

Abstract
The association of WW domain-containing oxidoreductase WWOX gene loss of function with central nervous system (CNS) related pathologies is well documented. These include spinocerebellar ataxia, epilepsy and mental retardation (SCAR12, OMIM: 614322) and early infantile epileptic encephalopathy (EIEE28, OMIM: 616211) syndromes. However, there is complete lack of understanding of the pathophysiological mechanisms at play. In this study, using a Wwox knockout (Wwox KO) mouse model (2 weeks old, both sexes) and stereological studies we observe that Wwox deletion leads to a significant reduction in the number of hippocampal GABA-ergic (γ-aminobutyric acid) interneurons. Wwox KO mice displayed significantly reduced numbers of calcium-binding protein parvalbumin (PV) and neuropeptide Y (NPY) expressing interneurons in different subfields of the hippocampus in comparison to Wwox wild-type (WT) mice. We also detected decreased levels of Glutamic Acid Decarboxylase protein isoforms GAD65/67 expression in Wwox null hippocampi suggesting lower levels of GABA synthesis. In addition, Wwox deficiency was associated with signs of neuroinflammation such as evidence of activated microglia, astrogliosis, and overexpression of inflammatory cytokines Tnf-a and Il6. We also performed comparative transcriptome-wide expression analyses of neural stem cells grown as neurospheres from hippocampi of Wwox KO and WT mice thus identifying 283 genes significantly dysregulated in their expression. Functional annotation of transcriptome profiling differences identified ‘neurological disease’ and ‘CNS development related functions’ to be significantly enriched. Several epilepsy-related genes were found differentially expressed in Wwox KO neurospheres. This study provides the first genotype-phenotype observations as well as potential mechanistic clues associated with Wwox loss of function in the brain.
Keywords: Wwox, Epilepsy, Hippocampus, GABA-ergic interneurons, Microgliosis, Astrogliosis
1. Introduction
WW domain-containing oxidoreductase (WWOX) is a member of the short-chain dehydrogenase/reductase (SDR) protein superfamily (Bednarek et al., 2000). It spans the common fragile site FRA16D in chromosome region ch16q23.1–23.2 (Bednarek et al., 2000; Ried et al., 2000). WWOX was originally described by our and other laboratories as a putative tumor suppressor gene associated tumor progression, and therapy resistance in multiple cancer types [Reviewed in (Aldaz et al., 2014; Schrock et al., 2017)]. In more recent studies, WWOX has been recognized for its role in a much wider array of human pathologies including metabolic conditions and central nervous system (CNS) related syndromes (Aldaz et al., 2014; Chang et al., 2014; Mallaret et al., 2014).
Findings of Wwox protein expression and distribution in embryonic and adult murine nervous system highlighted a previously unidentified role of this protein in the normal development and function of CNS (Chen et al., 2004). WWOX protein expression was also observed in various neuronal cell types and regions from the human brain and cerebellum (Nunez et al., 2006). In 2007 Gribaa et al. described a novel childhood onset recessive spinocerebellar ataxia, epilepsy and mental retardation syndrome (cataloged as SCAR12, OMIM: 614322) affecting children from a large consanguineous family and the defective locus was mapped to chromosomal region ch16q21-q23 (Gribaa et al., 2007). Later, in 2014 Mallaret et al. by using whole exome sequencing, identified WWOX as the culprit defective gene in SCAR12. All probands of the family described by Gribaa et al. and from an additional consanguineous family carrying homozygous loss of function germline mutations affecting WWOX suffered from SCAR12 (Mallaret et al., 2014). Soon thereafter, other groups reported new familial cases of WWOX homozygous loss of function associated with early infantile epileptic encephalopathy (EIEE28, OMIM: 616211) also known as WOREE syndrome (WWOX-related epileptic encephalopathy) (Abdel-Salam et al., 2014; Ben-Salem et al., 2015; Mignot et al., 2015; Tabarki et al., 2015; Valduga et al., 2015; Elsaadany et al., 2016). Most of these familial cases are characterized by homozygous missense, non-sense, or splice site mutations combined in some cases with deletions ultimately leading to WWOX loss of function (Mignot et al., 2015). In most cases of complete loss of WWOX function (e.g. homozygous non-sense mutations) patients presented severe brain structural defects and died within the first two years of life (Abdel-Salam et al., 2014; Ben-Salem et al., 2015; Tabarki et al., 2015; Valduga et al., 2015). Consistent with observations in humans, we reported that Wwox gene deletion leads to epileptogenesis and early death in mice, where all mice die by approximately 3 weeks of age (Ludes-Meyers et al., 2009; Mallaret et al., 2014). This is also in agreement with previous observations in rats spontaneously mutated at the Wwox locus (Suzuki et al., 2009). Notably, while the association of WWOX with CNS pathology is evident from the multiple human and rodent studies, there is complete lack of understanding on the pathophysiological mechanisms associated with WWOX loss of function in brain.
Clinical and experimental studies have demonstrated a clear association between certain types of epilepsy and hippocampal structural alterations (Schwartzkroin, 1994). Activated microglia and astrogliosis, are among the most evident deleterious changes affecting hippocampus function as a consequence of epileptic seizures (Devinsky et al., 2013). Thus, in this study, using Wwox KO mice we investigated the effects of Wwox deletion in affecting the abundance of hippocampal GABA-ergic inhibitory interneurons while investigating effects on inflammatory markers such as evidence of increased numbers or hypertrophy of glial fibrillary acidic protein (GFAP) positive astrocytes and ionized calcium binding adaptor molecule-1 (IBA-1) positive activated microglia. In an attempt to further understand the molecular effects of Wwox deletion in hippocampus we also performed RNA sequencing (RNA-Seq) analyses to compare the transcriptome profile of neural stem cell neurospheres generated from hippocampi of Wwox KO vs. WT mice.
2. Material and methods
2.1. Animal experiments
The protocol for the generation of full Wwox KO mice has been previously described by our group (Ludes-Meyers et al., 2009; Mallaret et al., 2014). Briefly, we crossed male Wwox flox/wt mice with female BK5-Cre + Wwox flox/wt mice (both in mixed 129SV/C57Bl/6 background). As described by Ramirez et al. Cre recombinase in BK5-Cre female breeder mice is activated in late oocytes with Cre protein persisting in the embryo leading to constitutive recombination and producing full knockout progeny in embryos that are flox/flox (Ramirez et al., 2004). As controls we used Cre+; Wwox wt/wt littermates (referred to as Wwox WT elsewhere in the text). Wwox KO mice (2 weeks old) and age-matched WT controls were used for comparative analysis. Mice from both sexes approximately 50% male, 50% female were used for all studies. All animals were bred and kept in a clean, modified-barrier animal facility, fed regular commercial mouse diet (Harlan Lab., Indianapolis, IN) under controlled light (12 L:12D) and temperature (20–24 °C). All animal research was conducted in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) at the University of Texas, MD Anderson Cancer Center, Science Park, following international guidelines and all research was approved by the corresponding Institutional Animal Care and Use Committee (IACUC).
2.2. Animal perfusion and tissue processing
Wwox KO and WT mice were subjected to terminal anesthesia with isoflurane and trans-cardiac perfusion with 4% paraformaldehyde solution in phosphate buffer (PB). Following this, the brain was carefully removed from the skull of each animal and post-fixed in 4% paraformaldehyde for ~16 h at 4 °C. The brain tissues were next treated with 30% sucrose solution in PB until it sank to the bottom and thirty-micrometer thick cryostat sections were cut coronally through the entire septo-temporal axis of the hippocampus. The sections were collected serially in 24-well plates filled with PBS. Every 20th section through the entire hippocampus was then selected from Wwox KO and WT mice and processed for immunohistochemistry, utilizing various antibodies for specific cell identification as described below.
2.3. Immunohistochemistry
Several sets of serial sections (every 15th or 20th) through the entire hippocampus were selected and processed for immunohistochemistry. The procedures used for immunostaining have been previously described (Mishra et al., 2015). Briefly, the sections were etched with PBS solution containing 20% methanol and 3% hydrogen peroxide for 20 min, rinsed thrice in PBS solution, and treated for 30 min in PBS solution containing 0.1% Triton-X 100 and an appropriate serum (10%) selected on the basis of the species in which the chosen secondary antibody was raised. The primary antibodies comprised anti-PV (P3088; Sigma-Aldrich), anti-NPY (T-4070; Peninsula Laboratories), anti-IBA-1 (ab5076; Abcam), or anti-GFAP (MAB360; Millipore). After an overnight incubation with the respective primary antibody solution, sections were washed thrice in PBS solution and incubated in an appropriate secondary antibody solution for 1 h. Biotinylated anti-rabbit (BA-1000; Vector Lab) and biotinylated anti-mouse (BA-2000; Vector Lab) were used as secondary antibodies in this study. The sections were washed thrice in PBS solution and treated with avidin-biotin complex reagent (PK-6100; Vector Lab) for 1 h. Peroxidase reaction was developed by using diaminobenzidine (SK-4100; Vector Lab) or vector SG (SK-4700; Vector Lab) as chromogens, and the sections were mounted on gelatin-coated slides, dehydrated, cleared, and cover slipped with Permount.
2.4. Stereological counting of PV+ and NPY+ GABA-ergic Interneurons
Numbers of PV+ or NPY+ interneurons were stereologically counted for the DG, CA1 and CA3 subfields of the hippocampus. All counts utilized every 15th or 20th section through the entire septo-temporal axis of the hippocampus and the optical fractionator method available in the StereoInvestigator system (Microbrightfield Inc.). The StereoInvestigator system consisted of a color digital video camera (Optronics Inc.) interfaced with a Nikon E600 microscope. A detailed protocol employed for cell counting using the optical fractionator has been previous described (Megahed et al., 2014).
2.5. Image analyses of IBA-1+, and GFAP+ immunoreactive structures in the hippocampus
The areas occupied by IBA-1+ microglia and GFAP+ astrocyte elements (area fractions) were measured per unit area of the tissue in the DG, CA1, and CA3 subfields of the hippocampus using Image J, as detailed in our previous reports (Kodali et al., 2015). In brief, images from the chosen subfields of the hippocampus were first digitized using 20× lens in a Nikon microscope outfitted with a digital video camera fixed to a computer and then saved in gray scale as a bitmap file. Using Image J, a binary image was created from each picture by opting a threshold that retained all IBA-1+ or GFAP+ immunopositive structures but no background. The area occupied by the IBA-1+ or GFAP+ structures (i.e., area fraction) in the binary image was quantified by selecting the Analyze command in the program. Area fraction of IBA-1+ or GFAP+ immunoreactive elements was calculated separately for DG, CA1, and CA3 subfields in each animal by using data from all chosen serial sections.
2.6. Statistical analysis
All the analysis (Wwox KO vs. WT) of the various cell subtypes was performed using statistical software GraphPad PRISM 7. Student’s two-tailed unpaired t-test was used to compare interneurons numbers and additional biomarkers. P values of < 0.05 were considered significant.
2.7. Immunoblot analysis
Total protein extracts were prepared from mice hippocampi. Protein isolation was done using RIPA buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 100 mM NaF, 1 mM Na3VO4, 1 mM EGTA, 1% Triton X-100, 10% Glycerol) with protease inhibitor cocktail (Roche). For western blotting, 20 μg of total protein were separated on 10% SDS-PAGE and transferred to a PVDF membrane. Membranes were blocked and antibodies were diluted in 5% dry milk/TBST. Primary antibodies used were: Gad65/67 (Millipore) 1:500 dilution, Wwox (using affinitypurified anti-WWOX rabbit polyclonal primary antibodies developed in our laboratory) 1:1000 dilution, HRP conjugated secondary antibodies were used followed by chemiluminescence (GE Healthcare). Actin was used as the protein loading control, and it was detected using monoclonal anti-actin antibody (Sigma) 1:5000 dilution and HRP conjugated anti-mouse secondary antibody 1:5000 dilution.
2.8. Generation of neurospheres
The detailed procedure for neural stem cells neurospheres generation and propagation is described elsewhere (Guo et al., 2012). Briefly, after euthanizing newborn Wwox KO or WT mice, the brain was collected in 20 ml cold HBSS (Hank’s Balanced Salt Solution; Invitrogen/Gibco) in a sterile petri dish. Hippocampus was dissected from the brain and carefully chopped into smaller pieces. Next, 1 ml Trypsin (0.5%) (SIGMA) was added, and tissue was incubated for 10–15 min at 37 °C to dissociate the cells. To remove undissipated tissue particles, the cell suspension was filtered through 70 μm strainer and centrifuged for 5 min at 3000 rpm. Cell pellet was suspended in neurobasal medium (NBM, Thermo Fisher Scientific) containing 20 ng/ml EGF, 10 ng/ml FGF, 2 mM glutamine, antibiotics (100 units/ml penicillin and 100 g/ml streptomycin), and 0.125 μg/ml fungizone, plated in 6-well plates and maintained at an atmosphere of 5% CO2 at 37 °C. After 4–7 days of incubation, neural stem cell formed neurospheres. Subcultures were prepared every 4–5 days by centrifugation of the neurospheres and dissociation of cells in trypsin; single-cell suspensions were re-plated in new culture dishes in fresh medium to obtain new neurospheres.
2.9. RNA-Seq and data analysis
RNA was isolated from neural stem cell neurospheres generated from Wwox KO and WT mouse hippocampi. To this end, we used TRIzol reagent (Invitrogen) and the RNeasy mini kit (Qiagen). RNA concentration and integrity were measured on an Agilent 2100 Bioanalyzer (Agilent Technologies). Only RNA samples with RNA integrity values (RIN) over 8.0 were considered for subsequent analysis. mRNA samples were processed for directional mRNA-seq library construction using the ScriptSeq v2 RNA-Seq Library Preparation Kit (Epicentre) according to the manufacturer’s protocol. We performed 76 nt paired-end sequencing using an Illumina HiSeq3000 platform and obtained ~40 million tags per sample. The short-sequenced reads were mapped to the mouse reference genome (mm10) by the splice junction aligner TopHat V2.0.10. We employed several R/Bioconductor packages to calculate gene expression abundance at the whole-genome level using the aligned records (BAM files) and to identify differentially expressed genes between Wwox KO and WT neurospheres. Briefly, to identify differentially expressed genes between Wwox KO and WT samples we computed fold change using the EdgeR Bioconductor package based on the normalized log2 based count per million values (Robinson et al., 2010). Data integration and visualization of differentially expressed transcripts between Wwox KO and WT mouse hippocampus derived neurospheres (log2 fold change [log2 FC] > ± 1, p-value < .005, false discovery rate [FDR] < 0.05) was done with the Multiexperiment Viewer Software. Functional enrichment analysis of dysregulated transcripts was performed using the Ingenuity Pathway Analysis (IPA) database (http://www.ingenuity.com/index.html).
2.10. Quantitative RT-PCR (qRT-PCR)
Total RNA from neurospheres was isolated as described above. Hippocampal tissue RNA was isolated using TRIzol reagent (Invitrogen) following manufacturer’s instructions. RNA was quantified, and cDNA was prepared using RevertAid First Strand cDNA Synthesis Kit (ThermoFisher, Scientific) following manufacturer’s instructions. The relative expression level for specific genes was determined in triplicate by qRT-PCR using the SYBR Green-based method. After normalization to Gapdh expression, the average fold change between Wwox KO and WT samples was calculated using the 2^-(ΔΔCt) method described elsewhere (Livak and Schmittgen, 2001). Student’s two-tailed unpaired t-test was used to compare gene expression values, p-values of < 0.05 were considered significant. Primer sequences of the genes tested by qRT-PCR are shown in Supplementary Table 1.
3. Results
3.1. Wwox KO mice hippocampi exhibit a decrease in PV+ and NPY+ inhibitory interneurons
Inhibitory signals from various subpopulations of GABA-ergic interneurons to principal neurons are critical to maintain the stability of the neuronal circuitry. The loss of GABA-ergic interneurons alters the balance of excitatory and inhibitory synaptic transmission inducing hippocampal hyperexcitability potentially leading to epileptiform activity and affecting cognitive functions (Liu et al., 2014). We first examined the distribution of fast-spiking parvalbumin expressing (PV+) GABA-ergic interneurons in the DG (dentate gyrus) and the CA1 and CA3 (cornu ammonis) hippocampal subfields of Wwox KO (n = 7) and WT (n = 7) mice. To this end, we used immunohistochemical staining of coronal brain sections with an antibody against PV followed by stereological quantification (Fig. 1). In the DG region of Wwox WT mice, PV+ interneurons were clearly observed (Fig. 1 A1). In contrast, the DG region of Wwox KO mice displayed a significant reduction in number of PV+ interneurons. Some hippocampal areas in Wwox KO mice showed a complete absence of such cells (Fig. 1 B1). A significantly low number of PV+ interneurons in Wwox KO mice was also evident in the CA1 and CA3 subfields (Figs. 1 A2–A3, B2–B3, and C2–C3). Stereological quantification indicated that Wwox KO mice displayed astatistically significant reduction in the total number of PV+ interneurons in the DG region (p < .005, Fig. 1 C1) and CA1 subfield (p < .05, Figs. 1 C2), in comparison to control mice. The overall reduction of PV+ interneurons in Wwox KO mice amounted to 44% when the hippocampus was taken in its entirety (p < .005, Fig. 1 D).
Fig. 1. |. Wwox KO mice hippocampi exhibit a decrease in PV+ interneurons.
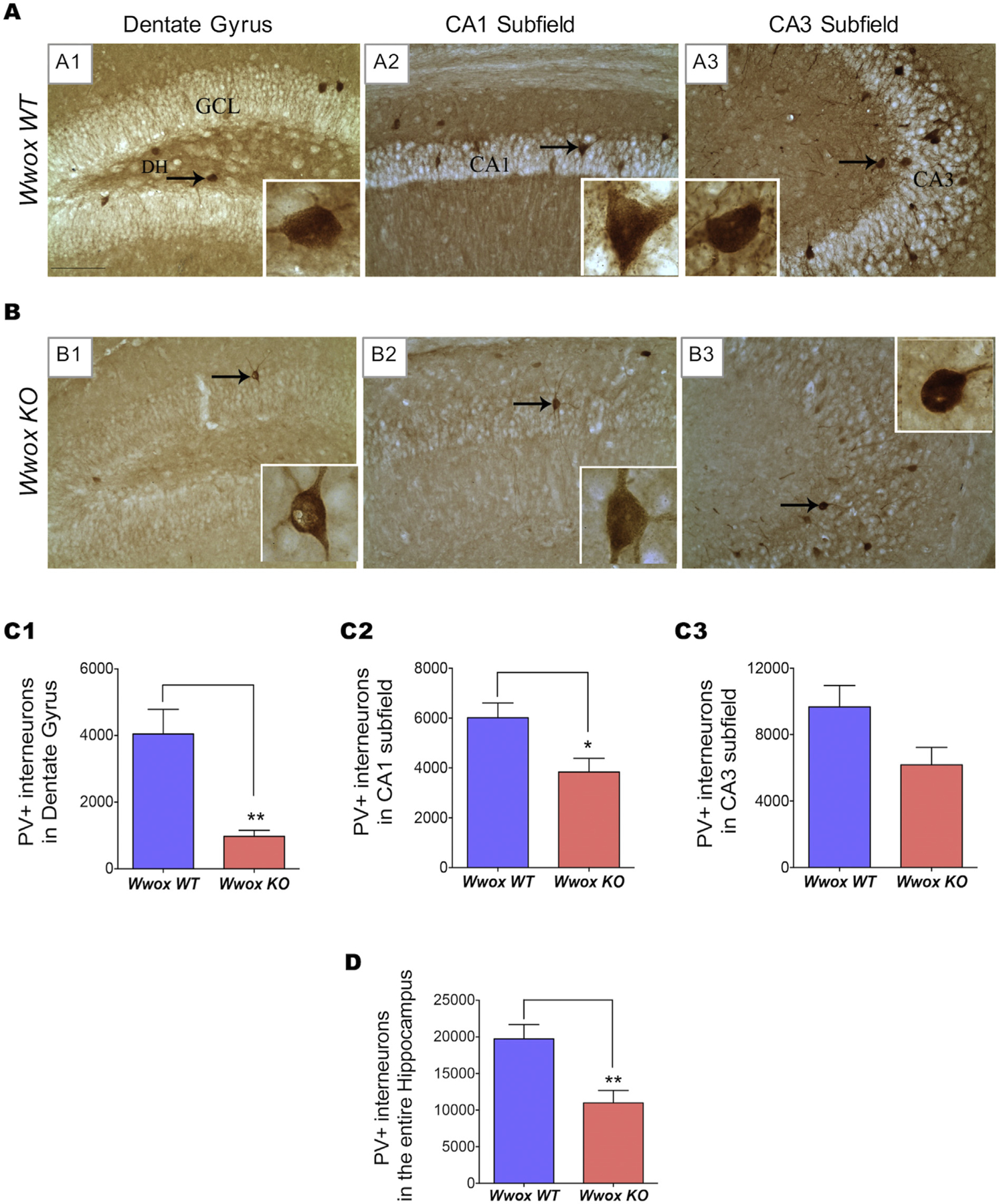
Panels (A) and (B) show representative microphotographs illustrating the distribution of PV+ interneurons in DG (dentate gyrus) and the CA1 and CA3 (cornu ammonis) subfields of the hippocampus of 2 wk. old Wwox WT(A1–A3) and Wwox KO(B1–B3) mice. The insets show magnified images of single interneuron from each subfield (marked with arrows). A paucity of PV+ neuronal cell bodies and processes can be appreciated in the various hippocampal regions of Wwox KO mice (B1–B3)versus comparable regions from Wwox WT control counterparts. Bar charts (C1–C3 and D) show stereological results comparing the number of PV+ interneurons in DG (C1), CA1 subfield (C2), CA3 subfield (C3), and the entire hippocampus (D). Bar graphs represent the average values of Wwox WT (n = 7) and Wwox KO (n = 7) mice as indicated. Error bars represent ± SEM, *p < .05; **p < .005, two tailed unpaired Student’s t-test. DH, dentate hilus; GCL, granule cell layer.
We also analyzed the abundance of a second subpopulation of GABA-ergic interneurons, the NPY+ interneurons. The neuropeptide Y released from NPY+ GABA-ergic interneurons is an endogenous anticonvulsant neurotransmitter that modulates the activity of excitatory neurons (Vezzani et al., 1999). We calculated the abundance of these interneurons in hippocampal subfields of Wwox KO (n = 6) and WT (n = 6) mice by immunostaining of coronal brain sections using a NPY antibody followed by stereological analysis (Fig. 2). In the DG region of WT mice, significant numbers of NPY+ cells were clearly observed, especially in the dentate hilus region (Fig. 2 A1), in contrast the DG region of Wwox KO mice displayed a 30% reduction in the total number of NPY+ interneurons when compared to control WT mice (p < .05) (Figs. 2 B1 and C1). However, the CA1 and CA3 subfields, did not exhibit any obvious difference in the number of NPY+ interneurons between Wwox KO and WT mice (Figs. 2 A2–A3, B2–B3, and C2–C3). Number of NPY+ interneurons in the entire hippocampus is shown in Fig. 2 D.
Fig. 2. |. Wwox KO mice hippocampi exhibit a decrease in NPY+ interneurons.
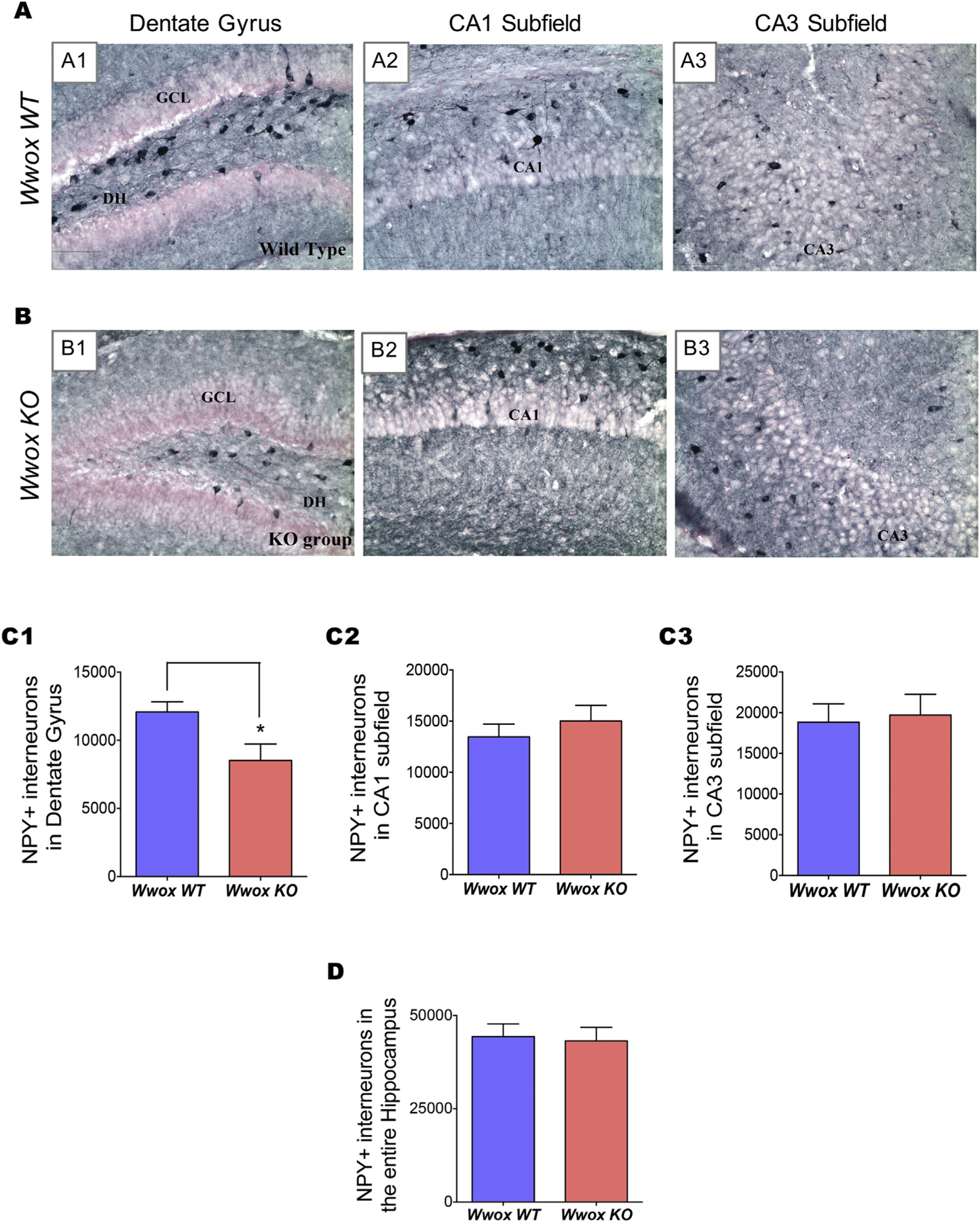
Panels (A) and (B) show representative images illustrating the distribution of NPY+ interneurons in DG and the CA1 and CA3 subfields of the hippocampus of 2 wk. old Wwox WT(A1–A3) and Wwox KO(B1–B3) mice. A significant reduction in the number of NPY+ cells is apparent in the DG subfield of a Wwox KO mouse (B1) in comparison to the same region from a Wwox WT counterpart (A1). Bar charts (C1–C3 and D) compare the average number of NPY+ interneurons in DG (C1), CA1 subfield (C2), CA3 subfield (C2), and the entire hippocampus (D) from Wwox WT (n = 6) and Wwox KO (n = 6) mice as indicated. Error bars represent ± SEM, *p < .05, two tailed unpaired Student’s t-test. DH, dentate hilus; GCL, granule cell layer.
In summary, Wwox KO mice exhibit significant reductions in the abundance of both PV+ and NPY+ GABA-ergic interneurons in the various hippocampal subfields.
3.2. Wwox KO mice hippocampi display evidence of microgliosis
Pathogenic insults to the CNS generate a non-specific reactive response affecting glial cells termed gliosis (Burda and Sofroniew, 2014). Gliosis involves hypertrophy and activation of different types of glial cells including microglia and astrocytes and is often the result of a reactive inflammatory response (Devinsky et al., 2013).Microgliosis (activation of microglia) is commonly the first observed stage of gliosis. We investigated microglia activation by immunohistochemical staining for ionized calcium binding adaptor molecule-1 (IBA-1), also known as allograft inflammatory factor expressed in activated macrophages, both in WT (n = 6) and Wwox KO (n = 6) mice (Fig. 3). The area fraction (AF) occupied by IBA-1+ cells was marginally higher in the DG region (Figs. 3 A1, B1 and C1), however the CA1 and CA3 subfields displayed a significant increase (p < .05) in the AF occupied by IBA-1+ structures in Wwox KO mice and when the hippocampus (p < .005) is considered in its entirety (Figs. 3 A2–A3, B2–B3, C2–C3, and D). Importantly, IBA-1+ cells in CA1 and CA3 subfields of Wwox KO were less ramified, displayed shorter and thicker processes and larger soma, characteristic of activated microglia (see Figs. 3 B2–B3 insets).
Fig. 3. |. Wwox KO mice hippocampi exhibit increase in IBA-1+ cells indicating microglia activation.
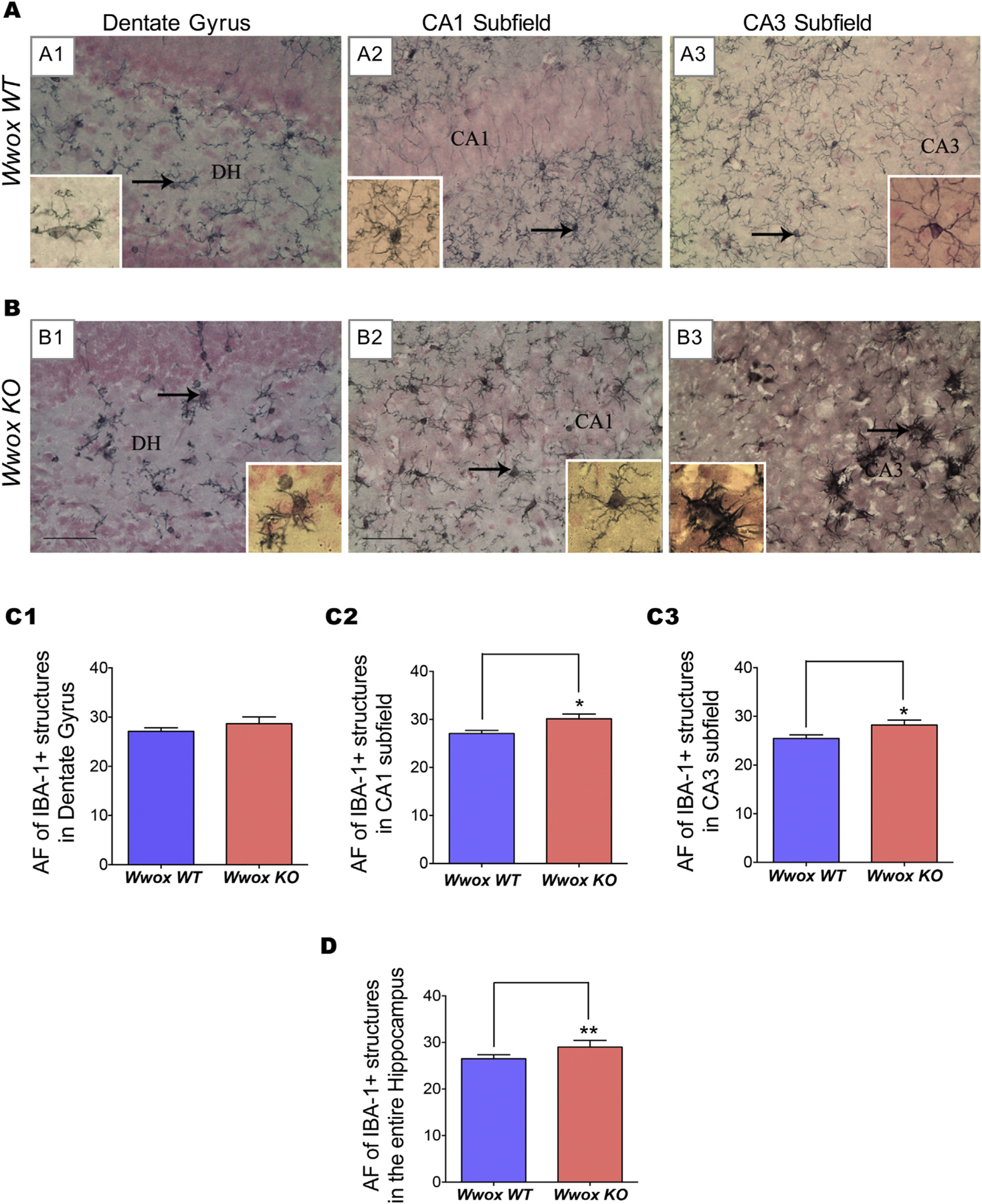
Panels (A) and (B) display representative images illustrating the distribution and morphology of IBA-1+ microglial cells in the DG and CA1 and CA3 subfields of the hippocampus of 2 wk. old Wwox WT(A1–A3) and Wwox KO(B1–B3) mice. A significant increase in the area fraction (AF) occupied by IBA-1+ structures in CA1 and CA3 subfield as well as entire hippocampus can be observed in Wwox KO mouse. Bar charts (C1–C3 and D) compared the average AF occupied by IBA-1+ astrocytes in DG (C1), CA1 subfield (C2), CA3 subfield (C3), and the entire hippocampus (D) from Wwox WT (n = 6) and Wwox KO (n = 6) mice as indicated. Error bars represent ± SEM, *p < .05, two tailed unpaired Student’s t-test. DH, dentate hilus.
3.3. Wwox KO mice hippocampi display evidence of astrogliosis and increased inflammatory cytokines expression
We also investigated for evidence of astrogliosis (activation of astrocytes) by means of glial fibrillary acidic protein (GFAP) immunostaining and area fraction analysis using Image J. We observed a significant increase in the area occupied by astrocytic elements in hippocampi from Wwox KO mice (n = 8) in comparison to WT mice (n = 6) (Fig. 4). Astrocytes displayed hypertrophy as well as hyperplasia in the hippocampus of Wwox KO mice. As a consequence, in Wwox KO mice, the area fraction (AF) occupied by GFAP+ soma and processes of astrocytes in the CA1 and CA3 subfields displayed a very significant increase of 36% and 53%, respectively (p < .05 Figs. 4 A2–A3, B2–B3, and C2–C3), whereas in the DG region the increment was marginal (Figs. 4 A1, B1 and C1). When the hippocampus was taken in its entirety, the overall increase in GFAP+ structures was 30% in Wwox KO mice (p < .05, Fig. 4 D).
Fig. 4. |. Wwox KO mice hippocampi exhibit increase in GFAP+ astroglial activation as evidence of increase gliosis.
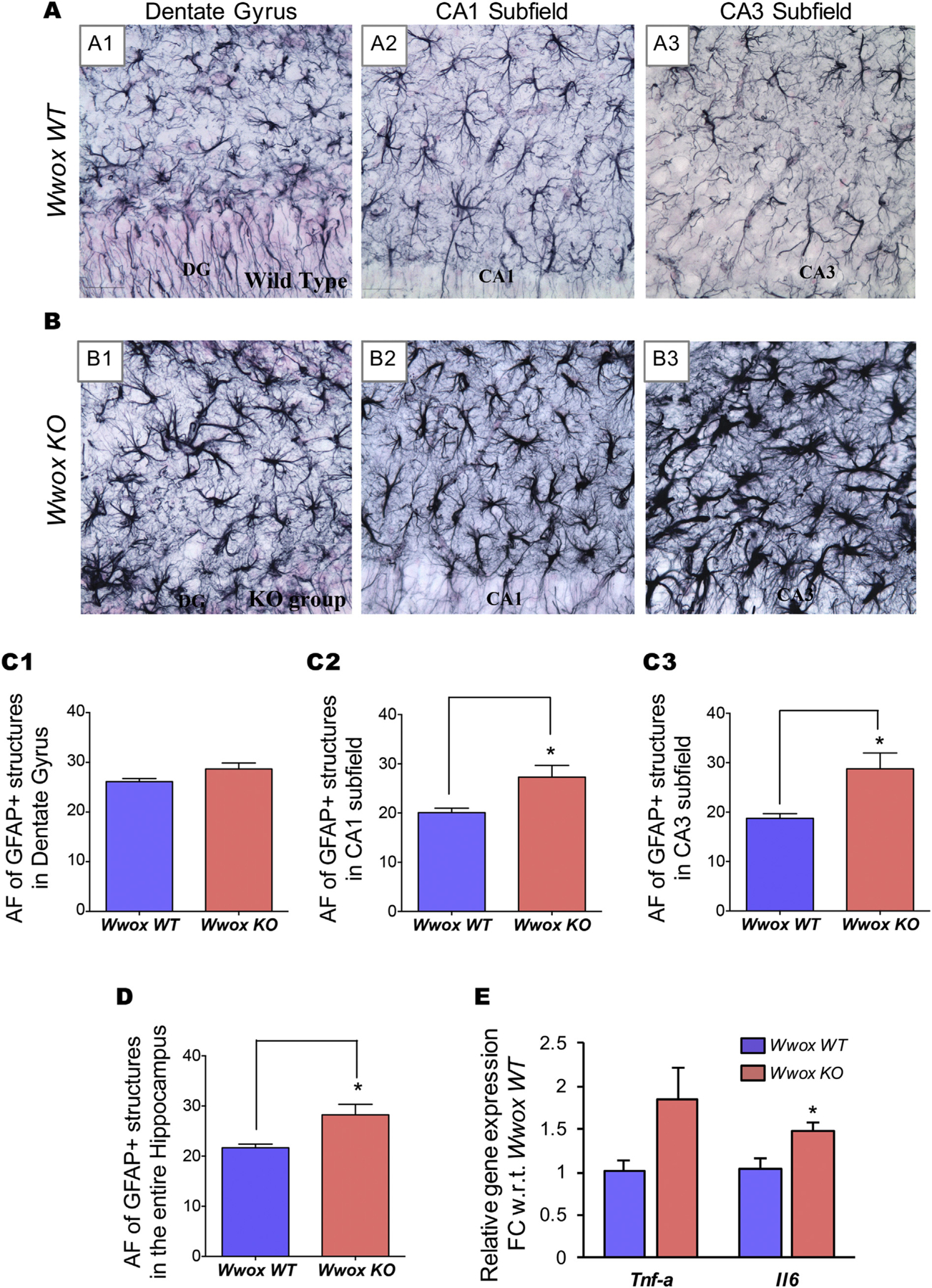
Panels (A) and (B) display representative microphotographs illustrating the distribution and morphology of GFAP+ astrocytes in the various hippocampal subfields of 2 wk. old Wwox WT(A1–A3) and Wwox KO mice (B1–B3). A considerable increase in the AF occupied by GFAP+ structures in the CA1 and CA3 subfields as well as the entire hippocampus is observed in Wwox KO mice. Bar graphs (C1–C3 and D) compare the average AF occupied by GFAP+ astrocytes in DG (C1), CA1 subfield (C2), CA3 subfield (C3), and the entire hippocampus (D) of Wwox WT (n = 6) and Wwox KO (n = 8) as indicated. Error bars represent ± SEM, *p < .05, two tailed unpaired Student’s t-test. (E) Expression of Tnf-a and Il6 in Wwox KO mice hippocampi (n = 4). Y axis in the graph represents relative linear FC values with respect (w.r.t) to Wwox WT mice hippocampi (n = 4); error bars represent ± SEM, *p value < .05, two tailed unpaired Student’s t-test.
Since we observed evidence of neuroinflammation in the form of activated microglia and reactive astrocytes in Wwox KO mice, we further investigated the expression of well known neuroinflammatory cytokines Il6 and Tnf-a in hippocampal tissue by qRT-PCR. We observed > 1.5 fold upregulation of both cytokines (Il6 significant at p < .05) in Wwox KO (n = 4) mice in comparison to Wwox WT (n = 4) mice (Fig. 4 E).
3.4. Wwox KO mice hippocampi show reduced GAD65/67 protein expression
GABA, the major inhibitory neurotransmitter in the mammalian brain, is synthesized by two glutamate decarboxylase isoforms, GAD65 and GAD67. GAD65/67 protein expression was shown to accurately reflect levels of GABA synthesis and abundance (Lindefors, 1993), therefore decreased expression of these proteins would also indicate lower GABA levels. As observed in Fig. 5A, Western blot analysis of hippocampal protein extracts obtained from Wwox KO mice (n = 5) compared with WT mice (n = 5) revealed significantly lower levels of GAD65/67 protein expression. Quantitative densitometry analysis demonstrates a significant reduction in GAD65/67 expression in Wwox KO mice (p < .05, Fig. 5B).
Fig. 5. |. Wwox KO mice hippocampi show reduced expression of Gad65/67 isoforms.
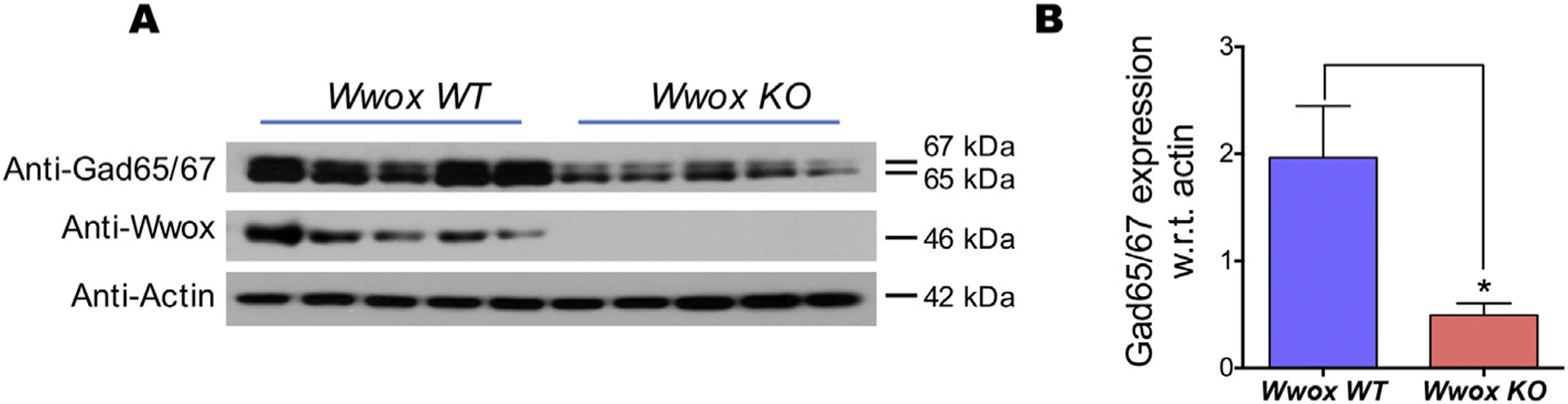
(A) Immunoblot of glutamic acid decarboxylase (Gad) protein isoforms in Wwox WT and Wwox KO mice hippocampi tissue lysates as indicated. (B) Bar graphs representing the average densitometry immunoblot bands representing Gad65/67 protein expression, normalized to Actin loading control, in Wwox WT (n = 5) and Wwox KO (n = 5) mice. Error bars represent ± SEM, *p value < .05, two tailed unpaired Student’s t-test.
3.5. Comparative transcriptome profiling of hippocampus derived neural stem cell neurospheres
To gain some insight on the molecular effects of Wwox deletion, we performed RNA-Seq analyses for comparing the gene expression profiles of neural stem cell (NSC) neurospheres generated from hippocampi of Wwox KO and WT mice. Neurospheres are an excellent model system for the study of NSCs in vitro (Guo et al., 2012). Our aim for performing transcriptome profiling on RNA isolated from neurospheres is to determine the effect of Wwox deletion in comparable pure populations of progenitor cells capable of differentiating into diverse CNS cell subtypes while avoiding the confounding factors of tissue heterogeneity that would arise from analyzing total hippocampus RNA.
Unsupervised analysis of RNA-Seq data demonstrates clear segregation of Wwox KO and WT control mice samples (Fig. 6 A). Comparative transcriptome analysis allowed us to identify 283 differentially expressed genes between both groups (log2 FC > ± 1, p value < .005, FDR < 0.05) (Fig. 6 A). Out of the 283 differentially expressedgenes, 184 transcripts were up-regulated and 99 were down-regulated in Wwox KO samples when compared with control samples (Supplementary Table 2). Next, we performed functional enrichment analysis using the Ingenuity Pathway Analysis (IPA) platform. Interestingly, based on the behavior of dysregulated transcripts in the Wwox KO group, IPA identified ‘neurological disease’ among the top diseases (p-value range: 7.10E-05 – 7.74E-20) and ‘nervous system development and function’ as the top physiological biofunction (p-value range: 6.82E-05 – 9.06E-26). Within the neurological disease subgroup, ‘epilepsy’, ‘seizure disorder’, ‘cognitive impairment’ and ‘neurodegeneration’ showed a positive Z score value implying that these disorders are positively associated with Wwox deletion (Fig. 6 B).
Fig. 6. |. Gene expression profile of Wwox KO vs. WT mice hippocampal neural stem cells neurospheres.
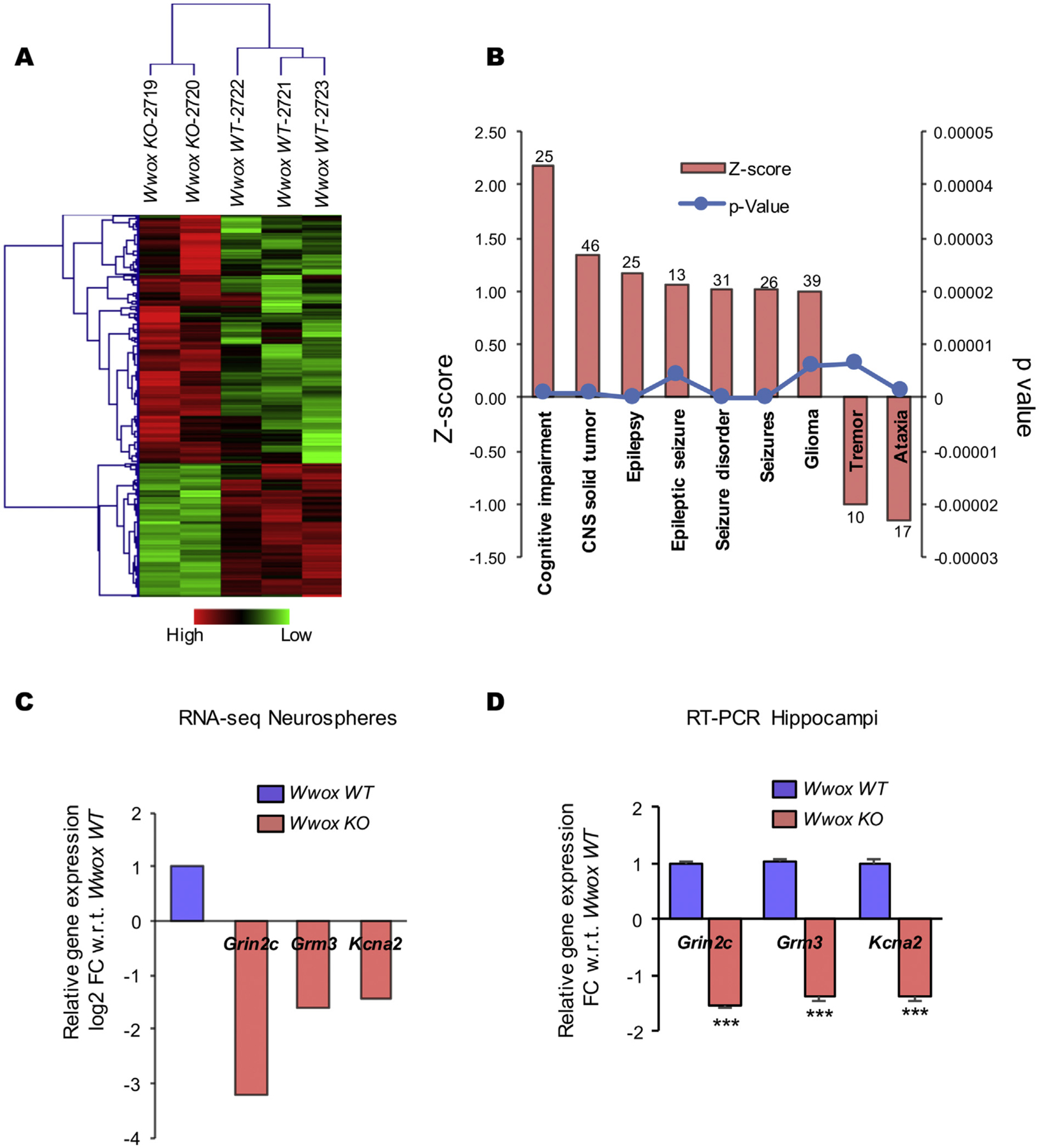
(A) Unsupervised clustering and heatmap for the 283 differentially expressed genes with log2 FC > ± 1, p-value < .005, FDR < 0.05 comparing Wwox KO and Wwox WT mice hippocampi derived neurospheres. Red or green colors indicate differentially up or downregulated genes, respectively. The mean signals were background corrected and transformed to the log2 scale. (B) Bar graph representing functional annotations indicating enrichment for cognitive impairment, epilepsy and seizure related disorders as consequence of dysregulated gene expression in NSCs from Wwox KO mice with Z scores > 1 and p-value < .00001, numbers on the top of each bar represents the number of genes enriched in that specific category as per IPA analysis. (C, D) Relative expression of Grin2C, Grm3, and Kcna2 as per RNA-Seq data, Wwox KO mice (n = 2) relative to Wwox WT (n = 3) (C), and as per qRT-PCR on RNA obtained from Wwox KO (n = 4) relative to Wwox WT (n = 4) hippocampi (D). Y axis in the RNA-Seq data graph (C) represents relative log2 FC values while in hippocampi graph (D) it represents relative linear FC values, with respect to Wwox WT mice; error bars represent ± SEM, ***p < .0001, two tailed unpaired Student’s t-test.
Next, from the list of 283 differentially expressed genes we identified a group of transcripts on the basis of either one of the following two criteria: (i) the transcript falls within a panel of 172 genes implicated in epilepsy as reported by Rim et al.; this dataset is based on an extensive literature review and the Online Mendelian Inheritance in Man (OMIM) database search (Rim et al., 2018), or (ii) the gene is identified by IPA as a member of the set of dysregulated genes that identify with epilepsy and/or seizure related disorders as per functional enrichment analyses. Table 1 shows the list of 16 genes that were short-listed based on the described criteria. Also shown in Table 1 is the function and potential relevance of these genes with epilepsy supported by available literature (Table 1). All these RNA-Seq observations were validated by qRT-PCR on RNA isolated from neurospheres (data not shown).
Table 1. Dysregulated genes as per RNA-Seq analysis in Wwox KO derived neurospheres with relevance to epilepsy.
Complete list of 283 differentially expressed genes (log2 FC > ±1, p value<.005, FDR < 0.05) between Wwox WT and Wwox KO groups is given in Supplementary Table 2.
| Gene | Description | Log2 FC | p value | FDR | Function/relevance | References |
|---|---|---|---|---|---|---|
| Gabbr2 | GABA B Receptor, 2 | 1.72 | 5.91E-10 | 1.98E-07 | Heterodimerizes with GABBR1 participates in sustained inhibitory GABA-ergic neurotransmission | (Karlsson et al., 1992; Han et al., 2012) |
| Fgf1 | Fibroblast Growth Factor 1 | 1.30 | 2.92E-10 | 3.71E-07 | Involved with morpho-functional alterations in brain circuitries associated with epileptogenesis | (Riva et al., 1995; Paradiso et al., 2013) |
| Fzd9 | Frizzled Receptor 9 | 1.2 | 4.80E-07 | 8.20E-04 | Wnt signaling receptor in the brain, associated with Williams Beuren Syndrome | (Zhao et al., 2005) |
| Gabra2 | GABA A Receptor, Alpha 2 | 1.15 | 7.01E-04 | 3.45E-02 | Mutations in this gene are associated with epilepsy | (Hung et al., 2013; Srivastava et al., 2014; Orenstein et al., 2018) |
| Chl1 | Cell Adhesion Molecule L1 Like | 1.01 | 6.53E-11 | 1.04E-07 | Neural recognition molecule involved in signal transduction pathways; deletions of this gene likely responsible for mental defects in 3p syndrome | (Angeloni et al., 1999; Frints et al., 2003) |
| Kcna2 | Voltage-Gated Potassium Channel | −1.45 | 4.68E-09 | 4.68E-09 | Role in neuronal repolarization; loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy | (Pena and Coimbra, 2015; Syrbe et al., 2015; Corbett et al., 2016; Hundallah et al., 2016; Masnada et al., 2017; Sachdev et al., 2017) |
| Stx1b | Syntaxin 1B | −1.47 | 1.60E-04 | 1.07E-02 | Role in excitatory pathway of synaptic transmission; known to be implicated in epilepsy and seizure disorder | (Lerche et al., 2001; Schubert et al., 2014) |
| Gabbr1 | GABA B Receptor, 1 | −1.48 | 1.49E-06 | 2.09E-04 | Heterodimerizes with GABBR2 participates in sustained inhibitory GABA-ergic neurotransmission | (Karlsson et al., 1992; Han et al., 2012) |
| Kcnj10 | Potassium Channel, Inwardly Rectifying Subfamily J Member 10 | −1.54 | 2.76E-04 | 1.60E-02 | Responsible for the potassium buffering action of glial cells; mutations are associated with seizure and epilepsy syndromes | (Reichold et al., 2010; Sala-Rabanal et al., 2010; Freudenthal et al., 2011) |
| Alpl | Alkaline Phosphatase | −1.56 | 5.12E-08 | 1.04E-05 | Tissue non-specific alkaline phosphatase; knock out in mice induces fatal seizure disorder | (Waymire et al., 1995) |
| Grm3 | Glutamate Metabotropic Receptor 3 | −1.61 | 8.21E-10 | 2.66E-07 | Glutamate receptor; deregulation of GluR signaling functions may occur in some forms of epilepsy | (Meldrum, 1994; Ure et al., 2006) |
| Grin2b | Glutamate Ionotropic Receptor NMDA Subunit 2B | −2.14 | 2.75E-06 | 3.60E-04 | Mutations are associated with early infantile epileptic encephalopathy | (Lemke et al., 2014; Platzer et al., 2017) |
| Arx | Aristaless Related Homeobox | −2.33 | 8.89E-06 | 1.01E-04 | Homeodomain transcription factor; crucial role in cerebral development, loss of function mutations cause epilepsy and mental retardation | (Scheffer et al., 2002; Stromme et al., 2002; Gronskov et al., 2004) |
| Apoe | Apolipoprotein E | −2.64 | 9.36E-32 | 9.11E-28 | Role in the fat metabolism; associated with Alzheimer’s disease and epilepsy | (Busch et al., 2007) |
| Atp1a2 | ATPase Na+/K+ Transporting Subunit Alpha 2 | −3.15 | 6.42E-16 | 5.68E-13 | Role in establishing and maintaining the electrochemical gradients of Na and K ions across the plasma membrane | (Jurkat-Rott et al., 2004; Wilbur et al., 2017) |
| Grin2c | Glutamate Ionotropic Receptor NMDA Subunit 2C | −3.22 | 6.55E-06 | 7.73E-04 | NMDA receptors are crucial for neuronal communication, known to be involved in various neurological and psychiatric disorders | (Paoletti et al., 2013) |
To further investigate whether any of the gene expression changes observed affecting progenitor cells in vitro would be observable in vivo, we analyzed expression of all genes on RNA isolated from hippocampi freshly dissected from a separate set of Wwox KO (n = 4) and WT (n = 4) mice. From this in vitro – in vivo comparison the most striking changes behaving in the same direction in both conditions was the very significant downregulation in expression of the NMDA receptor Grin2c, glutamate receptor Grm3, and voltage-gated potassium channel Kcna2 genes in Wwox KO mice when compared to WT controls (Figs. 6 C and D).
4. Discussion
This study provides the first pathophysiological evidence on the effects of Wwox loss of function in the brain. Importantly, we observed that Wwox deletion leads to a significant reduction in the overall number of PV+ and NPY+ GABA-ergic inhibitory interneurons in mouse hippocampus. Additionally, we also detected evidence of active neuroinflammation and gliosis in the brains of Wwox KO mice.
Loss of GABA-ergic inhibitory function have been observed in several human epileptiform disorders and experimental animal models of epilepsy and is postulated to be a susceptibility factor or trigger of some genetic forms of human epilepsy (Powell, 2013). Genetic mutations resulting in functional changes in GABA receptors including the selective loss, or functional impairment, of GABA-ergic interneurons may disrupt the regulation of local excitatory circuits, resulting in hyperexcitability of neuronal networks thus contributing to epileptogenesis (Williams and Battaglia, 2013). Among the most vulnerable interneurons in human and animal models of epilepsy are those expressing PV. We observed significantly lower numbers of PV+ interneurons in the hippocampi of Wwox KO mice and it was described that reduced number of PV+ interneurons can lead to hippocampal hyperexcitability (Schwaller et al., 2004). Furthermore, studies in epilepsy models have shown that loss of PV+ interneurons in the DG greatly reduces the inhibition of dentate granule cells thus contributing to epileptogenesis (Kobayashi and Buckmaster, 2003). Our analysis also shows that the overall number of NPY+ interneurons is reduced in the DG region of Wwox KO mice. Like PV+ interneurons, NPY+ interneurons also play an important role in regulating the activity of hippocampal circuitry. The NPY released from these interneurons modulates the activity of excitatory neurons by consistently hyperpolarizing and reducing their spike frequency (Fu and van den Pol, 2007) and by inhibiting the glutamate release to principal hippocampal neurons (Colmers and Bleakman, 1994). NPY is an endogenous anti-seizure compound, which is evident from its upregulation in conditions such as status epilepticus, and reduced epileptiform activity in neurons upon NPY treatment (Vezzani et al., 1999). It has also been shown that adult Npy null mice show increased sensitivity and poor outcomes after chemically induced seizures (Erickson et al., 1996). Thus, the substantial loss of PV+ and NPY+ interneurons in Wwox null mice may induce hippocampal hyperexcitability and be, at least in part, responsible for the epileptic phenotype observed in these mice. Furthermore, substantial loss of PV+ interneurons in the hippocampus can considerably impair theta rhythm involved in cognitive function (Klausberger et al., 2005). Reduced NPY can also impair learning and memory function (Zaben and Gray, 2013). In this context, learning and memory deficits in conditions such as autism may be linked at least partially to alterations affecting WWOX.
Also, we observed a reduction in levels of the two glutamate decarboxylase (GAD) isoforms Gad65 and Gad67 in Wwox null hippocampi. GAD65 and GAD67 are the principal enzymes that synthesize GABA from glutamate in the mammalian brain (Lindefors, 1993). Expression of GADs has been reported to accurately reflect levels of GABA synthesis and activity (Lindefors, 1993). For instance, selective elimination of Gad65 has been shown to elicit spontaneous seizures that results in increased mortality in Gad65 −/− mice (Kash et al., 1997). Thus, decreased expression of Gad65/67 in the hippocampi of Wwox null mice goes hand in hand with the described reduction in numbers of GABA-ergic interneurons and suggest the existence of a hippocampal hyperexcitable state.
Clinical and experimental observations have provided strong evidence that inflammatory processes within the brain might constitute a crucial mechanism in the pathophysiology of seizures and epilepsy (Vezzani et al., 2011). We observed that Wwox null mice display signs of neuroinflammation as suggested by the increased detection of IBA-1+ cells (i.e., activated microglia) and GFAP+ astrocytes as well as increased expression of inflammatory cytokines Tnf-a and Il6. TNF-a and IL6 are the most extensively studied prototypical cytokines in the brain which are first expressed in activated microglia and astrocytes as consequence of pathogenic insults such as trauma, infection or neurodegenerative diseases (Glass et al., 2010). Notably, pro-inflammatory molecules, microgliosis, astrogliosis, and other indicators of inflammation have been found in the resected hippocampi of patients with different forms of epilepsy and also in rodent models of epilepsy (Shapiro et al., 2008; Vezzani et al., 2011). The finding of biomarkers of neuroinflammation in Wwox KO mice brains supports the idea that inflammation might play a role in Wwox related epilepsy. However, further investigation is needed to gather additional mechanistic insight on what is cause or consequence upon Wwox loss of function.
We also performed RNA-Seq to profile the transcriptome wide effects of Wwox deletion in neural progenitor cells. Interestingly, the comparative analysis of transcriptional profiles from NSC derived from Wwox KO vs. WT mice reveled significant differences reflecting alterations that identify with CNS related disorders and in particular ‘cognitive impairment’, ‘seizures’, ‘seizure disorders’ and ‘epilepsy’ among the most prevalent functional annotations. Importantly, this is in close agreement with human familial syndromes associated with WWOX mutations and loss of function. At the specific gene level, we observed a significant down-regulation of Glutamate Ionotropic Receptor NMDA Subunit 2C (Grin2c), Glutamate Metabotropic Receptor 3 (Grm3), and Potassium Voltage-Gated Channel Subfamily A Member 2 (Kcna2) in Wwox KO NSCs as well as in whole hippocampal tissue.
GRIN2C is a subunit of the glutamate-gated ion channels N-methyl-d-aspartate receptors (NMDAR). NMDARs are crucial for neuronal communication and are capable of converting specific patterns of neuronal activity into long-term changes in synapse structure and function. Dysfunction of NMDARs including GRIN2A and GRIN2B has been reported in various neurological and psychiatric disorders (Paoletti et al., 2013). Interestingly, GRIN2A loss of function mutations, either through nonsense-mediated mRNA decay or premature termination resulting in the synthesis of dysfunctional protein, have been associated with focal epilepsy and speech disorder with or without mental retardation (Endele et al., 2010). In line, GRIN2B loss of function mutations have been associated with mental retardation and autism (Endele et al., 2010), while in cases of early infantile epileptic encephalopathy both loss and gain of GRIN2B function mutations have been reported (Platzer et al., 2017). Therefore, it is logical to speculate that Grin2C downregulation may also lead to abnormal NMDAR receptor function by altering receptor subunit composition and indeed this has been speculated to be a key variable and potential cause of epileptogenesis (Paoletti et al., 2013).
KCNA2 is a member of voltage-gated potassium channel family that are expressed in the CNS and plays an important role in neuronal excitability and neurotransmitter release (Lai and Jan, 2006). Many de novo missense and loss- or gain-of-function mutations in KCNA2 have been associated with severe early-onset epileptic encephalopathy, ataxia, cognitive and language impairment, and behavioral disorders (Masnada et al., 2017). In most of the cases, the onset of epilepsy occurred within the first to second year of life, while the loss- and gain-of-function subgroups show the earliest, often neonatal onset. Mice harboring Kcna2 mutations are also reported to display chronic motor incoordination, ataxia, increased seizure, and premature death due to loss of Kcna2 function (Xie et al., 2010). Early-onset epilepsy and premature death observed in KCNA2 mutant humans and mice are similar phenotypes to those observed in our Wwox KO mouse model which makes Kcna2 an important candidate for future studies.
We also observed a decrease in Grm3 expression in Wwox KO mice. Grm3 is a G protein-coupled metabotropic glutamate receptor and acts in part as an inhibitory autoreceptor, influencing synaptic plasticity (Niswender and Conn, 2010). A role of this receptor has been associated with the pathophysiology and therapy of many neuropsychiatric disorders including bipolar disorders, Alzheimer’s disease, anxiety disorders and in particular schizophrenia (Moreno et al., 2009). A potential role for metabotropic glutamate receptors has also been suggested in epilepsy (Aronica et al., 2000).
In conclusion, our observations may explain, at least in part, what is occurring in the brains of patients affected by WWOX loss of function pathologies. One notable observation regarding WWOX related epileptic disorders is that cases characterized by complete WWOX loss of function mutations, as in early infantile epileptic encephalopathies (EIEE28, OMIM #616211) also described as WOREE syndrome, are always accompanied by mental retardation, usually display severe brain structural abnormalities, and patients die within the first two years of life (Abdel-Salam et al., 2014; Ben-Salem et al., 2015; Mignot et al., 2015; Tabarki et al., 2015; Valduga et al., 2015), thus displaying a very short lifespan similar to our mouse Wwox KO model where all mice die by approximately 3 weeks of age (Ludes-Meyers et al., 2009). However, in other cases presented with missense mutations that lead to expression of a defective WWOX protein (i.e. hypomorphs) premature death was not reported (Mallaret et al., 2014; Mignot et al., 2015). In addition to epilepsy and seizure disorders, alterations affecting WWOX have also been associated with other neuropathologies including Alzheimer’s disease (Sze et al., 2004; Wang et al., 2012),autism (Bartnik et al., 2012; Leppa et al., 2016) and other intellectual disabilities (Alkhateeb et al., 2016) and interestingly the WWOX locus was also found associated to infant brain volumes (Xia et al., 2017).
Importantly, in recent studies, Wwox has been identified as one of a very limited set of long neural genes characterized by harboring recurrent double strand break clusters and undergoing genomic rearrangements in primary neural stem/progenitor cells as part of critical processes likely associated with the specialized behavior of brain cells (Wei et al., 2016). Strinkingly, most ofthese genes have been associated to CNS pathologies and cancer (Wei et al., 2016). These observations further stress the critical relevance of WWOX in normal CNS development.
The observations reported here fill a gap of knowledge providing clues to start understanding the consequences and potential mechanisms at play in the CNS of patients affected by WWOX loss of function conditions. Although these human pathologies are rare, it stimulates to further pursue studies aimed at understanding the clearly important role of WWOX in normal CNS development and function.
Supplementary Material
Acknowledgements
This work was supported by CPRIT Core Facility Support Grant (RP120348); The Virginia Harris Cockrell Foundation to C.M.A. and The Department of Defense (CDMPRP Grant) and Department of Veterans Affairs (Merit Award) to A.K.S. We want to thank The University of Texas MD Anderson Cancer Center Animal Support Facility in Smithville (P30 NIH CA16672) for providing necessary infrastructure and facilities for conducting this work. The authors are also grateful to Yue Lu and Kevin Lin for bioinformatic support.
Abbreviations:
- WWOX
WW domain containing oxidoreductase
- EIEE
Early infantile epileptic encephalopathy
- PV
Parvalbumin
- NPY
Neuropeptide Y
- GAD65/67
Glutamic Acid Decarboxylase protein isoforms
- GABA
γ-aminobutyric acid
- WOREE
WWOX-related epileptic encephalopathy
- IBA-1
Ionized calcium binding adaptor molecule-1-positive
- GFAP
Glial fibrillary acidic protein
- DG
Dentate gyrus
- DH
Dentate hilus
- CA1 and CA3
Cornu ammonis subfields
- GCL
Granule cell layer
- GRIN2C
Glutamate Ionotropic Receptor NMDA Subunit 2C
- GRM3
Glutamate Metabotropic Receptor 3
- KCNA2
Potassium Voltage-Gated Channel Subfamily A Member 2
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2018.09.026.
Conflict of interest
The authors declare no competing financial interests.
References
- Abdel-Salam G, Thoenes M, Afifi HH, Korber F, Swan D, Bolz HJ, 2014. The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet J Rare Dis 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldaz CM, Ferguson BW, Abba MC, 2014. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim. Biophys. Acta 1846, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhateeb AM, Aburahma SK, Habbab W, Thompson IR, 2016. Novel mutations in WWOX, RARS2, and C10orf2 genes in consanguineous Arab families with intellectual disability. Metab. Brain Dis 31, 901–907. [DOI] [PubMed] [Google Scholar]
- Angeloni D, Lindor NM, Pack S, Latif F, Wei MH, Lerman MI, 1999. CALL gene is haploinsufficient in a 3p- syndrome patient. Am. J. Med. Genet 86, 482–485. [DOI] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA, 2000. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur. J. Neurosci 12, 2333–2344. [DOI] [PubMed] [Google Scholar]
- Bartnik M, et al. , 2012. Application of array comparative genomic hybridization in 102 patients with epilepsy and additional neurodevelopmental disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet 159B, 760–771. [DOI] [PubMed] [Google Scholar]
- Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM, 2000. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3–24.1, a region frequently affected in breast cancer. Cancer Res. 60, 2140–2145. [PubMed] [Google Scholar]
- Ben-Salem S, Al-Shamsi AM, John A, Ali BR, Al-Gazali L, 2015. A novel whole exon deletion in WWOX gene causes early epilepsy, intellectual disability and optic atrophy. J. Mol. Neurosci 56, 17–23. [DOI] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV, 2014. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch RM, Lineweaver TT, Naugle RI, Kim KH, Gong Y, Tilelli CQ, Prayson RA, Bingaman W, Najm IM, Diaz-Arrastia R, 2007. ApoE-epsilon4 is associated with reduced memory in long-standing intractable temporal lobe epilepsy. Neurology 68, 409–414. [DOI] [PubMed] [Google Scholar]
- Chang HT, Liu CC, Chen ST, Yap YV, Chang NS, Sze CI, 2014. WW domain-containing oxidoreductase in neuronal injury and neurological diseases. Oncotarget 5, 11792–11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ST, Chuang JI, Wang JP, Tsai MS, Li H, Chang NS, 2004. Expression of WW domain-containing oxidoreductase WOX1 in the developing murine nervous system. Neuroscience 124, 831–839. [DOI] [PubMed] [Google Scholar]
- Colmers WF, Bleakman D, 1994. Effects of neuropeptide Y on the electrical properties of neurons. Trends Neurosci. 17, 373–379. [DOI] [PubMed] [Google Scholar]
- Corbett MA, Bellows ST, Li M, Carroll R, Micallef S, Carvill GL, Myers CT, Howell KB, Maljevic S, Lerche H, Gazina EV, Mefford HC, Bahlo M, Berkovic SF, Petrou S, Scheffer IE, Gecz J, 2016. Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology 87, 1975–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA, 2013. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 36, 174–184. [DOI] [PubMed] [Google Scholar]
- Elsaadany L, El-Said M, Ali R, Kamel H, Ben-Omran T, 2016. W44X mutation in the WWOX gene causes intractable seizures and developmental delay: a case report. BMC Med. Genet 17, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endele S, et al. , 2010. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet 42, 1021–1026. [DOI] [PubMed] [Google Scholar]
- Erickson JC, Clegg KE, Palmiter RD, 1996. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature 381, 415–421. [DOI] [PubMed] [Google Scholar]
- Freudenthal B, Kulaveerasingam D, Lingappa L, Shah MA, Brueton L, Wassmer E, Ognjanovic M, Dorison N, Reichold M, Bockenhauer D, Kleta R, Zdebik AA, 2011. KCNJ10 mutations disrupt function in patients with EAST syndrome. Nephron Physiol. 119, 40–48. [DOI] [PubMed] [Google Scholar]
- Frints SG, Marynen P, Hartmann D, Fryns JP, Steyaert J, Schachner M, Rolf B, Craessaerts K, Snellinx A, Hollanders K, D’Hooge R, De Deyn PP, Froyen G, 2003. CALL interrupted in a patient with non-specific mental retardation: gene dosage-dependent alteration of murine brain development and behavior. Hum. Mol. Genet 12, 1463–1474. [DOI] [PubMed] [Google Scholar]
- Fu LY, van den Pol AN, 2007. GABA excitation in mouse hilar neuropeptide Y neurons. J. Physiol 579, 445–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH, 2010. Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribaa M, Salih M, Anheim M, Lagier-Tourenne C, H’Mida D, Drouot N, Mohamed A, Elmalik S, Kabiraj M, Al-Rayess M, Almubarak M, Betard C, Goebel H, Koenig M, 2007. A new form of childhood onset, autosomal recessive spinocerebellar ataxia and epilepsy is localized at 16q21-q23. Brain 130, 1921–1928. [DOI] [PubMed] [Google Scholar]
- Gronskov K, Hjalgrim H, Nielsen IM, Brondum-Nielsen K, 2004. Screening of the ARX gene in 682 retarded males. Eur. J. Hum. Genet 12, 701–705. [DOI] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, Zhao X, 2012. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat. Protoc 7, 2005–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HA, Cortez MA, Snead OC, 2012. GABAB Receptor and Absence Epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (Eds.), Jasper’s Basic Mechanisms of the Epilepsies; Bethesda, MD. [Google Scholar]
- Hundallah K, Alenizi A, AlHashem A, Tabarki B, 2016. Severe early-onset epileptic encephalopathy due to mutations in the KCNA2 gene: Expansion of the genotypic and phenotypic spectrum. Eur. J. Paediatr. Neurol 20, 657–660. [DOI] [PubMed] [Google Scholar]
- Hung CC, Chen PL, Huang WM, Tai JJ, Hsieh TJ, Ding ST, Hsieh YW, Liou HH, 2013. Gene-wide tagging study of the effects of common genetic polymorphisms in the alpha subunits of the GABA(A) receptor on epilepsy treatment response. Pharmacogenomics 14 (203), 1849–1856. [DOI] [PubMed] [Google Scholar]
- Jurkat-Rott K, Freilinger T, Dreier JP, Herzog J, Gobel H, Petzold GC, Montagna P, Gasser T, Lehmann-Horn F, Dichgans M, 2004. Variability of familial hemiplegic migraine with novel A1A2 Na+/K+-ATPase variants. Neurology 62, 1857–1861. [DOI] [PubMed] [Google Scholar]
- Karlsson G, Kolb C, Hausdorf A, Portet C, Schmutz M, Olpe HR, 1992. GABAB receptors in various in vitro and in vivo models of epilepsy: a study with the GABAB receptor blocker CGP 35348. Neuroscience 47, 63–68. [DOI] [PubMed] [Google Scholar]
- Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S, 1997. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U. S. A 94, 14060–14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, O’Neill J, Huck JH, Dalezios Y, Fuentealba P, Suen WY, Papp E, Kaneko T, Watanabe M, Csicsvari J, Somogyi P, 2005. Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J. Neurosci 25, 9782–9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS, 2003. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J. Neurosci 23, 2440–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali M, Parihar VK, Hattiangady B, Mishra V, Shuai B, Shetty AK, 2015. Resveratrol prevents age-related memory and mood dysfunction with increased hippocampal neurogenesis and microvasculature, and reduced glial activation. Sci. Rep 5, 8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HC, Jan LY, 2006. The distribution and targeting of neuronal voltage-gated ion channels. Nat. Rev. Neurosci 7, 548–562. [DOI] [PubMed] [Google Scholar]
- Lemke JR, Hendrickx R, Geider K, Laube B, Schwake M, Harvey RJ, James VM, Pepler A, Steiner I, Hortnagel K, Neidhardt J, Ruf S, Wolff M, Bartholdi D, Caraballo R, Platzer K, Suls A, De Jonghe P, Biskup S, Weckhuysen S, 2014. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann. Neurol 75, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppa VM, Kravitz SN, Martin CL, Andrieux J, Le Caignec C, Martin-Coignard D, Dybuncio C, Sanders SJ, Lowe JK, Cantor RM, Geschwind DH, 2016. Rare Inherited and De Novo CNVs Reveal complex Contributions to ASD Risk in Multiplex families. Am. J. Hum. Genet 99, 540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche H, Weber YG, Baier H, Jurkat-Rott K, Kraus de Camargo O, Ludolph AC, Bode H, Lehmann-Horn F, 2001. Generalized epilepsy with febrile seizures plus: further heterogeneity in a large family. Neurology 57, 1191–1198. [DOI] [PubMed] [Google Scholar]
- Lindefors N, 1993. Dopaminergic regulation of glutamic acid decarboxylase mRNA expression and GABA release in the striatum: a review. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 17, 887–903. [DOI] [PubMed] [Google Scholar]
- Liu YQ, Yu F, Liu WH, He XH, Peng BW, 2014. Dysfunction of hippocampal interneurons in epilepsy. Neurosci. Bull 30, 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Ludes-Meyers JH, Kil H, Parker-Thornburg J, Kusewitt DF, Bedford MT, Aldaz CM, 2009. Generation and characterization of mice carrying a conditional allele of the Wwox tumor suppressor gene. PLoS One 4, e7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallaret M, Synofzik M, Lee J, Sagum CA, Mahajnah M, Sharkia R, Drouot N, Renaud M, Klein FA, Anheim M, Tranchant C, Mignot C, Mandel JL, Bedford M, Bauer P, Salih MA, Schule R, Schols L, Aldaz CM, Koenig M, 2014. The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain 137, 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masnada S, et al. , 2017. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 140, 2337–2354. [DOI] [PubMed] [Google Scholar]
- Megahed T, Hattiangady B, Shuai B, Shetty AK, 2014. Parvalbumin and neuropeptide Y expressing hippocampal GABA-ergic inhibitory interneuron numbers decline in a model of Gulf War illness. Front. Cell. Neurosci 8, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS, 1994. The role of glutamate in epilepsy and other CNS disorders. Neurology 44, S14–S23. [PubMed] [Google Scholar]
- Mignot C, et al. , 2015. WWOX-related encephalopathies: delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J. Med. Genet 52, 61–70. [DOI] [PubMed] [Google Scholar]
- Mishra V, Shuai B, Kodali M, Shetty GA, Hattiangady B, Rao X, Shetty AK, 2015. Resveratrol Treatment after Status Epilepticus Restrains Neurodegeneration and Abnormal Neurogenesis with suppression of Oxidative stress and Inflammation. Sci. Rep 5, 17807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JL, Sealfon SC, Gonzalez-Maeso J, 2009. Group II metabotropic glutamate receptors and schizophrenia. Cell. Mol. Life Sci 66, 3777–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ, 2010. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez MI, Ludes-Meyers J, Aldaz CM, 2006. WWOX protein expression in normal human tissues. J. Mol. Histol 37, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein N, Goldberg-Stern H, Straussberg R, Bazak L, Weisz Hubshman M, Kropach N, Gilad O, Scheuerman O, Dory Y, Kraus D, Tzur S, Magal N, Kilim Y, Shkalim Zemer V, Basel-Salmon L, 2018. A de novo GABRA2 missense mutation in severe early-onset epileptic encephalopathy with a choreiform movement disorder. Eur. J. Paediatr. Neurol 22, 516–524. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q, 2013. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Paradiso B, Zucchini S and Simonato M, Implication of fibroblast growth factors in epileptogenesis-associated circuit rearrangements. Front. Cell. Neurosci 7, 2013, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena SD, Coimbra RL, 2015. Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: proposal for a new channelopathy. Clin. Genet 87, e1–e3. [DOI] [PubMed] [Google Scholar]
- Platzer K, et al. , 2017. GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet 54, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell EM, 2013. Interneuron development and epilepsy: early genetic defects cause long-term consequences in seizures and susceptibility. Epilepsy Curr 13, 172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Page A, Gandarillas A, Zanet J, Pibre S, Vidal M, Tusell L, Genesca A, Whitaker DA, Melton DW, Jorcano JL, 2004. A keratin K5Cre transgenic line appropriate for tissue-specific or generalized Cre-mediated recombination. Genesis 39, 52–57. [DOI] [PubMed] [Google Scholar]
- Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R, 2010. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. U. S. A 107, 14490–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, Woollatt E, Kremmidiotis G, Gardner A, Venter D, Baker E, Richards RI, 2000. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet 9, 1651–1663. [DOI] [PubMed] [Google Scholar]
- Rim JH, Kim SH, Hwang IS, Kwon SS, Kim J, Kim HW, Cho MJ, Ko A, Youn SE, Kim J, Lee YM, Chung HJ, Lee JS, Kim HD, Choi JR, Lee ST, Kang HC, 2018. Efficient strategy for the molecular diagnosis of intractable early-onset epilepsy using targeted gene sequencing. BMC Med. Genet 11, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva MA, Fumagalli F, Blom JM, Donati E, Racagni G, 1995. Adrenalectomy reduces FGF-1 and FGF-2 gene expression in specific rat brain regions and differently affects their induction by seizures. Brain Res. Mol. Brain Res 34, 190–196. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdev M, Gainza-Lein M, Tchapyjnikov D, Jiang YH, Loddenkemper T, Mikati MA, 2017. Novel clinical manifestations in patients with KCNA2 mutations. Seizure 51, 74–76. [DOI] [PubMed] [Google Scholar]
- Sala-Rabanal M, Kucheryavykh LY, Skatchkov SN, Eaton MJ, Nichols CG, 2010. Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10). J. Biol. Chem 285, 36040–36048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Wallace RH, Phillips FL, Hewson P, Reardon K, Parasivam G, Stromme P, Berkovic SF, Gecz J, Mulley JC, 2002. X-linked myoclonic epilepsy with spasticity and intellectual disability: mutation in the homeobox gene ARX. Neurology 59, 348–356. [DOI] [PubMed] [Google Scholar]
- Schrock MS, Batar B, Lee J, Druck T, Ferguson B, Cho JH, Akakpo K, Hagrass H, Heerema NA, Xia F, Parvin JD, Aldaz CM, Huebner K, 2017. Wwox-Brca1 interaction: role in DNA repair pathway choice. Oncogene 36, 2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert J, et al. , 2014. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet 46, 1327–1332. [DOI] [PubMed] [Google Scholar]
- Schwaller B, Tetko IV, Tandon P, Silveira DC, Vreugdenhil M, Henzi T, Potier MC, Celio MR, Villa AE, 2004. Parvalbumin deficiency affects network properties resulting in increased susceptibility to epileptic seizures. Mol. Cell. Neurosci 25, 650–663. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, 1994. Role of the hippocampus in epilepsy. Hippocampus 4, 239–242. [DOI] [PubMed] [Google Scholar]
- Shapiro LA, Wang L, Ribak CE, 2008. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 49 (Suppl. 2), 33–41. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Cohen J, Pevsner J, Aradhya S, McKnight D, Butler E, Johnston M, Fatemi A, 2014. A novel variant in GABRB2 associated with intellectual disability and epilepsy. Am. J. Med. Genet. A 164A, 2914–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SM, Bruyere H, Lutcherath V, Gedeon AK, Wallace RH, Scheffer IE, Turner G, Partington M, Frints SG, Fryns JP, Sutherland GR, Mulley JC, Gecz J, 2002. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet 30, 441–445. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Katayama K, Takenaka M, Amakasu K, Saito K, Suzuki K, 2009. A spontaneous mutation of the Wwox gene and audiogenic seizures in rats with lethal dwarfism and epilepsy. Genes Brain Behav. 8, 650–660. [DOI] [PubMed] [Google Scholar]
- Syrbe S, et al. , 2015. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet 47, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, Su M, Pugazhenthi S, Jambal P, Hsu LJ, Heath J, Schultz L, Chang NS, 2004. Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer’s disease. J Biol Chem 279, 30498–30506. [DOI] [PubMed] [Google Scholar]
- Tabarki B, Alhashem A, Alshahwan S, Alkuraya FS, Gedela S, Zuccoli G, 2015. Severe CNS involvement in WWOX mutations: Description of five new cases. Am. J. Med. Genet. A 167A, 3209–3213. [DOI] [PubMed] [Google Scholar]
- Ure J, Baudry M, Perassolo M, 2006. Metabotropic glutamate receptors and epilepsy. J. Neurol. Sci 247, 1–9. [DOI] [PubMed] [Google Scholar]
- Valduga M, Philippe C, Lambert L, Bach-Segura P, Schmitt E, Masutti JP, Francois B, Pinaud P, Vibert M, Jonveaux P, 2015. WWOX and severe autosomal recessive epileptic encephalopathy: first case in the prenatal period. J. Hum. Genet 60, 267–271. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Sperk G, Colmers WF, 1999. Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci. 22, 25–30. [DOI] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T, Baram TZ, 2011. The role of inflammation in epilepsy. Nat. Rev. Neurol 7, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Juo LI, Lin YT, Hsiao M, Lin JT, Tsai CH, Tzeng YH, Chuang YC, Chang NS, Yang CN, Lu PJ, 2012. WW domain-containing oxidoreductase promotes neuronal differentiation via negative regulation of glycogen synthase kinase 3beta. Cell Death Differ. 19, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waymire KG, Mahuren JD, Jaje JM, Guilarte TR, Coburn SP, MacGregor GR, 1995. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet 11, 45–51. [DOI] [PubMed] [Google Scholar]
- Wei PC, Chang AN, Kao J, Du Z, Meyers RM, Alt FW, Schwer B, 2016. Long Neural Genes Harbor Recurrent DNA break Clusters in Neural Stem/Progenitor Cells. Cell 164, 644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbur C, Buerki SE, Guella I, Toyota EB, Evans DM, McKenzie MB, Datta A, Michoulas A, Adam S, Van Allen MI, Nelson TN, Farrer MJ, Connolly MB, Demos M, 2017. An Infant With Epilepsy and Recurrent Hemiplegia due to Compound Heterozygous Variants in ATP1A2. Pediatr. Neurol 75, 87–90. [DOI] [PubMed] [Google Scholar]
- Williams CA, Battaglia A, 2013. Molecular biology of epilepsy genes. Exp. Neurol 244, 51–58. [DOI] [PubMed] [Google Scholar]
- Xia K, Zhang J, Ahn M, Jha S, Crowley JJ, Szatkiewicz J, Li T, Zou F, Zhu H, Hibar D, Thompson P, Consortium E, Sullivan PF, Styner M, Gilmore JH, Knickmeyer RC, 2017. Genome-wide association analysis identifies common variants influencing infant brain volumes. Transl. Psychiatry 7, e1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC, 2010. A new Kv1.2 channelopathy underlying cerebellar ataxia. J. Biol. Chem 285, 32160–32173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaben MJ, Gray WP, 2013. Neuropeptides and hippocampal neurogenesis. Neuropeptides 47, 431–438. [DOI] [PubMed] [Google Scholar]
- Zhao C, Aviles C, Abel RA, Almli CR, McQuillen P, Pleasure SJ, 2005. Hippocampal and visuospatial learning defects in mice with a deletion of frizzled 9, a gene in the Williams syndrome deletion interval. Development 132, 2917–2927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.