

Abstract
It is well documented that the rate of aging can be slowed, but it remains unclear to which extent aging-associated conditions can be reversed. How the interface of immunity and metabolism impinges upon the diabetes pandemic is largely unknown. Here we show that NLRP3, a pattern recognition receptor, is modified by acetylation in macrophages and is deacetylated by SIRT2, an NAD+-dependent deacetylase and a metabolic sensor. We have developed a cell-based system that models aging-associated inflammation, a defined co-culture system that simulates the effects of inflammatory milieu on insulin resistance in metabolic tissues during aging, and aging mouse models, and demonstrate that SIRT2 and NLRP3 deacetylation prevent, and can be targeted to reverse, aging-associated inflammation and insulin resistance. These results establish the dysregulation of the acetylation switch of the NLRP3 inflammasome as an origin of aging-associated chronic inflammation and highlight the reversibility of aging-associated chronic inflammation and insulin resistance.
Graphical Abstract
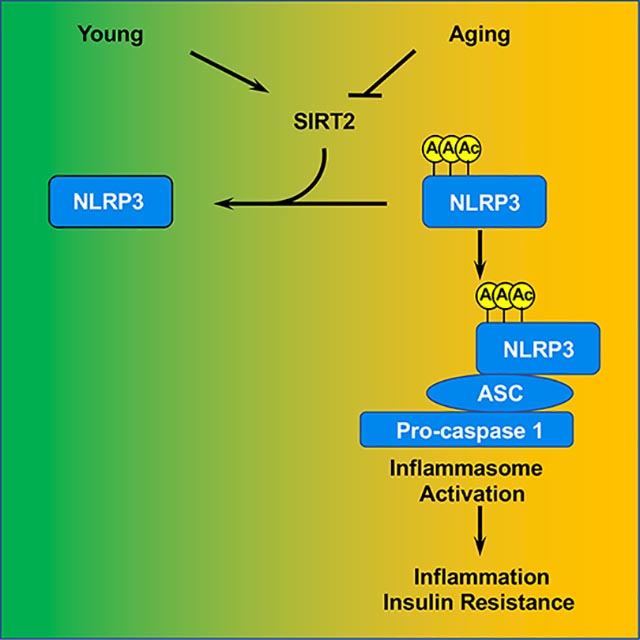
eTOC
He et al. show that SIRT2 deacetylates NLRP3 and inactivates the NLRP3 inflammasome. This interplay prevents, and can be targeted to reverse, aging-associated inflammation and insulin resistance. This study uncovers a mechanism underlying inflammaging and highlights the reversibility of aging-associated conditions.
INTRODUCTION
Aging is a systematic degenerative process that was long perceived as passive wear and tear. However, compelling evidence favors the view that aging is a regulated process amenable for genetic and environmental manipulations resulting in lifespan and healthspan extension (Kenyon, 2010; Lopez-Otin et al., 2013). Evidence is also emerging to suggest that aging-associated conditions may be reversed. Prominently, aging-associated stem cell deterioration and tissue degeneration have been shown to be reversed by genetic regulators (Brown et al., 2013; Leeman et al., 2018; Luo et al., 2019; Mohrin et al., 2015). The extent to which aging-associated conditions can be reversed is unknown but has profound implications for enhancing healthspan in the world with a growing aging population.
The immune system has evolved to launch robust yet acute responses necessary to effectively eliminate pathogens and safeguard tissue integrity. Chronic low-grade inflammation has no survival advantage to the host, yet epidemiological studies showed a marked increase in proinflammatory cytokines in aged (Chung et al., 2006; Fagiolo et al., 1993; Ferrucci et al., 2005; Forsey et al., 2003; Hager et al., 1994; Mysliwska et al., 1998) or obese individuals (Hotamisligil, 2010; Vandanmagsar et al., 2011). Numerous studies have suggested a connection between chronic inflammation and age- or overnutrition-related conditions (Barbieri et al., 2003; Beasley et al., 2009; Cartier et al., 2009; Cesari et al., 2004; Hsu et al., 2009; Kania et al., 1995; Maggio et al., 2006; Maggio et al., 2005; Pfeilschifter et al., 2002; Pou et al., 2007; Schaap et al., 2009) and diseases (Aggarwal et al., 2006; Cesari et al., 2003; Cushman et al., 2005; Kalogeropoulos et al., 2010; Pickup et al., 2000; Pradhan et al., 2001; Ravaglia et al., 2007; Rodondi et al., 2010; Tracy et al., 1997; Weaver et al., 2002; Yaffe et al., 2003), but the origins of aging- and overnutrition-associated chronic inflammation are largely unknown.
The NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome is a unique innate immune sensor that can be activated by a diverse array of endogenous metabolic signals to induce inflammation in the absence of overt infection (Martinon et al., 2009; Place and Kanneganti, 2018; Strowig et al., 2012). Aberrant activation of the NLRP3 inflammasome leads to the production of inflammatory cytokines IL-1β and IL18, and contributes to pathological inflammation in sterile inflammatory diseases, such as Alzheimer’s disease, Parkinson’s disease, obesity, diabetes, multiple sclerosis, and cancer (Duewell et al., 2010; Guo et al., 2015; Heneka et al., 2013; Inoue et al., 2012; Jourdan et al., 2013; Yan et al., 2015). In this study, we found that NLRP3 is modified by acetylation in macrophages and is targeted by sirtuin 2 (SIRT2), a cytosolic deacetylase, for deacetylation. We demonstrate that acetylation of NLRP3 facilitates the assembly and activation of the NLRP3 inflammasome. We provide evidence that the acetylation switch of NLRP3 inflammasome is physiologically relevant and regulates aging-associated inflammation and glucose homeostasis.
RESULTS
NLRP3 is Modified by Acetylation and is Targeted by SIRT2 for Deacetylation
To understand the regulation of the NLRP3 inflammasome, we investigated the posttranslational modifications of NLRP3. We immunopurified NLRP3 from NG5 cells, an immortalized NLRP3 knockout (KO) macrophage line stably expressing Flag-NLRP3 (Py et al., 2013). Mass spectrometry analyses revealed lysine residues modified by acetylation (Figure 1A, B, S1A). Western analyses further confirmed that NLRP3 was modified by acetylation in macrophages (Figure 1C).
Figure 1. NLRP3 is modified by acetylation in macrophages and is deacetylated by SIRT2.
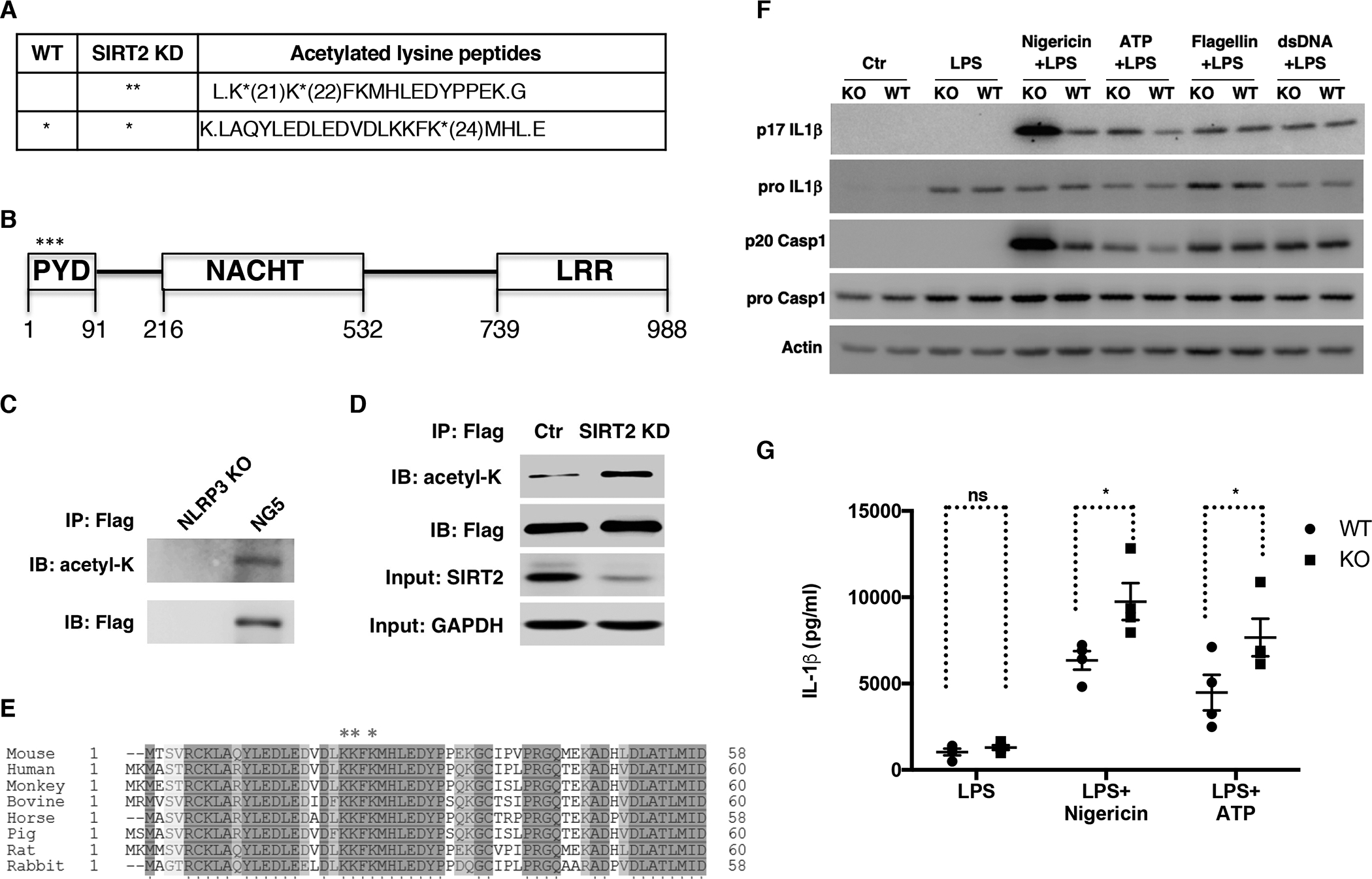
(A) NLRP3-Flag was immunopurified from NG5 cells treated with control or SIRT2 siRNA and analyzed by mass spectrometry. Acetylation sites are marked with *.
(B) Domain structure of NLRP3. Acetylation sites are marked with *.
(C) NLRP3-Flag was immunopurified from NG5 cells followed by Western analyses. NLRP3 KO cells were used as a negative control.
(D) NLRP3-Flag was immunopurified from NG5 cells treated with SIRT2 or control siRNA followed by Western analyses.
(E) The acetylation sites on NLRP3 are conserved across species. Sequence alignment of NLRP3 from various mammalian species is shown. Acetylated lysine residues are labeled with *.
(F, G) Bone marrow derived macrophages (BMDM) isolated from WT or SIRT2 KO mice were primed with LPS and then stimulated with nigericin or ATP (NLRP3 inducers), flagellin (NLRC4 inducer), or dsDNA (AIM2 inducer). Cell lysates were used for Western analyses for pro IL-1β and pro caspase 1 and culture supernatants were used for p17 IL-1β and p20 caspase 1 Western analyses (F). IL-1β in culture supernatants was quantified using ELISA (G).
Error bars represent SE. * p<0.05. ns: p>0.05. Student’s t test.
See also Figure S1.
Sirtuins are NAD+-dependent deacylases that regulate diverse metabolic processes (Du et al., 2011; Finkel et al., 2009; Jiang et al., 2011; Jing et al., 2016; Liu et al., 2008; Walker et al., 2010; Wang et al., 2010; Zhao et al., 2010). To determine whether sirtuins target NLRP3 for deacetylation, we knocked down sirtuins in NG5 cells via small interfering RNA (siRNA). Knocking down SIRT1 in NG5 cells did not have an effect on the acetylation level of NLRP3 (Figure S1B, C). In contrast, knocking down SIRT2 in NG5 cells increased the acetylation level of NLRP3 determined by Western analyses (Figure 1D). These lysine residues targeted for acetylation are highly conserved in mammals (Figure 1E). Mutating these lysine residues reduced the acetylation levels of NLRP3 (Figure S1D). These results suggest that SIRT2 targets NLRP3 for deacetylation in macrophages.
Because SIRT2 deacetylates NLRP3, we investigated whether SIRT2 regulates the NLRP3 inflammasome activation. We isolated bone marrow derived macrophages from wild type (WT) and SIRT2 KO mice and stimulated the cells for inflammasome activation. The expression of pro-IL1β in stimulated WT and SIRT2 KO macrophages was comparable (Figure 1F). SIRT2 KO macrophages had increased production of IL-1β and cleaved caspase 1 compared to WT controls in response to NLRP3 inducers, such as nigericin and ATP (Figure 1F). However, there was no difference in IL-1β production and caspase 1 cleavage between WT and SIRT2 KO macrophages in response to flagellin, a NLRC4 inflammasome inducer, or dsDNA, an AIM2 inflammasome inducer (Figure 1F). Consistent with a role of SIRT2 in suppressing NLRP3 inflammasome activation, pharmacological inhibition of SIRT2 also enhances caspase 1 activation in response to NLRP3 induction (Misawa et al., 2013). Together, these results suggest that SIRT2 represses the NLRP3 inflammasome activity.
Acetylation of NLRP3 Facilitates the Assembly and Activation of the NLRP3 Inflammasome
To investigate whether acetylation regulates the activity of the NLRP3 inflammasome, we treated NG5 cells with inducers for inflammasomes. Upon priming with lipopolysaccharides (LPS) followed by ATP stimulation to activate the NLRP3 inflammasome, the acetylation level of NLRP3 was increased (Figure 2A). However, treatment with LPS and dsDNA, an AIM2 inflammasome inducer, did not change the level of NLRP3 acetylation. The PYD domain mediates the interaction between NLRP3 and apoptosis-associated speck-like protein containing a CARD (ASC), and the formation of the inflammasome (Lu et al., 2014). We mutated the lysine residues at the acetylation sites of NLRP3 to arginine to mimic the constitutively deacetylated state and reconstituted NLRP3 KO macrophages with WT or mutant NLRP3 (K21/22/24R) via retroviral transduction. WT and mutant NLRP3 were expressed to comparable levels (Figure 2B). Upon treatment with LPS and ATP, cells reconstituted with WT NLRP3 processed IL-1β (Figure 2B). In contrast, cells reconstituted with NLRP3 mutant had significantly reduced level of IL-1β cleavage (Figure 2B). NLRP3 complexes with ASC to form inflammasome (Martinon et al., 2009; Strowig et al., 2012). In contrast to WT NLRP3, which triggered the formation of speck-like foci containing ASC, NLRP3 mutant was compromised in forming speck-like foci with ASC (Figure S2A, B), indicating that acetylation of NLRP3 facilitates the assembly and activation of the NLRP3 inflammasome.
Figure 2. Acetylation of NLRP3 enhances the NLRP3 inflammasome assembly and activity.
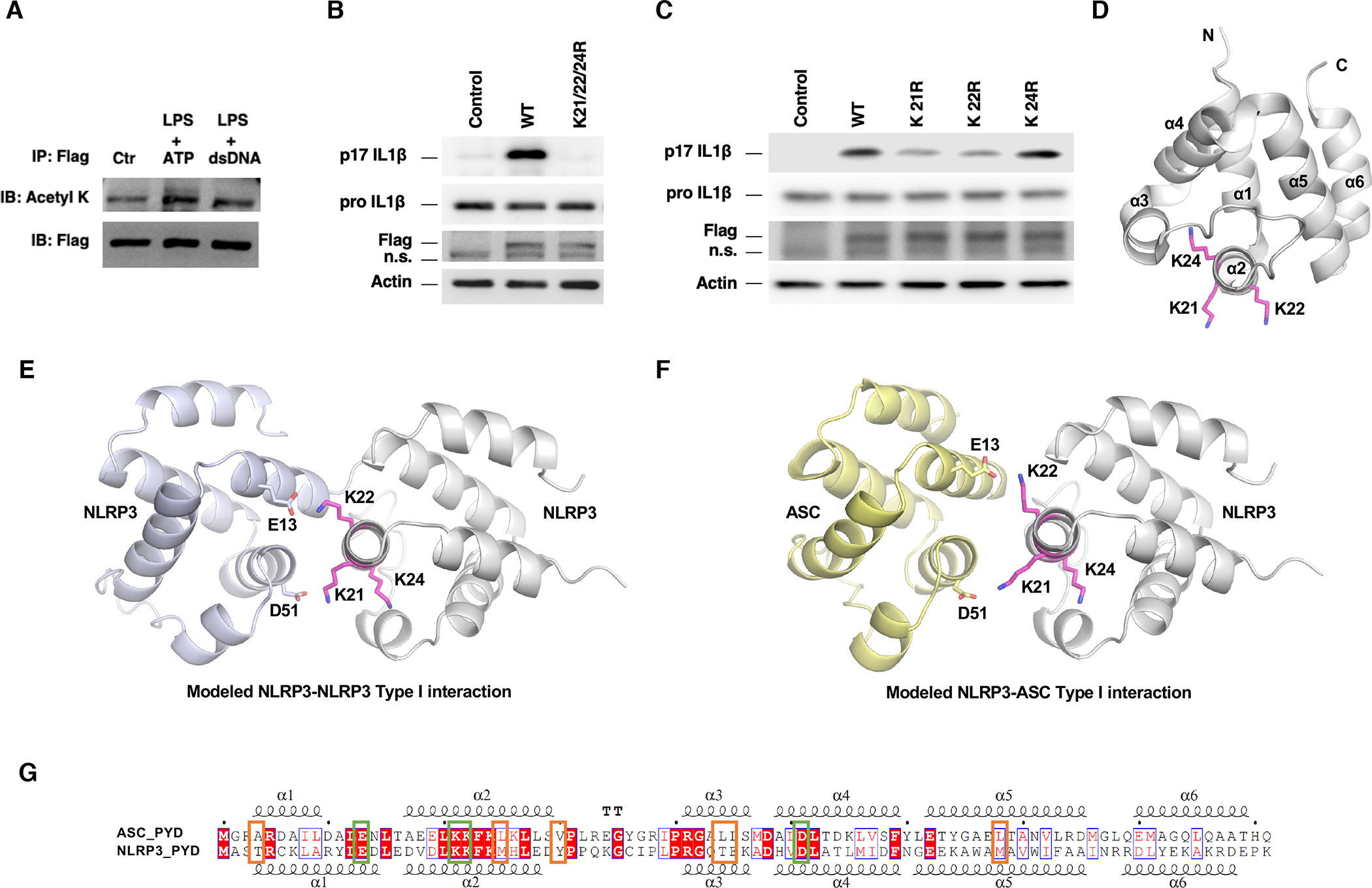
(A) NG5 cells were primed with LPS and treated with ATP or dsDNA. NLRP3-Flag was immunopurified followed by Western analyses.
(B, C) NLRP3 KO macrophages were reconstituted with NLRP3 mutants and WT NLRP3 control by retroviral transduction and were stimulated with LPS and ATP. Cell lysates were used for Western analyses for pro IL-1β. Culture supernatants were used for p17 IL-1β Western analyses. n.s., non-specific band.
(D) Position of K21, K22 and K24 residues in NLRP3 PYD domain structure (PDB code: 2NAQ).
(E, F) K21 and K22 are involved in Type I interactions in modeled NLRP3-NLRP3 interaction (E) and NLRP3-ASC interaction (F).
(G) Sequence alignment of the PYD domain of NLRP3 and ASC indicates conserved charged residues (green box) and less conserved hydrophobic residues (orange box) in Type I interface. See also Figure S2 and Table S1.
We next made and tested single mutations of lysine residues to arginine. K21R and K22R mutants, but not K24R mutant, resulted in reduced NLRP3 inflammasome activation and compromised formation of speck-like foci with ASC (Figure 2C, S2C), consistent with the model that K21 and K22 mediate the PYD-PYD interaction (Lu et al., 2014). Structural analyses showed that K24 was partially buried in the interior of the PYD, while K21 and K22 pointed to the exterior of the domain (Figure 2D). There was sufficient space surrounding K24 to accommodate a R mutation in the structure, consistent with the lack of effect of K24R mutant on inflammasome activity (Figure 2C). Similarly, the space was sufficient to accommodate an acetyl group, supporting a minimal effect from acetylation on K24.
The PYD interactions between NLRP3 and ASC, or ASC and ASC, or NLRP3 and NLRP3 use similar binding interfaces that are observed in the ASC filament structure (Lu et al., 2014; Oroz et al., 2016). To elucidate the potential structural mechanism for the effect of modification and mutation on K21 and K22, we modeled the NLRP3-NLRP3 and the NLRP3-ASC interactions by aligning the NLRP3 PYD structure (PDB code: 2NAQ) into the ASC oligomer (PDB code: 3J63) followed by energy minimization using the YASARA server (Krieger et al., 2009). K21 and K22 of NLRP3 were located at the Type I interface close to D51 and E13 of neighboring NLRP3 or ASC (Figure 2E, F), in keeping with disruption of charged interactions and impairment of ASC polymerization by K21E and K22E mutations (Lu et al., 2014). All these residues are conserved in the PYD domains of NLRP3 and ASC (Figure 2G, green box). Several hydrophobic residues in the PYD of ASC are also involved in oligomerization (Lu et al., 2014; Oroz et al., 2016), although they are less conserved in NLRP3 (Figure 2G, orange box). Collectively, the structural analyses suggest that acetylation at K21 and K22 of NLRP3 might alter both the charged interactions and the hydrophobic interactions.
To quantify the consequence of changing to arginine or acetyl-lysine (ALY) by acetylation at K21 and K22 of NLRP3, we modeled and energy minimized six pairs of type I PYD dimer structures: WT NLRP3 or K21/22R or K21/22ALY in complex with WT NLRP3 or WT ASC. Structural analyses by PDBePISA (http://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver) of the modeled complexes indicated that acetylation at K21 and K22 improved Type I interface stability through much enhanced hydrophobic interactions (Table S1). K21/22R mutations modestly enhanced the charged interactions but decreased overall interface stability due to loss of hydrophobic interactions. Furthermore, based on the energy calculation, acetylation and mutation at 21 and 22 positions of NLRP3 may affect the NLRP3-NLRP3 interaction more significantly than the NLRP3-ASC interaction (Table S1).
SIRT2 Prevents Aging- and Overnutrition-associated Chronic Inflammation and Insulin Resistance
We next determined the physiological relevance of SIRT2 regulation of the NLRP3 inflammasome by assessing the systematic inflammation and metabolic homeostasis of SIRT2 KO mice. Young SIRT2 KO mice were phenotypically unremarkable. Compared to the WT littermates, SIRT2 KO mice fed a chow diet for 6 months had normal body fat, similar levels of plasma IL-18 and glucose, and responded comparably in a glucose tolerance test (Figure 3). Because the NLRP3 inflammasome can be activated by endogenous metabolic signals associated with obesity and aging (Wen et al., 2011; Youm et al., 2012), we characterized SIRT2 KO mice fed a high fat diet for 6 months or fed a chow diet for 2 years. Compared to WT control mice, SIRT2 KO mice fed a high fat diet accumulated more body fat (Figure S3A), had increased levels of plasma glucose (Figure S3B) and insulin (Figure S3C). Glucose tolerance tests and insulin tolerance tests showed compromised glucose metabolism and insulin sensitivity in SIRT2 KO mice (Figure S3D, E, F, G), suggesting that SIRT2 prevents diet-induced obesity and insulin resistance. The plasma IL-18 level was increased in SIRT2 KO mice fed a high fat diet compared to the WT controls (Figure S3H), consistent with increased activation of the NLRP3 inflammasome under the condition of diet-induced obesity in the absence of SIRT2. SIRT2 KO mice had increased infiltration of stromal vascular fraction cells and macrophages in the adipose tissue, as well as increased expression of inflammatory cytokines, such as IL-6 (Figure S3I, J, K).
Figure 3. Young SIRT2 KO mice fed a chow diet are metabolically normal.
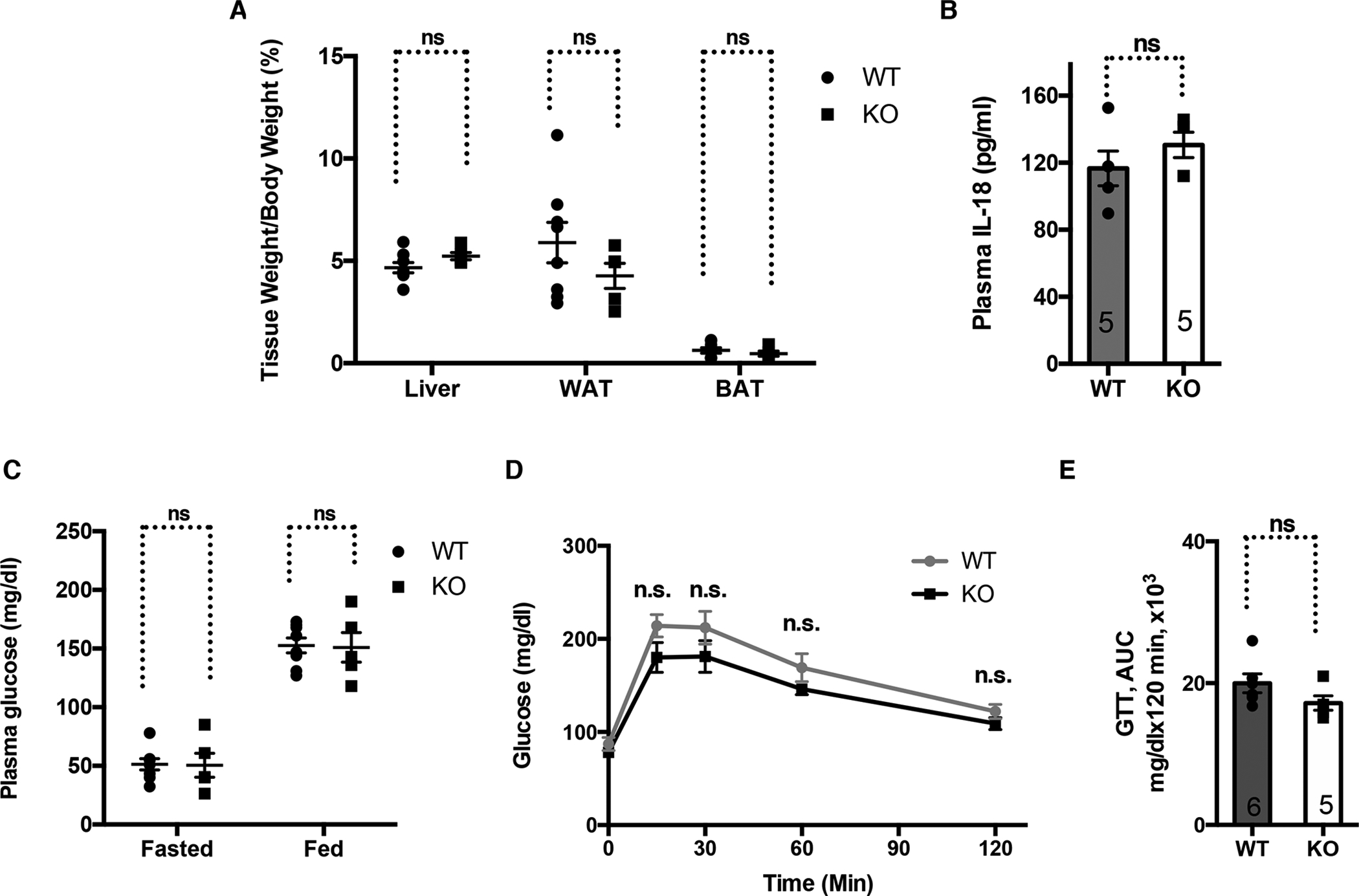
Young WT and SIRT2 KO mice fed a chow diet for 6 months were compared.
(A) Tissue weight.
(B) Plasma IL-18.
(C) Plasma glucose.
(D, E) Glucose tolerance test.
Error bars represent SE. n.s.: p>0.05. Student’s t test.
See also Figure S3.
At 2 years old, SIRT2 KO mice fed a chow diet also exhibited increased levels of plasma glucose and insulin (Figure 4A, B). Aged SIRT2 KO mice could not clear glucose as effectively as their WT control mice in glucose tolerance tests (Figure 4C, D) and showed impaired insulin signaling in insulin responsive tissues, as evidenced by reduced phosphorylation of Akt after the mice were infused with insulin (Figure 4E), indicating that SIRT2 is also required to maintain insulin sensitivity during physiological aging. Aged SIRT2 KO mice showed increased levels of plasma IL-18 (Figure 4F), suggesting that SIRT2 represses aging-associated NLRP3 inflammasome activation.
Figure 4. SIRT2 prevents aging-associated chronic inflammation and insulin resistance.
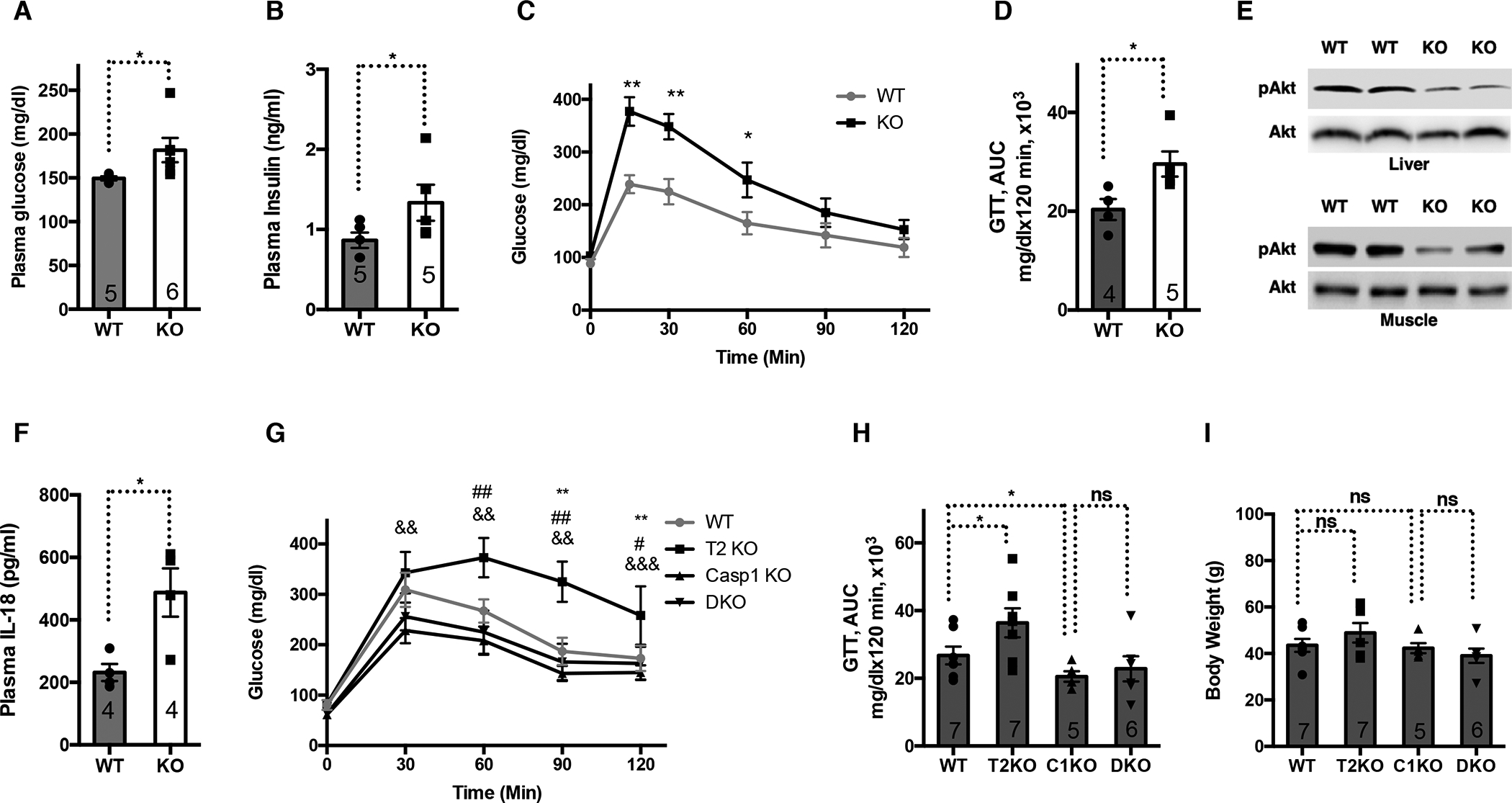
2-year-old WT and SIRT2 KO mice fed a chow diet were compared.
(A) Plasma glucose.
(B) Plasma insulin.
(C) Glucose tolerance test.
(D) Area under the curve for (C).
(E) Western blots of AKT and phosphorylated AKT in the livers and muscles after the mice were infused with insulin.
(F) Plasma IL-18.
(G) Glucose tolerance test for aged WT, SIRT2 KO (T2 KO), caspase 1 KO (C1 KO), and SIRT2/caspase 1 double KO mice (DKO). *: comparing WT and SIRT2 KO. #: comparing WT and Caspase 1 KO. &: comparing SIRT2 KO and DKO.
(H) Area under the curve for (G).
(I) Body Weight for mice used in (G).
Error bars represent SE. *, #: p<0.05. **, ##, &&: p<0.01. ***, &&&: p<0.001. n.s.: p>0.05. Student’s t test.
See also Figure S4.
To assess whether the inflammation and glucose metabolism effects of SIRT2 is attributable to the hematopoietic origin, we reconstituted the hematopoietic system of lethally irradiated recipient mice with hematopoietic stem cells (HSCs) isolated from old WT and SIRT2 KO mice. 4 months post-transplantation, mice reconstituted with SIRT2 KO HSCs had higher levels of plasma glucose, insulin, and IL-18 than mice reconstituted with WT HSCs (Figure S4). Thus, hematopoietic SIRT2 functions to repress aging-associated inflammation and maintains glucose homeostasis.
To determine whether SIRT2 maintains glucose homeostasis during physiological aging by repressing the NLRP3 inflammasome, we crossed SIRT2+/− and Caspase 1+/− to generate SIRT2−/−Caspase 1−/−, aged them for 2 years, and performed glucose tolerance tests. While aged SIRT2−/− mice showed glucose intolerance compared to WT control mice, SIRT2−/−Caspase 1−/− mice and Caspase 1−/− mice had comparable glucose tolerance (Figure 4G, H). No body weight difference was noted due to either genetic manipulation (Figure 4I). Together, these data support a causal relationship between the inflammasome and SIRT2-mediated prevention of aging-related glucose intolerance.
SIRT2 and NLRP3 Deacetylation Reverse Aging-associated Inflammation and Insulin Resistance ex vivo
qPCR analyses of macrophages isolated from young (3 months old) and old mice (2 years old) showed that the expression of SIRT2 was decreased in old macrophages (Figure 5A). To test whether the NLRP3 inflammasome is activated in old macrophages, we stimulated bone marrow derived macrophages from young and old mice with inflammasome inducers. Old macrophages showed increased caspase 1 and IL-1β cleavage compared to young macrophages upon the stimulation with NLRP3 inducers (nigericin and ATP) but not NLRC4 (flagellin) or AIM2 inducer (dsDNA) (Figure 5B). Compared to young mice, plasma IL-18 level was increased in old mice (Figure 3B, 4F). Together, these data suggest that the NLRP3 inflammasome is specifically activated in macrophages during physiological aging and contributes to the increased inflammatory milieu.
Figure 5. A cell-based system that models aging-associated inflammation.
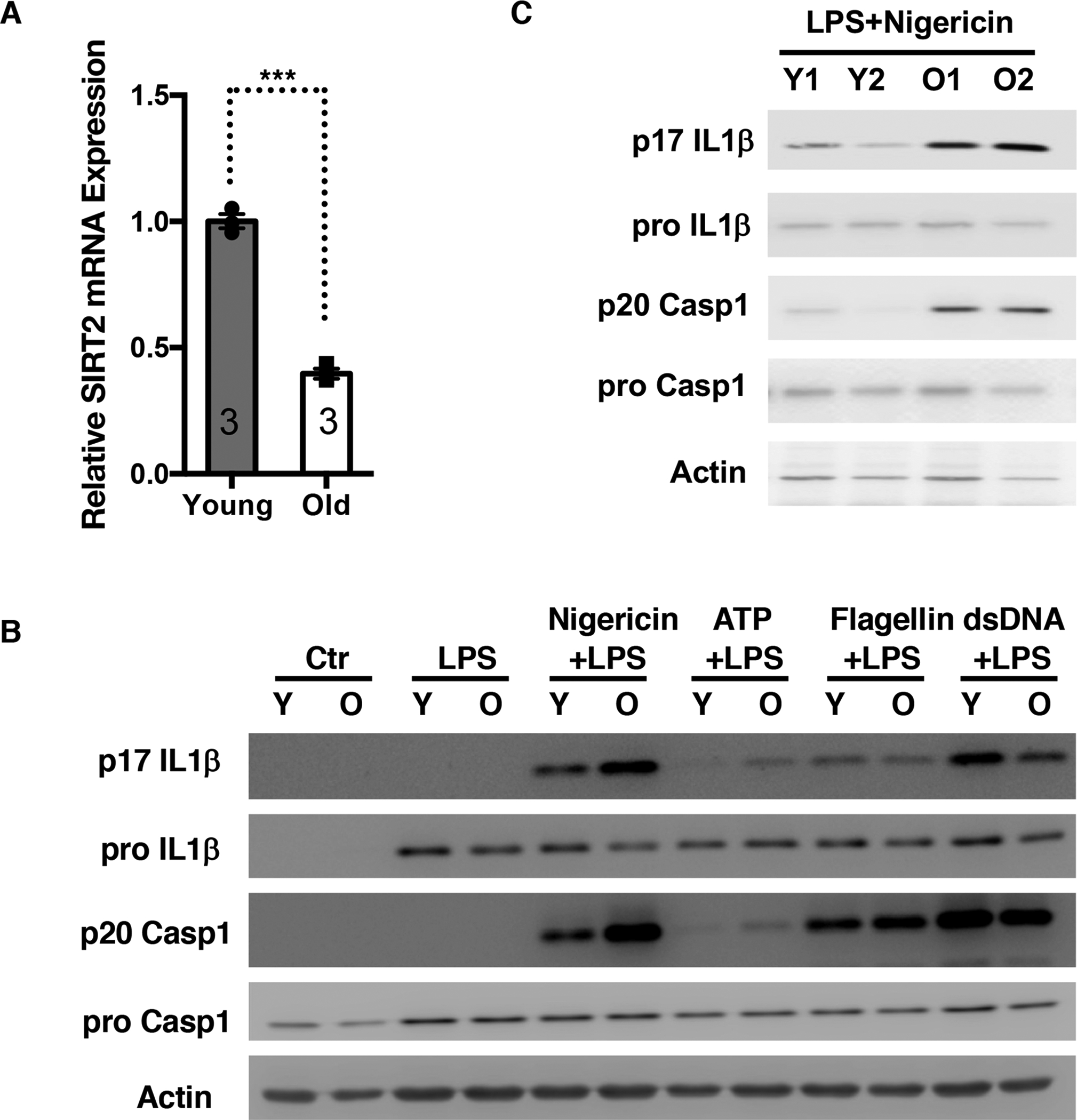
(A) SIRT2 mRNA levels in bone marrow derived macrophages (BMDMs) isolated from young and old mice were quantified by qPCR.
(B, C) BMDMs were isolated from young and old mice (B). Myeloid progenitors were isolated from bone marrow of young and old mice, immortalized with ER-Hoxb8, and differentiated into macrophages (C). Macrophages were primed with LPS, and stimulated with nigericin, ATP, flagellin, or dsDNA. Cell lysates were used for Western analyses for pro IL-1β and pro caspase
1. Culture supernatants were used for p17 IL-1β and p20 caspase 1 Western analyses. Error bars represent SE. ***: p<0.001. Student’s t test.
We asked whether the SIRT2-NLRP3 axis can be targeted to reverse aging-associated inflammation. Primary macrophages are not amenable for long-term culture and genetic manipulation. To circumvent these limitations, we immortalized the myeloid progenitors isolated from young and old mice using ER-Hoxb8 retrovirus (Wang et al., 2006) before induction to differentiation. Macrophages derived from immortalized progenitors of old mice also showed higher NLRP3 activity than those of young mice (Figure 5C), indicating that this cell-based system derived from immortalized progenitors models NLRP3 activation in primary macrophages isolated from young and old mice. We infected immortalized progenitors of old mice with SIRT2 lentivirus and control virus, which was followed by induction to differentiation and inflammasome stimulation (Figure 6A). SIRT2 overexpression reduced the production of IL-1β and cleavage of caspase 1 in response to the stimulation with a NLRP3 inducer (ATP) but not a NLRC4 inducer (flagellin) in macrophages derived from old mice (Figure 6B). Furthermore, we tested the effects of NLRP3 acetylation on aging-associated NLRP3 activation. We transduced immortalized progenitors of old mice with WT or constitutively deacetylated mutant (K21/22/24R) NLRP3. Upon selection, differentiation, and inflammasome stimulation, old macrophages reconstituted with the constitutively deacetylated mutant NLRP3 showed decreased production of IL-1β and cleavage of caspase 1 (Figure 6C). These data suggest that SIRT2 inactivation and NLRP3 acetylation underlie aging-associated NLRP3 activation in macrophages and can be targeted to reverse aging-associated inflammation.
Figure 6. SIRT2 and NLRP3 deacetylation reverse aging-associated inflammation and insulin resistance.
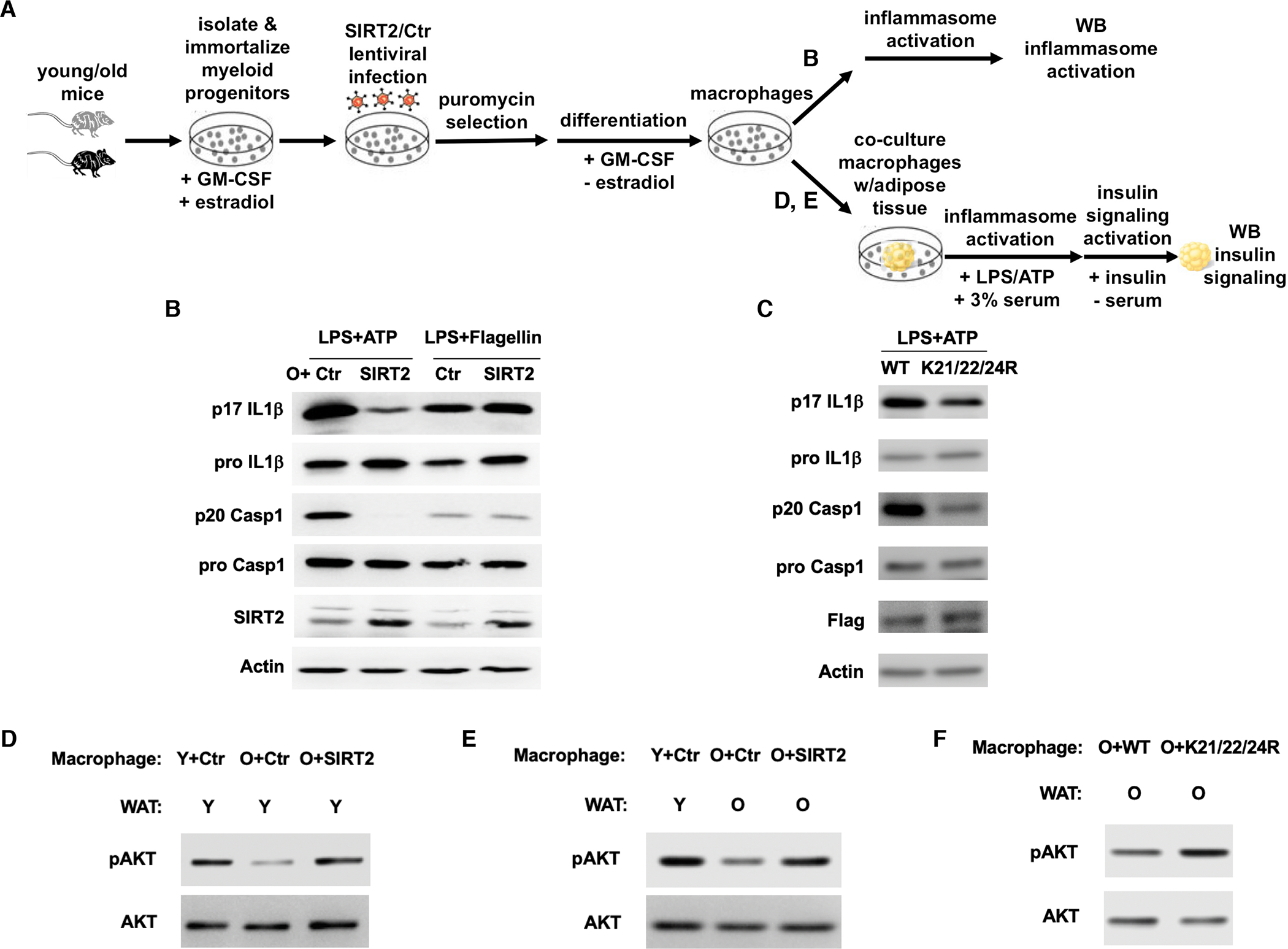
(A) Experimental design.
(B, C) Immortalized myeloid progenitors from old WT mice were transduced with control or SIRT2 lentivirus (B), WT or constitutively deacetylated mutant NLRP3 virus (C), selected, differentiated into macrophages, and treated for inflammasome activation. Cell lysates were used for Western analyses for pro IL-1β and pro caspase 1. Culture supernatants were used for p17 IL-1β and p20 caspase 1 Western analyses.
(D, E, F) Macrophages derived from immortalized myeloid progenitors from young or old mice with specified transduction were co-cultured with a piece of intact white adipose tissue from young or old mice as indicated, stimulated for NLRP3 inflammasome activation, followed by insulin signaling activation. The insulin signaling in the white adipose tissues was assessed by Western analyses.
We sought to determine whether the SIRT2-NLRP3 axis can be targeted to reverse aging-associated insulin resistance. To this end, we took advantage of the cell-based system we established that models NLRP3 activation in primary macrophages from young and old mice and devised a defined in vitro co-culture system to simulate the effects of the inflammatory milieu derived from the NLRP3 inflammasome on insulin resistance in metabolic tissues (Figure 6A). We co-cultured the macrophages derived from the immortalized myeloid progenitors of young or old mice with a piece of intact white adipose tissue from young or old mice, induced NLRP3 inflammasome activation followed by insulin signaling activation, before the white adipose tissues were analyzed for insulin signaling by Western analyses. We first compared the effect of overexpressing SIRT2 in old macrophages on white adipose tissues of young mice. Compared to young macrophages, co-culturing old macrophages with young white adipose tissues reduced insulin signaling, suggesting that the aberrant activation of the NLRP3 inflammasome in old macrophages is sufficient to induce insulin resistance in metabolic tissues (Figure 6D). Co-culturing old macrophages overexpressing SIRT2 with young white adipose tissues enhanced insulin signaling (Figure 6D), consistent with a role of SIRT2 in repressing the NLRP3 inflammasome in old macrophages (Figure 6B).
We next co-cultured young macrophages with young white adipose tissues (simulating young animals), old macrophages with old white adipose tissues (simulating old animals), and old macrophages overexpressing SIRT2 with old white adipose tissues, which enable us to assess whether SIRT2 activation in old macrophages can improve insulin resistance in old animals. We also co-cultured old macrophages transduced with WT or constitutively deacetylated mutant NLRP3 with old white adipose tissues to determine the effects of NLRP3 acetylation in old macrophages on insulin resistance in old animals. Compared to co-culture of young macrophages with young white adipose tissues, co-culture of old macrophages with old white adipose tissues resulted in reduced insulin signaling (Figure 6E). Overexpression of SIRT2 in old macrophages enhanced the insulin signaling in old white adipose tissues to the level comparable to young white adipose tissues co-cultured with young macrophages (Figure 6E). Compared to WT NLRP3, old macrophages reconstituted with constitutively deacetylated mutant NLRP3 (K21/22/24R) improved the insulin signaling in old white adipose tissues (Figure 6F). Thus, aging-associated insulin resistance can be reversed by quenching the inflammatory milieu, such as targeting the SIRT2-NLRP3 axis in macrophages.
NLRP3 Deacetylation Improves Aging-associated Glucose Homeostasis in vivo
To determine whether the NLRP3 acetylation switch in macrophages regulates aging-associated glucose metabolism in vivo, we generated aged mouse models reconstituted with the hematopoietic system expressing WT or K21/22/24R mutant NLRP3. We transduced HSCs isolated from aged mice with WT or K21/22/24R mutant NLRP3 virus, which were transplanted into lethally irradiated aged mice. 6 weeks post-transplantation, glucose tolerance tests showed that aged mice reconstituted with the hematopoietic system expressing K21/22/24R mutant NLRP3 cleared glucose more effectively than the mice reconstituted with the hematopoietic system expressing WT NLRP3 (Figure 7A, B). Plasma level of IL-18 was lower in the aged mice reconstituted with the hematopoietic system expressing K21/22/24R mutant NLRP3 than the control mice expressing WT NLRP3 (Figure 7C), while there was no body weight difference between these two groups (Figure 7D). Together, these data suggest a functional role of NLRP3 acetylation in regulating aging-associated glucose homeostasis in vivo.
Figure 7. NLRP3 Deacetylation Improves Aging-associated Glucose Homeostasis in vivo.
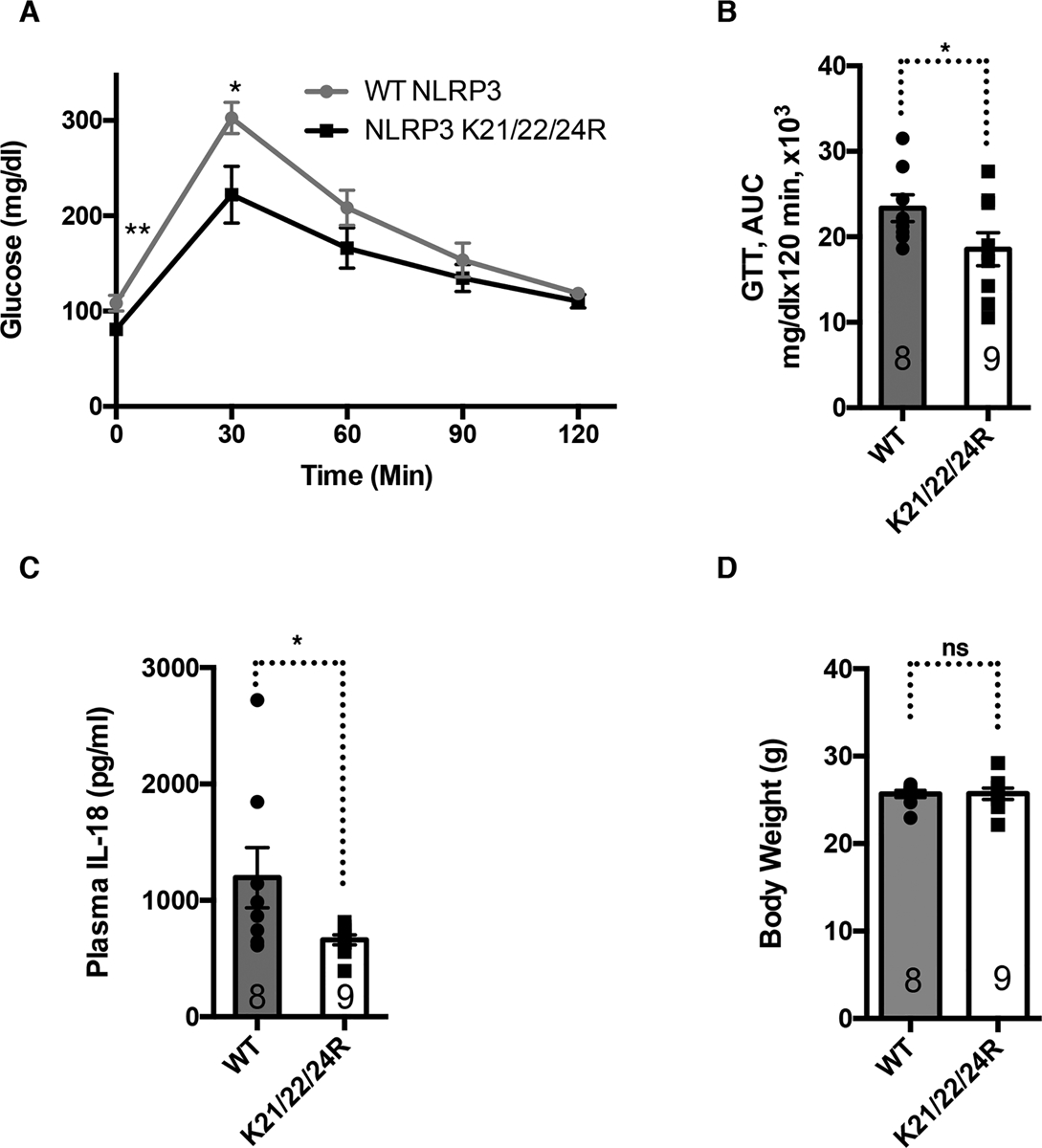
Hematopoietic stem cells from aged WT mice were transduced with WT or K21/22/24R mutant NLRP3 and transplanted into lethally irradiated aged WT mice to reconstitute their hematopoietic system. Mice were analyzed 6 weeks post-transplantation for glucose tolerance tests (A, B), plasma IL-18 (C), and body weight (D). Error bars represent SE. *: p<0.05. n.s.: p>0.05. Student’s t test.
DISCUSSION
Collectively, our work establishes a defined in vitro system that models aging-associated inflammation and insulin resistance, and enables further studies for feasibility, mechanistic basis, as well as therapeutic applications of reversing aging-associated inflammation and insulin resistance. Our studies identify an acetylation switch of the NLRP3 inflammasome in macrophages as a regulatory mechanism to ensure robust yet acute immune responses. Dysregulation of the acetylation switch of the NLRP3 inflammasome underlies chronic low-grade inflammation associated with aging and overnutrition, and perpetuates the development of insulin resistance. Importantly, this regulatory mechanism can be targeted to reverse aging-associated inflammation and insulin resistance.
Dysregulation of the NLRP3 inflammasome is implicated in numerous pathological conditions (Duewell et al., 2010; Guo et al., 2015; Heneka et al., 2013; Inoue et al., 2012; Jourdan et al., 2013; Yan et al., 2015). Its activity must be kept in check to prevent untoward physiological consequences and is subjected to multilayered regulation at the transcriptional (Bauernfeind et al., 2009) and posttranscriptional levels, such as phosphorylation (Song et al., 2017), ubiquitination (Py et al., 2013), and acetylation (Figure 1). The existence of the multilayered regulation may be essential to ensure an on or off switch of this powerful inflammatory machinery. We demonstrate that such fine regulation may be compromised under certain physiological conditions, such as aging and overnutrition (Figure 4, 6, 7, S3).
Advances in sirtuin biology suggest that sirtuins can modify multiple proteins with related functions to exert a concerted cellular function (Luo et al., 2017). Our new finding and study by Misawa (Misawa et al., 2013) indicate that SIRT2 regulates at least two downstream targets (NLRP3 and tubulin) to suppress two critical steps of the NLRP3 inflammasome activation (NLRP3 inflammasome assembly and transport), and suggest inactivation of the NLRP3 inflammasome an important cellular function of SIRT2.
Sirtuins regulate a diverse array of cellular pathways and physiological events. Because sirtuins possess NAD+-dependent deacylase activities, a number of histone and non-histone substrates for sirtuins have been identified (Finkel et al., 2009; Jiang et al., 2011; Liu et al., 2008; Qiu et al., 2010; Shin et al., 2011; Someya et al., 2010; Tao et al., 2010; Wang et al., 2010; Zhao et al., 2010). Although it is appreciated that acylation modulates protein biochemistry and regulates cellular functions, direct evidence supporting acylation impacts physiological function is lacking. Using an ex vivo co-culture system (Figure 6) and an in vivo mouse model reconstituted with the hematopoietic system expressing a constitutively deacetylated mutant (Figure 7), we provide evidence that acetylation impacts an aspect of physiology.
Aging is associated with the accumulation of somatic mutations in the hematopoietic system and expansion of the mutated blood cells (Busque et al., 2012; Genovese et al., 2014; Jaiswal et al., 2014; McKerrell et al., 2015; Xie et al., 2014). Individuals with clonal hematopoiesis are at higher risk for not only blood diseases but also diseases in distant tissues and earlier mortality (Bonnefond et al., 2013; Goodell and Rando, 2015; Jaiswal et al., 2014). These observations support the notion that aging-associated defects in HSCs can be propagated in their progeny, thereby having detrimental effects on distant tissues and organismal health span (Goodell and Rando, 2015) and raise the question of the impact of HSC aging on the development of aging-related diseases (Chen and Kerr, 2019). HSC aging is regulated by a mitochondrial metabolic checkpoint that is monitored by the SIRT2-NLRP3 axis (Brown et al., 2013; Chen and Kerr, 2019; Luo et al., 2019; Mohrin and Chen, 2016; Mohrin et al., 2015). Mouse models reconstituted with SIRT2 KO HSCs exhibited aging-associated chronic inflammation and compromised glucose homeostasis (Figure S4) while animals reconstituted with mutant NLRP3 HSCs showed improved inflammation and glucose homeostasis (Figure 7). Our findings suggest that HSC aging contributes to aging-associated chronic inflammation and loss of glucose homeostasis.
Limitations of Study
The effects of SIRT2 and NLRP3 acetylation on inflammation and glucose homeostasis were tested in C57BL/6 mouse strain only. It will be valuable to test these effects in other genetic backgrounds.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
NLRP3 mutant plasmids and immortalized myeloid progenitors from young and old mice were generated in this study and are available upon request. Please contact the Lead Contact, Danica Chen (danicac@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
SIRT2 knockout mice and caspase 1 knockout mice on C57BL6 background have been described (Bobrowska et al., 2012; Kuida et al., 1995). For co-culture experiments and transplant experiments, wild type mice on C57BL6 background were provided by the National Institute on Aging. Male mice were used in the study. Ages of the mice are specified in the description of each experiment and included in the figure legends of the corresponding experiments. Young mice were 3–5 months old and aged mice were 2 years old. Mice were housed on a 12:12 hr light:dark cycle at 25°C with ad libitum access to water and standard laboratory chow diet provided by LabDiet (0007688). The high-fat diet was provided by OpenSource Diets (D12079B). All animal procedures were in accordance with the animal care committee at the University of California, Berkeley. Health status checks were carried out by vets and euthanasia were performed if mice showed clinically apparent bleeding, or inability to move, eat or drink, or neurological/psychological changes, or clinically apparent spontaneous tumor formation, or mutilation. For insulin signaling, mice were fasted for 5 hr, injected with either phosphate-buffered saline or insulin (Sigma) at 2 mU/g body weight for 15 min (Lu et al., 2012).
METHOD DETAILS
Cell culture and RNAi
293T cells, NG5 cells (sex unknown), NLRP3 KO cells (sex unknown) were cultured in DMEM with 10% FBS at 37 °C with 5% CO2. Double-stranded siRNAs were purchased from Qiagen and were transfected into cells via RNAiMax (Invitrogen) according to manufacturer’s instructions. Mouse SIRT2 siRNA targeting sequence is 5’-CCAGAATAAGGCATTTCTCTA-3’. Mouse SIRT1 siRNA targeting sequence is AAGCGGCTTGAGGGTAATCAA. The control siRNA is non-targeting control (Qiagen). To induce caspase 1 activation, macrophages were primed with 100ng/ml LPS for 12 hours and then stimulated with ATP (3 mM) for 30mins, Nigericin (1.5 μM) for 1 hr, flagellin (0.5 μg/ml) for 1 hr, poly(dA:dT) (0.5 μg/ml) for 1 hr. Proteins from culture media were trichloroacetic acid (TCA) precipitated for Western analyses of p17 IL-1β and p20 caspase 1. Proteins from cell lysates were analyzed for pro IL-1β and pro caspase 1. For NLRP3 acetylation, cells were glucose starved for 6 hours before immunoprecipitation.
Pyroptosome formation
Pyroptosome formation was performed as previously described (Fernandes-Alnemri et al., 2009). Briefly, 5×105 293T cells were co-transfected with 0.8μg ASC-EGFP and 1.2 μg WT or mutant NLRP3 or control vector via Lipofectamine 2000 (Invitrogen). 48 hours posttransfection, cells were observed under fluorescence microscope for foci formation.
Immunoprecipitations
Immunoprecipitations were performed as previously described (Shin et al., 2013). Proteins were extracted in lysis buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 10% glycerol, 2 mM MgCl2, 1 mM DTT, 1% NP40, 1mM PMSF, and protease inhibitor). Protein extracts were subjected to centrifugation at 14,000 rpm for 10 min. Protein lysates were precleared with protein A/G beads (Santa Cruz Biotechnology) for 30 min before immunoprecipitation with Flag-resin (Sigma) overnight. Immunoprecipitates were extensively washed with lysis buffer and eluted with either Flag peptide (Sigma) for western analyses or 100mM Glycine solution (pH 3) for mass spectrometry analyses.
Mapping of acetylation on lysine residues
Mass spectrometry was performed by the Vincent J.Coates Proteomics/Mass Spectrometry Laboratory at UC Berkeley. Protein samples were cut with trypsin or trypsin and chymotrypsin. A nano LC column was packed in a 100 μm inner diameter glass capillary with an emitter tip. The column consisted of 10 cm of Polaris c18 5 μm packing material (Varian), followed by 4 cm of Partisphere 5 SCX (Whatman). The column was loaded by use of a pressure bomb and washed extensively with buffer A (see below). The column was then directly coupled to an electrospray ionization source mounted on a Thermo-Fisher LTQ XL linear ion trap mass spectrometer. An Agilent 1200 HPLC equipped with a split line so as to deliver a flow rate of 300 nl/min was used for chromatography. Peptides were eluted using a 4-step MudPIT procedure (Washburn et al., 2001). Buffer A was 5% acetonitrile/ 0.02% heptafluorobutyric acid (HBFA); buffer B was 80% acetonitrile/ 0.02% HBFA. Buffer C was 250 mM ammonium acetate/ 5% acetonitrile/ 0.02% HBFA; buffer D was same as buffer C, but with 500 mM ammonium acetate.
Protein identification was done with Integrated Proteomics Pipeline (IP2, Integrated Proteomics Applications, Inc. San Diego, CA) using ProLuCID/Sequest, DTASelect2 and Census (Cociorva et al., 2007; Park et al., 2008; Tabb et al., 2002; Xu et al., 2015). Tandem mass spectra were extracted into ms1 and ms2 files from raw files using RawExtractor (McDonald et al., 2004). Data was searched against the NCBI Mus Musculus protein database supplemented with sequences of common contaminants, concatenated to a decoy database in which the sequence for each entry in the original database was reversed (Peng et al., 2003). LTQ data was searched with 3000.0 milli-amu precursor tolerance and the fragment ions were restricted to a 600.0 ppm tolerance. All searches were parallelized and searched on the VJC proteomics cluster. Search space included all fully tryptic or chymotryptic peptide candidates with no missed cleavage restrictions. Carbamidomethylation (+57.02146) of cysteine was considered a static modification. We required 1 peptide per protein and both tryptic termini or chymotryptic for each peptide identification. The ProLuCID search results were assembled and filtered using the DTASelect program (Cociorva et al., 2007; Tabb et al., 2002) with a peptide false discovery rate (FDR) of 0.001 for single peptides and a peptide FDR of 0.005 for additional peptides for the same protein. Under such filtering conditions, the estimated false discovery rate was always less than 1% for the datasets used.
mRNA analysis
RNA was isolated from cells using Trizol reagent (Invitrogen). cDNA was generated using the qScript™ cDNA SuperMix (Quanta Biosciences). Gene expression was determined by real time PCR using Eva qPCR SuperMix kit (BioChain Institute) on an ABI StepOnePlus system. All data were normalized to β-Actin expression. PCR primers used for real time PCR were SIRT2-forward:TGGGCTGGATGAAAGAGAA. SIRT2-reverse:GGTCCACCTTGGAGAAGTCTG. SIRT1-forward:GCAACAGCATCTTGCCTGAT. SIRT1-reverse: GTGCTACTGGTCTCACTT. β-Actin-forward: GATCTGGCACCACACCTTCT. β-Actin-reverse: GGGGTGTTGAAGGTCTCAAA.
Isolation and immortalization of myeloid progenitors
Immortalization of myeloid progenitors was performed as described (Wang et al., 2006). Briefly, bone marrow was isolated from the femurs of mice after ammonium-chloride-potassium lysis of red blood cells and centrifugation onto a cushion of Ficoll-Paque. Ficoll-purified progenitors were infected with ER-Hoxb8 retrovirus and cultured in myeloid progenitor culture medium (RPMI-1640 with 10% FBS, 1% pen-strep-glutamine, 20ng/ml GM-CSF, 30μM beta mercaptoethanol, and 1μM estrogen). Immortalized myeloid progenitors were selected by moving nonadherent progenitor cells every 3 days to a new culture well for 3 weeks. Differentiation to macrophages was performed by removal of estrogen from the culture medium.
Lentiviral and retroviral transduction of macrophages
SIRT2 was cloned into the pFUGw lentiviral construct using BamH1 and Age1 sites. Lentivirus was produced as described (Qiu et al., 2010), concentrated by centrifugation, and resuspended with culture medium described above for NLRP3 KO cells or macrophages derived from myeloid progenitor cells. NLRP3 mutant constructs were generated using pMSCVgfp retroviral construct expressing WT NLRP3 and QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technology). Retrovirus was generated by transfecting 293T cells (ATCC) with pMSCVgfp retroviral constructs as well as VSV-G and gag/pol expression vectors using Lipofectamine 2000 transfection kit (Invitrogen). 48 hours posttransfection, culture supernatant was filtered through 0.45-mm-pore cellulose acetate filters, supplemented with 10 μg/ml of polybrene, and was applied to macrophages. Cells were subjected to another cycle of infection on the next day.
Co-culture of macrophages and white adipose tissues
The protocol is modified from (Miao et al., 2014). Briefly, macrophages were differentiated from the immortalized myeloid progenitors. A piece of intact epididymal white adipose tissue isolated from a wild type mouse was put into a well, in which macrophages were pre-cultured. The co-culture system was treated with 100 ng/ml LPS for 24 hours and then stimulated with 3 mM ATP for 30mins, then stimulated with 100 nM insulin for 30 min. The white adipose tissues were analyzed by Western analyses.
ELISA test
IL-1β and IL-18 concentration were measured using the ELISA kits (MBL international). Insulin concentration was measured using the insulin ELISA kit (Crystal Chem).
Glucose tolerance test and insulin tolerance test
Glucose tolerance test was performed on mice fasted for 12 hours by giving an intraperitoneal injection of D-glucose (1–2 g glucose/kg body weight, Sigma). Insulin tolerance test was performed on fasted mice by giving an intraperitoneal injection of insulin (0.5U insulin/kg body weight, Sigma). Glucose concentrations were measured with a glucometer (Bayer Contour) at the indicated time points.
Transplantation Assays
250 sorted hematopoietic stem cells from donor mice were mixed with helper bone marrow cells and injected into lethally irradiated recipient mice. A split lethal dose of irradiation of 900 rad (500 rad + 400 rad) was used to irradiate the recipient mice. The source of the helper bone marrow cells was the same as the recipient mice. Recipient mice were analyzed for glucose metabolism and inflammation.
Retroviral Transduction of HSCs
As previously described (Zhao et al., 2009), sorted HSCs were prestimulated for 5–10 hr in a 96 well U bottom dish in StemSpan SFEM (Stem Cell Technologies) supplemented with 10% FBS (Stem Cell Technologies), 1% Penicillin/Streptomycin (Invitrogen), IL3 (20ng/ml), IL6 (20ng/ml), TPO (50ng/ml), Flt3L (50ng/ml), and SCF (100ng/ml) (Peprotech). Retrovirus was produced as described above, concentrated by centrifugation, and resuspended with supplemented StemSpan SFEM media. The retroviral media were added to HSCs in a 96 well plate, spinoculated for 90 min at 270g in the presence of 8ug/ml polybrene. This process was repeated 24 hr later with a fresh batch of retroviral media.
Structure modeling and analysis
Structure modeling was performed using existing NLRP3 PYD monomer structure (PDB code: 2NAQ) (Oroz et al., 2016) and ASC PYD filament structure (PDB code: 3J63) (Lu et al., 2014). NLRP3 PYD structure was aligned to one or two ASC molecules in the filament structure by Coot (Emsley and Cowtan, 2004) to generate the initial NLRP3-ASC and NLRP3-NLRP3 dimer models. The dimer structures were modified at K21/22 positions in Coot before going through the YASARA energy minimization server (Krieger et al., 2009) to produce the final structure models. The interface analyses were performed in PDBePISA server (Krissinel and Henrick, 2007). Structure images were generated using the PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC (Delano, 2002).
QUANTIFICATION AND STATISTICAL ANALYSIS
Mice were randomized to groups and analysis of mice and tissue samples was performed by investigators blinded to the treatment or the genetic background of the animals. Sample size (n) can be found on the bar graphs representing the biological replicates (the number of mice), except for Figure S2, where n represents the technical replicates (the number of images). No statistical methods were used to predetermine sample sizes or test for normal distribution. The number of biological replicates was chosen based on the nature of the experiments and published papers describing similar experiments. Statistical analysis was performed with Student’s t-test (Excel). Data are presented as means and error bars represent standard errors. In all corresponding figures, * represents p<0.05. ** represents p<0.01. *** represents p<0.001. ns represents p>0.05.
DATA AND CODE AVAILABILITY
This study did not generate unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Actin antibody produced in rabbit | Sigma-Aldrich | Cat# A2066; RRID: AB_476693 |
| Caspase 1 Monoclonal Antibody (5B10), eBioscience(TM) | Thermo Fisher Scientific | Cat# 14-9832; RRID: AB_2016691 |
| SIRT2 antibody | Santa Cruz | Cat# SC 20966; RRID: AB_2188598 |
| SIRT2 antibody | Proteintech | Cat# 15345-1-AP |
| Monoclonal ANTI-FLAG antibody | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 |
| rabbit IgG antibody | Santa Cruz | Cat# sc-2027, RRID:AB_737197 |
| GAPDH Antibody | Santa Cruz | Cat# sc-25778, RRID:AB_10167668 |
| anti-Acetylated Lysine antibody | Biolegend | Cat# 623402, RRID:AB_315968 |
| Caspase 1 Monoclonal Antibody | Thermo Fisher Scientific | Cat# 14-9832-80, RRID:AB_2016624 |
| Mouse IL-1 beta/IL-1F2 Polyclonal antibody | R&D Systems | Cat# AF-401-NA, RRID:AB_416684 |
| AKT antibody | Cell Signaling | Cat# 9272, RRID:AB_329827 |
| Phospho-Akt (Ser473) antibody | Cell Signaling | Cat# 4060, RRID:AB_2315049 |
| Streptavidin APC-Cy7 | Biolegend | 405208 |
| CD3 Biotin | Biolegend | 100304 145-2C11 |
| B220 Biotin | Biolegend | 103204 RA3-6B2 |
| Gr1 Biotin | Biolegend | 108404 RB6-8C5 |
| CD8a Biotin | Biolegend | 100704 536–6.8 |
| Mac1 Biotin | Biolegend | 101204 M1/70 |
| Ter119 Biotin | Biolegend | 116204 TER-119 |
| CD4 Biotin | Biolegend | 100404 GK1.5 |
| CD48 FITC | Biolegend | 103404 HM48-1 |
| CD150 PE | Biolegend | 115904 TC15-12F12.2 |
| c-Kit APC | Biolegend | 105812 2B8 |
| Sca1 Pacific Blue | Biolegend | 108120 D7 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| LPS | Invivogen | Cat# tlrl-eklps |
| Nigericin | Sigma | Cat# N7143 |
| ATP | Invivogen | Cat#tlrl-atpl |
| Flagellin | Invivogen | Cat#tlrl-epstfla |
| dsDNA | Invivogen | Cat# tlrl-patn |
| D-glucose | Sigma | Cat# G8270 |
| Insulin | Sigma | Cat# I0516 |
| Dulbecco’s Modification of Eagle’s Medium | Invitrogen | Cat# 11965092 |
| Fetal Bovine Serum | Invitrogen | Cat#10437-028 |
| Penicillin Streptomycin Solution (100x) | Invitrogen | Cat# 15140122 |
| 0.25% Trypsin | Invitrogen | Cat# 25200056 |
| TRIzol Reagent | Invitrogen | Cat# 15596026 |
| RNAiMax | Invitrogen | Cat# 13778150 |
| ANTI-FLAG® M2 Affinity Gel | Sigma | Cat# A2220 |
| Protein A/G PLUS-Agarose | Santa Cruz | Cat# sc-2003 |
| Protease inhibitor | Thermo | Cat# A32963 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| trichloroacetic acid (TCA) | Sigma | Cat# T0699 |
| CD117 (c-kit) MicroBeads, mouse | Miltenyi Biotec | Cat# 130-094-224 |
| Stemspan SFEM | Stemcell technologies | Cat# 09600 |
| ES-Cult FBS | Stemcell technologies | Cat# 06952 |
| Murine IL3 | Peprotech | Cat# 213–13 |
| Murine IL6 | Peprotech | Cat# 216–16 |
| Murine Flt3 ligand | Peprotech | Cat# 250–31L |
| Murine TPO | Peprotech | Cat# 315–14 |
| Murine SCF | Peprotech | Cat# 250–03 |
| Murine GM-CSF | Peprotech | Cat# 315–03 |
| Estradiol | Sigma | E8875 |
| Puromycin | Sigma | P9620 |
| Ficoll-Paque plus | GE-healthcare Life Science | 17144002 |
| Critical Commercial Assays | ||
| Mouse IL-18 ELISA Kit | MBL international | Cat#7625 |
| Mouse Insulin ELISA Kit | Crystal Chem | Cat#90080 |
| Eva qPCR SuperMix kit | BioChain Institute | Cat# K5052200 |
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 210518 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-3216 |
| NLRP3 KO macrophages | (Py et al., 2013) | N/A |
| NG5 | (Py et al., 2013) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: SIRT2 KO | (Bobrowska et al., 2012) | N/A |
| Mouse: Caspase1 KO | (Kuida et al., 1995) | N/A |
| Mouse: C57BL/6J | National Institute on Aging | |
| Oligonucleotides | ||
| SIRT2 qPCR forward TGGGCTGGATGAAAGAGAA |
Elim | N/A |
| SIRT2 qPCR reverse GGTCCACCTTGGAGAAGTCTG |
Elim | N/A |
| SIRT1 qPCR forward GCAACAGCATCTTGCCTGAT |
Elim | N/A |
| SIRT1 qPCR reverse GTGCTACTGGTCTCACTT |
Elim | N/A |
| Actin qPCR forward GATCTGGCACCACACCTTCT |
Elim | N/A |
| Actin qPCR reverse GGGGTGTTGAAGGTCTCAAA |
Elim | N/A |
| SIRT2 siRNA CCAGAATAAGGCATTTCTCTA |
Qiagen | N/A |
| SIRT1 siRNA AAGCGGCTTGAGGGTAATCAA |
Qiagen | N/A |
| Recombinant DNA | ||
| pMSCVgfp-NLRP3 | (Fernandes-Alnemri et al., 2009) | |
| pMSCV-ASC-EGFP | (Fernandes-Alnemri et al., 2009) | |
| pFUGw-SIRT2 | (Luo et al., 2019) | |
| pMSCV-Hoxb8 | (Wang et al., 2006) | |
| EcoPac | (Wang et al., 2006) | |
| Software and Algorithms | ||
| Coot | (Emsley and Cowtan, 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| YASARA energy minimization server | (Krieger et al., 2009) | http://www.yasara.org/minimizationserver.htm |
| PDBePISA server | (Krissinel and Henrick, 2007) | http://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver |
| PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC | (Delano, 2002) | https://pymol.org/2/ |
| Other | ||
| N/A | ||
Highlights.
NLRP3 is modified by acetylation and is deacetylated by SIRT2.
NLRP3 acetylation facilitates the activation of the NLRP3 inflammasome.
SIRT2 prevents and reverses aging-associated inflammation and insulin resistance.
NLRP3 deacetylation regulates aging-associated inflammation and insulin resistance.
Context and Significance.
It is well documented that the rate of aging can be slowed, but it remains unclear to which extent aging-associated conditions can be reversed. Understanding the reversibility of aging-associated conditions has important implications in treating aging-related diseases. Here researchers at the University of California, Berkeley discovered that the SIRT2 enzyme regulates the NLRP3 inflammasome, a cellular machinery that produces inflammatory cytokines, and prevents inflammation and insulin resistance in aged but not young mice. The researchers demonstrated that SIRT2 and NLRP3 modulation can be targeted to reverse inflammation and insulin resistance in aged mice. These findings demonstrate an underlying cause of aging-associated inflammation and highlight the reversibility of aging-associated conditions.
ACKNOWLEDGMENTS
We thank R. Vance and H. Mao for discussion, L. Kohlstaedt for mass spectrometry, B. Py, J. Yuan, E. Alnemri for NG5 and NLRP3 KO macrophages, NLRP3 and ASC constructs, M. Kamps for MSCV-HoxB8 construct. Supported by NIH R01DK 117481 (D.C.), R01DK101885 (D.C.), R01AG063404 (D.C.), R01AG 063389 (D.C.), DP1HD087988 and R01Al124491 (H.W.), National Institute of Food and Agriculture (D.C.), France-Berkeley Fund (D. C.), Glenn/AFAR Scholarship (H.L.), James C.Y. Soong Fellowship (H. C., W.-C. M.), Government Scholarship for Study Abroad (GSSA) from Taiwan (W.-C. M.), the ITO Scholarship (R.O.), and the Honjo International Scholarship (R.O.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and one table.
REFERENCES
- Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, and Sethi G (2006). Inflammation and cancer: how hot is the link? Biochem Pharmacol 72, 1605–1621. [DOI] [PubMed] [Google Scholar]
- Barbieri M, Ferrucci L, Ragno E, Corsi A, Bandinelli S, Bonafe M, Olivieri F, Giovagnetti S, Franceschi C, Guralnik JM, et al. (2003). Chronic inflammation and the effect of IGF-I on muscle strength and power in older persons. Am J Physiol Endocrinol Metab 284, E481–487. [DOI] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183, 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley LE, Koster A, Newman AB, Javaid MK, Ferrucci L, Kritchevsky SB, Kuller LH, Pahor M, Schaap LA, Visser M, et al. (2009). Inflammation and race and gender differences in computerized tomography-measured adipose depots. Obesity (Silver Spring) 17, 1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrowska A, Donmez G, Weiss A, Guarente L, and Bates G (2012). SIRT2 ablation has no effect on tubulin acetylation in brain, cholesterol biosynthesis or the progression of Huntington’s disease phenotypes in vivo. PLoS One 7, e34805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefond A, Skrobek B, Lobbens S, Eury E, Thuillier D, Cauchi S, Lantieri O, Balkau B, Riboli E, Marre M, et al. (2013). Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat Genet 45, 1040–1043. [DOI] [PubMed] [Google Scholar]
- Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, and Chen D (2013). SIRT3 reverses aging-associated degeneration. Cell Rep 3, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al. (2012). Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 44, 1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier A, Cote M, Lemieux I, Perusse L, Tremblay A, Bouchard C, and Despres JP (2009). Age-related differences in inflammatory markers in men: contribution of visceral adiposity. Metabolism 58, 1452–1458. [DOI] [PubMed] [Google Scholar]
- Cesari M, Penninx BW, Newman AB, Kritchevsky SB, Nicklas BJ, Sutton-Tyrrell K, Rubin SM, Ding J, Simonsick EM, Harris TB, et al. (2003). Inflammatory markers and onset of cardiovascular events: results from the Health ABC study. Circulation 108, 2317–2322. [DOI] [PubMed] [Google Scholar]
- Cesari M, Penninx BW, Pahor M, Lauretani F, Corsi AM, Rhys Williams G, Guralnik JM, and Ferrucci L (2004). Inflammatory markers and physical performance in older persons: the InCHIANTI study. J Gerontol A Biol Sci Med Sci 59, 242–248. [DOI] [PubMed] [Google Scholar]
- Chen D, and Kerr C (2019). The Epigenetics of Stem Cell Aging Comes of Age. Trends Cell Biol. [DOI] [PMC free article] [PubMed]
- Chung HY, Sung B, Jung KJ, Zou Y, and Yu BP (2006). The molecular inflammatory process in aging. Antioxid Redox Signal 8, 572–581. [DOI] [PubMed] [Google Scholar]
- Cociorva D, T. D,L, and Yates JR (2007). Validation of tandem mass spectrometry database search results using DTASelect. Curr Protoc Bioinformatics Chapter 13, Unit 13 14. [DOI] [PubMed]
- Cushman M, Arnold AM, Psaty BM, Manolio TA, Kuller LH, Burke GL, Polak JF, and Tracy RP (2005). C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 112, 25–31. [DOI] [PubMed] [Google Scholar]
- Delano WL (2002). The PyMol Molecular Graphics System.
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. (2011). Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fagiolo U, Cossarizza A, Scala E, Fanales-Belasio E, Ortolani C, Cozzi E, Monti D, Franceschi C, and Paganelli R (1993). Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol 23, 2375–2378. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Datta P, Wu J, and Alnemri ES (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, Guralnik JM, and Longo DL (2005). The origins of age-related proinflammatory state. Blood 105, 2294–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Deng CX, and Mostoslavsky R (2009). Recent progress in the biology and physiology of sirtuins. Nature 460, 587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsey RJ, Thompson JM, Ernerudh J, Hurst TL, Strindhall J, Johansson B, Nilsson BO, and Wikby A (2003). Plasma cytokine profiles in elderly humans. Mech Ageing Dev 124, 487–493. [DOI] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell MA, and Rando TA (2015). Stem cells and healthy aging. Science 350, 1199–1204. [DOI] [PubMed] [Google Scholar]
- Guo H, Callaway JB, and Ting JP (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature medicine 21, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager K, Machein U, Krieger S, Platt D, Seefried G, and Bauer J (1994). Interleukin-6 and selected plasma proteins in healthy persons of different ages. Neurobiol Aging 15, 771–772. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu FC, Kritchevsky SB, Liu Y, Kanaya A, Newman AB, Perry SE, Visser M, Pahor M, Harris TB, Nicklas BJ, et al. (2009). Association between inflammatory components and physical function in the health, aging, and body composition study: a principal component analysis approach. J Gerontol A Biol Sci Med Sci 64, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Williams KL, Gunn MD, and Shinohara ML (2012). NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America 109, 10480–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Wang S, Xiao M, Lin Y, Zhou L, Lei Q, Xiong Y, Guan KL, and Zhao S (2011). Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell 43, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H, Hu J, He B, Negron Abril YL, Stupinski J, Weiser K, Carbonaro M, Chiang YL, Southard T, Giannakakou P, et al. (2016). A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 29, 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, Tam J, Han T, Mukhopadhyay B, Skarulis MC, et al. (2013). Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nature medicine 19, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, Liu Y, Hoffmann U, Bauer DC, Newman AB, et al. (2010). Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol 55, 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kania DM, Binkley N, Checovich M, Havighurst T, Schilling M, and Ershler WB (1995). Elevated plasma levels of interleukin-6 in postmenopausal women do not correlate with bone density. J Am Geriatr Soc 43, 236–239. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ (2010). The genetics of ageing. Nature 464, 504–512. [DOI] [PubMed] [Google Scholar]
- Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, and Karplus K (2009). Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 77 Suppl 9, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, and Flavell RA (1995). Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267, 2000–2003. [DOI] [PubMed] [Google Scholar]
- Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA, Dulken BW, Zhao X, Yeo RW, Ho TT, et al. (2018). Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359, 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J 3rd, et al. (2008). A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 456, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, and Egelman EH (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR, and Birnbaum MJ (2012). Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 18, 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Chiang HH, Louw M, Susanto A, and Chen D (2017). Nutrient Sensing and the Oxidative Stress Response. Trends Endocrinol Metab 28, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Mu WC, Karki R, Chiang HH, Mohrin M, Shin JJ, Ohkubo R, Ito K, Kanneganti TD, and Chen D (2019). Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep 26, 945–954 e944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio M, Basaria S, Ble A, Lauretani F, Bandinelli S, Ceda GP, Valenti G, Ling SM, and Ferrucci L (2006). Correlation between testosterone and the inflammatory marker soluble interleukin-6 receptor in older men. J Clin Endocrinol Metab 91, 345–347. [DOI] [PubMed] [Google Scholar]
- Maggio M, Basaria S, Ceda GP, Ble A, Ling SM, Bandinelli S, Valenti G, and Ferrucci L (2005). The relationship between testosterone and molecular markers of inflammation in older men. J Endocrinol Invest 28, 116–119. [PubMed] [Google Scholar]
- Martinon F, Mayor A, and Tschopp J (2009). The inflammasomes: guardians of the body. Annu Rev Immunol 27, 229–265. [DOI] [PubMed] [Google Scholar]
- McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, Graumann J, Johnson JR, Cociorva D, and Yates JR 3rd (2004). MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun Mass Spectrom 18, 2162–2168. [DOI] [PubMed] [Google Scholar]
- McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, Understanding Society Scientific G, Crawley C, Craig J, Scott MA, et al. (2015). Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell reports 10, 1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Ou J, Ma Y, Guo F, Yang Z, Wiggins M, Liu C, Song W, Han X, Wang M, et al. (2014). Macrophage CGI-58 deficiency activates ROS-inflammasome pathway to promote insulin resistance in mice. Cell Rep 7, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, and Akira S (2013). Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature immunology 14, 454–460. [DOI] [PubMed] [Google Scholar]
- Mohrin M, and Chen D (2016). The mitochondrial metabolic checkpoint and aging of hematopoietic stem cells. Curr Opin Hematol 23, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, and Chen D (2015). Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysliwska J, Bryl E, Foerster J, and Mysliwski A (1998). Increase of interleukin 6 and decrease of interleukin 2 production during the ageing process are influenced by the health status. Mech Ageing Dev 100, 313–328. [DOI] [PubMed] [Google Scholar]
- Oroz J, Barrera-Vilarmau S, Alfonso C, Rivas G, and de Alba E (2016). ASC Pyrin Domain Self-associates and Binds NLRP3 Protein Using Equivalent Binding Interfaces. J Biol Chem 291, 19487–19501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Venable JD, Xu T, and Yates JR 3rd (2008). A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods 5, 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ, and Gygi SP (2003). Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2, 43–50. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Koditz R, Pfohl M, and Schatz H (2002). Changes in proinflammatory cytokine activity after menopause. Endocr Rev 23, 90–119. [DOI] [PubMed] [Google Scholar]
- Pickup JC, Chusney GD, Thomas SM, and Burt D (2000). Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci 67, 291–300. [DOI] [PubMed] [Google Scholar]
- Place DE, and Kanneganti TD (2018). Recent advances in inflammasome biology. Curr Opin Immunol 50, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pou KM, Massaro JM, Hoffmann U, Vasan RS, Maurovich-Horvat P, Larson MG, Keaney JF Jr., Meigs JB, Lipinska I, Kathiresan S, et al. (2007). Visceral and subcutaneous adipose tissue volumes are cross-sectionally related to markers of inflammation and oxidative stress: the Framingham Heart Study. Circulation 116, 1234–1241. [DOI] [PubMed] [Google Scholar]
- Pradhan AD, Manson JE, Rifai N, Buring JE, and Ridker PM (2001). C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286, 327–334. [DOI] [PubMed] [Google Scholar]
- Py BF, Kim MS, Vakifahmetoglu-Norberg H, and Yuan J (2013). Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49, 331–338. [DOI] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, and Chen D (2010). Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 12, 662–667. [DOI] [PubMed] [Google Scholar]
- Ravaglia G, Forti P, Maioli F, Chiappelli M, Montesi F, Tumini E, Mariani E, Licastro F, and Patterson C (2007). Blood inflammatory markers and risk of dementia: The Conselice Study of Brain Aging. Neurobiol Aging 28, 1810–1820. [DOI] [PubMed] [Google Scholar]
- Rodondi N, Marques-Vidal P, Butler J, Sutton-Tyrrell K, Cornuz J, Satterfield S, Harris T, Bauer DC, Ferrucci L, Vittinghoff E, et al. (2010). Markers of atherosclerosis and inflammation for prediction of coronary heart disease in older adults. Am J Epidemiol 171, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaap LA, Pluijm SM, Deeg DJ, Harris TB, Kritchevsky SB, Newman AB, Colbert LH, Pahor M, Rubin SM, Tylavsky FA, et al. (2009). Higher inflammatory marker levels in older persons: associations with 5-year change in muscle mass and muscle strength. J Gerontol A Biol Sci Med Sci 64, 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, Nabavi N, Mohrin M, Wojnoonski K, et al. (2013). SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep 5, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Zhang D, and Chen D (2011). Reversible acetylation of metabolic enzymes celebration: SIRT2 and p300 join the party. Mol Cell 43, 3–5. [DOI] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, and Prolla TA (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, Liu X, Huang YJ, Cai H, Zhan XY, et al. (2017). NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell 68, 185–197 e186. [DOI] [PubMed] [Google Scholar]
- Strowig T, Henao-Mejia J, Elinav E, and Flavell R (2012). Inflammasomes in health and disease. Nature 481, 278–286. [DOI] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, and Yates JR 3rd (2002). DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et al. (2010). Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy RP, Lemaitre RN, Psaty BM, Ives DG, Evans RW, Cushman M, Meilahn EN, and Kuller LH (1997). Relationship of C-reactive protein to risk of cardiovascular disease in the elderly. Results from the Cardiovascular Health Study and the Rural Health Promotion Project. Arterioscler Thromb Vasc Biol 17, 1121–1127. [DOI] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, and Dixit VD (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, Boss O, Hirsch ML, Ribich S, Smith JJ, et al. (2010). Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev 24, 1403–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, and Kamps MP (2006). Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nature methods 3, 287–293. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. (2010). Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, and Yates JR 3rd (2001). Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19, 242–247. [DOI] [PubMed] [Google Scholar]
- Weaver JD, Huang MH, Albert M, Harris T, Rowe JW, and Seeman TE (2002). Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology 59, 371–378. [DOI] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, and Ting JP (2011). Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12, 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. (2014). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature medicine 20, 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015). ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Lindquist K, Penninx BW, Simonsick EM, Pahor M, Kritchevsky S, Launer L, Kuller L, Rubin S, and Harris T (2003). Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology 61, 76–80. [DOI] [PubMed] [Google Scholar]
- Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, and Zhou R (2015). Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73. [DOI] [PubMed] [Google Scholar]
- Youm YH, Kanneganti TD, Vandanmagsar B, Zhu X, Ravussin A, Adijiang A, Owen JS, Thomas MJ, Francis J, Parks JS, et al. (2012). The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell reports 1, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, et al. (2009). Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. (2010). Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate unique datasets or code.