

Abstract
Targeted therapy against the programmed cell death ligand-1 (PD-L1) blockade holds considerable promise for the treatment of different tumor types; however, little effect has been observed against gliomas thus far. Effective glioma therapy requires a delivery vehicle that can reach tumor cells in the central nervous system, with limited systemic side effect. In this study, we developed a cyclic peptide iRGD (CCRGDKGPDC)-conjugated solid lipid nanoparticle (SLN) to deliver small interfering RNAs (siRNAs) against both epidermal growth factor receptor (EGFR) and PD-L1 for combined targeted and immunotherapy against glioblastoma, the most aggressive type of brain tumors. Building on recent studies showing that radiation therapy alters tumors for enhanced nanotherapeutic delivery in tumor-associated macrophage-dependent fashion, we showed that low-dose radiation primes targeted SLN uptake into the brain tumor region, leading to enhanced downregulation of PD-L1 and EGFR. Bioluminescence imaging revealed that radiation therapy followed by systemic administration of targeted SLN leads to a significant decrease in glioblastoma growth and prolonged mouse survival. This study combines radiation therapy to prime the tumor for nanoparticle uptake along with the targeting effect of iRGD-conjugated nanoparticles to yield a straightforward but effective approach for combined EGFR inhibition and immunotherapy against glioblastomas, which can be extended to other aggressive tumor types.
Keywords: glioblastoma, targeted therapy, solid lipid nanoparticle, PD-L1, radiation, immunotherapy
Graphical Abstract
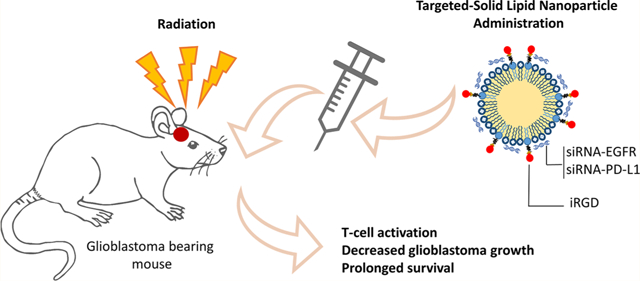
Glioblastoma (GBM) is the most common primary malignant brain tumor in adults with poor prognosis.1 Despite all treatment efforts including surgical resection combined with radiation and chemotherapy, median survival of patients with GBM is less than 15 months with a 5 year survival rate of 4–5%.1,2 Although many therapeutic approaches have been developed against GBM, the possibility of using these in the clinic has been limited by the lack of safe and efficacious delivery systems with ability to cross the blood–brain barrier (BBB) and to deliver the therapeutic to the tumor and/or microenvironment.3,4 A number of receptors known to modulate tumor cell proliferation, viability, and differentiation have been explored for targeted therapies against cancer in general and GBM in particular, but unfortunately, they could not achieve the desired success in the clinic.5 For instance, the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase overexpressed in several solid tumors and in up to 60% of GBMs,6 has been targeted with different strategies including small molecule inhibitors, monoclonal antibodies, ribonucleic acid (RNA)-based agents, and vaccines, however, with limited efficacy.7,8 Recent results have shown that one’s immune response has a considerable potential to promote immune-mediated tumor eradication and to improve long-term survival in several types of cancers including GBM.9,10 Immunotherapeutic strategies currently under investigation include immune checkpoint therapies, engineered T-cells, monoclonal antibodies, and cancer vaccines.9 The goal of checkpoint inhibition is to enhance anticancer immune activation by downregulating inhibitory pathways such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or programmed cell death ligand 1 (PD-L1).11–13
PD-L1 is a transmembrane protein that inhibits T-cell-mediated immune attack through binding to its receptor PD-1 on tumor-specific T-cells. PD-L1 has been reported to be overexpressed in several cancer types,12 and recent studies showed that blocking PD-1/PD-L1 pathway or decreasing their expression in tumor cells plays an important role in the adaptive immune response.9,14 Interestingly, some studies reported an association between EGFR activation and PD-L1 upregulation;15–17 thus, reducing the expression of both EGFR and PD-L1 may result in an increased therapeutic effect.
Delivery of small interfering RNA (siRNA) to block genes responsible for tumor development/proliferation is a promising strategy for cancer therapy. The use of therapeutic naked siRNA is not efficient for several reasons including their hydrophilic structure, high molecular weights, and anionic charges, preventing them from crossing cell membranes to reach the action sites.18–20 Various strategies have been explored to develop delivery systems able to transport nucleic acids to the brain including nanoparticles carrying targeting ligand on their surface to bypass the BBB and thus enhance uptake in gliomas.21,22 Over the past decade, a number of synthetic tumor-penetrating peptides have also been identified and shown to have high affinity to both angiogenic vessels and tumor cells.23,24 The cyclic peptide iRGD (CCRGDKGPDC), which exhibits high affinity to αv integrins present on the vascular endothelial cells followed by a proteolytic cleavage, converting the internal CendR motif, and binding to neuropilin-1 (NRP-1) to achieve efficient tumor cellular uptake, is a promising tumor-penetrating peptide.25–27
Solid lipid nanoparticles (SLNs), an alternative nonviral gene delivery system to liposomes or polymeric nanoparticles, have attracted increased attention due to their biocompatibility, biodegradability, stability, low cost, ease of large-scale production, and versatility with respect to charge and size.28 SLNs can be produced in nanosize, containing cationic moieties for siRNA incorporation, and can also be modified with a targeting ligand, allowing them to penetrate the brain29 and promoting retention at the target site.30,31 Recent studies have shown that short bursts of radiation therapy can prime tumors for enhanced accumulation and intratumoral distribution of nanotherapeutics in tumor-associated macrophage-dependent fashion,32,33 as well as activating neutrophil infiltration to mediate the transport of nanotherapeutics across the tumor vessel barrier.34 Thus, the use of radiation therapy to improve targeting efficiency of SLNs may offer an exciting improvement in GBM therapy.
Here, we developed an iRGD-conjugated SLN for delivery of siRNAs against EGFR and PD-L1 for combined targeted therapy and immunotherapy against GBM. We demonstrated that a short burst of radiation therapy primes GBMs for enhance uptake of this targeted SLN, leading to activation of immune response, inhibition of tumor growth, and improved mouse survival. Our findings suggest a radiation-induced nanoparticle-based combined targeted and immunotherapy against glioblastomas, which could also be extended to other aggressive tumor types.
RESULTS
Formation and Characterization of Nanoparticles.
We developed an SLN using the microemulsion dilution method and characterized it by pseudoternary phase diagram construction studies.35,36 For formation of the w/o microemulsion system, the hydrophilic lipophilic balance of the system was adjusted to >10 with the selected nontoxic surfactants.37 Lipid, surfactant, and cosurfactant mixtures were first heated above the lipid melting temperature (~65 °C). Stable and clear oil in water (o/w) microemulsion formation was determined by titration of ultrapure water into the oily mixture (Figure 1a). By diluting the obtained hot o/w microemulsion into cold water, SLN was formed (Figure 1b). We then functionalized SLN [f(SLN)] by incorporating DSPE-PEG(2000)-DBCO into the formulation (Figure 1b,c). To obtain targeted SLN, tumor-penetrating peptide iRGD was conjugated to f(SLN), generating f(SLN)-iRGD via copper-free click chemistry between DBCO in SLN and azide groups in iRGD. The final concentration for SLN, f(SLN), and f(SLN)-iRGD was 5 mg/mL, with respect to solid lipids. Finally, siRNAs were electrostatically bound to the outer surface of the formulation, generating f(SLN)-iRGD:siRNA (Figure 1c).
Figure 1.
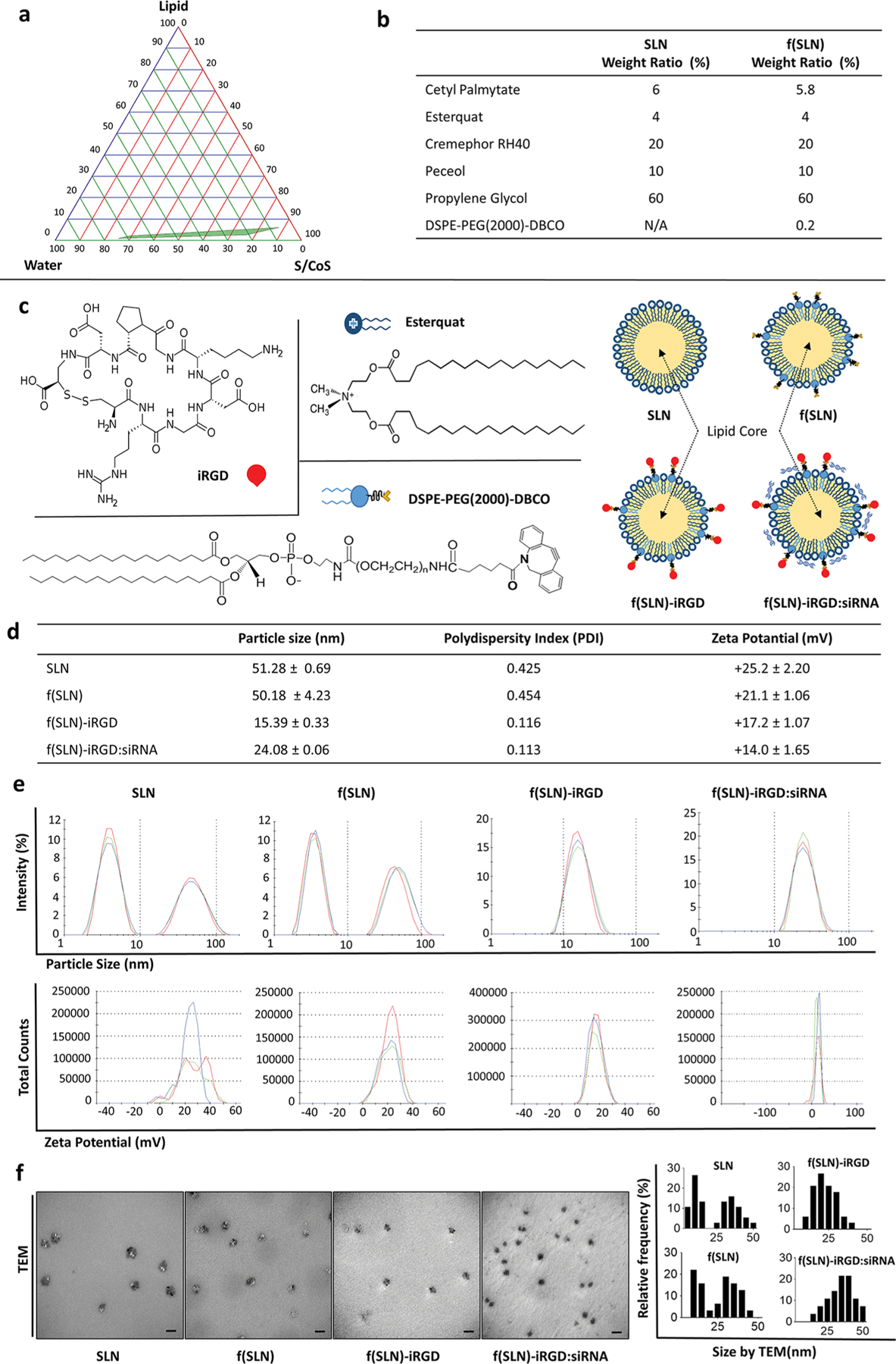
Construction and characterization of f(SLN)-iRGD:siRNA for targeted glioblastoma therapy. (a) Pseudoternary phase diagram for the microemulsion system formed with lipid (cetyl palmitate), surfactant/cosurfactant (S/CoS; cremephor RH 40+peceol/propylene glycol), and water (UPH2O); green area represents transparent o/w microemulsion formation region. (b) Weight ratio of the different components of SLN and f(SLN). (c) Chemical structure of DSPE-PEG(2000)-DBCO, Esterquat, and iRGD peptide with schematic overview of SLN, DSPE-PEG(2000)-DBCO containing functionalized SLN [f(SLN)], iRGD-targeted f(SLN) [f(SLN)-iRGD], and targeted f(SLN) complexed to siRNA [f(SLN)-iRGD:siRNA]. (d) Characterization of SLN, f(SLN), f(SLN)-iRGD, and f(SLN)-iRGD:siEGFR. (e) Size and ζ-potential distribution of SLN, f(SLN), f(SLN)-iRGD, and f(SLN)-iRGD:siEGFR; blue, green, and red represent different measurements for the same nanoparticle. (f) Transmission electron micrograph (left) and size distribution of each nanoparticle analyzed by ImageJ (right); scale bar, 100 nm.
In order to determine the particle size and ζ-potential of all nanoparticles, we performed dynamic light scattering (DLS) measurements. The mean average diameter of SLN was measured to be 51.3 nm, which is not significantly different than the f(SLN) having a size of 50.2 nm (Figure 1d,e). By incorporating iRGD to the surface of f(SLN), the size and size distribution of the nanoparticles were both decreased with an average diameter of 15.4 nm, and the size distribution of the nanoparticles became monodisperse (Figure 1e). This reduction in size may be due to the surface stabilizer effect of short chain peptides on nanoparticles.38 After binding of siRNA, the f(SLN)-iRGD:siRNA complex had a diameter of 24.1 nm. Transmission electron microscopy images also support the particle size measurement data obtained through DLS method and showed that f(SLN)-iRGD and f(SLN)-iRGD:siRNA have a particle size smaller than that of SLN and f(SLN) (Figure 1f). The ζ-potential measurements showed that all prepared nanoparticles have a positive surface charge (Figure 1d,e), probably due to the Esterquat, a frequently used cationic lipid in SLN studies.35 This positive charge allows binding of siRNAs to the outer surface of SLN by electrostatic interaction.39,40 Additionally, with the help of the polyethylene glycol (PEG) contained in the functional lipid DSPE-PEG(2000)-DBCO, the nanoparticles are more resistant to the neutralizing effects of serum proteins.41–43 To examine the nucleic acid binding and protection ability of obtained nanoparticles, we performed a gel retardation assay,44,45 with different ratios of SLN:siRNA and showed that SLN, f(SLN), and f(SLN):iRGD have the ability to complex with siRNA. Specifically, the migration of siRNA was completely retarded when the ratio of nanoparticles to siRNA reached 105:1 (w/w) for all formulations, equivalent to 1320 nmol of siRNA per gram of nanoparticle and 2.25 Esterquat molecule for every phosphate group on the siRNA (Figure 2a). Thus, this ratio was used to prepare SLN:siRNA, f(SLN):siRNA, and f(SLN)-iRGD:siRNA complexes in all subsequent experiments. Furthermore, the obtained f(SLN)-iRGD:siRNA complex effectively protected the siRNA against serum nuclease degradation up to 6 h, whereas the naked siRNA was completely degraded within 3 h as analyzed by agarose gel electrophoresis (Figure 2b). The smear represents external factors other than siRNA, such as f(SLN)-iRGD, serum, and sodium dodecyl sulfate (SDS) solution (Figure 2b).
Figure 2.

In vitro evaluation of developed SLNs. (a) Agarose gel photograph of complexes containing constant amounts of siRNA against EGFR and increasing amounts of SLN, f(SLN), or f(SLN)-iRGD with ratios of 35:1, 70:1, 105:1, 140:1, SLNs/siRNA, w/w [A2–5: SLN:siRNA complexes; A6–9: f(SLN):siRNA complexes; B2–5: f(SLN)-iRGD:siRNA complexes, respectively; A1, B1: naked siRNA control]. (b) Serum stability of siRNA incubated in mouse serum at 37 °C for different time points (0, 0.5, 1, 3, 6 h). Naked siRNA (top row) and released siRNA from f(SLN)-iRGD:siRNA complex (bottom row) are shown. f(SLN)-iRGD+serum+SDS were loaded into the first well of both rows as a control. (c) U87 and GL261 cell viability after incubation with different concentration of SLN, f(SLN), or f(SLN)-iRGD; data shown as mean ± SD (n = 4). (d–g) SLN-mediated knockdown of EGFR and/or PDL-1. Relative EGFR mRNA expression in human U87 cells treated with different concentrations of f(SLN)-iRGD:siEGFR (d), relative EGFR and PDL-1 mRNA expression in GL261 cells treated with different amounts of f(SLN)-iRGD complexed to either siEGFR or siPDL1 (e), siCTRL (f), or both siEGFR/PDL1 (g); ***P < 0.001 mock vs f(SLN)-iRGD:siEGFR/PDL1 for EGFR, ###P < 0.001 for PD-L1; *P < 0.05, **P < 0.01, ***P < 0.001; β-actin as housekeeping gene. Data are expressed as mean ± SD (n = 3).
Evaluation of Targeted Nanoparticle in Culture and in Vivo.
We first tested cytotoxicity of different concentration of SLN, f(SLN), and f(SLN)-iRGD formulations on cultured cells and observed that all three formulations had no significant cytotoxicity on U87 cells (up to 300 μg/mL) or GL261 cells (up to 400 μg/mL) (Figure 2c). These results were attributed to the biocompatibility of the developed nanoparticles. We next evaluated the ability of targeted SLN in delivering siRNAs to glioma cells. f(SLN)-iRGD carrying siRNA against human EGFR (siEGFR) was applied at increasing doses on human U87 glioma cells and resulted in dose-dependent knockdown, where the 40 nM dose led to 54 ± 2.6% decrease in EGFR expression (***P < 0.001) (Figure 2d). Similarly, GL261 mouse glioma cell line treated with f(SLN)-iRGD carrying either siRNA against mouse EGFR or PD-L1 showed 49 ± 4.5 and 47 ± 2.3% decrease at the 40 nM dose, respectively (***P < 0.001) (Figure 2e). When we complexed both siRNAs to the same f(SLN)-iRGD, similar knockdown efficiency was observed for both genes (at 40 nM, 54.7 ± 8.9% for EGFR and 58.6 ± 4.8% for PD-L1, ***P < 0.001), whereas the same SLN carrying a control scrambled siRNA (siCTRL) showed no significant effect (Figure 2f,g).
To confirm targeted efficiency, we conjugated nontargeted f(SLN) and targeted f(SLN)-iRGD to Cy5.5 and evaluated their uptake on glioma cells by flow cytometry. The uptake of f(SLN)-iRGD by αvβ3 integrin positive U87 or GL261 cells was significantly higher than that of f(SLN) (P = 0.0011, f(SLN) vs f(SLN)-iRGD on U87; P = 0.0046, f(SLN) vs f(SLN)-iRGD on GL261; Figure 3a). Moreover, when we preincubated the cells with iRGD peptide, the nanoparticle uptake was significantly blocked (as compared to scrambled iRGD control), suggesting that the observed uptake of f(SLN)-iRGD is iRGD-mediated (Figure 3a; **P < 0.01). To corroborate these results with another assay, we repeated the same experiment using f(SLN)-iRGD:siEGFR and evaluated knockdown efficiency of EGFR in U87 and GL261 cells using quantitative reverse transcription polymerase chain reaction (qRT-PCR) and observed that preincubation with iRGD peptide reversed EGFR knockdown (Figure 3b). These results suggest that our method effectively conjugates the iRGD peptide onto the surface of SLN and that the resulting f(SLN)-iRGD exhibits high affinity/specificity to cells expressing integrin αvβ3 and NRP-1 receptor.
Figure 3.

f(SLN)-iRGD specific targeting of glioblastoma cells. (a) Human U87 or mouse GL261 glioma cells were preincubated (or not) with free iRGD or scriRGD (20 μM for competitive binding) for 30 min and treated with 50 μg/mL of either f(SLN)-Cy5.5, f(SLN)-iRGD-Cy5.5, or f(SLN)-scriRGD-Cy5.5. Three hours post-treatment, cells were washed and analyzed by flow cytometry. Representative flow cytometry charts (left) and relative percentage of Cy5.5-positive cells as compared to untreated mock group (right) are shown, **P < 0.01 (n = 3). (b) Similar experimental setup to (a) but using siEGFR-complexed SLN. Twenty-four hours post-treatment, RNA was extracted and analyzed by qRT-PCR for EGFR mRNA (β-actin as housekeeping gene) (*P < 0.05, **P < 0.01, ***P < 0.001). (c) Mice bearing GL261 tumors were retro-orbital injected with f(SLN)-iRGD:siEGFR/PDL1 or PBS control on days 8, 9, and 11 post-tumor implantation. Twenty-four hours after the last injection, brain tumors were removed, RNA was extracted and analyzed for EGFR and PD-L1 mRNA levels by qRT-PCR. Data expressed as mean ± SD (*P < 0.05, n = 5).
Finally, we assessed the ability of f(SLN)-iRGD carrying both siEGFR and siPDL1 (f(SLN)-iRGD:siEGFR/PDL1) in penetrating the brain and targeting and delivering its content to GBMs in vivo. Following three systemic (retro-orbital) injections of 75 μg (in 100 μL of PBS) of f(SLN)-iRGD:siEGFR/PDL1 (on days 8, 9, and 11 post-tumor implantation), qRT-PCR on tumor tissue lysate revealed a significant decrease in both EGFR and PD-L1 mRNA levels as compared to untreated control mice (*P < 0.05) (Figure 3c).
Radiation Therapy Primes Glioblastoma for f(SLN)-iRGD Uptake.
Recent studies have shown that radiation therapy alters the tumor for enhanced nanotherapeutic delivery in tumor-associated macrophage-dependent fashion.32,33 We therefore sought to evaluate this phenomenon for targeted delivery of f(SLN)-iRGD to glioblastomas. Mice bearing GL261 tumors expressing firefly luciferase (Fluc) were irradiated with 5 Gy and 3 days later, retro-orbitally injected with 75 μg (in 100 μL of PBS) Cy5.5-labeled f(SLN)-iRGD. We used this time frame because the nanoparticle uptake by tumors was shown to be maximized at 72 h after irradiation.33 Twenty-four hours later, tumor volume was monitored by bioluminescence imaging, and brains were dissected and imaged for Cy5.5. Interestingly, mice that received radiation had a significantly higher Cy5.5 signal in their brain compared to nonirradiated mice (*P < 0.05) (Figure 4a), indicating that radiation therapy primes glioblastomas for enhanced SLN delivery.
Figure 4.
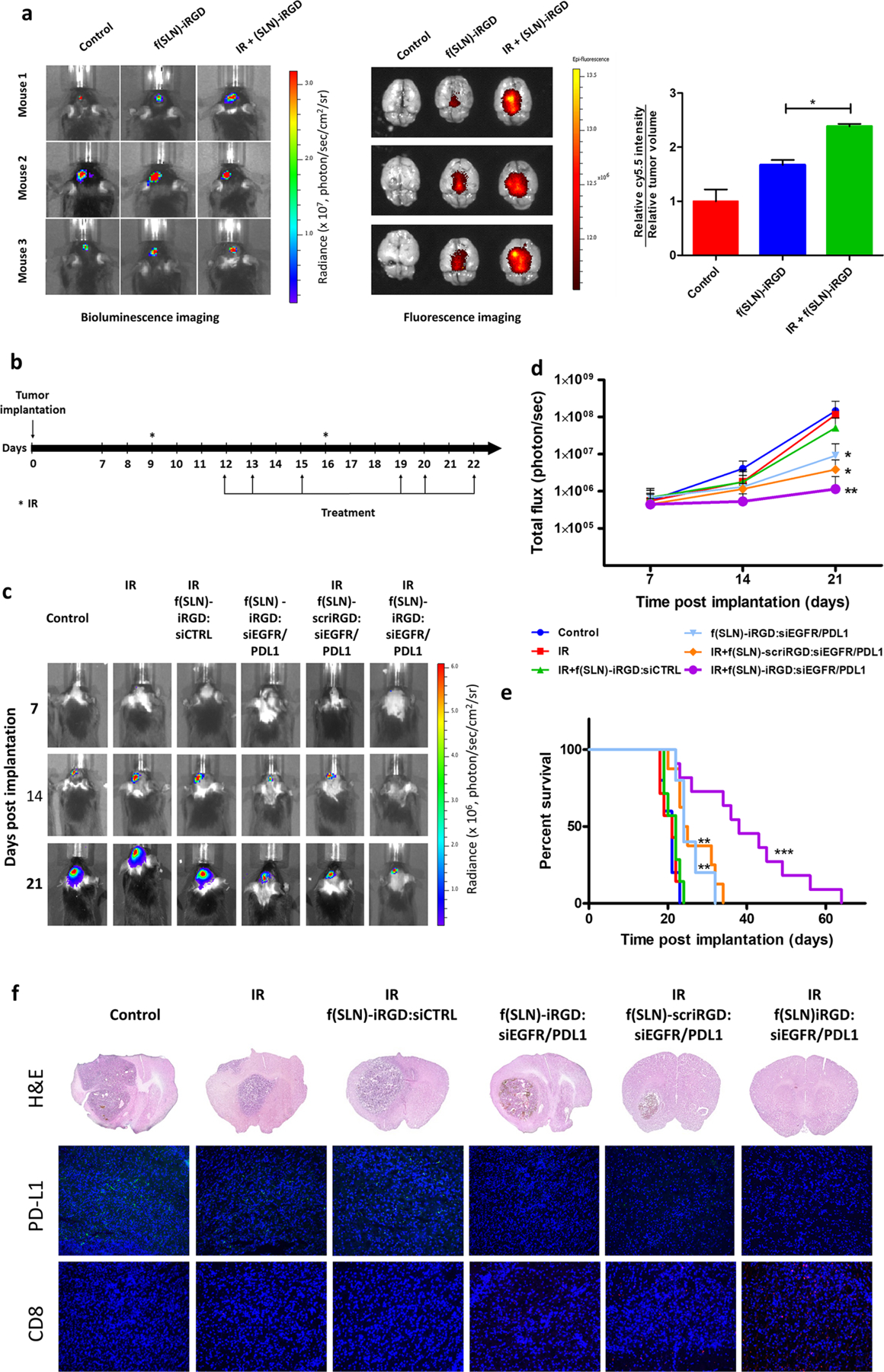
Radiation primes glioblastoma for SLN targeted delivery. (a) Mice bearing GL261-Fluc tumors were irradiated (or not as control) and 3 days later received retro-orbital administration of f(SLN)-iRGD:Cy5.5 or PBS control. Twenty-four hours postinjection, tumor volume was first evaluated by Fluc imaging (left), and brains were removed and imaged ex vivo for Cy5.5 (middle). The mean fluorescence intensity was calculated and normalized to tumor volume (right; n = 3, *P < 0.05). (b–f) Mice bearing GL261-Fluc tumors were irradiated (or not as a control) and retro-orbitally injected with either f(SLN)-iRGD:siCTRL, f(SLN)-iRGD:siEGFR/PDL1, or f(SLN)-scriRGD:siEGFR/PDL1 according to the scheme in (b). Tumor growth was monitored weekly by Fluc imaging, and survival was recorded. Images from a representative mouse from each group is shown over time (c). Quantification of tumor-associated Fluc radiance intensity with data presented as mean ± SD; *P < 0.05 control vs f(SLN)-iRGD:siRNA and control vs IR+f(SLN)-scriRGD:siRNA; **P < 0.01 control vs IR+f(SLN)-iRGD:siRNA by ANOVA (d). Kaplan–Meier survival curves are shown (n = 5–12); **P < 0.01 control vs f(SLN)-iRGD:siRNA; **P < 0.01 control vs IR+f(SLN)-scriRGD:siRNA; ***P < 0.001 control vs IR+f(SLN)-iRGD:siRNA; **P < 0.01 IR+f(SLN)-scriRGD:siRNA vs IR+f(SLN)-iRGD:siRNA; by Mantel–Cox (log-rank) test (e). H&E staining and immunohistological analysis using anti-PD-L1 and anti-CD8 antibodies on brain sections of a representative mouse from each group (DAPI, blue; PD-L1, green; and CD8, red) (f).
Radiation-Induced Antitumoral Activity of f(SLN)-iRGD:siEGFR/siPDL1 in Orthotopic Glioblastoma Xenograft Model.
Finally, we evaluated the ability for f(SLN)-iRGD to deliver siRNAs for combined targeted EGFR and anti-PD-L1 therapies to glioblastomas, primed with radiation. C57BL/6 mice bearing GL261 xenografts expressing Fluc were irradiated with 5 Gy (or not irradiated as a control) and 3 days later were retro-orbitally injected with 75 μg (in 100 μL of PBS) of either f(SLN)-iRGD:siCTRL, a targeted nanoparticle carrying control scrambled siRNA, f(SLN)-iRGD:siEGFR/ PDL1, a targeted nanoparticle carrying siRNAs against both EGFR and PD-L1, or f(SLN)-scriRGD:siEGFR/PDL1, a nontargeted nanoparticle modified with scrambled iRGD and carrying both siRNAs. Radiation therapy and nanoparticle injection were repeated according to the scheme in Figure 4b, and tumor growth was monitored by Fluc bioluminescence imaging over time (Figure 4c,d). Radiation alone or together with f(SLN)-iRGD:siCTRL did not have a significant effect on tumor growth or mouse survival as compared to control group (median survival was 21, 21, and 22 respectively, Figure 4c–e). On the other hand, f(SLN)-iRGD:siEGFR/PDL1 without radiation had a moderate effect on tumor growth (on day 21, total flux was 9.1 ± 0.9 × 106 versus 14.3 ± 0.8 × 107 for control, *P < 0.05, Figure 4c,d) and mouse survival (median survival 24 days versus 21 days for control, **P = 0.0072, Figure 4e). Similarly, radiation plus nontargeted f(SLN)-scriRGD:siEGFR/PDL1 also has a moderate effect on tumor growth (at day 21, total flux was 3.8 ± 0.3 × 106, *P < 0.05 versus control) and mouse median survival (24.5 days, **P = 0.0070 versus control). Importantly, mice that received radiation therapy and injected with f(SLN)-iRGD:siEGFR/PDL1 had the most therapeutic benefit with decreased tumor growth (at day 21, total flux was 1.1 ± 0.1 × 106, **P < 0.01 versus control) and increased mouse survival (median survival 38 days, ***P = 0.0001 versus control, **P = 0.0040 versus radiation + f(SLN)-scriRGD:siEGFR/PDL1 (Figure 4d,e)). These results were confirmed by ex vivo histological examination with hematoxylin and eosin (H&E) staining (Figure 4f). In addition, immunohistochemical staining of brain slices demonstrated that radiation + f(SLN)-iRGD:-siEGFR/PDL1 decreased expression of PD-L1 while increasing recruitment of CD8+ T-cells, in line with tumor growth and survival analysis above (Figure 4f). These data collectively suggest that radiation therapy indeed primes glioblastomas for SLN-based siRNA delivery for targeted EGFR and anti-PD-L therapy and leads to slower tumor growth and enhanced mouse survival.
DISCUSSION
The transport of therapeutics across the BBB represents one of the main challenges for an effective treatment of CNS tumors. In this study, using the microemulsion dilution technique, we developed an iRGD-conjugated targeted SLNs to provide a strategy to deliver therapeutics to GBMs.29,46 SLNs are attractive candidates for brain delivery due to their low cytotoxicity, lipophilic properties, and their simple modification with specific ligands for targeted delivery.35 The obtained targeted nanoparticles are smaller than 20 nm, contain PEG, and have a positive ζ-potential, which have shown great promise for gene delivery applications through the BBB.47,48 Due to their positive ζ-potential, we complexed these SLNs with two siRNAs against EGFR and PD-L1 at their outer surface via electrostatic interactions. EGFR is a well-known receptor responsible for aggressive tumor growth and metastases in many types of cancers, including glioblastomas, and is an attractive candidate for targeted therapy.7,8,49 On the other hand, PD-L1 is a ligand that activates the programmed cell death protein 1 (PD-1) and able to inhibit the proliferation and activation of T-cells, eventually leading to immune suppression.11,12,50 PD-L1/PD-1 blockade (such as durvalumab, atezolizumab, avelumab, novolumab) showed a great promise in the clinic and became the therapy of choice for many different tumor types in a fast manner, either alone or in combination with other therapies.51–54 Interestingly, some studies reported an association between EGFR activation and PD-L1 upregulation;15–17 thus, reducing the expression of both EGFR and PD-L1 may result in an increased therapeutic effect. Unfortunately, both targeted therapy and PD-L1 blockade showed limited efficacy against brain tumors so far.49,55 In that context, the developed f(SLN)-iRGD:siEGFR/PDL1 has several advantages for targeted and immunotherapy against glioblastomas. This nanoparticle delivers small siRNAs against EGFR and PDL-1, overcoming size limitations of these therapies. Further, our SLN provides an increased stability of these siRNAs in serum with dose-dependent gene silencing effect in glioma cells. Further, the chosen iRGD targeting peptide has a circular nature and therefore could inhibit platelets aggregation more effectively, compared to its linear counterpart, due to the inducing effect of disulfide bond conformational stability.56
Tumor-penetrating peptides have been used for several years to deliver diagnostics and therapeutics into the extravascular tumor parenchyma. Different delivery systems such as viral vectors, exosomes, or polymeric nanoparticles have used the iRGD peptide for targeted delivery to tumors.57–59 Our study supports glioblastoma targeting by iRGD through the αvβ3 integrin and then the NRP-1 receptor binding ability. Following receptor-mediated endocytosis, the siRNA-loaded nanoparticles likely encounter sequential pH drop in the endosome and lysosome by the acidic character of the endosomal media.60 Protonation of SLNs at endosomal pH might facilitate their interaction with negatively charged endosomal lipid membrane and cause the release of siRNA into the cytoplasm. In addition, cationic lipids carrying amino groups may fuse with the endosomal membrane and disturb the bilayer organism, contributing to endosomal destabilization and thus release of siRNA into the cytoplasm.61,62 Besides increasing the targeting ability and cellular uptake through the iRGD, these solid lipid nanoparticles have several advantages including lipophilicity and positive charge which allow them to pass through the BBB. Thus, the developed targeted nanoparticle is a promising alternative for tumor imaging and therapeutic delivery.
Radiation therapy has been used against cancer for decades; however, the use of radiation as a targeting enhancer for nanoparticle delivery is a more recent phenomenon. Recent studies have shown that radiation therapy improves nanoparticle delivery by changing endothelial architecture, permeability, and/or decreased tumor interstitial fluid pressure32,63 or affecting tumor-associated macrophages role in peripheral tumors33 and tumor-specific vascular disruption.64 Furthermore, it has been suggested that radiation activates neutrophil infiltration that contributes to the delivery of nanotherapeutics to the tumoral tissue.34 Higher radiation doses (≥15 Gy) have been linked to fibrosis, infertility, and bowel damage. On the other hand, the lower 5 Gy dose was found to be effective in enhancing nanoparticle delivery.33 Although a high dose of radiation has been shown to disrupt the BBB,65 our imaging and functional studies revealed that 5 Gy radiation therapy enhanced transportation of SLN to glioblastomas and led to an increase in therapeutic effect and mouse survival. Targeted SLN through iRGD showed the most therapeutic effect in preirradiated mice (as compared to nontargeted SLN, or targeted SLN with no radiation), despite previous studies claiming that active targeting does not show drastic improvement,66–68 and proving further that the observed effect is not due to potential BBB disruption by radiation. Hence, combining nanoparticle-based targeted delivery systems with radiation may lead to promising therapy in a clinical setting. In this study, we have used GL261 mouse glioma cells, a well-established model to evaluate immunotherapy against gliomas; however, it would be important to validate these findings in other immune-competent models and perform full immune cell analysis in vivo before clinical translation.
CONCLUSION
In summary, the targeted nanoparticle-based gene delivery system that we developed maintains a protective structure for siRNAs against the harsh biological environment and provides an opportunity to directly target glioblastomas by passing the BBB. Moreover, radiation therapy primed glioblastomas for targeted delivery of these nanoparticles, yielding activation of immune response, a slower tumor growth, and prolonged mouse survival. This delivery system is not limited to glioblastoma but can be extended to other tumors, and the siRNAs can be diverse to target different molecular pathways. The radiation-induced targeted nanoparticle-based gene delivery strategy highlights an effective treatment approach for aggressive tumor types such as glioblastomas.
EXPERIMENTAL SECTION
Materials.
Cetyl palmitate and Cremephor RH40 were purchased from BASF (Ludwigshafen, Germany). Esterquat was obtained from Gerbu Biotechnik (Gaiberg, Germany). Peceol was kindly provided by Gattefosse (Saint Priest, France). Propylene glycol was obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). DSPE-PEG(2000)-DBCO was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Custom-designed iRGD and scrambled iRGD (scriRGD) peptides with an azide group were synthesized by China Peptides (Shanghai, China). For the preparation of fluorescent-labeled nanoparticles, sulfo-cyanine 5.5 azide was purchased from Lumiprobe Co (Cockeysville, MD, USA).
All siRNAs were provided from Invitrogen (Carlsbad, CA, USA). siRNA sequences were as follows for sense and antisense strands respectively: against human EGFR: 5′-GGAGCUGCCCAUGAGAAAUtt-3′ and 5′-AUUUCUCAUGGGCAGCUCCtt-3′; mouse EGFR: 5′-GGAAAAGAAAGUCUGCCAAtt-3′ and 5′-UUGGCAGACUUUCUUUUCCtt-3′; mouse PD-L1:5′-GCGAAUCACGCUGAAAGUCtt-3′ and 5′-GACUUUCAGCGUGAUUCGCtt-3′; scrambled sequence: 5′-ACCAUACGGAAGCUUGGtt-3′ and 5′-CCAAGCUUCCUCGUAUGGUtt-3′.
Cell Culture.
Human glioblastoma cell line (U87) was obtained from the American Type Culture Collection (ATCC). The mouse glioblastoma cell line (GL261) was kindly provided by Dr. Xandra Breakefield, Massachusetts General Hospital, Boston, MA. Both of these cell lines express integrin αvβ3 and NRP-1.58,69–71 Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and 1% penicillin–streptomycin. All cell lines were tested to be mycoplasma-free and routinely maintained in a humidified atmosphere at 37 °C and 5% CO2. For bioluminescence imaging studies, cells were transduced with a lentivirus vector carrying an expression cassette for firefly luciferase (Fluc) as previously described.72,73
Preparation of the Solid Lipid Nanoparticles.
SLNs were prepared using a modified protocol of the melt-emulsification technique.35,37,46,74 First, an oil in water (o/w) microemulsion system was formed using cetyl palmitate as internal oil phase, Cremephor RH 40, Peceol as surfactants (S), and propylene glycol as cosurfactant (CoS). To impart cationic character to SLNs, Esterquat was incorporated into the oil phase of the microemulsion.35 These components were weighed and heated above the lipid melting temperature (~65 °C). Hot ultrapure water was then slowly added onto the mixture with approximately 2% of total weight increment each time. Transparent regions belonging to o/w microemulsion on the phase diagrams were determined. Following construction of the phase diagram, a microemulsion consisting of 5.5% oil, 49.5% surfactant/cosurfactant, and 45% water was found to yield a transparent formulation and was selected for further studies. Then, the obtained hot o/w microemulsion was dispersed in cold ultrapure distilled water (0–4 °C) at a ratio of 1:10 (v/v) under magnetic stirring at 1000 rpm. SLNs were formed when hot microemulsion droplets were met with cold water. The final nanoparticle concentration of all SLNs was calculated to be 5 mg/mL, with respect to solid lipids.
Functionalization of SLNs and Conjugation of iRGD to SLNs.
To obtain functional SLNs [f(SLN)], a part of cetyl palmitate was replaced with DSPE-PEG(2000)-DBCO (dibenzylcyclootyne) at a weight ratio of 29:1 [CP:DSPE-PEG(2000)-DBCO, w/w] at the SLN preparation step. The DBCO-conjugated f(SLN) was then ready for conjugation to azide-containing molecules via copper-free click chemistry as we and others have previously described.58,75,76 Briefly, iRGD or scrambled iRGD peptide (scr-iRGD) were commercially synthesized with an azide group at the lysine residue and then conjugated to the reactive DBCO groups on f(SLN) using 1:1 molar ratio of iRGD/scriRGD:f(SLN) and incubating at a rotating mixer for 2 h at room temperature. The unconjugated iRGD/scriRGD was then removed by four washing steps on 100 kDa ultrafiltration tubes (Millipore, USA). We have previously shown that this method yields a peptide binding efficiency of around 87.2%.58
Characterization of the Developed Systems.
The particle size, polydispersity index, and surface charge of the prepared nanoparticles [SLN, f(SLN), f(SLN)-iRGD, and f(SLN)-iRGD:siEGFR] were determined using dynamic light scattering (NanoZS, Malvern). For morphology characterization, a JEM-2100 transmission electron microscope (JEOL, Tokyo, Japan) was employed.
Preparation of SLN:siRNA Complexes.
To determine the optimum complexation protocol, different weight ratios of nanoparticles to siRNA were used (w/w, 0:1, 35:1, 70:1, 105:1, 140:1) and allowed to incubate on a benchtop shaker for 30 min at room temperature to complete the binding of siRNA onto the nanoparticles via electrostatic interaction. Complexation efficiency of siRNAs was analyzed by a gel retardation assay as previously described using 2% gel.40,77 The SLN:siRNA complexes were freshly prepared before each use in all studies.
Serum Stability Assay.
Serum was isolated from fresh blood obtained from 8 week old female C57BL/6 mice without anticoagulants. To precipitate the coagulation factors, fibrinogen and blood cells, whole blood samples were allowed to coagulate overnight at 4 °C. Subsequently, the tubes were centrifuged for 10 min at 2700g, and the supernatant consisting of serum was carefully collected and used freshly. f(SLN)-iRGD:siRNA (105:1, w/w) complexes were prepared, and serum was added at a final concentration of 50%.41 As a control, naked siRNA was treated with the same concentration of serum. After the predetermined incubation period (0, 0.5, 1, 3, 6 h) at 37 °C, SDS solution was added to each tube (2% final concentration) and incubated at room temperature for 5 min to terminate the enzymatic digestion and to release siRNA from the complexes. siRNA degradation was observed by agarose gel electrophoresis as described above. As a control, the same amount of f(SLN)-iRGD, serum, and SDS solution mixture was loaded on the same gel.
Cytotoxicity Analysis.
The in vitro cytotoxicity of developed nanoparticles was investigated on U87 and GL261 cell lines. Cells were plated in 96-well plates at a density of 5 × 103 cells per well in 100 μL and treated with SLN, f(SLN), or f(SLN)-iRGD at different concentrations (100, 200, 300, 400, 500 μg/mL, with respect to solid lipids) for 24 h. The proportion of viable cells was evaluated by Alamar Blue cell viability assay (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Cell viability was calculated by normalizing the fluorescence of media from treated cells to untreated cells.78
Evaluation of SLNs for siRNA Delivery in Vitro.
Human U87 cells were seeded in 24-well plates at 1 × 104 cells/well and treated with different amounts of f(SLN)-iRGD:siEGFR targeting the human gene (10, 20, 40, 80 nM, with respect to siRNA). Similarly, mouse GL261 cells were plated and treated with either f(SLN)-iRGD:siEGFR, f(SLN)-iRGD:siPDL1, or f(SLN)-iRGD:siEGFR/PDL1 targeting the mouse genes. Scrambled siRNA was used as a negative control (siCTRL). Twenty-four hours later, cells were washed with PBS and incubated in culture medium for another 24 h. Cells were then harvested, and RNA was isolated and analyzed by qRT-PCR using primer pairs specific to EGFR, PD-L1, and β-actin as a housekeeping gene. Results were analyzed with the ΔΔCT method with untreated cells as a control. Each experiment was repeated in triplicate and at least three times.
Evaluation of Nanoparticle Targeting Ability.
To monitor nanoparticles uptake by cells, we conjugated cyanin 5.5 (Cy5.5; synthesized with an azide group) to the DBCO strand of nanoparticle by click chemistry. The unconjugated Cy5.5 was removed by Amicon ultra 0.5 mL centrifugal filters (Ultracell 100 K, cellulose 100000).58 To examine binding of nanoparticles in vitro, 50 μg/mL (with respect to solid lipids) of Cy5.5-labeled f(SLN), f(SLN)-iRGD, or f(SLN)-scriRGD was incubated with integrin αvβ3 and NRP-1-positive U87 and GL261 cells at 37 °C for 3 h.58,69–71 For blocking experiments, cells were preincubated with iRGD or scriRGD peptide at 20 μM for 30 min prior to nanoparticle addition. Subsequently, the cells were harvested by trypsinization, washed three times, and resuspended in PBS. Untreated cells were included as a mock control. The fluorescence intensity of 20000 cells from each sample was then measured by flow cytometry (Accuri C6 cytometer, BD Biosciences, San Jose, CA, USA) and analyzed using the FlowJo software. The percentage of Cy5.5-positive cells was determined by comparison to the mock group. To confirm iRGD-dependent targeting ability of these nanoparticles, the same experiment was repeated using f(SLN)-iRGD:siEGFR (40 nM with respect to siRNA) on U87 and GL261 cells, and EGFR knockdown efficiency was evaluated by qRT-PCR, as described above.
In Vivo GBM Model.
All animal studies were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care following guidelines set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Orthotopic glioblastoma xenograft model was established with GL261 cell line in 8–10 week old C57BL/6 female mice. GL261 cell line was selected as it is a well-accepted model to study glioblastoma immunotherapy,79 does not require a deficient immune system, and may mimic closely the growth and immune response of human GBM.80 Mice were anesthetized with 3% isoflurane under oxygen and stereotactically implanted with 50000–250000 GL261 cells expressing Fluc in 2 μL of PBS using a 30 gauge Hamilton syringe and the following coordinates: 2.5 mm lateral, 0.5 mm anterior to bregma, and 2.5 mm depth from the skull surface. Bioluminescence imaging was performed using the Xenogen IVIS 200 imaging system (PerkinElmer, Waltham, MA) as we previously described.72 Briefly, mice were injected intraperitoneally with 150 μg per gram body weight of d-luciferin and transferred into the imaging chamber. Imaging was acquired 10 min post-luciferin injection, and the signal intensity was quantitated using the Living Image software 3.0 from Xenogen Imaging Technologies (PerkinElmer, Waltham, MA).81
For evaluation of gene silencing efficiency in vivo, 75 μg (with respect to solid lipids) of f(SLN)-iRGD-siEGFR/PDL1 in 100 μL or the same volume of PBS vehicle control was injected retro-orbitally in mice bearing GL261-Fluc tumors, on days 8, 9, and 11 post-tumor implantation (n = 5). Twenty-four hours after the last injection, mice were sacrificed, the tumor was harvested, and total RNA was isolated and analyzed by qRT-qPCR using murine-specific β-actin, EGFR, and PD-L1 primers.
Radiation Therapy Effect on SLN GBM Uptake.
To investigate the effect of radiation on nanoparticle uptake by GBM, 1 week postimplantation of 25 × 104 GL261-Fluc cells in C57BL/6 mice, each group (n = 4) of mice received intravenous (through retro-orbital route) injection of either PBS, f(SLN)-iRGD:Cy5.5 (75 μg in 100 μL PBS), or ionizing radiation (IR; 5 Gy) followed by the same amounts of f(SLN)-iRGD:Cy5.5 3 days later. Twenty-four hours later, mice were sacrificed by cervical dislocation under anesthesia, and brains were dissected out immediately. Fluorescence signals of Cy5.5 in the dissected brains were imaged using the Xenogen IVIS 200 imaging system (PerkinElmer) and a 1 min exposure. The fluorescence intensity was expressed as the mean radiant efficiency [p/s/cm2/sr]/[mW/cm2] and normalized to tumor volume obtained from bioluminescence imaging. PBS injected mice were used as a negative control.
Evaluation of f(SLN)-iRGD:siEGFR/PDL1 for GBM Therapy.
GL261-Fluc cells (5 × 104) were stereotactically injected in the striatum of 8–10 weeks old C57BL/6 mice. Once tumors were formed, as analyzed by Fluc imaging, mice were randomized into six groups and treated with either (1) PBS (n = 6), (2) IR (n = 8), (3) IR + f(SLN)-iRGD:siCTRL (n = 8), (4) f(SLN)-iRGD:siEGFR/PDL1 (n = 6), (5) IR + f(SLN)-scriRGD:siEGFR/PDL1 (n = 9), (6) IR + f(SLN)-iRGD:siEGFR/PDL1 (n = 12). IR (5 Gy) was performed at days 9 and 16, and nanoparticles (75 μg of all formulations in a total volume of 100 μL of PBS through retro-orbital route) were administered on days 12, 13, 15, 19, 20, and 22 post-tumor implantation (for a total of six injections). Bioluminescence imaging was performed once a week to follow tumor growth, and survival was recorded.
Immuno/histological Analysis.
After the last administration of SLN, one mouse from each group was sacrificed and perfused with cold PBS followed by 4% paraformaldehyde (PFA). Brains were carefully isolated, fixed for an additional 24 h with 4% PFA, embedded in OCT compound (Sakura, Japan), and sectioned into 14 μm sections using a microtome. Brain sections were stained with H&E to visualize the tumor. Sections were also stained with 6-diamidino-2-phenylindole (DAPI) for nuclei and anti-PD-L1 (1:100; Clone 10F.9G2, Millipore Sigma, USA) or anti-CD8 (1:100; CT-CD8a, Thermo Fisher Scientific, USA) antibodies, followed by corresponding secondary antibody.
Statistical Analysis.
GraphPad Prism v6.01 software was used for statistical analysis of all data. P < 0.05 was considered to be statistically significant. For analysis between multiple groups, a one-way ANOVA was performed followed by Sidak’s multiple comparison test to compare differences between two groups. An unpaired two-tailed student t test was used for the comparison of two samples. Survival was analyzed using Kaplan–Meier curves and log-rank (Mantel–Cox) tests.
ACKNOWLEDGMENTS
This work was supported by grant from NIH/NCI P01CA069246 (B.A.T., E.A.C., and R.W.) and NIH/NINDS P30NS04776 (B.A.T.). G.E.A. was supported by TUBITAK (The Scientific and Technological Research Council of Turkey) 2214/A scholarship. The authors would like to thank Michael F. Cuccarese from the Center for Systems Biology at the Massachusetts General Hospital for his help with DLS measurement experiments, the MGH Neuroscience Image Analysis Core (for confocal microscopy), and the MGH Vector Core (for producing the viral vector), and Ellen Sapp at the MGH EM core (supported by NIH/NINDS P30NS04776) as well as 1S10RR025504 Shared Instrumentation Grant for the IVIS imaging system.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Stupp R; Mason WP; van den Bent MJ; Weller M; Fisher B; Taphoorn MJB; Belanger K; Brandes AA; Marosi C; Bogdahn U; Curschmann J; Janzer RC; Ludwin SK; Gorlia T; Allgeier A; Lacombe D; Cairncross JG; Eisenhauer E; Mirimanoff RO Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med 2005, 352, 987– 996. [DOI] [PubMed] [Google Scholar]
- (2).Stupp R; Hegi ME; Mason WP; van den Bent MJ; Taphoorn MJ; Janzer RC; Ludwin SK; Allgeier A; Fisher B; Belanger K; Hau P; Brandes AA; Gijtenbeek J; Marosi C; Vecht CJ; Mokhtari K; Wesseling P; Villa S; Eisenhauer E; Gorlia T; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol 2009, 10, 459–466. [DOI] [PubMed] [Google Scholar]
- (3).Carlsson SK; Brothers SP; Wahlestedt C Emerging Treatment Strategies for Glioblastoma Multiforme. EMBO Mol. Med 2014, 6, 1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tamborini M; Locatelli E; Rasile M; Monaco I; Rodighiero S; Corradini I; Comes Franchini M; Passoni L; Matteoli MA Combined Approach Employing Chlorotoxin-Nanovectors and Low Dose Radiation to Reach Infiltrating Tumor Niches in Glioblastoma. ACS Nano 2016, 10, 2509–2520. [DOI] [PubMed] [Google Scholar]
- (5).Touat M; Idbaih A; Sanson M; Ligon KL Glioblastoma Targeted Therapy: Updated Approaches from Recent Biological Insights. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 2017, 28, 1457–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Liu F; Hon GC; Villa GR; Turner KM; Ikegami S; Yang H; Ye Z; Li B; Kuan S; Lee AY; Zanca C; Wei B; Lucey G; Jenkins D; Zhang W; Barr CL; Furnari FB; Cloughesy TF; Yong WH; Gahman TC; et al. EGFR Mutation Promotes Glioblastoma through Epigenome and Transcription Factor Network Remodeling. Mol. Cell 2015, 60, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yewale C; Baradia D; Vhora I; Patil S; Misra A Epidermal Growth Factor Receptor Targeting in Cancer: A Review of Trends and Strategies. Biomaterials 2013, 34, 8690–8707. [DOI] [PubMed] [Google Scholar]
- (8).Yang J; Yan J; Liu B Targeting EGFRvIII for Glioblastoma Multiforme. Cancer Lett 2017, 403, 224–230. [DOI] [PubMed] [Google Scholar]
- (9).Del Paggio JC Immunotherapy: Cancer Immunotherapy and the Value of Cure. Nat. Rev. Clin. Oncol 2018, 15, 268–269. [DOI] [PubMed] [Google Scholar]
- (10).Hwang WL; Pike LRG; Royce TJ; Mahal BA; Loeffler JS Safety of Combining Radiotherapy with Immune-Checkpoint Inhibition. Nat. Rev. Clin. Oncol 2018, 15, 477–494. [DOI] [PubMed] [Google Scholar]
- (11).Ribas A; Wolchok JD Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sharpe AH; Pauken KE The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol 2017, 18, 153–167. [DOI] [PubMed] [Google Scholar]
- (13).Jacquelot N; Roberti MP; Enot DP; Rusakiewicz S; Ternès N; Jegou S; Woods DM; Sodre AL; Hansen M; Meirow Y; Sade-Feldman M; Burra A; Kwek SS; Flament C; Messaoudene M; Duong CPM; Chen L; Kwon BS; Anderson AC; Kuchroo VK; et al. Predictors of Responses to Immune Checkpoint Blockade in Advanced Melanoma. Nat. Commun 2017, 8, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Antonios JP; Soto H; Everson RG; Orpilla J; Moughon D; Shin N; Sedighim S; Yong WH; Li G; Cloughesy TF; Liau LM; Prins RM PD-1 Blockade Enhances the Vaccination-Induced Immune Response in Glioma. JCI Insight 2016, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen N; Fang W; Zhan J; Hong S; Tang Y; Kang S; Zhang Y; He X; Zhou T; Qin T; Huang Y; Yi X; Zhang L Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol 2015, 10, 910–923. [DOI] [PubMed] [Google Scholar]
- (16).Zhang M; Li G; Wang Y; Zhao S; Haihong P; Zhao H; Wang Y Expression in Lung Cancer and Its Correlation with Driver Mutations: A Meta-Analysis. Sci. Rep 2017, 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li X; Lian Z; Wang S; Xing L; Yu J Interactions between EGFR and PD-1/PD-L1 Pathway: Implications for Treatment of NSCLC. Cancer Lett 2018, 418, 1–9. [DOI] [PubMed] [Google Scholar]
- (18).Sato Y; Note Y; Maeki M; Kaji N; Baba Y; Tokeshi M; Harashima H Elucidation of the Physicochemical Properties and Potency of SiRNA-Loaded Small-Sized Lipid Nanoparticles for siRNA Delivery. J. Controlled Release 2016, 229, 48–57. [DOI] [PubMed] [Google Scholar]
- (19).Cui J; Qin L; Zhang J; Abrahimi P; Li H; Li G; Tietjen GT; Tellides G; Pober JS; Mark Saltzman W Ex Vivo Pretreatment of Human Vessels with siRNA Nanoparticles Provides Protein Silencing in Endothelial Cells. Nat. Commun 2017, 8, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wu C; Li J; Wang W; Hammond PT Rationally Designed Polycationic Carriers for Potent Polymeric siRNA-Mediated Gene Silencing. ACS Nano 2018, 12, 6504–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Banks WA From Blood-Brain Barrier to Blood-Brain Interface: New Opportunities for CNS Drug Delivery. Nat. Rev. Drug Discovery 2016, 15, 275–292. [DOI] [PubMed] [Google Scholar]
- (22).Huse JT; Holland EC Targeting Brain Cancer: Advances in the Molecular Pathology of Malignant Glioma and Medulloblastoma. Nat. Rev. Cancer 2010, 10, 319–331. [DOI] [PubMed] [Google Scholar]
- (23).Kauffman WB; Fuselier T; He J; Wimley WC Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci 2015, 40, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Guidotti G; Brambilla L; Rossi D Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci 2017, 38, 406–424. [DOI] [PubMed] [Google Scholar]
- (25).Ruoslahti E Tumor Penetrating Peptides for Improved Drug Delivery. Adv. Drug Delivery Rev 2017, 110–111, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yang Y; Wang X; Liao G; Liu X; Chen Q; Li H; Lu L; Zhao P; Yu Z iRGD-Decorated Red Shift Emissive Carbon Nanodots for Tumor Targeting Fluorescence Imaging. J. Colloid Interface Sci 2018, 509, 515–521. [DOI] [PubMed] [Google Scholar]
- (27).Wang K; Zhang X; Liu Y; Liu C; Jiang B; Jiang Y Tumor Penetrability and Anti-Angiogenesis Using IRGD-Mediated Delivery of Doxorubicin-Polymer Conjugates. Biomaterials 2014, 35, 8735–8747. [DOI] [PubMed] [Google Scholar]
- (28).Feng L; Mumper RJA Critical Review of Lipid-Based Nanoparticles for Taxane Delivery. Cancer Lett 2013, 334, 157–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tapeinos C; Battaglini M; Ciofani G Advances in the Design of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Targeting Brain Diseases. J. Controlled Release 2017, 264, 306–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ruan S; Hu C; Tang X; Cun X; Xiao W; Shi K; He Q; Gao H Increased Gold Nanoparticle Retention in Brain Tumors by in Situ Enzyme-Induced Aggregation. ACS Nano 2016, 10, 10086–10098. [DOI] [PubMed] [Google Scholar]
- (31).Doolittle E; Peiris PM; Doron G; Goldberg A; Tucci S; Rao S; Shah S; Sylvestre M; Govender P; Turan O; Lee Z; Schiemann WP; Karathanasis E Spatiotemporal Targeting of a Dual-Ligand Nanoparticle to Cancer Metastasis. ACS Nano 2015, 9, 8012–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Stapleton S; Milosevic M Radiation Effects on the Tumor Microenvironment: Implications for Nanomedicine Delivery. Adv. Drug Delivery Rev 2017, 109, 119–130. [DOI] [PubMed] [Google Scholar]
- (33).Miller MA; Chandra R; Cuccarese MF; Pfirschke C; Engblom C; Stapleton S; Adhikary U; Kohler RH; Mohan JF; Pittet MJ; Weissleder R Radiation Therapy Primes Tumors for Nanotherapeutic Delivery via Macrophage-Mediated Vascular Bursts. Sci. Transl. Med 2017, 9, No. eaal0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chu D; Dong X; Zhao Q; Gu J; Wang Z Photo-sensitization Priming of Tumor Microenvironments Improves Delivery of Nanotherapeutics via Neutrophil Infiltration. Adv. Mater 2017, 29, 1701021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).de Jesus MB; Zuhorn IS Solid Lipid Nanoparticles as Nucleic Acid Delivery System: Properties and Molecular Mechanisms. J. Controlled Release 2015, 201, 1–13. [DOI] [PubMed] [Google Scholar]
- (36).Erel G; Kotmakcı M; Akbaba H; Sözer Karadaglı, S.; Kantarcı AG Nanoencapsulated Chitosan Nanoparticles in Emulsion-Based Oral Delivery System: In Vitro and in Vivo Evaluation of Insulin Loaded Formulation. J. Drug Delivery Sci. Technol 2016, 36, 161–167. [Google Scholar]
- (37).Lawrence MJ; Rees GD Microemulsion-Based Media as Novel Drug Delivery Systems. Adv. Drug Delivery Rev 2012, 64, 175–193. [DOI] [PubMed] [Google Scholar]
- (38).Ahumada M; Jacques E; Andronic C; Comer J; Poblete H; Alarcon EI Novel Specific Peptides as Superior Surface Stabilizers for Silver Nano Structures: Role of Peptide Chain Length. J. Mater. Chem. B 2017, 5, 8925–8928. [DOI] [PubMed] [Google Scholar]
- (39).Liao ZX; Ho YC; Chen HL; Peng SF; Hsiao CW; Sung HW Enhancement of Efficiencies of the Cellular Uptake and Gene Silencing of Chitosan/SiRNA Complexes via the Inclusion of a Negatively Charged Poly(γ-Glutamic Acid). Biomaterials 2010, 31, 8780–8788. [DOI] [PubMed] [Google Scholar]
- (40).Lei Y; Tang L; Xie Y; Xianyu Y; Zhang L; Wang P; Hamada Y; Jiang K; Zheng W; Jiang X Gold Nanoclusters-Assisted Delivery of NGF SiRNA for Effective Treatment of Pancreatic Cancer. Nat. Commun 2017, 8, 15130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Patil ML; Zhang M; Minko T Multifunctional Triblock Nanocarrier (PAMAM-PEG-PLL) for the Efficient Intracellular siRNA Delivery and Gene Silencing. ACS Nano 2011, 5, 1877–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ni D; Zhang J; Bu W; Xing H; Han F; Xiao Q; Yao Z; Chen F; He Q; Liu J; Zhang S; Fan W; Zhou L; Peng W; Shi J Dual-Targeting Upconversion Nanoprobes across the Blood-Brain Barrier for Magnetic Resonance/Fluorescence Imaging of Intracranial Glioblastoma. ACS Nano 2014, 8, 1231–1242. [DOI] [PubMed] [Google Scholar]
- (43).Ojea-Jimenez I; Garcia-Fernandez L; Lorenzo J; Puntes VF Facile Preparation of Cationic Gold Nanoparticle-Bioconjugates for Cell Penetration and Nuclear Targeting. ACS Nano 2012, 6, 7692–7702. [DOI] [PubMed] [Google Scholar]
- (44).Yao H; Wang K; Wang Y; Wang S; Li J; Lou J; Ye L; Yan X; Lu W; Huang R Enhanced Blood–brain Barrier Penetration and Glioma Therapy Mediated by a New Peptide Modified Gene Delivery System. Biomaterials 2015, 37, 345–352. [DOI] [PubMed] [Google Scholar]
- (45).Kwak G; Kim D; Nam GH; Wang SY; Kim IS; Kim SH; Kwon IC; Yeo Y Programmed Cell Death Protein Ligand-1 Silencing with Polyethylenimine-Dermatan Sulfate Complex for Dual Inhibition of Melanoma Growth. ACS Nano 2017, 11, 10135–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dal Magro R; Ornaghi F; Cambianica I; Beretta S; Re F; Musicanti C; Rigolio R; Donzelli E; Canta A; Ballarini E; Cavaletti G; Gasco P; Sancini G ApoE-Modified Solid Lipid Nanoparticles: A Feasible Strategy to Cross the Blood-Brain Barrier. J. Controlled Release 2017, 249, 103–110. [DOI] [PubMed] [Google Scholar]
- (47).Zheng M; Tao W; Zou Y; Farokhzad OC; Shi B Nanotechnology-Based Strategies for siRNA Brain Delivery for Disease Therapy. Trends Biotechnol 2018, 36, 562. [DOI] [PubMed] [Google Scholar]
- (48).Sekerdag E; Lüle S; Bozdag Pehlivan S;Öztu N; Kara A; Kaffashi A; Vural I; Isıkay I; Yavuz B; Oguz KK; Söylemezoğlu F; Gürsoy-Özdemir Y; Mut MA Potential Non-Invasive Glioblastoma Treatment: Nose-to-Brain Delivery of Farnesylthiosalicylic Acid Incorporated Hybrid Nanoparticles. J. Controlled Release 2017, 261, 187–198. [DOI] [PubMed] [Google Scholar]
- (49).Gan HK; Van Den Bent M; Lassman AB; Reardon DA; Scott AM Antibody-Drug Conjugates in Glioblastoma Therapy: The Right Drugs to the Right Cells. Nat. Rev. Clin. Oncol 2017, 14, 695–707. [DOI] [PubMed] [Google Scholar]
- (50).Shen W; Patnaik MM; Ruiz A; Russell SJ; Peng KW Immunovirotherapy with Vesicular Stomatitis Virus and PD-L1 Blockade Enhances Therapeutic Outcome in Murine Acute Myeloid Leukemia. Blood 2016, 127, 1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).C Carlsson R; Issazadeh-navikas S PD-L1, Inflammation and Glioblastoma. Immunol. Clin. Res 2014, 2, 1–5. [Google Scholar]
- (52).Wein L; Luen SJ; Savas P; Salgado R; Loi S Checkpoint Blockade in the Treatment of Breast Cancer: Current Status and Future Directions. Br. J. Cancer 2018, 119, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).McDermott DF; Huseni MA; Atkins MB; Motzer RJ; Rini BI; Escudier B; Fong L; Joseph RW; Pal SK; Reeves JA; Sznol M; Hainsworth J; Rathmell WK; Stadler WM; Hutson T; Gore ME; Ravaud A; Bracarda S; Suarez C; Danielli R; et al. Clinical Activity and Molecular Correlates of Response to Atezolizumab Alone or in Combination with Bevacizumab versus Sunitinib in Renal Cell Carcinoma. Nat. Med 2018, 24, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lee HT; Lee JY; Lim H; Lee SH; Moon YJ; Pyo HJ; Ryu SE; Shin W; Heo YS Molecular Mechanism of PD-1/PD-L1 Blockade via Anti-PD-L1 Antibodies Atezolizumab and Durvalumab. Sci. Rep 2017, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Fukumura D; Kloepper J; Amoozgar Z; Duda DG; Jain RK Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat. Rev. Clin. Oncol 2018, 15, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Bogdanowich-Knipp SJ; Jois DSS; Siahaan TJ The Effect of Conformation on the Solution Stability of Linear vs. Cyclic RGD Peptides. J. Pept. Res 1999, 53, 523–529. [DOI] [PubMed] [Google Scholar]
- (57).Puig-Saus C; Rojas LA; Laborda E; Figueras A; Alba R; Fillat C; Alemany R iRGD Tumor-Penetrating Peptide-Modified Oncolytic Adenovirus Shows Enhanced Tumor Transduction, Intratumoral Dissemination and Antitumor Efficacy. Gene Ther 2014, 21, 767–774. [DOI] [PubMed] [Google Scholar]
- (58).Tian T; Zhang HX; He CP; Fan S; Zhu YL; Qi C; Huang NP; Xiao ZD; Lu ZH; Tannous BA; Gao J Surface Functionalized Exosomes as Targeted Drug Delivery Vehicles for Cerebral Ischemia Therapy. Biomaterials 2018, 150, 137–149. [DOI] [PubMed] [Google Scholar]
- (59).Sugahara KN; Teesalu T; Karmali PP; Kotamraju VR; Agemy L; Girard OM; Hanahan D; Mattrey RF; Ruoslahti E Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Resnier P; Montier T; Mathieu V; Benoit JP; Passirani C A Review of the Current Status of siRNA Nanomedicines in the Treatment of Cancer. Biomaterials 2013, 34, 6429–6443. [DOI] [PubMed] [Google Scholar]
- (61).Kim HJ; Kim A; Miyata K; Kataoka K Recent Progress in Development of siRNA Delivery Vehicles for Cancer Therapy. Adv. Drug Delivery Rev 2016, 104, 61–77. [DOI] [PubMed] [Google Scholar]
- (62).Kanasty R; Dorkin JR; Vegas A; Anderson D Delivery Materials for siRNA Therapeutics. Nat. Mater 2013, 12, 967–977. [DOI] [PubMed] [Google Scholar]
- (63).Miller MA; Zheng YR; Gadde S; Pfirschke C; Zope H; Engblom C; Kohler RH; Iwamoto Y; Yang KS; Askevold B; Kolishetti N; Pittet M; Lippard SJ; Farokhzad OC; Weissleder R Tumour-Associated Macrophages Act as a Slow-Release Reservoir of Nano-Therapeutic Pt(IV) pro-Drug. Nat. Commun 2015, 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Kunjachan S; Detappe A; Kumar R; Ireland T; Cameron L; Biancur DE; Motto-Ros V; Sancey L; Sridhar S; Makrigiorgos GM; Berbeco RI Nanoparticle Mediated Tumor Vascular Disruption: A Novel Strategy in Radiation Therapy. Nano Lett 2015, 15, 7488–7496. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (65).Van Vulpen M; Kal HB; Taphoorn MJB; El Sharouni SY Changes in Blood-Brain Barrier Permeability Induced by Radiotherapy: Implications for Timing of Chemotherapy? Oncol. Rep 2002, 9, 683–688. [PubMed] [Google Scholar]
- (66).Wilhelm S; Tavares AJ; Dai Q; Ohta S; Audet J; Dvorak HF; Chan WCW Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater 2016, 1, 16014. [Google Scholar]
- (67).LoRusso P; Krop I; Miller K; Ma C; Siegel BA; Shields AF; Molnar I; Wickham T; Reynolds J; Campbell K; Hendriks B; McClure T; Moyo V; Munster P Abstract CT234: A Phase I Study of MM-302, a HER2-Targeted PEGylated Liposomal Doxorubicin, in Patients with HER2+ Metastatic Breast Cancer. Cancer Res 2015, 75, CT234–CT234. [Google Scholar]
- (68).Von Hoff DD; Mita MM; Ramanathan RK; Weiss GJ; Mita AC; Lorusso PM; Burris HA; Hart LL; Low SC; Parsons DM; Zale SE; Summa JM; Youssoufian H; Sachdev JC Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors. Clin. Cancer Res 2016, 22, 3157–3163. [DOI] [PubMed] [Google Scholar]
- (69).Chen L; Miao W; Tang X; Zhang H; Wang S; Luo F; Yan J Inhibitory Effect of Neuropilin-1 Monoclonal Antibody (NRP-1 MAb) on Glioma Tumor in Mice. J. Biomed. Nanotechnol 2013, 9, 551–558. [DOI] [PubMed] [Google Scholar]
- (70).Miyauchi JT; Chen D; Choi M; Nissen JC; Shroyer KR; Djordevic S; Zachary IC; Selwood D; Tsirka SE Ablation of Neuropilin 1 from Glioma-Associated Microglia and Macrophages Slows Tumor Progression. Oncotarget 2016, 7, 9801–9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Silginer M; Weller M; Ziegler U; Roth P Integrin Inhibition Promotes Atypical Anoikis in Glioma Cells. Cell Death Dis 2014, 5, No. e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Wurdinger T; Badr C; Pike L; de Kleine R; Weissleder R; Breakefield XO; Tannous BA A Secreted Luciferase for Ex Vivo Monitoring of In Vivo Processes. Nat. Methods 2008, 5, 171–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Mizrak A; Bolukbasi MF; Ozdener GB; Brenner GJ; Madlener S; Erkan EP; Ströbel T; Breakefield XO; Saydam O Genetically Engineered Microvesicles Carrying Suicide MRNA/ Protein Inhibit Schwannoma Tumor Growth. Mol. Ther 2013, 21, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Kotmakc M; Akbaba H; Erel G; Ertan G; Kantarcı G Improved Method for Solid Lipid Nanoparticle Preparation Based on Hot Microemulsions: Preparation, Characterization, Cytotoxicity, and Hemocompatibility Evaluation. AAPS PharmSciTech 2017, 18, 1355–1365. [DOI] [PubMed] [Google Scholar]
- (75).Tamura M; Yanagawa F; Sugiura S; Takagi T; Sumaru K; Kanamori T Click-Crosslinkable and Photodegradable Gelatin Hydrogels for Cytocompatible Optical Cell Manipulation in Natural Environment. Sci. Rep 2015, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Bzymek KP; Puckett JW; Zer C; Xie J; Ma Y; King JD; Goodstein LH; Avery KN; Colcher D; Singh G; Horne DA; Williams JC Mechanically Interlocked Functionalization of Monoclonal Antibodies. Nat. Commun 2018, 9, 1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Gooding M; Malhotra M; McCarthy DJ; Godinho BMDC; Cryan JF; Darcy R; O’Driscoll CM Synthesis and Characterization of Rabies Virus Glycoprotein-Tagged Amphiphilic Cyclodextrins for SiRNA Delivery in Human Glioblastoma Cells: In Vitro Analysis. Eur. J. Pharm. Sci 2015, 71, 80–92. [DOI] [PubMed] [Google Scholar]
- (78).Wang Y; Malcolm DW; Benoit DSW Controlled and Sustained Delivery of SiRNA/NPs from Hydrogels Expedites Bone Fracture Healing. Biomaterials 2017, 139, 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Garofalo S; D’Alessandro G; Chece G; Brau F; Maggi L; Rosa A; Porzia A; Mainiero F; Esposito V; Lauro C; Benigni G; Bernardini G; Santoni A; Limatola C Enriched Environment Reduces Glioma Growth through Immune and Non-Immune Mechanisms in Mice. Nat. Commun 2015, 6, 6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Jacobs VL; Valdes PA; Hickey WF; De Leo JA Current Review of In Vivo GBM Rodent Models: Emphasis on the CNS-1 Tumour Model. ASN Neuro 2011, 3, No. AN20110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Tannous B Gaussia Luciferase Reporter Assay for Monitoring of Biological Processes in culture and in vivo. Nat. Protoc 2009, 4, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]