, a small population of EdU-positive cells labeled at E14.5 were detected in the intermediate zone of E18.5 embryonic cortices (Fig. 4I). These observations confirm that abnormal cortical layering in KIF2A+/H321D embryos is mainly caused by post-mitotic mechanisms of neuronal positioning.
Increased cell death but no anomalies in proliferation
To investigate developmental processes underlying microcephaly observed in KIF2A+/H321D mice, we studied proliferation of neural progenitors and cell survival rates at different developmental stages. First, we analyzed the presence of PH3 (Phosphorylation of histone 3), a marker of highly condensed chromatin during mitosis, especially prophase and metaphase (16) in proliferative cell in the ventricular zone in order to determine their mitotic index. No difference in the population of mitotic progenitors was observed between KIF2A+/H321D Nestincre and WT embryos, at neither of the studied embryonic stages (E12.5, E13.5, E14.5, E16.5; Supplementary Material, Fig. S6A). Next, to investigate whether the KIF2A mutation affects the balance between proliferative and neurogenic divisions of progenitors, we performed Pax6/Tbr2/Tuj1 immunolabeling at different developmental stages. We observed no alteration in the relative percentage of apical (Pax6+/Tbr2−) and basal (Pax6−/Tbr2+) progenitors. The proportion of newly generated neurons (Pax6−/Tbr2−/Tuj1+) also remained the same between mutant and WT mice (Supplementary Material, Fig. S6B). Regarding our previous observation with overexpression of p.His321Asp mutation by in utero electroporation (6), we additionally quantified the length of primary cilia and the percentage of ciliated cells in brain cortices and mouse embryonic fibroblasts; however, we observed no modifications in those parameters (Supplementary Material, Fig. S6C and D). Combined, these observations show no major effect of the KIF2A p.His321Asp variant on progenitor proliferation or primary cilia in knock-in embryos.
We then assessed cell death by cleaved caspase 3 immunolabeling. In KIF2A+/H321D Nestincre embryos, cleaved caspase 3 levels were strikingly upregulated, especially at E12.5 and E13.5 (Fig. 5A and B). At these early developmental stages, cleaved caspase 3 positive cells were located mainly in the IZ and the developing CP within layers labeled by TBR2 and TUJ1 (Fig. 5D and E), suggesting cell death affects both progenitors and newborn neurons. The number of stained cells then progressively decreased at E14.5 and E16.5 (Fig. 5C and D) and no apoptosis was observed after birth at P2 (Supplementary Material, Fig. S6D). Interestingly, protein extracts from KIF2A+/H321D Nestincre embryos at E12 revealed an increase in p53 levels compared to the control embryos (Fig. 5G), indicating a link between cell death and the p53 pathway.
Figure 5.
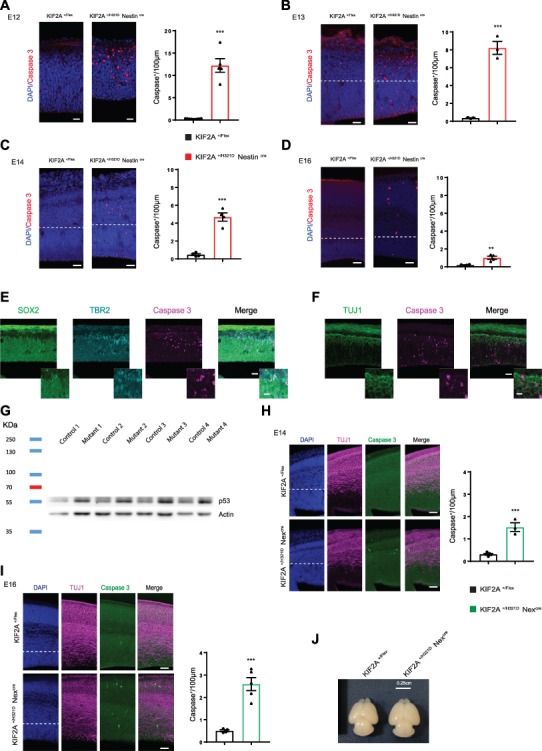
KIF2A His321Asp variant causes p53-related cell death. (A–D) Immunofluorescence staining of cleaved caspase 3 (red) on embryonic cortices at E12, E13, E14 and E16 (respectively A–D) from KIF2A+/H321DNestinCre and control mice, scale bars A—40 μm, B and C—40 μm, D—80 μm. Histograms show the number of Caspase 3+ cells per 100 μm. Data from at least three embryos per group are presented as mean ± SEM, two-tailed t-test. (E, F) Immunofluorescence staining of SOX2 (green), TBR2 (cyan) and Caspase 3 (magenta) (E); and TUJ1 (green) and Caspase 3 (magenta) (F) on E12 embryonic cortices from KIF2A+/H321DNestinCre and control mice, scale bar 30 and 5 μm for insets. (G) p53 protein expression level in brain extracts from KIF2A +/Flex and KIF2A+/H321DNestinCre embryos a E12 detected by immunoblotting. (H, I) Cell death studies in KIF2A+/H321DNex miceCre. Images show coronal sections of E14 (G) and E16 (H) embryonic cortices from KIF2A+/H321DNestinCre and control mice, stained for Caspase 3 (green), TUJ1 (magenta) and counterstained with DAPI (blue), scale bar: H—60 μm, I—60 μm. Histograms show the number of Caspase 3+ cells per 100 μm. Data from at least three embryos per group are presented as mean ± SEM, two-tailed t-test. (J) Brains from KIF2A+/H321DNexCre and corresponding controls at P2. ***A: p Value = 0.0001; ***B: p Value = 0.0004; ***C: p Value = 0,0001; **D: p Value = 0,0039; ***H: p Value = 0,0002; ***I: p Value = 0,0004.
In KIF2A+/H321D Nexcre embryos, increased cell death was still detectable at E14.5 and E16.5 (Fig. 5H and I); however, the extent of the phenotype was significantly lower in comparison with KIF2A+/H321D Nestincre embryos. Additionally, no visible reduction in brain size was detected in P2 pups (Fig. 5J). Therefore, it is possible to deduce that extensive cell death during early embryonic stages (E12.5–E13.5) is likely the main contributing factor for the microcephaly in KIF2A+/H321D Nestincre mice. In KIF2A+/H321D Nexcre animals, slightly increased apoptotic levels appear insufficient to cause a significant reduction in brain size.
KIF2A p.His321Asp mutation affects microtubule dynamics and depolymerization rates
Finally, we aimed to investigate the intracellular mechanisms at the origin of neurodevelopmental disorders in KIF2A+/H321D mice. Since KIF2A is described as a MT depolymerizing kinesin (4,5), we focused our studies on MT dynamics. We conducted a MT depolymerization assay in fibroblasts derived from the subject in which the mutation was identified. By incubating the cells with the MT depolymerizing agent nocodazole and visualizing MTs at different time points, we show that the KIF2A pathogenic variant causes MT overstability. For all three unrelated controls, around 50% of the cells present with completely depolymerized MTs after 20 min of incubation with nocodazole, while for fibroblasts carrying the KIF2A p.His321Asp variant this population is reduced to 25% (Fig. 6A). This MTs depolymerization resistance to nocodazole was also showed in mouse embryonic fibroblasts derived from the mouse model with a significant increase of the percentage of cells with polymerized MT after 10, 15 and 20 min of incubation with nocodazole (Supplementary Material, Fig. S7A). We confirmed these results by monitoring live nocodazole-induced MT depolymerization in transfected HeLa cells Tubulin GFP stable line. Even after 30 min of incubation with nocodazole, a complex MT network was observed in HeLa cells transfected with the p.His321Asp variant, while almost no MTs were detected in cells expressing an empty vector or KIF2A-WT (Supplementary Material, Fig. S7A and Supplementary Material, Videos 3–5). We further assessed MT plus end dynamics in fibroblasts by videomicroscopy after transfection with the plus-end binding protein EB3 fused to GFP. A significant increase in plus-end dynamics was observed for fibroblasts carrying the p.His321Asp variant, compared to all three unrelated controls (Fig. 6B and Supplementary Material, Videos 6–9). Similar results were observed in mouse embryonic fibroblasts, with a tendency to increased mean speed of EB3 in cells derived from KIF2A+/H321D Rosacre mice. Together these data show that the KIF2A mutation modifies the polymerization–depolymerization rates and prevents KIF2A from depolymerizing MTs. To gain insight into the activity difference of wild-type Kif2A and the His321Asp mutant, we expressed both proteins as monomeric motor domain constructs that include the kinesin-13-specific neck (Kif2A-NM; amino acids 153–553) (Fig. 6C), which is required to catalytically depolymerize MTs in vitro (17–19). Both proteins were purified to homogeneity from Escherichia coli and exhibited the same secondary structure content and stability by circular dichroism (Supplementary Material, Fig. S7A–C). According to the distribution of tubulin between supernatant and pellet fractions obtained by centrifugation of reactions in which each protein was mixed with taxol-stabilized MTs, the His321Asp mutant has severely diminished ability to depolymerize MTs in comparison with wild-type Kif2A (Fig. 6D and E). This is to be expected considering the proximity of histidine 321 to the adenine moiety of the nucleotide and the strict requirement of ATP turnover for catalytic MT depolymerization (Fig. 6F).
Figure 6.
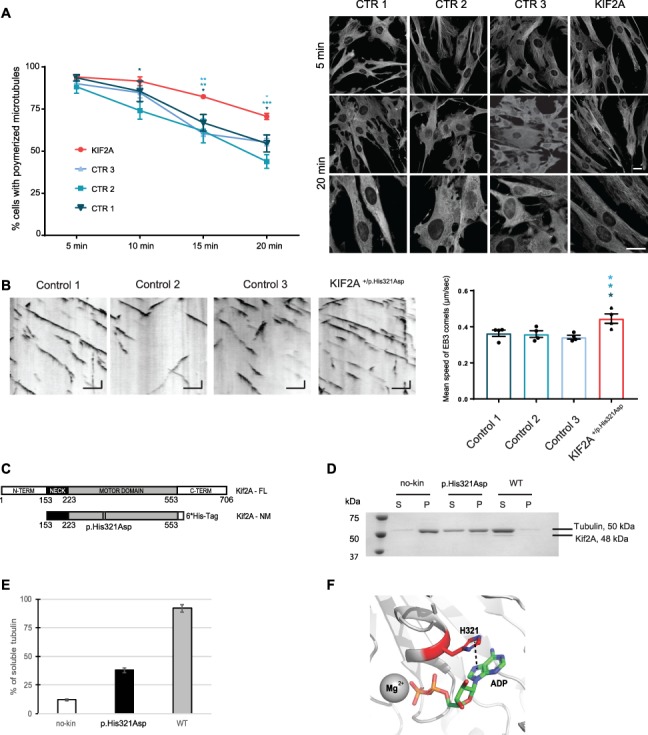
KIF2A His321Asp variant effects on microtubule stability and dynamics. (a) Depolymerization assay in fibroblasts derived from a subject bearing the KIF2A His321Asp variant and three controls. Left panel: Graph represents the percentage of cells with polymerized microtubules over 20 min time after nocodazole treatment. Data are presented as mean ± SEM, n = 3 biologically independent experiments. Two way ANOVA with Tukey’s post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, compared to controls. Right: representative images of cells at 5 and 20 min of nocodazole treatment, after immunofluorescence staining of tubulin (gray), scale bar 20 μm. (B) Microtubule dynamics in fibroblasts derived from a subject bearing the heterozygous KIF2A p.His321Asp variant and three controls. Left panel: Representative kymographs of dynamic EB3 comets in different controls and mutant fibroblasts. Kymographs are calibrated in time (y, s) and space (x, μm). Scale bar Y = 10 s, X = 5 μm. Right panel: Quantification of EB3 mean velocity. Data are presented as mean ± SEM, n = 4 biologically independent experiments, one-way ANOVA with Dunnett’s post hoc test, *P < 0.05 compared to controls. (C) Biochemical analysis of WT and p.His321Asp mutant of Kif2A. (C) Schematic representation of domain composition of human Kif2A and its constructs used in this study. Location of the H321D mutation is shown. (D) MT depolymerization activity of WT and H321D Kif2A. Taxol-stabilized MTs (1.5 μM) were incubated with WT, H321D (0.075 μM), and no kinesin for 10 min at 25°C. Soluble tubulin and MT polymers were separated into supernatant (S) and pellet (P) by sedimentation. Equal volume of fractions was analyzed on SDS-PAGE gel. (E) Percentage of soluble tubulin in each depolymerization reactions was calculated by densitometry of the bands using ImageJ software. (F) View of the nucleotide-binding site of Kif2A (PDB ID 2GRY). H321 residue (red, sticks) and its pi-interaction with the adenosine moiety of the nucleotide (dash line) are shown. Figure was generated with PyMOL.
Discussion
Spatial- and temporal-restricted knockout mouse models can be generated using Cre/lox recombination system. However, expression of a point mutation, irrespective of its position in a given gene, in a tissue- and time-restricted manner is still a challenging issue and efficient strategies remain lacking.
Here, we have used the FLExII switch technology to generate and characterize a conditional knock-in mouse model for MCD related to KIF2A mutations. Our objective was to better understand the pathophysiological mechanisms of the p.His321Asp variant, which leads to neurodevelopmental disorders associated with epilepsy. We have previously investigated pathogenic KIF2A mutations by the means of in utero electroporation and in vitro cellular studies (6) and have now aimed to complete our previous study with an in vivo model that mimics physiological conditions in subjects with Kif2A-related MCD.
We have crossed the conditional model with three different types of Cre: RosaCre mice to constitutively express the mutation, NestinCre mice to express the mutation in neuronal progenitors and NexCre mice to exclusively express the mutation in post mitotic neurons. We used KIF2A+/H321DROSACre mice to mimic the patient condition and showed indeed that this model recapitulates several phenotypes identified in human patients with KIF2A mutations. Namely, KIF2A+/H321DROSACre mice present with neuroanatomical defects and significant reduction in brain size, correlating with the microcephaly and subcortical heterotopia described in subjects (1,2). Additionally, these mice displayed hyperactivity and memory deficit, which are a hallmark of mouse models of intellectual disability (20). Finally, KIF2A+/H321DROSACre mice have an increased susceptibility to PTZ-induced epileptic seizures. Together, these traits make the KIF2A+/H321D mice a model, highly representative of the human disease and a powerful tool for future studies on therapeutic strategies.
Though the knock-in model herein described recapitulated several features of the human disease, it is worth mentioning that hippocampal heterotopia does not appear to be a common abnormality in human patients. In addition, the absence of spontaneous seizures and the mild behavior phenotype highlighted in the mouse model contrast with the severity of patients phenotype. These differences are likely to be related to the lack of appropriate monitoring tests and tools that allow rational comparison between human and mice.
Interestingly, hippocampal heterotopia was reported in several mouse models for MCD-related genes, though its extent and localization may differ between mouse models (21–23), and this developmental abnormality was always evidenced by histopathological approaches. In patients, subtle abnormalities of the hippocampus may exist but their detection for the moment may escape commonly used brain imaging techniques (24).
We then used KIF2A+/H321D NestinCre and KIF2A+/H321D NexCre mice to further decipher the developmental processes contributing to the phenotype. We demonstrate that abnormal cortical layering and neuronal positioning are mainly caused by anomalies in post-mitotic mechanisms, since these phenotypes were detected in both KIF2A+/H321D NestinCre and KIF2A+/H321D NexCre mice. No significant differences were observed between the migration dynamics of WT and mutant embryos; however, we detected alterations in the multipolar-to-bipolar morphology transition, which could be at the origin of the aforementioned abnormalities. In fact, neurons, unable to adopt a bipolar morphology, would not succeed in initiating glial-guided locomotion and would remain in the intermediate zone of the cortex or hippocampus (23–25). Similar results were obtained in our previous study based on the in utero electroporation assay, where we showed that introducing the pathogenic variant during cortical development disrupts multipolar-to-bipolar transition in migrating neurons leading to their accumulation in the IZ. (25,26). It is possible to hypothesize a link between an aberrant multipolar-to-bipolar transition and the described function of KIF2A in MT depolymerization (27,28). This morphological transition is highly dependent on MT dynamics and MT-dependent intracellular transport to specific site, mechanical forces and local signaling (29).
We further show that the microcephaly in KIF2A+/H321DROSACre and KIF2A+/H321D Nestincre mice is related to a major increase in cell death during early stages of brain development (E12.5–E13.5). Very high levels of apoptotic cells were detected during these early stages in the entire cortex of KIF2A+/H321D Nestincre but not in KIF2A+/H321D Nexcre embryos, which do not present with reduced brain size. We also demonstrate that high cell death correlates with an increased p53 levels. So far, several studies have linked KIF2A inactivation to cell death in cancer tissue (30,31). They show that apoptosis is linked to a decrease of AKT and PI3K levels after inactivation of KIF2A by siRNA, which subsequently leads to a downregulation of the PI3K-AKT pathway and activation of apoptosis. There is no evidence that KIF2A is a direct upstream regulator of the PI3K/Akt pathway, but it is possible to imagine that this alteration is indirectly linked to a defect in MT dynamics. KIF2A also plays a major role during mitosis and chromosome segregation. Perturbation during these processes can lead to an aberrant number of chromosomes linked to centrosome defect in cell after division and finally a p53-mediated apoptosis without proliferation defects (32,33).
Interestingly, we found no proliferation defects or primary cilium abnormalities. This is in contrast with our previously published in utero investigations (6). Indeed, by overexpressing mutant KIF2A during development, we have shown defective cell cycle and ciliogenesis. Since in the KIF2A+/H321D mouse model, the variant is at physiological concentrations, while with the in utero electroporation approach the variant is intensely overexpressed and can lead to aberrant dose-dependent effects and the observed difference.
KIF2A plays a major role in MTs dynamics and a disruption of MT depolymerization can interfere with multiple processes requiring MT during cortical development and in particular morphology changes accompanying newborn neurons locomotion. In this line of thoughts, our study raises the possibility that the p.His321Asp variant prevents the KIF2A protein from promoting MTs depolymerization. We have shown that overexpression of mutant KIF2A induces MT stability and halts MT depolymerization. Additionally, we showed by EB3 comets study that KIF2A mutation is associated with an accelerated MT polymerization rate possibly due to an imbalance in polymerization/depolymerization dynamics. All these results converge toward a MT depolymerizing defect linked to an inability of KIF2A to depolymerize MTs. Several studies have shown that KIF2A needs to bind and hydrolyze ATP in order to be able to depolymerize MT and release the tubulin dimer (19,27,34). All mutations in patient with MCD linked to KIF2A are located in ATP-binding domain and lead to a sequestration of KIF2A to the MTs (1,3). Convergent evidence indicates that mutant KIF2A proteins are not able to bind or hydrolyze ATP, to depolymerize and release MT leading therefore to the apparent abnormal very stable interaction between KIF2A and MTs.
As KIF2A-related MCD is caused by heterozygous de novo missense variants, development of knock-in models appears the most appropriate strategy to mimic the human genetic molecular conditions. However, it is worth mentioning that insights have been also gained through investigations of constitutive and conditional Kif2a homozygous knockout (Kif2a −/−) models reported by Homma and coworkers (28,35).
Besides the early death of Kif2a−/− mice (within 1 day of birth) that is not observed in the knock-in Kif2a His321Asp model, the other potential difference corresponds to the increase in neuronal death during early stages of cortical development that was only observed in the Kif2a+/H312D model. That being said, it is important to mention that brain and hippocampal developmental abnormalities, as well as neuronal morphological changes, and MT dynamics defects are strikingly similar between both models. At first sight, these similarities might sound coherent with the molecular mechanisms underlined by the dominant negative effect of Kif2a His321Asp heterozygous missense variant and Kif2a−/− genotype. Indeed, as Kif2a functions as homodimer, a significant fraction of Kif2a wild-type polypeptide will be prevented from being normally functional because of its association with the mutant protein, mimicking therefore Kif2a loss–of-function resulting from inactivation of both alleles in Kif2a−/− condition.
Homma and colleagues also investigated, through tamoxifen-inducible Kif2a conditional knockout (Kif2a-cKO) mice, consequences of postnatal Kif2a loss-of-function in the brain. Interestingly, they showed that despite exhibiting no significant defects in neuronal proliferation or migration, tamoxifen-treated Kif2a-cKO mice (at the third postnatal week) showed weight loss, hyperactivity and seizures. Comparative electrophysiological recording between WT and Kif2a-cKO mice using electrodes inserted into the hippocampus and the cortex allowed to show the aberrant spikes and paroxysmal EEG events occur in the hippocampus but not in the cortex. Interestingly, these findings suggest that postnatal KIF2A expression is also required for normal brain function and neuroanatomical abnormalities resulting from Kif2a dysfunction during early stages of development are maybe not the only defect that underlies patients’ behavioral and epileptogenic phenotypes.
Finally, it should be pointed out that our work additionally demonstrates the functionality of the FLExII Cre-dependent genetic switch technique and confirms it is now possible to generate a conditional knock-in regardless of the position of the point mutation in a given gene. So far and with the exceptions of cKI mouse models targeting last exons and the study reported by Obeng et al. (36) most attempts to generate cKI mouse models remained unsuccessful (37). By degenerating the mutated exon while conserving the same protein sequence except for the mutation, it is possible to significantly reduce the homology between the WT and mutated exons and ensure successful inversion/excision of the recombination allele, as well as stability and bona fide splicing of transcripts bearing the construct.
In view of the findings, we can conclude that by using this new generation of KIF2A conditional knock-in mouse model we were able to modelize the patient phenotypes and dissect spatially the impact of the p.His321Asp mutation on each developmental process. As a perspective, it would be interesting to take advantage of the conditional property of this model to study consequences of postnatal expression of the mutation in mice expected to be free of major neuroanatomical abnormalities and assess whether such late expression contribute to the pathophysiological mechanisms underlying the above highlighted susceptibility to epilepsy.
Materials and Methods
Mice generation
Mice were handled according to national and international guidelines (authorization number 2017022316297963, French MESR) and the procedures followed were in accordance with the ethical standards of the responsible committee on mouse experimentation [Comité d’éthique pour l’expérimentation animale (Strasbourg, France)]. Knock-in KIF2A mice with conditional expression of the point mutation were generated in the Institut Clinique de la Souris (Celphedia, Phenomin, ICS, Illkirch). The KIF2A locus was engineered as follows. A 688 bp wild-type mouse genomic fragment comprising exon 10 and surrounding intronic sequences was PCR amplified and subcloned between LoxP and Lox511 sites in an ICS proprietary vector. The basic vector already contains all lox sites in the correct orientation as well as a NeoR cassette surrounded by FRT sites. In a second cloning step, a 529 bps synthetic fragment (String DNA fragment ordered from GeneArt) comprising the degenerated human exon 10 and surrounding human intronic sequences was cloned in an inversed orientation. Both 5′ (4.3 kb) and 3′ (3.2 kb) homology arms were cloned successively. The final construct was linearized and electroporated in proprietary derived C57Bl/6 N ES cells. Positive clones were selected by long-range PCR and further validated by Southern blot using both a Neo probe and a 3′ external probe. The fully validated ES cell clone 28, which did not show any abnormalities by ddPCR and karyotype spreading, was microinjected in BALB/cN blastocysts; chimeras were obtained and germline transmission of the recombinant allele was achieved in a C57BL/6 N pure genetic background.
KIF2AFlex/Flex mice were crossed with Rosa26Deleter-Cre+/− (Soriano et al., 1999) to express ubiquitously the KIF2A mutation. KIF2A+/Flex mice were crossed with Nestin-Cre+/− (Graus-Porta et al., 2001; Tronche et al., 1999) to express KIF2A mutation specifically in neuronal progenitors. KIF2A+/Flex mice were crossed with Nex-Cre+/− mice (Goebbels et al., 2006) to express KIF2A mutation only in post-mitotic neuronal cells. For staging of embryos, the day of vaginal plug was considered as E0.5 and embryos used for comparative analyses are littermates.
Genotyping
To verify the correct recombination and excision step of the KIF2A FLEx II construction, we have performed PCR on tail fragment with FastStart PCR Master (Roche) and the following primers: Lf: 5′CAGGAAGGGCCCAGTTGTCTATTATACC3′; Mr: 5′CCCTTGCTTTGGGATTATACAAATGCC3′; Mf: 5′CGTGTGTTTGTGAATGTAGAGGCCAGTC3′; Lxr: 5′CGGGGATCCTCTAGAGCTTATAACTTCG3′; Er: 5′GCTCCCTATAGAGATGTCCTTCTCGAAG3′; Ef: 5′GCTCGAAGATGGTCTCGACCAACG3′. The position of the different primers used is represented in Supplementary Material, Figure 1D.
To determine the genotype, the presence or absence and size of PCR fragments were analyzed to detect the presence of exon 10 (Lf/Mr), the presence of distal LoxP (Lf/Mr), LoxP-specific PCR (Mf/Lxr), the presence of distal Lox511 (Mf/Er) and the excision of the floxed exon (Lf/Ef). The presence of the point mutation was confirmed by Sanger sequencing on the first F1 animals. For each crosses with different types of Cre, the presence or absence of the Cre was genotyped by a qPCR with internal primers at Cre recombinase (82 bp): Forward: 5’CGCAAGAACCTGATGGACATG3’, Reverse: 5’ ACCGGCAAACGGACAGAA3’.The PCR cycling conditions were 1 cycle at 95°C for 4 min, 34 cycles at 94°C for 30 s, 62°C for 30 s, 72°C during 1 min, 1 cycle at 72°C for 7 min and 1 cycle at 20°C for 5 min. After, PCR products were analyzed by agarose gel electrophoresis.
RT-qPCR
Brain cortices of 10-week-old mice were grinded using Precellys Lysing Kit (Bertin Technologies) and total RNA were extracted using TRI Reagent (Molecular Research center, Inc.) following manufacturer’s instructions. The purity and the quality of RNA were confirmed by defining the ratio of absorbance at 260 and 280 nm wavelengths (NanoDrop® ND1000, ThermoScientific). cDNA samples were synthetized with Transcriptor First Strand cDNA Synthesis Kit (Roche) and following the manufacturer’s instructions. qRT-PCR was performed in a LightCycler PCR instrument (Roche) using SYBR Green Master Mix (Qiagen) and the following primers for full KIF2A exon 9–11: 5′-ccttcgatgactcagctcct-3’(Forward) and 5′-agtggaaaggtgtttgacttact-3′ (Reverse); KIF2A exon 10 5′-aatatttgaaagggggccatgg-3′ (Forward) and 5′-gcagactggaagtgggaaaa-3′ (Reverse). Reaction conditions were carried out for 50 cycles (10 min initial denaturation 95°C, 15 s at 95°C, 20s at 59° and 25 s at 72°C); each qRT-PCR reaction was performed in triplicates and relative mRNA expression of KIF2A was normalized to actin mRNA expression for the mouse model and to RPLPO for ES cells. The relative transcript expression of mRNA levels was calculated by the Ct method and the area of the peak for KIF2A exon 9–11 primers. The correct rearrangement of the construction was verified by PCR on cDNA samples using Taq DNA Polymerase (Roche) using the same primers as those used for qPCR. PCR products were sent to GATC biotech for Sanger sequencing.
Western blotting
Brain cortices were grinded using Precellys Lysing Kit (Bertin Technologies) and lysed in RIPA buffer (Tris HCl 1 M, pH 7.7; NaCl 5 M; EDTA 0.5 M; Triton X-100) supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Sigma Aldrich). Protein concentration was measured with Bio-Rad protein assay reagent (Biorad Laboratories, CA). Samples were denatured at 95°C for 10 min in loading buffer, then resolved in SDS-PAGE and transferred onto nitrocellulose membrane. Membranes were blocked in 5% non-fat milk in TBS buffer, 0.1% Tween and then immunoblotted using anti-KIF2A (Ab37005, rabbit, Abcam, 1:10 000) or anti-p53 (ab26, mouse, 1:1000, Abcam) and anti-beta actin coupled HRP (A3854, Sigma-Aldrich, 1:50000) antibodies.
KIF2A+/Flex ES cells generation and validation
In order to generate KIF2A+/H321D ES cell clone, KIF2A+/Flex ES cell clone was incubated with HTN-Cre (6 μM; Excellgen Ref EG-1001). RT-qPCR was performed as previously described in the ‘RT-qPCR’ part, and the correct rearrangement of the construction was verified as previously with Sanger sequencing. For immunofluorescence, ES cells were plated on glass coverslips, fixed with 4% paraformaldehyde solution for 20 min. Coverslips were then blocked by 30 min incubation with 0.3% Triton-X100 and 2% newborn calf serum in PBS. Primary antibodies were incubated overnight at 4°C. Slides were then incubated with the corresponding Alexa Fluor-conjugated secondary antibodies (Life Technologies) in a 1/500 dilution in 1X PBS for 30 min at room temperature. Slides were mounted with a DAPI containing fluoromount-G mounting medium (Interchim). Images were acquired using TCS SP8 confocal microscope (Leica Microsystems).
Neuroanatomical study
Neuroanatomical study was carried out using six male mice at precisely 16-week old (n = 3 per group). Mice were euthanized in a CO2 chamber and brains were dissected and fixed in 4% buffered formalin for 48 h and then transferred to 70% ethanol.
Samples were embedded in paraffin using an automated embedding machine (Sakura Tissue-Tek VIP) and cut at a thickness of 5 μm with a microtome in order to obtain sagittal brain region at lateral +0.60 mm. The sections were then stained with 0.1% Luxol Fast Blue (Solvent Blue 38; Sigma-Aldrich) and 0.1% Cresyl violet acetate (Sigma-Aldrich) and scanned using Nano-zoomer 2.0HT, C9600 series at 20× resolution. A total of 166 brain parameters, made of area and length measurements as well as cell level features, were taken blind to the genotype across one sagittal section. Data were analyzed using a two-tailed Student’s t-test assuming equal variance to determine whether a brain region is associated with neuroanatomical defect or not.
Behavioral tests
Rotarod test
This test measures the ability of an animal to maintain balance on a rotating rod (Bioseb, Chaville, France). Mice were given three testing trials during which the rotation speed accelerated from 4 to 40 rpm in 5 min. Trials were separated by 5- to 10-min interval. The average latency was used as index of motor coordination performance.
Grip test
This test measures the maximal muscle strength (g) using an isometric dynamometer connected to a grid (Bioseb). Mice were allowed to grip the grid with all its paws, and then they were pulled backwards until they released it. Each mouse was submitted to three consecutive trials immediately after the modified SHIRPA procedure. The maximal strength developed by the mouse before releasing the grid was recorded and the average value of the three trials adjusted to body weight.
Crenelated beam
This test allows gait analysis, which is relatively specific for evaluation of proprioceptive sensitivity. The performance in this test depends also on the integrity of motor coordination and vestibular function. Animals require an accurate paw placement to succeed in this test. The apparatus is a 1.7 cm wide and 63 cm long crenelated wooden beam, elevated 18 cm above the bench surface. Crenels are 2 cm long and 2 cm deep. The two extremities of the beam are not crenelated and used as starting and goal points during testing. A home cage is placed at the goal point to allow mice being motivated to perform the task. Animals were submitted to three testing trials during which they were placed at the starting extremity and allowed to walk the beam distance. The latency to cross the beam and the number of hind paws fails (when one or both hind paws misses the merlon and slip in the crenellated parts of the beam) were measured.
Object recognition task
Mice were tested in automated open fields (Panlab, Barcelona, Spain), each virtually divided into central and peripheral regions. The open fields were placed in a room homogeneously illuminated at 70 Lux at the level of each open field. The objects to be discriminated were a glass marble (2.5 cm diameter) and a plastic dice (2 cm). Animals were first habituated to the open field for 30 min. Each mouse was placed in the periphery of the open field and allowed to explore freely the apparatus for 30 min. The distance traveled, the number of rears and time spent in the central and peripheral regions were recorded over the test session. The number of entries and the percentage of time spent in center area are used as index of emotionality/anxiety. The next day, mice were tested for object recognition in the same open-field arenas under 70 Lux illumination. They were submitted to a 10-min acquisition trial during which they were placed in the open field in the presence of sample objects (A and A’) (2.5 cm diameter marble or 2 cm edge plastic dice). The time the animal took to explore the samples (when the animal’s snout was directed towards the object) was manually recorded. A 10-min retention trial was performed 3 h later. During this trial, one of the samples A and another object B were placed in the open field, and the times tA and tB the animal takes to explore the two objects were recorded. A recognition index (RI) is defined as (tB/(tA + tB)) x 100.
Y-maze spontaneous alternation
The apparatus is a Y-maze made of Plexiglas and has three identical arms (40 x 9 x 16 cm) placed at 120° from each other. Each arm has walls with specific motifs allowing distinguish it from the others. Each mouse was placed at the end of one arm; the head directed to the walls and allowed to explore freely the apparatus for 5 min, with the experimenter out of the animal’s sight. The percentage of spontaneous alternations was used as index of working memory performance. The number of arms visited and the latency to exit the starting arm were also scored as an index of ambulatory activity and emotionality in the Y-maze, respectively.
Immunohistochemistry
Adult mice were anesthetized with a mix of Ketamine (130 mg/kg), Xylazine (13 mg/kg) and NaCl 0.9% and then perfused intracardially with paraformaldehyde 4% in 0.1 M phosphate buffer using a peristaltic pump. Brains were dissected and post-fixed in paraformaldehyde 4% in 0.1 M phosphate buffer overnight at 4°C. Embryo or pup brains were dissected and fixed in paraformaldehyde 4% in 0.1 M phosphate buffer overnight at 4°C. Brains were placed in a solution of 4% low-melting agarose (Bio-Rad) and cut into coronal sections (80 μm) using a vibrating-blade microtome (Leica VT1000S, Leica microsystems). Sections were blocked in PBS with 0.3% Triton X-100 and 2% of newborn calf serum (Life Technologies) at room temperature for 1 h. All primary antibodies were incubated overnight at 4°C in PBS with 0.3% Triton X-100 and 2% of newborn calf serum. After washing in 1X PBS, sections were incubated with corresponding Alexa Fluor-conjugated secondary antibodies in a 1:500 dilution in 1X PBS for 1 h 30 min at room temperature. After washing in 1X PBS, sections were incubated with Hoechst 33342 trihydrochloride trihydrate (Life Technologies) in a 1:2000 dilution for 30 min at room temperature. After washing, sections were mounted with Fluoromount-G mounting medium (Interchim). Microscopy was performed on a TCS SP8 confocal microscope (Leica Microsystems) and image stitching was made by the LAS X core software (Leica Microsystems). Image analyses were achieved using ImageJ software (NIH) and images were assembled with Adobe Illustrator CS6 16.0.5 (Adobe Systems). All experiments are carried on littermate mice and all analyzed brain slices are selected with brain atlas to be comparable.
Antibodies
The following antibodies were used for immunohistochemistry: anti-phospho Histone H3 (06–570, rabbit, 1: 500, Millipore), anti-Tbr2 (14–4875-80, rat, 1: 200, eBiosciences), anti-Pax6 (PRB- 278P, rabbit, 1: 200, Covance), anti-Kif2a (ab37005, rabbit, 1: 6000, Abcam), anti-NeuN (MAB377, mouse, 1: 100, Millipore),anti-Ctip2 (ab18465, rat, 1:500, Abcam), anti-Tbr1 (ab31940, rabbit, 1:500, Abcam), anti-Tbr1 (Ab183032, rabbit, 1:1000, Abcam), anti-Satb2 (ab51502, mouse, 1:400, Abcam), anti-nestin (ab6142, mouse, 1:500, Abcam), anti-Tuj1 (MMS-435p, mouse, 1:500, Eurogentec), anti-Sox-2 (sc-365 823, mouse, 1:200, Santa Cruz Biotechnology), anti-cleaved caspase 3 (AF835, rabbit, 1:500, R&D System), anti-V5 (R96025, mouse, 1:500, Invitrogen), anti-RFP (5F8, rat, 1:1000, Cromotech), anti-Arl13B (17711–1-AP, rabbit, 1:500, ProteinTech), anti-tubulin [YL1/2] (ab6160, rat, 1:1000, Abcam), anti-GFP (ThermoFisher, chicken, A10262) and anti-p53 (ab26, mouse, 1:1000, Abcam).
EdU
For neuronal positioning experiments, E14.5 pregnant mice were intraperitoneally injected with EdU (5-ethynyl-2′-deoxyuridine solution, Invitrogen) diluted in NaCl 0.9% at the dose of 40 mg EdU/kg of body weight and embryos were harvested and fixed at E18.5. EdU staining was done using Click-iT EdU Alexa Fluor 647 Imaging Kit (Invitrogen) according to manufacturer’s protocol.
DNA constructs
Commercially available Human untagged KIF2A cDNA (NM_004520) cloned in pCMV6-XL5 vector (SC117315) were purchased from Origene. Variant p.His321Asp was introduced by site-directed mutagenesis using QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies). pDEST40-Arl13b11 expressing V5 tagged Arl13b was purchased from Addgene (#40873). BLBP-GFP plasmid was kindly provided by N. Heintz (The Rockefeller University, NY, USA); NeuroD-GFP vector was kindly provided by J. Godin (IGBMC, Strasbourg, France) and pSilencer-CAG-Venus-2 vectors were kindly provided by J. Courchet (Institut NeuroMyoGene, Lyon). All plasmid DNAs were prepared using the EndoFree plasmid purification kit (Macherey Nagel).
In utero electroporation
In utero electroporation was performed as described previously (38) using timed pregnant mice at E14.5. Animal experimentations were performed at the IGBMC animal facilities. At E14.5, the pregnant mice were anesthetized with isoflurane (2 l/min of oxygen, 3% isoflurane during induction and 2.5% during surgery). The uterine horns were exposed, and a lateral ventricle of each embryo was injected using pulled glass capillaries (Harvard Apparatus) with Fast Green (2 μg/ml; Sigma) combined with the DNA constructs prepared with EndoFree plasmid purification kit (Macherey Nagel) and a reporter vector at 1 μg/μl each. Plasmids were further electroporated into the neuronal progenitors adjacent to the ventricle by delivering five electric pulses at 35 V for 50 ms at 950 ms intervals using a CUY21EDIT electroporator (Sonidel). After electroporation, embryos were placed back in the abdominal cavity and development was allowed to continue until E15 or E18. Embryo brains were dissected and fixed in 4% paraformaldehyde in PBS (Phosphate buffered saline) overnight and then sectioned at 80 μm slices using a VT1000S vibratome (Leica Biosystems).
Electroencephalograms
Implantation of electrodes was done under general anesthesia (Propofol, Fentanyl, Domitor mixture 100 μl/10 g). KIF2A+/H321D (n = 3) and KIF2A+/Flex wild-type littermate mice (n = 3) aged postnatal 12 weeks were implanted with stainless steel wire electrodes (Phymep, France). For each mouse, five single-contact electrodes were placed over the left and right frontoparietal cortices. The electrodes were secured into the skull and soldered to a microconnector that was fixed to the skull by acrylic cement. An electrode over the surface of the cerebellum served as ground for all derivations. All mice were allowed to recover for a period of 1 week before EEG recordings. Freely moving mice were then recorded in their housing transparent cage and were connected to a recording system. EEG signals were amplified with a band-pass filter setting of 0.1–70 Hz with a 64-channel system (Coherence, Natus) and sampled at 256 Hz. Recordings were performed during 1 h for evidence of spontaneous convulsive seizure. At the end of the recording period, animals were injected intraperitoneally with a convulsive dose of 10 mg/kg of pentylenetetrazole (PTZ; Sigma-Aldrich, Co), a GABAA receptor antagonist, to evaluate seizure threshold. EEG was recorded during 30 min after PTZ. Video EEGs were reviewed offline for electrographic seizures.
Cell culture
HeLa Tubulin GFP, kindly provided by Patrick Schultz, were cultured in DMEM (Gibco) supplemented with 1 g/l of glucose, 5% fetal calf serum (FCS) and gentamycine (DMEM–1 g/l glucose–5% FCS–gentamycine). Subject’s human fibroblast was cultured in DMEM (Gibco) supplemented with 1 g/l of glucose, 10% FCS and gentamycine (DMEM–1 g/l glucose–10% FCS–gentamycine). All cell lines were grown at 37°C in 5% CO2. p.His321Asp fibroblasts are the same that those reported by Poirier et al. (1) and Broix et al. (6). Control fibroblasts are the same that those reported by Chantrel-Groussard et al. in Human Molecular Genetics in 2001 (39). For mouse embryonic fibroblast primary culture, embryos at E14.5 from a cross between KIF2AFlex/Flex mice and Rosa26Deleter-Cre+/− were isolated. The head and the internal organs were removed. The embryos body were minced and trypsinized with 0.05% trypsin during 30 min, passed through a 70 μm nylon cell strainer (Falcon) and cultured into a 30 mm2 petri dish in DMEM (4.5 g/l glucose) with GLUTAMAX-I + 10% FCS ES tested (Fisher) + β mercaptoethanol (0.1 M), AA non-essentials 1X, Na Pyruvate (1 mm), Penicillin (100 U/ml) and Streptomycin (100 μg/ml). The cells were split in a T75 when they were freshly confluent and used for experiment at passages two and three (40).
EB3 Tracking analyses
Human and mouse embryonic fibroblasts were cultured in fluorodish™ FD35-100 (World Precision Instruments) and transfected with EB3-GFP plasmid using Lipofectamine 2000 (Invitrogen) following manufacturer instructions. Live video-microscopy recording was performed using a Confocal Spinning disk– NikonTi–Roper iLas FRAP system equipped with a PLAN APO VC 60x objective (Nikon Instrument, Inc., Melville, USA) and driven by Metamorph 7.0 (Molecular Devices, LLC). Samples were maintained at 37°C and 5% CO2 during acquisition. Images were collected with Photometrics Prime 95B™ Scientific CMOS Camera (PHOTOMETRICS) every 200 ms during 2 min. Movie assembly and time projections were done using ImageJ software (http://rsb.info. nih.gov/ij/, NIH). For generation and analyses of kymographs, Image J plug-in kymotoolbox (Available on demand on http://www.bic.u-bordeaux.fr) was used. Kymographs were calibrated in time (y in s) and space (x in μm) and analyses were performed manually by following EB3 comet trajectories. Mean speed was extracted by the plug-in.
Microtubule depolymerization kinetics
For microtubule depolymerization kinetics, human and mouse embryonic fibroblasts were grown on glass coverslips. At t = 0 medium was replaced with pre-warmed medium containing 10 μM of Nocodazole (Sigma-Aldrich, Co). Cells were then maintained at 37°C. At the indicated time points (0, 5, 10, 15 and 20 min), cells were fixed in 4% paraformaldehyde in 0.1 M PBS for 20 min at room temperature. Cells were then blocked in 5% donkey serum in PBS-0.3% Triton-X and then incubated for 1 h with the primary antibody. Alexa-coupled secondary antibody (Thermo Fischer) was used at 1/500. Coverslips were mounted with Fluoromount-G mounting medium with DAPI (Interchim). Images were acquired using a confocal microscope TCS SP8 X (Leica microsystems) and analyzed using ImageJ software (NIH).
Time-lapse microtubule depolymerization
HeLa Tubulin GFP were cultured in fluorodish™ FD35-100 (World Precision Instruments) and transfected with pCMV6 empty, pCMV6 KIF2A WT or pCMV6 KIF2A p.his321Asp plasmids using Lipofectamine 2000 (Invitrogen) following manufacturer instructions. Live video-microscopy recording was performed 24 h after transfection using a Confocal Spinning disk Inverted Leica DMI8 system equipped with a HC PL APO 100x objective (Leica Biosystems) and driven by Metamorph 7.0 (Molecular Devices, LLC). At t = 0, medium was removed and replaced with fresh medium with 10 μM of Nocodazole (Sigma-Aldrich, Co). Samples were maintained at 37°C and 5% CO2 during acquisition. Images were collected with Orca Flash 4.0 (Hamamatsu) every 10 s during 30 min. Movie assembly was done using ImageJ software (http://rsb.info. nih.gov/ij/, NIH).
Time-lapse videomicroscopy
Time-lapse videomicroscopy of migrating neurons was performed as described previously (41). At E16.5, embryo brains electroporated 2 days earlier were dissected, embedded in 3% low-melt agarose (BioRad) diluted in HBSS (Hank’s Balanced Salt Solution, ThermoFisher Scientific) and sliced (300 μm) with a vibratome (Leica VT1000S, Leica Microsystems). Brain slices were cultured 16–24 h in semi-dry conditions (Millicell inserts, Merck Millipore) in a humidified incubator at 37°C in a 5% CO2 atmosphere in wells containing Neurobasal medium supplemented with 2% B27, 1% N2 and 1% penicillin/streptomycin (Gibco, Life Technologies). Slice cultures were placed in a humidified and thermoregulated chamber maintained at 37°C on the stage of an inverted confocal microscope. Time-lapse imaging was performed with a Leica SP8 X scanning confocal microscope equipped with a 25X objective. About 40–50 successive ‘z’ optical planes were acquired every 1.5 μm and every 30 min during 10 h. Sequences were analyzed using Image J and the ‘Manual Tracking’ Plugin.
Protein expression and purification
The wild-type human Kif2A protein construct containing the neck and the motor domain (Kif2A-NM; residues 153–553) was expressed and purified as described previously (Trofimova 2018, Nat. Comm). Kif2A p.His321Asp mutation (residues 153–553) was introduced by site-directed mutagenesis using QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) and primers 5′-gtcaccacccatagtatcagtttttccacttccag-3′ and 5′-ctggaagtggaaaaactgatactatgggtggtgac-3′ and was cloned into the pET28a vector to incorporate a C-terminal 6xHis tag. The plasmid was transformed into BL21 (DE3) Rosetta®(DE3) E. coli cells (Novagene) and recombinant protein expression was induced by addition of 0.5 mm IPTG to the LB culture media, in which cells were grown at 16°C overnight. Cells were lysed in buffer A (20 mm Potassium phosphate, pH 7.2, 300 mm NaCl, 1 mm MgCl2, 1 mm EGTA, 5 mm β-mercaptoethanol) supplemented with 0.2 mg/ml Lysozyme, 0.2 mm ATP and protease inhibitors (Roche) for 30 min on ice and then sonicated. The lysate was centrifuged and the supernatant fraction was loaded on pre-equilibrated Ni-NTA resin (Qiagen) for affinity-based protein purification. Unbounded proteins were washed away with buffer A supplemented with 20 mm imidazole. Kif2A H321D protein was eluted with an increasing linear gradient of buffer B (buffer A supplemented with 300 mm imidazole) and eluted fractions containing Kif2A H321D protein were pooled and dialyzed against HEPES buffer (20 mm HEPES, 1 mm MgCl2, 150 mm NaCl, 0.2 mm ATP, 1 mm DTT, pH 7.2). Proper folding of the Kif2A H321D mutant protein was confirmed by Circular Dichroism using a Chirascan CD spectrometer (Applied Photophysics Ltd). No significant changes in secondary structure were observed in comparison with wild-type Kif2A (Supplementary Material, Fig. S9C). The Kif2A H321D mutant protein solution was concentrated to 1 mg/ml using an Amicon® Ultra-4 filter (Millipore) and then flash-frozen in liquid N2.
Tubulin was isolated from bovine brain and purified by two cycles of polymerization–depolymerization in a high-molarity PIPES buffer as described by Castoldi (42). Purified tubulin was flash-frozen in liquid N2 and stored at −80°C until use.
MT depolymerization assay
MT preparation and sedimentation-based MT depolymerization assays are essentially the same as described previously (19). Briefly, 0.75 μM of Kif2A WT or Kif2A H321D were mixed with 20 μM taxol-stabilized MT in BRB80-based depolymerization buffer (80 mm PIPES pH 6.8, 1 mm MgCl2, 1 mm EGTA, 20 μM taxol, 75 mm KCl, 1 mm DTT, 0.02% Tween), supplemented with 1 mm ATP. Reactions were incubated at room temperature for 10 min. Free tubulin in solution was separated from remaining MT pellet by centrifugation at 120 000g for 10 min at 25°C. The supernatant fraction was retrieved from the sedimentation mixture and added to 1/2 volume of 2 × SDS loading buffer (Laemmli buffer). The remaining pellet was resuspended in equal volume of BRB80-based buffer and incubated on ice for 10 min, vortexed and then mixed with equal volume of 2 × SDS loading buffer. Equal portions of the supernatant and pellet samples were resolved on 10% SDS-PAGE. The gel was stained with Coomassie blue for comparison of tubulin quantities. ImageJ software was used for quantification of intensity of the tubulin bands.
Statistics
All experiments were done in at least three independent replicates and analyses were performed blinded to genotype or condition. All statistics were calculated with GraphPad Prism 6. Data are presented as mean ± SEM. The experimental n, the test used and the statistical significance are indicated in the figure legends. Time-lapse recordings were performed in a minimum of three electroporated embryos per group. All histological quantifications were performed on three brain sections per brain of at least three different animals per condition. For morphometric analysis of the mice, a minimum of three animals were studied and analyzed in a single-specific brain section. Experiments performed in HeLa tubulin GFP cells were done independently repeated at least three times. For EB3 comets, in each experiment, 10 cells were analyzed and in each cell 15–30 comets were quantified. For depolymerization, around 100 cells were analyzed per condition in three independent experiments. For behavioral analysis, 10 WT male mice and 8 mutant mice were used. The behavioral data were analyzed using unpaired Student’s t-test or repeated measures analysis of variance with one between factor (genotype) and one within factor (time). A one group t-test was also used to compare the object recognition index of each group to the chance level, and paired t-test was used to compare the duration of exploration between the novel and familiar objects for each group during the retention trial. Qualitative parameters (e.g. clinical observations) were analyzed using χ2 test. The level of significance was set at P < 0.05. Three mice of each genotype were used for EEG recordings and challenged with PTZ.
Authors’ Contributions
J.G.G. and E.L.I. wrote the manuscript. J.G. conceived and designed the experiments; performed experiments and analyzed the data of immunohistochemistry studies in mice, mouse model validation, mouse embryonic fibroblasts culture, cellular studies in human and mouse fibroblasts, qPCR studies and WB and revised the manuscript. E.I. performed experiments and analyzed the data of IUE, time-lapse, cellular studies in human fibroblasts. D.T. performed and analyzed in vitro experiments. H.M. performed and analyzed behavioral studies. G.R. performed and analyzed EEG studies and revised the manuscript. L.B. provided assistance with validation in ES cells and reviewed the manuscript. N.D. and V.S. provided technical assistance and prepared reagents. Jer. C. performed qPCR studies. P.V. provided assistance with WB experiments. N.B.-B. provided patient fibroblasts. B.Y. designed experiments related to neuroanatomical studies. M.-C.B participated in the development of the mouse model. M.-V.H provided assistance, contributed with suggestions and reviewed the manuscript. B.H.K and J.S.A. designed experiments related to in vitro studies, provided assistance, contributed with suggestions and reviewed the manuscript. Ja.C. coordinated and supervised the study.
Funding
EU Seventh Framework Programme FP7 under the projects GENCODYS (COLLABORATION PROJECT-2009-2.1.1-1/241995 to Ja.C.); DESIRE (grant agreement no. 602531 to Ja.C.); the French state funds through the ‘Agence Nationale de la Recherche’ under the projects ANR-15-CE16–0018-01 and the programme Investissements d’Avenir labeled (ANR-10-IDEX-0002-02, ANR-10-LABX-0030-INRT to B.Y. and Ja.C.).
Supplementary Material
Acknowledgements
B.H.K. acknowledges the funding support from Canadian Institutes of Health Research (CIHR Project Grant) and the Fonds de recherche du Québec—Santé (FRQS). The funders had no role in the study design, data collection and analysis, decision to publish or manuscript preparation. We are grateful to the patient and their parents who have accepted the establishment of cultures of fibroblasts. We thank the Imaging Center of IGBMC (ici.igbmc.fr), in particular Elvire Guiot and Erwan Grandgirard for their assistance in the microscope imaging experiments. We are grateful to Julien Couchet for providing us pSilencer-CAG-Venus-2 plasmids. We are grateful to the staff of the mouse facilities of the Institut Clinique de la souris in particular Chadia Toubari and Nadine Banquart-Ott (ICS) and the Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) for their kind help. We thank Christophe Mittelhauser for his implication in behavioral studies. We would like to thank Dr Juliette Godin for her careful reading of previous versions of the manuscript and constructive comments.
Conflict of Interest statement
The authors declare no competing financial interests.
References
- 1. Poirier K., Lebrun N., Broix L., Tian G., Saillour Y., Boscheron C., Parrini E., Valence S., Saint Pierre B., Oger M. et al. (2013) Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet, 45, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cavallin M., Bijlsma E.K., El Morjani A., Moutton S., Peeters E.A.J., Maillard C., Pedespan J.M., Guerrot A.-M., Drouin-Garaud V., Coubes C. et al. (2017) Recurrent KIF2A mutations are responsible for classic lissencephaly. Neurogenetics, 18, 73–79. doi: 10.1007/s10048-016-0499-8. [DOI] [PubMed] [Google Scholar]
- 3. Tian G., Cristancho A.G., Dubbs H.A., Liu G.T., Cowan N.J. and Goldberg E.M. (2016) A patient with lissencephaly, developmental delay, and infantile spasms, due to de novo heterozygous mutation of KIF2A. Mol. Genet. Genomic Med., 4, 599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desai A., Verma S., Mitchison T.J. and Walczak C.E. (1999) Kin I Kinesins are microtubule-destabilizing enzymes. Cell, 96, 69–78. [DOI] [PubMed] [Google Scholar]
- 5. Walczak C.E., Gayek S. and Ohi R. (2013) Microtubule-depolymerizing kinesins. Annu. Rev. Cell Dev. Biol., 29, 417–441. [DOI] [PubMed] [Google Scholar]
- 6. Broix L., Asselin L., Silva C.G., Ivanova E.L., Tilly P., Gilet J.G., Lebrun N., Jagline H., Muraca G., Saillour Y. et al. (2017) Ciliogenesis and cell cycle alterations contribute to KIF2A-related malformations of cortical development. Hum. Mol. Genet., 27, 224–238. [DOI] [PubMed] [Google Scholar]
- 7. Schnütgen F., Doerflinger N., Calléja C., Wendling O., Chambon P. and Ghyselinck N.B. (2003) A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol., 21, 562–565. [DOI] [PubMed] [Google Scholar]
- 8. Friedrich G. and Soriano P. (1991) Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev., 5, 1513–1523. [DOI] [PubMed] [Google Scholar]
- 9. Dubois N.C., Hofmann D., Kaloulis K., Bishop J.M. and Trumpp A. (2006) Nestin-Cre transgenic mouse line Nes-Cre 1 mediates highly efficient Cre/lox P mediated recombination in the nervous system, kidney, and somite-derived tissues. Genesis, 44, 355–360. [DOI] [PubMed] [Google Scholar]
- 10. Goebbels S., Bormuth I., Bode U., Hermanson O., Schwab M.H. and Nave K.-A. (2006) Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis, 44, 611–621. [DOI] [PubMed] [Google Scholar]
- 11. Collins S.C., Wagner C., Gagliardi L., Kretz P.F., Fischer M.-C., Kessler P., Kannan M. and Yalcin B. (2018) A method for parasagittal sectioning for neuroanatomical quantification of brain structures in the adult mouse. Curr. Protoc. Mouse Biol., 8, e48. [DOI] [PubMed] [Google Scholar]
- 12. Sukoff Rizzo S.J. and Crawley J.N. (2017) Behavioral Phenotyping assays for genetic mouse models of neurodevelopmental, neurodegenerative, and psychiatric disorders. Annu. Rev. Anim. Biosci., 5, 371–389. [DOI] [PubMed] [Google Scholar]
- 13. Loscher W. (2011) Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure, 20, 359–368. [DOI] [PubMed] [Google Scholar]
- 14. Kitazawa A., Kubo K., Hayashi K., Matsunaga Y., Ishii K. and Nakajima K. (2014) Hippocampal pyramidal neurons switch from a multipolar migration mode to a novel “climbing” migration mode during development. J. Neurosci., 34, 1115–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Docampo-Seara A., Santos-Durán G.N., Candal E. and Rodríguez Díaz M.Á. (2019) Expression of radial glial markers (GFAP, BLBP and GS) during telencephalic development in the catshark (Scyliorhinus canicula). Brain Struct. Funct., 224, 33–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sawicka A. and Seiser C. (2012) Histone H3 phosphorylation – a versatile chromatin modification for different occasions. Biochimie, 94, 2193–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maney T., Wagenbach M. and Wordeman L. (2001) Molecular dissection of the microtubule depolymerizing activity of mitotic centromere-associated Kinesin. J. Biol. Chem., 276, 34753–34758. [DOI] [PubMed] [Google Scholar]
- 18. Hertzer K.M., Ems-McClung S.C., Kline-Smith S.L., Lipkin T.G., Gilbert S.P. and Walczak C.E. (2006) Full-length dimeric MCAK is a more efficient microtubule depolymerase than minimal domain monomeric MCAK. Mol. Biol. Cell, 17, 700–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trofimova D., Paydar M., Zara A., Talje L., Kwok B.H. and Allingham J.S. (2018) Ternary complex of Kif2A-bound tandem tubulin heterodimers represents a kinesin-13-mediated microtubule depolymerization reaction intermediate. Nat. Commun., 9, 2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Verma V., Paul A., Amrapali Vishwanath A., Vaidya B. and Clement J.P. (2019) Understanding intellectual disability and autism spectrum disorders from common mouse models: synapses to behaviour. Open Biol., 9, 180265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stouffer M.A., Golden J.A., Francis F., Umrs I., Universités S. and Pierre U. (2015) Neurobiology of disease neuronal migration disorders : focus on the cytoskeleton and epilepsy. Neurobiol. Dis., 92, 18–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Belvindrah R., Nosten-Bertrand M. and Francis F. (2014) Neuronal migration and its disorders affecting the CA3 region. Front. Cell. Neurosci., 8, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li G. and Pleasure S.J. (2014) The development of hippocampal cellular assemblies. Wiley Interdiscip. Rev. Dev. Biol., 3, 165–177. [DOI] [PubMed] [Google Scholar]
- 24. Kuchukhidze G., Koppelstaetter F., Unterberger I., Dobesberger J., Walser G., Zamarian L., Haberlandt E., Maier H., Ortler M., Gotwald T. et al. (2010) Hippocampal abnormalities in malformations of cortical development: MRI study. Neurology, 74, 1575–1582. [DOI] [PubMed] [Google Scholar]
- 25. Ohshima T., Hirasawa M., Tabata H., Mutoh T., Adachi T., Suzuki H., Saruta K., Iwasato T., Itohara S., Hashimoto M. et al. (2007) Cdk5 is required for multipolar-to-bipolar transition during radial neuronal migration and proper dendrite development of pyramidal neurons in the cerebral cortex. Development, 134, 2273–2282. [DOI] [PubMed] [Google Scholar]
- 26. Falnikar A., Tole S., Liu M., Liu J.S. and Baas P.W. (2013) Polarity in migrating neurons is related to a mechanism analogous to cytokinesis. Curr. Biol., 23, 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ogawa T., Saijo S., Shimizu N., Jiang X. and Hirokawa N. (2017) Mechanism of catalytic microtubule depolymerization via KIF2-tubulin transitional conformation. Cell Rep., 20, 2626–2638. [DOI] [PubMed] [Google Scholar]
- 28. Homma N., Takei Y., Tanaka Y., Nakata T., Terada S., Kikkawa M., Noda Y. and Hirokawa N. (2003) Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell, 114, 229–239. [DOI] [PubMed] [Google Scholar]
- 29. Cooper J.A. (2014) Molecules and mechanisms that regulate multipolar migration in the intermediate zone. Front. Cell. Neurosci., 8, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang K., Lin C., Wang C., Shao Q., Gao W., Song B., Wang L., Song X., QU X. and Wei F. (2014) Silencing Kif2a induces apoptosis in squamous cell carcinoma of the oral tongue through inhibition of the PI3K/Akt signaling pathway. Mol. Med. Rep., 9, 273–278. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y., You X., Liu H., Xu M., Dang Q., Yang L., Huang J. and Shi W. (2017) High KIF2A expression predicts unfavorable prognosis in diffuse large B cell lymphoma. Ann. Hematol., 96, 1485–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lizarraga S.B., Margossian S.P., Harris M.H., Campagna D.R., Han A.P., Blevins S., Mudbhary R., Barker J.E., Walsh C.A. and Fleming M.D. (2010) Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development, 137, 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marthiens V., Rujano M.A., Pennetier C., Tessier S., Paul-Gilloteaux P. and Basto R. (2013) Centrosome amplification causes microcephaly. Nat. Cell Biol., 15, 731–740. [DOI] [PubMed] [Google Scholar]
- 34. Asenjo A.B., Chatterjee C., Tan D., DePaoli V., Rice W.J., Diaz-Avalos R., Silvestry M. and Sosa H. (2013) Structural model for tubulin recognition and deformation by kinesin-13 microtubule depolymerases. Cell Rep., 3, 759–768. [DOI] [PubMed] [Google Scholar]
- 35. Homma N., Zhou R., Naseer M.I., Chaudhary A.G., Al-Qahtani M.H. and Hirokawa N. (2018) KIF2A regulates the development of dentate granule cells and postnatal hippocampal wiring. elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Obeng E.A., Chappell R.J., Seiler M., Chen M.C., Campagna D.R., Schmidt P.J., Schneider R.K., Lord A.M., Wang L., Gambe R.G. et al. (2016) Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell, 30, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Capulli M., Costantini R., Sonntag S., Maurizi A., Paganini C., Monti L., Forlino A., Shmerling D., Teti A. and Rossi A. (2019) Testing the Cre-mediated genetic switch for the generation of conditional knock-in mice. PLoS One, 14, e0213660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tabata H. and Nakajima K. (2001) Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience, 103, 865–872. [DOI] [PubMed] [Google Scholar]
- 39. Chantrel-Groussard K., Geromel V., Puccio H., Koenig M., Munnich A., Rötig A. and Rustin P. (2001) Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet., 10, 2061–2067. [DOI] [PubMed] [Google Scholar]
- 40. Qiu L.Q., Lai W.S., Stumpo D.J. and Blackshear P.J. (2016) Mouse embryonic fibroblast cell culture and stimulation. Bio protoc. 6:e1859. doi: 10.21769/BioProtoc.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tielens S., Godin J.D. and Nguyen L. (2016) Real-time recordings of migrating cortical neurons from GFP and Cre Recombinase expressing mice. Curr. Protoc. Mouse Biol., 74, 3.29.1–3.29.23. [DOI] [PubMed] [Google Scholar]
- 42. Castoldi M. and Popov A.V. (2003) Purification of brain tubulin through two cycles of polymerization–depolymerization in a high-molarity buffer. Protein Expr. Purif., 32, 83–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.