

Abstract
Human pluripotent stem cell (hPSC) technology has revolutionized the field of biology through the unprecedented ability to study the differentiation of human cells in vitro. In the past decade, hPSCs have been applied to study development, model disease, develop drugs, and devise cell replacement therapies for numerous biological systems. Of particular interest is the application of this technology to study and treat optic neuropathies such as glaucoma. Retinal ganglion cells (RGCs) are the primary cell type affected in these diseases, and once lost, they are unable to regenerate in adulthood. This necessitates the development of strategies to study the mechanisms of degeneration as well as develop translational therapeutic approaches to treat early- and late-stage disease progression. Numerous protocols have been established to derive RGCs from hPSCs, with the ability to generate large populations of human RGCs for translational applications. In this review, the key applications of hPSCs within the retinal field are described, including the use of these cells as developmental models, disease models, drug development, and finally, cell replacement therapies. In greater detail, the current report focuses on the differentiation of hPSC-derived RGCs and the many unique characteristics associated with these cells in vitro including their genetic identifiers, their electrophysiological activity, and their morphological maturation. Also described is the current progress in the use of patient-specific hPSCs to study optic neuropathies affecting RGCs, with emphasis on the use of these RGCs for studying disease mechanisms and pathogenesis, drug screening, and cell replacement therapies in future studies.
Keywords: hPSCs, Retinal ganglion cells, Pluripotent stem cells, Retina, Optic neuropathies
Retinal ganglion cells (RGCs) play a crucial role in transmitting visual information from the eye to the brain. This transduction pathway can be severed due to disease or injury, which can inhibit light information from reaching the appropriate processing centers, and further result in loss of vision and blindness. Damage to the RGCs can occur in response to injury to the tissue, as well as following the onset of diseases known as optic neuropathies. Such debilitating conditions lead to the degeneration and eventual loss of RGCs, as these cells do not possess the capacity to regenerate in adulthood.
To date, no therapies exist to delay or halt the progression of RGC degeneration. Furthermore, by the time a clinical diagnosis has been delivered to a patient, a significant percentage of the RGC population has already been irreversibly lost [1]. This shortcoming necessitates the development of strategies to study the progression of RGC degeneration and pathogenesis as well as develop translational therapeutic approaches targeting RGCs. Human pluripotent stem cells (hPSCs) serve as an attractive model for such studies as they can be derived from patient somatic sources and can provide an unlimited source of cells that can be differentiated to any cell type of the body [2, 3]. As such, the utilization of hPSCs as a model system has revolutionized the field of developmental biology, translational disease modeling, and personalized medicine [4–7].
5.1. Applications of hPSCs
hPSCs can be used as an impactful and resourceful developmental model as they allow access to some of the earliest time points of embryonic development that would otherwise be unavailable. Before the discovery of hPSC technology, our understanding of retinal development was largely informed by animal models, with a limited option for studying the retina in humans through the use of human fetal or postmortem tissue. However, obtaining such samples was associated with numerous difficulties, as they were only accessible at limited developmental time points and ethical and legal issues limited their availability. Following the discovery of hPSCs, studies have effectively demonstrated their use as a novel model to study the major stages of human retinogenesis, including the primitive eye field giving rise to the evaginating optic vesicle, as well as the development of an optic cup-like structure deemed retinal organoids [8–11]. These hPSCs give rise to distinct populations of retinal neurons which not only follow the temporal sequence of embryonic retinal development, but also recapitulate the cellular mosaicism and lamination of the in vivo retina, allowing for a more bonafide model to study retinal development and disease [9, 11–16]. Furthermore, patient-specific hPSC-derived retinal neurons can be used for studying cell-specific mechanisms and have future implications for studying regeneration of retinal tissue following injury or disease [14, 17–20].
The studies of optic neuropathies caused by genetic determinants using hPSCs are of particular interest as they are the result of known mutations, which allow for a more direct connection of cellular changes to a particular phenotype [5, 7]. Patient-specific hPSCs can be differentiated into retinal cell types such as photoreceptors and RPE in a consistent and reproducible manner to study retinal degenerative disorders that cause damage to more outer retinal cell types, with the remarkable ability to use such cells for drug screening, cell replacement, and targeted therapeutics [11, 12, 20–27]. Such studies have conducted thorough and in-depth experiments that have identified specific cellular changes in outer retinal cell types such as oxidative and ER stress, autophagy deficits, alterations in protein trafficking, and phagocytotic defects associated with disease-causing mutations. Disorders in which inner retinal cell types, such as RGCs, are the primary affected cell type remain less explored, with a limited number of studies available which describe cell-specific disease deficits and pathogenesis [14, 28–35]. As such, the use of hPSCs to study RGC-specific diseases is critical to unveiling disease mechanisms that cause the degeneration and eventual death of these cells in various optic neuropathies.
With the ability to elucidate mechanisms of neurodegeneration, which underlie numerous retinal diseases, hPSCs can be used to differentiate and enrich large populations of cells for drug development, including those high-throughput assays that utilize large chemical libraries to identify potential targets and pathways for drug development [4–6, 36]. This includes screening enriched populations of cells for safety and toxicity purposes, as well as developing new drugs which are able to target specific cellular pathways and circuits, with the hope of using such drugs for future therapeutics to treat neurodegenerative diseases [5, 6]. More specifically, patient-derived hPSCs have been differentiated into retinal cells such as RPE and photoreceptors and utilized to effectively screen drugs for future therapeutic purposes [20, 25, 27, 37] with studies using hPSC-derived RGCs for drug screening applications remaining largely unexplored [14, 38]. Although these strategies are not targeted at late-stage disease progression, the development of these approaches will be effective in early and mid stages of progression where few retinal neurons have degenerated and those remaining neurons can be rescued by pharmacological approaches.
Lastly, hPSCs can be utilized for the development of cellular replacement strategies, with these approaches often targeting late-stage disease progression where the majority of the cell population has degenerated, and as such, cellular replacement is no longer an option. Cell replacement strategies require the ability to derive specific cell types in a timely and reproducible manner as well as the capacity to identify a given cell type in living cultures. hPSC replacement strategies have been most successful for those cell types with relatively short-distance synaptic targets such as cells of the outer retina including photoreceptors and RPE [17–19, 21, 39]. These studies have generated functional retinal cell types with the ability for these cells to perform as they normally would within the innate retina [11, 19, 26], allowing for successful transplantation following late-stage diagnosis of outer retinal diseases. However, cell replacement for projection neurons of the retina such as RGCs presents numerous difficulties, as these cells will need to extend axons over long distances and navigate through an unhealthy environment to connect with their proper synaptic targets in the brain. In order for hPSC-derived RGCs to be used as an effective cell replacement strategy, many obstacles remain including proper integration into the retina, the extension of long neurites that can find their appropriate synaptic targets, and the formation of proper and functional synapses in the brain.
5.2. RGC Differentiation from hPSCs
In order to efficiently derive RGCs from hPSCs, a clear understanding of retinal development is essential, as many of the same principles are translated from development in vivo to inform in vitro systems. For instance, RGCs are one of the earliest born cell types in the retina, followed closely by amacrine cells, cones, and horizontal cells. At later stages of retinal development, rods are generated, followed by bipolar cells and eventually Muller glia. Similarly, when hPSCs are directed to differentiate toward a retinal lineage, a similar temporal sequence of development is recapitulated, with RGCs being one of the first cell types specified, followed sequentially by later-born retinal cell types as predicted from in vivo studies [8, 10, 14, 20, 40–42].
RGCs serve as the final output of the retina by sending light to brain regions such as the lateral geniculate nucleus or superior colliculus [43, 44]. As such, RGCs tend to have much larger cell bodies and thicker axons, both of which are needed for long-distance propagation of action potentials [45–47]. These axons fasciculate together in the nerve fiber layer and form the optic nerve, which relays information to multiple brain regions. hPSC-derived RGCs have been known to display similar distinct morphologies (Fig. 5.1), with long neurites, fasciculated axons, and large three-dimensional cell bodies [14, 31, 32, 48–50]. These RGCs have also shown some degree of target specificity, with neurites preferentially targeting superior colliculus explants in vitro [51]. Additionally, the RGC layer occupies a distinct position in the retinal architecture, residing in the innermost layers [52–54]. Similarly, as hPSCs have shown the ability to self-organize into optic cup-like structures called retinal organoids that recapitulate in vivo retinal organization, RGCs reside within the innermost layer of these structures and photoreceptors occupy the more peripheral layers [8, 10, 11, 14].
Fig. 5.1.
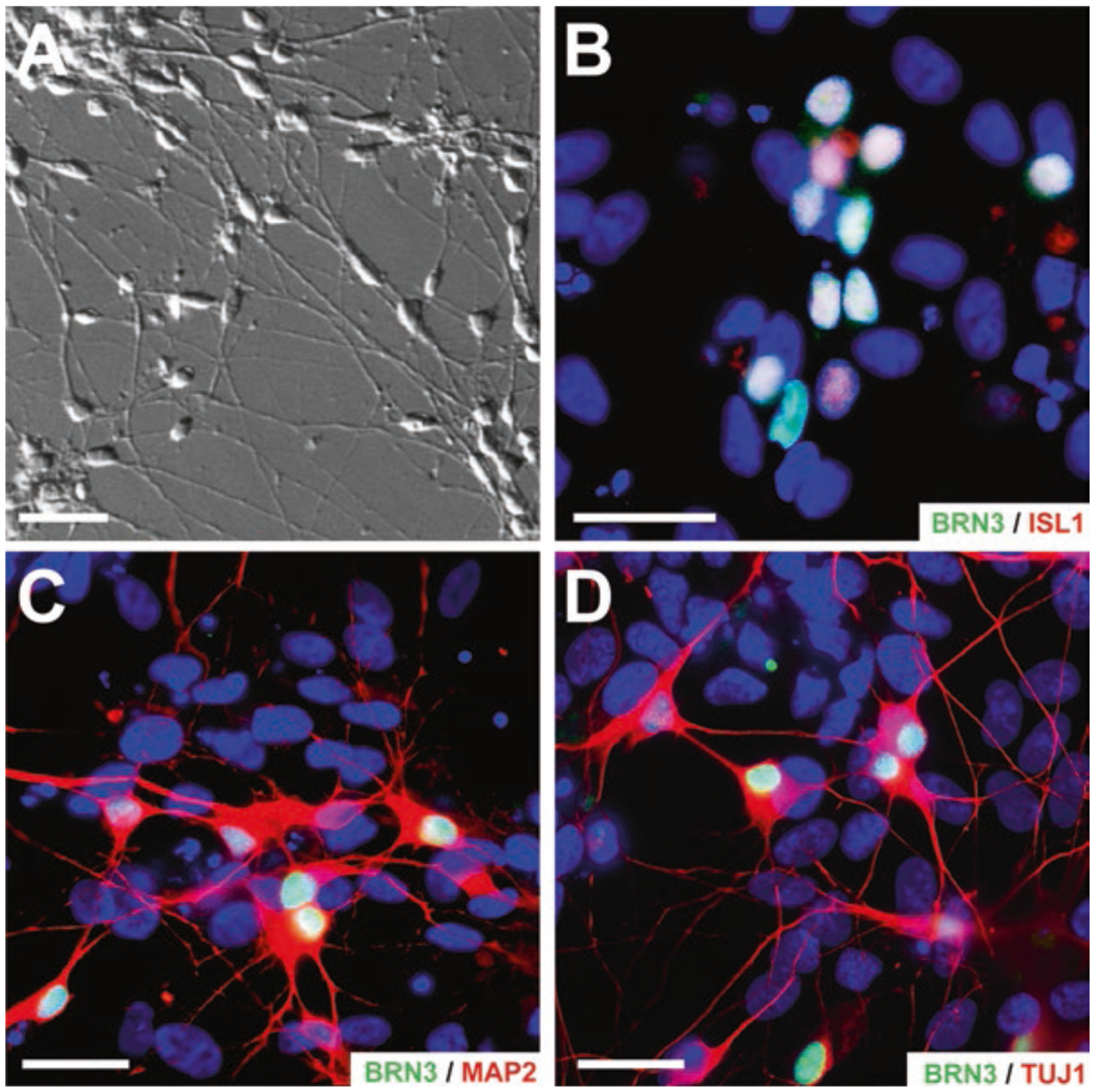
Common markers of hPSC-derived retinal ganglion cell. hPSC-derived RGCs exhibit transcriptional and morphological features. (a) DIC imaging of retinal cultures demonstrated RGC-like morphology with large, three-dimensional somas, and long neurite projections. (b, c) hPSC-derived RGCs can be identified by the expression of RGC-associated transcription factors such as ISL1 and BRN3. (c, d) Immunocytochemistry displayed unique morphological features of RGCs with BRN3-positive cells extending lengthy MAP2- and TUJ1-positive neurites. Scale bar for (a) is 50 and 100 μm for (b–d)
Within the retina, glutamate is used as the main excitatory neurotransmitter, and similar to their in vivo counterparts, it has been shown that hPSC-derived RGCs have the ability to respond to glutamate, recapitulating the presynaptic organization in vivo [55, 56]. These cells have also exhibited EPSCs [31], action potentials [14, 31, 56, 57], spontaneous calcium transients [32], and are sensitive to the voltage-gated potassium and sodium channel blockers TEA and TTX, respectively [14, 57]. While these features do not definitively distinguish RGCs from other neuronal cell types, RGCs are the predominant cell type within the retina that have the capacity to both respond to glutamatergic stimulation and conduct action potentials [58].
In order to identify RGCs apart from other cell types in differentiating cultures of hPSCs, a variety of genetic markers are often used to confirm the identity of these cells (Table 5.1). Early studies utilized TUJ1 as a common marker of RGC-like cells [40, 55], which is expressed by RGCs but does not necessarily confer any specificity, as many projection neurons throughout the central nervous system also express this marker [60, 61]. Therefore, more specific markers were needed in the field to identify RGCs when derived from hPSCs.
Table 5.1.
A summary of studies to date outlining the differentiation of retinal ganglion cells from human pluripotent stem cells
| Authors | ihc markers | Functional characteristics | Disease state modeled | References |
|---|---|---|---|---|
| Huang | Spiking activity, EPSCs | N/A | Riazifar [31], Chen [28] | |
| TUJ1 | ||||
| Fingert | N/A | TBK1-Normal Tension Glaucoma | Tucker [33] | |
| Thy1 | ||||
| Zack | Action potentials, responsive to AMPA/Kainate | N/A | Sluch et al. [56], Sluch [59] | |
| Thy1 | ||||
| Takahashi | N/A | N/A | Maekawa et al. [49], Kobayashi et al. [30] | |
| Thy1 | ||||
| Meyer | Action potentials, voltage-gated channels | E50K-normal tension glaucoma | Ohlemacher [14], Langer [48] | |
| DCX | ||||
| Wong | Voltage-gated channels, Axonal transport | Leber’s Hereditary Optic Neuropathy | Gill et al. [57], Wong [34] | |
| TUJ1 | ||||
| Ahmad | Voltage-gated channels, action potentials, calcium transients | Six6-primary open-angle glaucoma | Teotia et al. [32, 51] | |
| Thy1 | ||||
| Azuma | Voltage-gated channels, action potentials | N/A | Tanaka et al. [50], Yokoi et al. [35] | |
| NFH | ||||
| Ge | N/A | N/A | Huang et al. [29] | |
| Thy1 |
The most common transcription factor used to identity RGCs has been BRN3 (Fig. 5.1). BRN3 is expressed specifically by RGCs within the retina and is expressed shortly after RGC specification persisting into adulthood [62–64]. However, BRN3 expression is also present in other cells including somatosensory and auditory neurons [62, 63, 65, 66]. Thus, when starting with a population of hPSCs that can differentiate into any cell type of the body, caution must be taken not to rely solely on one marker as proof of identity. Instead, the combinatorial expression of genetic markers, morphological features, and functional characteristics must be combined to definitively identify a presumptive RGC in vitro.
A variety of additional markers have been used to identify RGCs, including ISLET1, HuC/D, and SNCG. Within the retina, these markers show a high degree of specificity for RGCs but are less reliable in identifying hPSC-derived RGCs as the expression of these markers can be found in other neuronal cell types [67–70]. More recently, RNA-binding protein with multiple splicing (RBMPS) has been shown to specifically label RGCs, although this expression often occurs later in development, with limited use of this protein to identify RGCs early in hPSC differentiation [71].
Current research regarding RGC development and maturation has highlighted the diverse nature of these cells, with the discovery and identification of more than 30 subtypes that differ in molecular, morphological, and physiological properties in animal models [72, 73]. These subtypes express specific molecular markers which categorize RGCs into distinct subtypes. The identification of RGC subtypes further enhances the understanding of RGC characteristics including their molecular signatures as well as their mosaicism in the retina, with recent studies identifying a number of RGC subtypes in hPSC-derived cells [48]. This ability to identify hPSC-derived RGC subtypes will allow for the ability to tailor future studies identifying RGC subtypes in human-derived cells.
5.3. Translational Applications of hPSC-Derived RGCs
Human pluripotent stem cells provide a fundamental and unique tool in the study of human retinal degenerative diseases, including optic neuropathies such as glaucoma, which explicitly target RGCs. As these cells provide the critical connection between the eye and the brain to transmit visual information, their degeneration results in vision loss and eventual blindness. Traditionally, the study of disease states has been limited to the development and use of animal models, which have led to significant advances in understanding retinal disease progression but sometimes fail when translated to humans in a clinical setting due to important differences between species [74, 75].
In order to address these obstacles, researchers have focused their work on the use of human stem cells to study the pathogenesis and treatment of human degenerative diseases. The development of hPSCs revolutionized the field of disease modeling with the ability to generate cells from patient-specific sources with disease-causing mutations [2, 3, 76]. Furthermore, the use of hPSCs allows for the high-throughput screening of thousands of compounds for their therapeutic efficacy, as well as providing critical safety and toxicity information in human cells to drug developers that cannot be fully elucidated with animal models.
Many hPSC-based genetic models of RGC degeneration, specifically glaucomatous degeneration, have been developed over the past few years [14, 32, 33], including mutations in SIX6, TANK Binding Kinase 1 (TBK1), and Optineurin (OPTN), all of which have been associated with forms of familial normal tension glaucoma (NTG). SIX6 is widely known for its role in eye development and morphogenesis, although mutations in this transcription factor have also been implicated as a contributor to NTG [77, 78]. Interestingly, hPSCs derived from patients with a missense mutation in SIX6 generated neural and retinal cells inefficiently, with reduced expression and dysregulation of key developmental genes [32]. In addition, SIX6-mutant patient RGCs displayed severe developmental, morphological, and electrophysiological deficits, with reduced neurite outgrowth and deficiency in the expression of axonal guidance molecules. Furthermore, SIX6-mutated RGCs demonstrated significantly higher levels of activated caspase-3. It is hypothesized that this missense mutation in SIX6 results in developmentally defective RGCs that might put these RGCs at higher risk for degeneration in adulthood.
When hPSCs are derived from a patient population with a known genetic basis of underlying retinal disease, the resulting cells recreate certain features of the disease phenotype and model the degeneration associated with retinal diseases. Duplications in TBK1 have been associated with development of NTG, although its exact role remains poorly understood. It is hypothesized that due to the close association of TBK1 with autophagy, duplications could disrupt the autophagic pathway, leading to RGC degeneration [79–82]. Interestingly, the generation of hPSCs from patient sources with the TBK1 duplication demonstrated decreased levels of autophagy activation when compared to control RGCs, thereby allowing for subsequent studies of disease mechanisms leading to degeneration of RGCs [33]. The accumulated results of studies such as these provide an important stepping stone towards the development of therapeutic approaches for retinal degenerative diseases.
The directed differentiation of RGCs from hPSCs provides a large quantity of cells for drug screening efforts, which can then specifically target RGCs to assess the ability of candidate compounds to rescue a disease phenotype. hPSC-derived RGCs allow for the screening of new drug compounds as well as the development of personalized treatment therapies, particularly when derived from patient-specific sources with a confirmed degenerative phenotype. With this in mind, recent studies have successfully recapitulated some of the degenerative process associated with optic neuropathies in hPSC-derived RGCs, with subsequent drug screening approaches identifying candidate treatment factors capable of rescuing RGC degeneration. Mutations in the OPTN protein have been associated with multiple types of neurodegeneration, including glaucoma and amyotrophic lateral sclerosis and glaucoma [38, 83]. The E50K missense mutation in OPTN has been associated with severe and early-onset NTG in a clinical setting [84]. Like TBK1, OPTN is closely associated with the autophagy pathway as it can act as an autophagy receptor [85, 86]. It is thought that OPTN mutations lead to degeneration through the dysregulation or blockage of the autophagy pathway. In a recent study, hPSC-derived RGCs derived from a patient with an E50K mutation in the OPTN gene displayed elevated levels of activated caspase-3 compared to control lines, with the ability to rescue these damaged RGCs following the treatment of these cells with neuroprotective factors such as BDNF and GDNF [14]. Therefore, the use of patient-derived hPSCs has provided an in-depth understanding of disease progression and mechanisms, which have subsequently enabled the identification of compounds to combat the degeneration of RGCs.
Postautosomal dominant optic atrophy (DOA) is the most common hereditary optic atrophy that culminates in degeneration of RGCs and eventual central vision loss [87]. Mutations in the OPA1 gene, which affect inner mitochondrial membrane proteins, are the most common cause of DOA, resulting in mitochondrial dysfunction, decreased ATP production, as well as mitochondrial fragmentation [88–90]. Consequently, generation of hPSCs from patients carrying an OPA1 mutation exhibited significantly more apoptosis and inefficiently differentiated into RGCs, suggesting that mutations in OPA1 mediate apoptosis and contribute to the pathogenesis of optic atrophy. In addition to modeling a disease, hPSC-derived RGCs can also be used to uncover neuroprotective agents that slow or even halt the progression of RGC degeneration. The addition of Noggin and estrogen to OPA1-mutated iPSCs promoted the differentiation of RGCs, representing potential therapeutic agents for OPA1-related optic atrophy. Taken together, these studies represent the first demonstration of disease modeling using hPSC-derived RGCs and are an important step forward in understanding disease mechanisms and identifying potential therapeutic interventions.
Traditional approaches to combat RGC degeneration have relied on treatment during early stages of the disease process when neuroprotection is still feasible. hPSC-derived RGCs can be used to effectively study early stages of retinal degenerative diseases, and subsequent drug screening approaches may aid in the development of neuroprotective strategies for degenerating RGCs. Unfortunately, a majority of optic neuropathy patients have experienced significant RGC loss by the time of diagnosis, and the large number of cells that are irreversibly lost at later stages in the disease renders neuroprotection futile. In such cases, the transplantation of healthy RGCs to replace degenerated cells remains the final option to restore visual function. However, there has been a lack of successful development of replacement strategies for RGCs due to obstacles such as the long-distance projection of RGC axons as well as the functional formation of synapses with appropriate postsynaptic targets. In spite of these obstacles, studies have demonstrated the ability of hPSC-derived RGCs to survive on a tissue-engineered scaffold following transplantation into the vitreous chamber of rabbits and rhesus monkeys [91].
Recent work has described the ability to elegantly combine the use of CRISPR gene editing and hPSC technology to elucidate potential therapeutic pathways involved in RGC degeneration and find suitable compounds to intervene within this pathway [36, 59]. In such studies, hPSC-derived RGCs have engineered to express a tdTomato-P2A-Thy1.2 reporter driven under the expression of RGC marker BRN3, with the use of the Thy1.2 surface receptor to further immunopurify and isolate RGCs. The resultant tdTomato-positive RGCs were treated with colchicine to simulate axonal injury allowing for the subsequent investigation of pathways that mediated RGC death. In particular, the dual leucine zipper kinase (DLK) pathway and its downstream partner leucine zipper kinase (LZK) pathway were discovered as mediators of RGC cell death and were proposed as possible targets for intervention to increase cell survival. Treatment of RGCs with Sunitinib, a FDA-approved drug that is known to interfere with the DLK and LZK pathways, was shown to enhance survival of injured hPSC-RGCs in a dose-dependent manner. This study provided integral insights into RGC degeneration, particularly the role of the DLK and LZK pathway in RGC injury, and demonstrated the first use of CRISPR-engineered hPSC-derived RGCs for drug screening applications.
Taken together, the generation of genetic disease models from hPSC-derived RGCs allows for the previously unattainable insight into early disease mechanisms and the development of tests for early detection in a clinical setting. In addition, hPSCs also allow for the generation of large populations of patient-specific cells for high-throughput drug screening. Finally, the derivation of patient-specific cells is a groundbreaking advancement in personalized medicine as the ability to screen compounds on a patient’s own cells could greatly optimize the process of discovering the most effective therapies for retinal degeneration.
5.4. Future Applications of hPSC-Derived RGCs
Efforts from the past decade have utilized hPSCs to study the development and disease pathologies of all different cell types of the retina. While most efforts have emphasized cells of the outer retina, the use of hPSCs to study RGC-specific development and disease is largely lacking, with important implications for future studies elucidating developmental and disease mechanisms within such cells. Future applications of hPSC-derived RGCs include the use of retinal organoids as a model for RGC development and disease, exploring RGC subtypes in human cells and how these subtypes may be differentially affected in disease states, studying cell replacement and drug screening therapies for treating optic neuropathies, and utilizing genome editing to enhance the study of human RGCs in vitro.
The term retinal organoid refers to a three-dimensional structure derived from stem cells, which recapitulates the temporal development and spatial lamination of the retina. As RGCs are one of the first cells to develop, retinal organoids display similar lamination to the in vivo retina, with RGCs found within inner layers and photoreceptors residing in more peripheral layers (Fig. 5.2). Recent efforts have utilized retinal organoids for studying cells and diseases of the outer retina including photoreceptors and RPE due to their short synaptic contacts and the ability for these cells to mature in this environment [11, 16, 26, 92]. However, there have been a limited number of studies focusing on RGC development within retinal organoids. As RGCs are one of the first to develop within the retina, the accessibility of studying RGC differentiation within retinal organoids provides a more feasible timeline than that of photoreceptors that take upward of 200 days to become fully mature. Although the timeline of development is considerably shorter, the ability for RGCs to fully mature into a bonafide cell type remains unclear. As the projection neurons of the retina, RGCs extend long axons out of the eye and into the brain to synapse with postsynaptic targets. Within retinal organoids, RGC axons are limited to the space and confinement of these structures, without the current accessibility to extend outward toward a specific target. Additionally, RGCs are one of the only cell types in the retina to fire action potentials in order to conduct their visual information to the brain. No studies currently exist displaying RGCs within retinal organoids to possess this capability, which is crucial for using cells grown in such a manner for reliable developmental and disease studies, although dissociated hPSC-derived RGCs have demonstrated functional properties [14, 31, 32, 51, 56, 59]. Future studies in this area may find the need to incorporate biomechanical engineering approaches in order to devise an exit for RGC axons confined within retinal organoids using an extracellular matrix mold or scaffolding device. Additionally, future studies will need to focus on expediting the maturation of RGCs within retinal organoids, including their ability to fire action potentials, in order for these cells to be used as a model which closely recapitulates the mechanisms of the human retina.
Fig. 5.2.
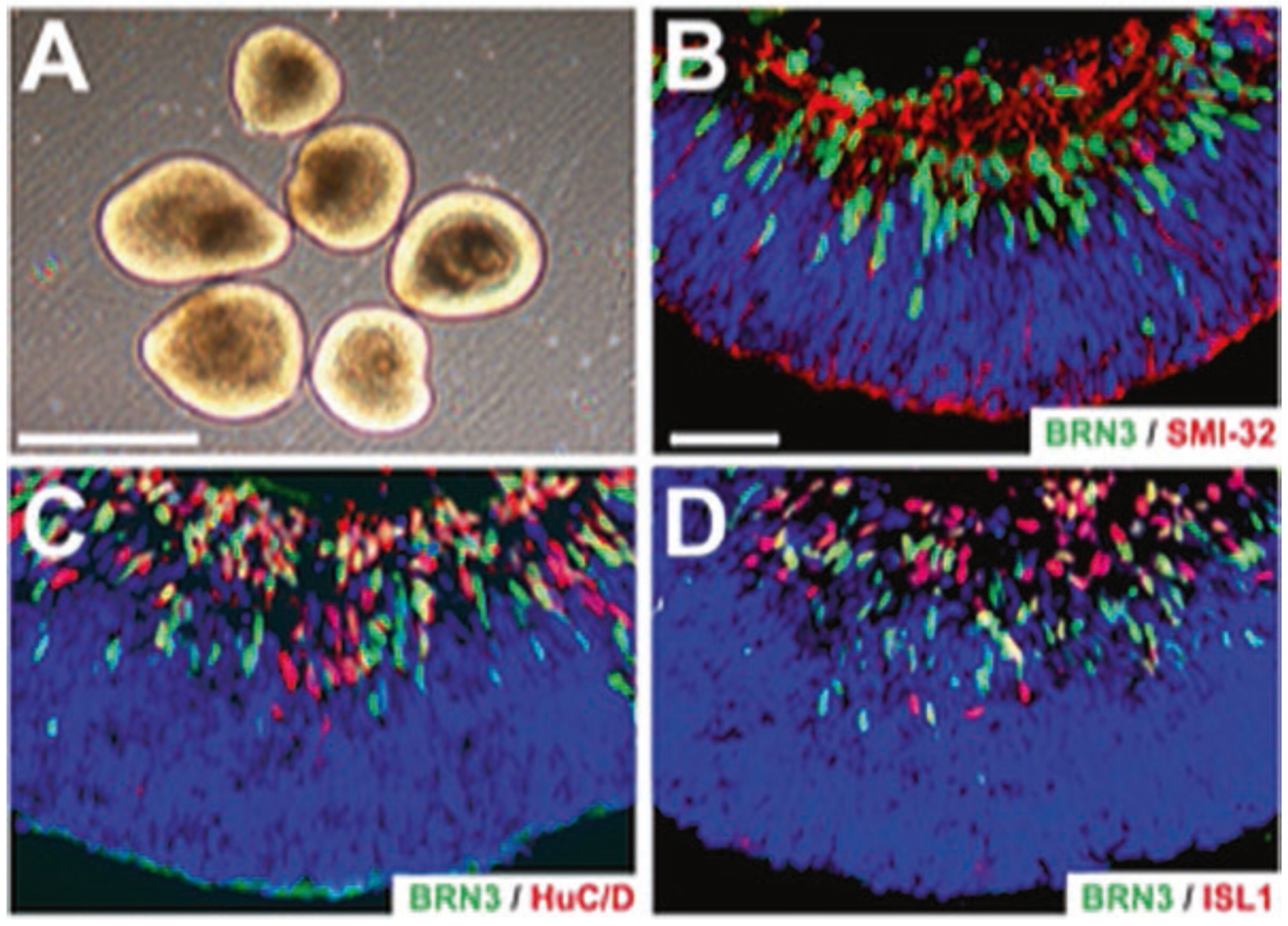
Retinal organoids sustain unique morphology and cellular lamination. (a) Retinal organoids exhibit a bright outer ring around the periphery indicating retinal organization and lamination. (b–d) Retinal organoids exhibit widespread expression of the RGC marker BRN3, neural markers HUC/D, and ISLET1, as well as cytoskeletal marker SMI-32 co-localized within apical layers of the organoid. Scale bars equal 500 μm for (a) and 50 μm for (b–d)
In recent years, the study of RGCs has become more complex with the discovery of more than 30 different subtypes of these cells, all of which possess varying molecular, structural, and functional characteristics [72, 73]. Although the majority of RGC subtypes have been characterized in animal models, more recently a variety of these subtypes have also been identified in hPSC-derived cells [48]. Future efforts will focus on the differentiation of specific subtypes from hPSCs as well as their ability to conduct similar functional characteristics in vitro as observed within the in vivo retina. Not only have these animal studies described the complex nature of these many subtypes, they have also described the differential survival and regeneration of different subtypes following acute injury or disease [93–99]. As such, this leaves opportunity to study this phenomenon within hPSC-derived RGCs in order to properly understand the degeneration of specific RGC subtypes in different optic neuropathies as well as provide a greater understanding of how to address degenerative cell loss in cell replacement studies and therapeutics in the future.
hPSC-derived RGCs also have the ability to be used for large-scale drug screening to address early disease progression as well as provide an opportunity for cell replacement strategies to address late-stage degeneration [5, 6]. In the future, hPSC-derived RGCs can be grown in large, reproducible quantities from numerous patient sources harboring disease mutations to utilize large chemical libraries and databases, with the hope of finding specific compounds that may provide rescue and therapeutic benefits to diseased RGCs. More so, the discovery of therapeutic compounds could be utilized for early disease diagnosis and progression in order to halt or rescue cell-specific deficits. To address late-stage disease progression, hPSCs provide an advantageous source for cell replacement strategies as these cells can be expanded indefinitely and differentiated into all cell types of the body, including RGCs [14, 31, 32, 35, 57, 59, 100]. In order to be able to use hPSC-derived RGCs for cell replacement strategies, many obstacles need to be addressed by future studies. To be used for cell replacement, hPSC-derived RGCs must possess the capacity to differentiate and function as an in vivo RGC would, including the ability fire repetitive action potentials, extend axons long distances to a proper target, and make functional synaptic connections. Each of these points will need to be addressed fully by future studies in order for hPSCs to be used as a reliable source for cell replacement in end-stage disease progression affecting RGCs.
Lastly, CRIPSR engineering is an emerging and fast-growing technology which can be used to better study RGC development and disease at high and in-depth capacities in future studies [101]. To study early RGC development, CRISPR technology has the ability to engineer transcriptional activators or repressors [102–104]. These allow for the conditional expression of genes at specific time points in development, with the ability to identify pathways which are essential for proper RGC maturation and may be dysfunctional in disease states. hPSC-derived RGCs can also be engineered to express epitope tags that allow for visualization of proteins that are especially difficult to study or for which no antibodies exist, including those involving apoptotic and autophagy pathways [105, 106]. More so, this technique would also allow a fluorescent reporter to tag a protein of interest, allowing for the identification and study of proteins in living cultures, including the ability to easily identify RGCs in vitro and study their electrophysiological properties [56, 59, 102, 105].
CRISPR engineering can also be used to create specific gene mutations [101, 107], including those known to cause various optic neuropathies including mutations in TBK1, OPTN, and SIX6. This technology allows for the generation of disease-harboring hPSCs for rare genetic determinants where patient sources are scarce and allows for the direct creation of an isogenic control which is important for identifying disease phenotypes across many cell lines. More so, CRISPR technology can be used to correct gene mutations in cases where patient samples are abundant and hPSCs can be readily reprogrammed and differentiated. The ability to correct these specific gene mutations allows for a more direct connection of disease mechanisms and phenotypes between healthy and diseased samples. CRISPR-engineered hPSCs also provide a personalized source of cells for cell replacement therapy with the ability to collect somatic cells from patients, correct gene-causing mutations, reprogram them into healthy hPSCs, and use patient-specific cells for replacement of degenerated RGCs [108, 109]. CRISPR engineering will be essential and necessary for future studies of hPSC-derived RGCs in regard to studying their proper development in vitro, how they are affected in disease states, and how these cells can be used for cell replacement strategies.
5.5. Conclusions
Over the past decade, hPSC technology has been utilized as a reliable tool to elucidate cell development and various neurodegenerative diseases when derived from patient-specific sources. hPSCs can be readily differentiated into all cell types of the retina in a manner which closely recapitulates retinogenesis, with a variety of cell types exhibiting characteristics of bonafide retinal cell types such as photoreceptors and RPE. The majority of these studies have observed outer retinal cell development and disease, with limited studies available looking at RGC differentiation and maturation and diseases that target RGCs such as glaucoma. A greater understanding of RGC-specific differentiation and maturation is needed in order to elucidate important development pathways and signaling cascades, which may be adversely affected in disease which target RGCs. CRISPR engineering has opened up the possibilities of studying the development of RGCs in vitro by the insertion of fluorescent reporters driven by important neural, retinal, and RGC-specific genes, allowing a more comprehensive technique for studying cell-specific differentiation. An enhanced understanding of RGC development will create the opportunity to develop a cell type that closely resembles native RGCs and can be used as a more reliable model for studying disease phenotypes as well as their ability to be used for cell replacement therapies for optic neuropathies in the future.
Acknowledgments
Grant support was provided by the National Eye Institute (R01 EY030986 and R21 EY031120), Indiana Department of Health Brain and Spinal Cord Injury Fund (JSM), an IU Collaborative Research Grant from the Office of the Vice President for Research (JSM), an award from the IU Signature Center for Brain and Spinal Cord Injury (JSM), a grant from Stark Neurosciences Research Institute, Eli Lilly and Company, and by the Indiana Clinical and Translational Sciences Institute, funded in part by grant # UL1TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences (SO) and an IUPUI Graduate Office First Year University Fellowship (KL), and the Purdue Research Foundation Fellowship (KL).
Contributor Information
Sarah K. Ohlemacher, Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA
Kirstin B. Langer, Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA
Clarisse M. Fligor, Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA
Elyse M. Feder, Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA
Michael C. Edler, Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA Department of Medical and Molecular Genetics, Indiana University, Indianapolis, IN, USA.
Jason S. Meyer, Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA Department of Medical and Molecular Genetics, Indiana University, Indianapolis, IN, USA; Stark Neurosciences Research Institute, Indiana University, Indianapolis, IN, USA.
References
- 1.Harwerth RS, Quigley HA (2006) Visual field defects and retinal ganglion cell losses in patients with glaucoma. Arch Ophthalmol 124:853–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872 [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R et al. (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920 [DOI] [PubMed] [Google Scholar]
- 4.Ebert AD, Svendsen CN (2010) Human stem cells and drug screening: opportunities and challenges. Nat Rev Drug Discov 9:367–372 [DOI] [PubMed] [Google Scholar]
- 5.Grskovic M, Javaherian A, Strulovici B, Daley GQ (2011) Induced pluripotent stem cells--opportunities for disease modelling and drug discovery. Nat Rev Drug Discov 10:915–929 [DOI] [PubMed] [Google Scholar]
- 6.Inoue H, Yamanaka S (2011) The use of induced pluripotent stem cells in drug development. Clin Pharmacol Ther 89:655–661 [DOI] [PubMed] [Google Scholar]
- 7.Marchetto MC, Brennand KJ, Boyer LF, Gage FH (2011) Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum Mol Genet 20:R109–R115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y (2011) Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472:51–56 [DOI] [PubMed] [Google Scholar]
- 9.Meyer JS, Shearer RL, Capowski EE, Wright LS, Wallace KA, McMillan EL, Zhang SC, Gamm DM (2009) Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc Natl Acad Sci U S A 106:16698–16703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, Saito K, Yonemura S, Eiraku M, Sasai Y (2012) Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10:771–785 [DOI] [PubMed] [Google Scholar]
- 11.Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao LH, Peters A, Park TS, Zambidis ET, Meyer JS et al. (2014) Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun 5:4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamao H, Mandai M, Okamoto S, Sakai N, Suga A, Sugita S, Kiryu J, Takahashi M (2014) Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep 2:205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohlemacher SK, Iglesias CL, Sridhar A, Gamm DM, Meyer JS (2015) Generation of highly enriched populations of optic vesicle-like retinal cells from human pluripotent stem cells. Curr Protoc Stem Cell Biol 32:1h.8.1–1h.820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohlemacher SK, Sridhar A, Xiao Y, Hochstetler AE, Sarfarazi M, Cummins TR, Meyer JS (2016) Stepwise Differentiation of Retinal Ganglion Cells from Human Pluripotent Stem Cells Enables Analysis of Glaucomatous Neurodegeneration. Stem Cells 34:1553–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reichman S, Slembrouck A, Gagliardi G, Chaffiol A, Terray A, Nanteau C, Potey A, Belle M, Rabesandratana O, Duebel J et al. (2017) Generation of storable retinal organoids and retinal pigmented epithelium from adherent human iPS cells in xeno-free and feeder-free conditions. Stem Cells 35:1176–1188 [DOI] [PubMed] [Google Scholar]
- 16.Volkner M, Zschatzsch M, Rostovskaya M, Overall RW, Busskamp V, Anastassiadis K, Karl MO (2016) Retinal organoids from pluripotent stem cells efficiently recapitulate retinogenesis. Stem Cell Rep 6:525–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carr AJ, Vugler AA, Hikita ST, Lawrence JM, Gias C, Chen LL, Buchholz DE, Ahmado A, Semo M, Smart MJ et al. (2009) Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS One 4:e8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.da Cruz L, Fynes K, Georgiadis O, Kerby J, Luo YH, Ahmado A, Vernon A, Daniels JT, Nommiste B, Hasan SM et al. (2018) Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat Biotechnol 36:328. [DOI] [PubMed] [Google Scholar]
- 19.Lamba DA, McUsic A, Hirata RK, Wang PR, Russell D, Reh TA (2010) Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PLoS One 5:e8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer JS, Howden SE, Wallace KA, Verhoeven AD, Wright LS, Capowski EE, Pinilla I, Martin JM, Tian S, Stewart R et al. (2011) Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 29:1206–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruczek K, Gonzalez-Cordero A, Goh D, Naeem A, Jonikas M, Blackford SJI, Kloc M, Duran Y, Georgiadis A, Sampson RD et al. (2017) Differentiation and transplantation of embryonic stem cell-derived cone photoreceptors into a mouse model of end-stage retinal degeneration. Stem Cell Rep 8:1659–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maruotti J, Wahlin K, Gorrell D, Bhutto I, Lutty G, Zack DJ (2013) A simple and scalable process for the differentiation of retinal pigment epithelium from human pluripotent stem cells. Stem Cells Transl Med 2:341–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mellough CB, Sernagor E, Moreno-Gimeno I, Steel DH, Lako M (2012) Efficient stage-specific differentiation of human pluripotent stem cells toward retinal photoreceptor cells. Stem Cells 30:673–686 [DOI] [PubMed] [Google Scholar]
- 24.Peng CH, Huang KC, Lu HE, Syu SH, Yarmishyn AA, Lu JF, Buddhakosai W, Lin TC, Hsu CC, Hwang DK et al. (2018) Generation of induced pluripotent stem cells from a patient with X-linked juvenile retinoschisis. Stem Cell Res 29:152–156 [DOI] [PubMed] [Google Scholar]
- 25.Singh R, Kuai D, Guziewicz KE, Meyer J, Wilson M, Lu J, Smith M, Clark E, Verhoeven A, Aguirre GD et al. (2015) Pharmacological modulation of photoreceptor outer segment degradation in a human iPS cell model of inherited macular degeneration. Mol Ther 23:1700–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wahlin KJ, Maruotti JA, Sripathi SR, Ball J, Angueyra JM, Kim C, Grebe R, Li W, Jones BW, Zack DJ (2017) Photoreceptor outer segment-like structures in long-term 3D retinas from human pluripotent stem cells. Sci Rep 7:766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida T, Ozawa Y, Suzuki K, Yuki K, Ohyama M, Akamatsu W, Matsuzaki Y, Shimmura S, Mitani K, Tsubota K et al. (2014) The use of induced pluripotent stem cells to reveal pathogenic gene mutations and explore treatments for retinitis pigmentosa. Mol Brain 7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Riazifar H, Guan MX, Huang T (2016) Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res Ther 7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang L, Chen M, Zhang W, Sun X, Liu B, Ge J (2018) Retinoid acid and taurine promote NeuroD1-induced differentiation of induced pluripotent stem cells into retinal ganglion cells. Mol Cell Biochem 438:67–76 [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi W, Onishi A, Tu HY, Takihara Y, Matsumura M, Tsujimoto K, Inatani M, Nakazawa T, Takahashi M (2018) Culture systems of dissociated mouse and human pluripotent stem cell-derived retinal ganglion cells purified by two-step immunopanning. Invest Ophthalmol Vis Sci 59:776–787 [DOI] [PubMed] [Google Scholar]
- 31.Riazifar H, Jia Y, Chen J, Lynch G, Huang T (2014) Chemically induced specification of retinal ganglion cells from human embryonic and induced pluripotent stem cells. Stem Cells Transl Med 3:424–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teotia P, Van Hook MJ, Wichman CS, Allingham RR, Hauser MA, Ahmad I (2017b) Modeling glaucoma: retinal ganglion cells generated from induced pluripotent stem cells of patients with SIX6 risk allele show developmental abnormalities. Stem Cells 35:2239–2252 [DOI] [PubMed] [Google Scholar]
- 33.Tucker BA, Solivan-Timpe F, Roos BR, Anfinson KR, Robin AL, Wiley LA, Mullins RF, Fingert JH (2014) Duplication of TBK1 stimulates autophagy in iPSC-derived retinal cells from a patient with normal tension glaucoma. J Stem Cell Res Ther 3:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong RCB, Lim SY, Hung SSC, Jackson S, Khan S, Van Bergen NJ, De Smit E, Liang HH, Kearns LS, Clarke L et al. (2017) Mitochondrial replacement in an iPSC model of Leber’s hereditary optic neuropathy. Aging 9:1341–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yokoi T, Tanaka T, Matsuzaka E, Tamalu F, Watanabe SI, Nishina S, Azuma N (2017) Effects of neuroactive agents on axonal growth and pathfinding of retinal ganglion cells generated from human stem cells. Sci Rep 7:16757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welsbie DS, Mitchell KL, Jaskula-Ranga V, Sluch VM, Yang Z, Kim J, Buehler E, Patel A, Martin SE, Zhang PW et al. (2017) Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase-dependent injury signaling in neurons. Neuron 94:1142–1154.e1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin ZB, Okamoto S, Osakada F, Homma K, Assawachananont J, Hirami Y, Iwata T, Takahashi M (2011) Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS One 6:e17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minegishi Y, Nakayama M, Iejima D, Kawase K, Iwata T (2016) Significance of optineurin mutations in glaucoma and other diseases. Prog Retin Eye Res 55:149–181 [DOI] [PubMed] [Google Scholar]
- 39.Assawachananont J, Mandai M, Okamoto S, Yamada C, Eiraku M, Yonemura S, Sasai Y, Takahashi M (2014) Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice. Stem Cell Rep 2:662–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirami Y, Osakada F, Takahashi K, Okita K, Yamanaka S, Ikeda H, Yoshimura N, Takahashi M (2009) Generation of retinal cells from mouse and human induced pluripotent stem cells. Neurosci Lett 458:126–131 [DOI] [PubMed] [Google Scholar]
- 41.Sridhar A, Ohlemacher SK, Langer KB, Meyer JS (2016) Robust differentiation of mRNA-reprogrammed human induced pluripotent stem cells toward a retinal lineage. Stem Cells Transl Med 5:417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sridhar A, Steward MM, Meyer JS (2013) Nonxenogeneic growth and retinal differentiation of human induced pluripotent stem cells. Stem Cells Transl Med 2:255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herrera E, Erskine L, Morenilla-Palao C (2017) Guidance of retinal axons in mammals. Semin Cell Dev Biol 85:48. [DOI] [PubMed] [Google Scholar]
- 44.Martersteck EM, Hirokawa KE, Evarts M, Bernard A, Duan X, Li Y, Ng L, Oh SW, Ouellette B, Royall JJ et al. (2017) Diverse central projection patterns of retinal ganglion cells. Cell Rep 18:2058–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolb H, Nelson R, Mariani A (1981) Amacrine cells, bipolar cells and ganglion cells of the cat retina: a Golgi study. Vision Res 21:1081–1114 [DOI] [PubMed] [Google Scholar]
- 46.Volgyi B, Chheda S, Bloomfield SA (2009) Tracer coupling patterns of the ganglion cell subtypes in the mouse retina. J Comp Neurol 512:664–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe M, Rodieck RW (1989) Parasol and midget ganglion cells of the primate retina. J Comp Neurol 289:434–454 [DOI] [PubMed] [Google Scholar]
- 48.Langer KB, Ohlemacher SK, Phillips MJ, Fligor CM, Jiang P, Gamm DM, Meyer JS (2018) Retinal ganglion cell diversity and subtype specification from human pluripotent stem cells. Stem Cell Rep 10:1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maekawa Y, Onishi A, Matsushita K, Koide N, Mandai M, Suzuma K, Kitaoka T, Kuwahara A, Ozone C, Nakano T et al. (2016) Optimized culture system to induce neurite outgrowth from retinal ganglion cells in three-dimensional retinal aggregates differentiated from mouse and human embryonic stem cells. Curr Eye Res 41:558–568 [DOI] [PubMed] [Google Scholar]
- 50.Tanaka T, Yokoi T, Tamalu F, Watanabe S, Nishina S, Azuma N (2015) Generation of retinal ganglion cells with functional axons from human induced pluripotent stem cells. Sci Rep 5:8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teotia P, Van Hook MJ, Ahmad I (2017a) A co-culture model for determining the target specificity of the de novo generated retinal ganglion cells. Bio Protoc 7:e2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curcio CA, Allen KA (1990) Topography of ganglion cells in human retina. J Comp Neurol 300:5–25 [DOI] [PubMed] [Google Scholar]
- 53.Dowling JE (1970) Organization of vertebrate retinas. Invest Ophthalmol 9:655–680 [PubMed] [Google Scholar]
- 54.Dowling JE, Boycott BB (1966) Organization of the primate retina: electron microscopy. Proc R Soc Lond B Biol Sci 166:80–111 [DOI] [PubMed] [Google Scholar]
- 55.Lamba DA, Karl MO, Ware CB, Reh TA (2006) Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A 103:12769–12774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sluch VM, Davis CH, Ranganathan V, Kerr JM, Krick K, Martin R, Berlinicke CA, Marsh-Armstrong N, Diamond JS, Mao HQ et al. (2015) Differentiation of human ESCs to retinal ganglion cells using a CRISPR engineered reporter cell line. Sci Rep 5:16595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gill KP, Hung SS, Sharov A, Lo CY, Needham K, Lidgerwood GE, Jackson S, Crombie DE, Nayagam BA, Cook AL et al. (2016) Enriched retinal ganglion cells derived from human embryonic stem cells. Sci Rep 6:30552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Velte TJ, Masland RH (1999) Action potentials in the dendrites of retinal ganglion cells. J Neurophysiol 81:1412–1417 [DOI] [PubMed] [Google Scholar]
- 59.Sluch VM, Chamling X, Liu MM, Berlinicke CA, Cheng J, Mitchell KL, Welsbie DS, Zack DJ (2017) Enhanced stem cell differentiation and immunopurification of genome engineered human retinal ganglion cells. Stem Cells Transl Med 6:1972–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharma RK, Netland PA (2007) Early born lineage of retinal neurons express class III beta-tubulin isotype. Brain Res 1176:11–17 [DOI] [PubMed] [Google Scholar]
- 61.Sullivan KF, Cleveland DW (1986) Identification of conserved isotype-defining variable region sequences for four vertebrate beta tubulin polypeptide classes. Proc Natl Acad Sci U S A 83:4327–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O’Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG (1996) Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature 381:603–606 [DOI] [PubMed] [Google Scholar]
- 63.Gan L, Xiang M, Zhou L, Wagner DS, Klein WH, Nathans J (1996) POU domain factor Brn-3b is required for the development of a large set of retinal ganglion cells. Proc Natl Acad Sci U S A 93:3920–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nadal-Nicolas FM, Jimenez-Lopez M, Sobrado-Calvo P, Nieto-Lopez L, Canovas-Martinez I, Salinas-Navarro M, Vidal-Sanz M, Agudo M (2009) Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci 50:3860–3868 [DOI] [PubMed] [Google Scholar]
- 65.Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, Bilsland J, Cao L, Stevens E, Whiting P et al. (2012) Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol 30:715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E (2013) Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 500:217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ericson J, Thor S, Edlund T, Jessell TM, Yamada T (1992) Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science 256:1555–1560 [DOI] [PubMed] [Google Scholar]
- 68.Genead R, Danielsson C, Andersson AB, Corbascio M, Franco-Cereceda A, Sylven C, Grinnemo KH (2010) Islet-1 cells are cardiac progenitors present during the entire lifespan: from the embryonic stage to adulthood. Stem Cells Dev 19:1601–1615 [DOI] [PubMed] [Google Scholar]
- 69.Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W et al. (2011) LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8:267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Phillips RJ, Hargrave SL, Rhodes BS, Zopf DA, Powley TL (2004) Quantification of neurons in the myenteric plexus: an evaluation of putative pan-neuronal markers. J Neurosci Methods 133:99–107 [DOI] [PubMed] [Google Scholar]
- 71.Rodriguez AR, de Sevilla Muller LP, Brecha NC (2014) The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J Comp Neurol 522:1411–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dhande OS, Stafford BK, Lim JA, Huberman AD (2015) Contributions of retinal ganglion cells to subcortical visual processing and behaviors. Ann Rev Vis Sci 1:291–328 [DOI] [PubMed] [Google Scholar]
- 73.Sanes JR, Masland RH (2015) The types of retinal ganglion cells: current status and implications for neuronal classification. Annu Rev Neurosci 38:221–246 [DOI] [PubMed] [Google Scholar]
- 74.Shanks N, Greek R, Greek J (2009) Are animal models predictive for humans? Philos Ethics Humanit Med 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shelley C (2012) Complex systems, evolution, and animal models. Stud Hist Philos Biol Biomed Sci 43:311. [DOI] [PubMed] [Google Scholar]
- 76.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ (2008) Disease-specific induced pluripotent stem cells. Cell 134:877–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carnes MU, Liu YP, Allingham RR, Whigham BT, Havens S, Garrett ME, Qiao C, Katsanis N, Wiggs JL, Pasquale LR et al. (2014) Discovery and functional annotation of SIX6 variants in primary open-angle glaucoma. PLoS Genet 10:e1004372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wiggs JL, Yaspan BL, Hauser MA, Kang JH, Allingham RR, Olson LM, Abdrabou W, Fan BJ, Wang DY, Brodeur W et al. (2012) Common variants at 9p21 and 8q22 are associated with increased susceptibility to optic nerve degeneration in glaucoma. PLoS Genet 8:e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fingert JH (2011) Primary open-angle glaucoma genes. Eye (Lond) 25:587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kawase K, Allingham RR, Meguro A, Mizuki N, Roos B, Solivan-Timpe FM, Robin AL, Ritch R, Fingert JH (2012) Confirmation of TBK1 duplication in normal tension glaucoma. Exp Eye Res 96:178–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, Dikic I (2013) Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126:580–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ryan TA, Tumbarello DA (2018) Optineurin: a coordinator of membrane-associated cargo trafficking and autophagy. Front Immunol 9:1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H et al. (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465:223–226 [DOI] [PubMed] [Google Scholar]
- 84.Aung T, Rezaie T, Okada K, Viswanathan AC, Child AH, Brice G, Bhattacharya SS, Lehmann OJ, Sarfarazi M, Hitchings RA (2005) Clinical features and course of patients with glaucoma with the E50K mutation in the optineurin gene. Invest Ophthalmol Vis Sci 46:2816–2822 [DOI] [PubMed] [Google Scholar]
- 85.Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19:983–997 [DOI] [PubMed] [Google Scholar]
- 86.Ying H, Yue BY (2016) Optineurin: the autophagy connection. Exp Eye Res 144:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen T, Wu H, Guo L, Liu L (2015) A modified rife algorithm for off-grid DOA estimation based on sparse representations. Sensors 15:29721–29733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barboni P, Savini G, Cascavilla ML, Caporali L, Milesi J, Borrelli E, La Morgia C, Valentino ML, Triolo G, Lembo A et al. (2014) Early macular retinal ganglion cell loss in dominant optic atrophy: genotype-phenotype correlation. Am J Ophthalmol 158:628–636.e623 [DOI] [PubMed] [Google Scholar]
- 89.Lodi R, Tonon C, Valentino ML, Iotti S, Clementi V, Malucelli E, Barboni P, Longanesi L, Schimpf S, Wissinger B et al. (2004) Deficit of in vivo mitochondrial ATP production in OPA1-related dominant optic atrophy. Ann Neurol 56:719–723 [DOI] [PubMed] [Google Scholar]
- 90.Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, Wissinger B, Pinti M, Cossarizza A, Vidoni S et al. (2008) OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 131:352–367 [DOI] [PubMed] [Google Scholar]
- 91.Li K, Zhong X, Yang S, Luo Z, Li K, Liu Y, Cai S, Gu H, Lu S, Zhang H et al. (2017) HiPSC-derived retinal ganglion cells grow dendritic arbors and functional axons on a tissue-engineered scaffold. Acta Biomater 54:117–127 [DOI] [PubMed] [Google Scholar]
- 92.Phillips MJ, Wallace KA, Dickerson SJ, Miller MJ, Verhoeven AD, Martin JM, Wright LS, Shen W, Capowski EE, Percin EF et al. (2012) Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Invest Ophthalmol Vis Sci 53:2007–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Daniel S, Clark AF, McDowell CM (2018) Subtype-specific response of retinal ganglion cells to optic nerve crush. Cell Death Dis 4:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Della Santina L, Ou Y (2017) Who’s lost first? Susceptibility of retinal ganglion cell types in experimental glaucoma. Exp Eye Res 158:43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Duan X, Qiao M, Bei F, Kim IJ, He Z, Sanes JR (2015) Subtype-specific regeneration of retinal ganglion cells following axotomy: effects of osteopontin and mTOR signaling. Neuron 85:1244–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.El-Danaf RN, Huberman AD (2015) Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types. J Neurosci 35:2329–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Majander A, Joao C, Rider AT, Henning GB, Votruba M, Moore AT, Yu-Wai-Man P, Stockman A (2017) The pattern of retinal ganglion cell loss in OPA1-related autosomal dominant optic atrophy inferred from temporal, spatial, and chromatic sensitivity losses. Invest Ophthalmol Vis Sci 58:502–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ou Y, Jo RE, Ullian EM, Wong RO, Della Santina L (2016) Selective vulnerability of specific retinal ganglion cell types and synapses after transient ocular hypertension. J Neurosci 36:9240–9252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Puyang Z, Gong HQ, He SG, Troy JB, Liu X, Liang PJ (2017) Different functional susceptibilities of mouse retinal ganglion cell subtypes to optic nerve crush injury. Exp Eye Res 162:97–103 [DOI] [PubMed] [Google Scholar]
- 100.Daniszewski M, Senabouth A, Nguyen QH, Crombie DE, Lukowski SW, Kulkarni T, Sluch VM, Jabbari JS, Chamling X, Zack DJ et al. (2018) Single cell RNA sequencing of stem cell-derived retinal ganglion cells. Sci Data 5:180013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. [DOI] [PubMed] [Google Scholar]
- 102.Dominguez AA, Lim WA, Qi LS (2016) Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol 17:5–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H et al. (2015) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517:583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI et al. (2016) CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell 18:541–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ratz M, Testa I, Hell SW, Jakobs S (2015) CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci Rep 5:9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R (2013) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154:1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sander JD, Joung JK (2014) CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32:347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stern JH, Temple S (2011) Stem cells for retinal replacement therapy. Neurotherapeutics 8:736–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wiley LA, Burnight ER, Songstad AE, Drack AV, Mullins RF, Stone EM, Tucker BA (2015) Patient-specific induced pluripotent stem cells (iPSCs) for the study and treatment of retinal degenerative diseases. Prog Retin Eye Res 44:15–35 [DOI] [PubMed] [Google Scholar]