

Abstract
A sensitive and specific method for the analysis of anisodamine and its metabolites in rat urine by liquid chromatography–electrospray ionization tandem mass spectrometry (LC–MS/MS) was developed. Various extraction techniques (free fraction, acid hydrolyses and enzyme hydrolyses) and their comparison were carried out for investigation of the metabolism of anisodamine. After extraction procedure the pretreated samples were injected on a reversed-phase C18 column with mobile phase (0.2 ml/min) of methanol/0.01% triethylamine solution (adjusted to pH 3.5 with formic acid) (60:40, v/v) and detected by MS/MS. Identification and structural elucidation of the metabolites were performed by comparing their changes in molecular masses (ΔM), retention-times and full scan MSn spectra with those of the parent drug. At least 11 metabolites (N-demethyl-6β-hydroxytropine, 6β-hydroxytropine, tropic acid, N-demethylanisodamine, hydroxyanisodamine, anisodamine N-oxide, hydroxyanisodamine N-oxide, glucuronide conjugated N-demethylanisodamine, sulfate conjugated and glucuronide conjugated anisodamine, sulfate conjugated hydroxyanisodamine) and the parent drug were found in rat urine after the administration of a single oral dose 25 mg/kg of anisodamine. Hydroxyanisodamine, anisodamine N-oxide and the parent drug were detected in rat urine for up 95 h after ingestion of anisodamine.
Keywords: Anisodamine, LC–MS/MS, Metabolite
1. Introduction
Drug metabolism experiment has played an important role in the drug discovery, drug design and drug clinical application. Therefore, fast and efficient ways to provide accurate information of drug metabolism on the target compounds and their major metabolites are required [1], [2], [3]. In the past, GC with electron capture detection or nitrogen phosphorus detection and HPLC with ultraviolet (UV) spectrophotometric detection, fluorescence detection or electrochemical detection (ECD) were the main methods to detect drugs and their major metabolites in vivo. But these technologies cannot provide high enough sensitivity, specificity and molecular structural information for the qualitative assay of drugs and their metabolites. The coupled GC–MS technology can overcome these insufficiencies, but it often requires time consuming derivatization of the target compound [4], [5]. So, this method is not suitable for the detection of drug metabolites, too.
Since the introduction of atmospheric pressure ionization (API) interfaces, LC–MS has been increasingly used to determine drugs and their metabolites for pre-clinical and clinical studies [6], [7], [8]. LC–MS system allows for the analyses of thermolabile, highly polar and non-volatile metabolites owing to its soft-ionization technique and high sensitivity, and the target compounds can be directly determined in mixtures without complicated sample preparation or derivatization. Compared with LC–MS, LC–MS/MS can give us the maximum amount of structural information and high specificity for qualitative analysis at trace levels. It has been proved to be a powerful approach for the metabolic identification of drugs [9], [10], [11], [12], [13]. So, LC–MS/MS technique is frequently the initial choice for metabolite detection and identification. Structural elucidation of drug metabolites using LC–MS/MS is based on the premise that metabolites retain the substructures of the parent drug molecule. MS/MS product ion spectrum of each metabolite provides detailed substructural information of its structure. So, using the product ion spectrum of parent drug as a substructural template, metabolites presented in crude mixtures may be rapidly identified and detected based on their changes in molecular masses (ΔM) and spectral patterns of product ions even without standards for each metabolite [14], [15], [16].
Anisodamine, tropic acid-6β-hydroxide-3α-tropic ester, was a kind of tropane alkaloids extracted from the leaves of traditional Chinese medicine Anisodus tanguticus (its synthetic form is called 654-2). It has widespread activities such as spasmolytic, anaesthetic, acesodyne and ophthalmic effects, and it is often used to treat transmissible shock, hepatitis, nephritis, sugar diabetes and so on [17], [18], [19]. In China, anisodamine was tentatively used to treat patients for Severe Acute Respiratory Syndrome (SARS) in 2003. Despite its important therapeutical values, its metabolism in vivo is not clear yet. Up till now, the works only focused on the quantitative determination of anisodamine in rabbit and human serum by means of thin layer chromatography [20], micellar liquid chromatography [21] and reversed phase-high performance liquid chromatography [22], [23].
In this paper, a highly sensitive and specific LC–MS/MS method was presented to identify anisodamine and its metabolites in rat urine. The LC–MS/MS analyses of urine sampled from healthy rats after administration of 25 mg/kg anisodamine revealed that the parent drug and its eleven metabolites existed in rat urine, which will be useful for future studies involving anisodamine, such as clinical therapy.
2. Experimental
2.1. Chemicals
Anisodamine was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (China). β-Glucuronidase (from Escherichia coli) were purchased from Sigma (St. Louis, MO, USA). Methanol was of HPLC grade (Fisher chemical Co. Inc., CA, USA). Distilled water, prepared from demineralised water, was used throughout the study. Other reagents used were of analytical grade.
2.2. Apparatus
LC–MS and LC–MS/MS experiments were performed on an LCQ Duo quadrupole ion trap mass spectrometer with a TSP4000 HPLC pump and a TSP AS3000 autosampler using both positive and negative electrospray as the ionization process (all components from Finnigan, Austin, TX, USA). The software Xcalibur version 1.2 (Finnigan) was applied for system operation and data collection. A high-speed desk centrifuge (TGL-16C, Shanghai Anting Scientific Instrument Factory, China) was used to centrifuge urine samples. The urine samples were extracted on ODS-18 solid-phase extraction cartridges (3 ml/ 200 mg, AccuBondII, Agilent Technologies, Palo Alto, CA, USA).
2.3. Sample preparation
2.3.1. Administration
Six wistar rats were obtained from Hubei Experimental Animal Research Center (China) 6 days before the experiment. The rats were provided standard laboratory food and water ad libitum. The rats weighed 197–205 g at the time of the experiment. The rats were housed in metabolic cages and fasted for 24 h but with access to water, then they were administered 25 mg/kg oral gavage doses of anisodamine. Urine samples were collected at different time-points: 1, 8, 22, 32, 47, 57, 71, 79, 95 and 100 h and centrifuged at 3000 × g for 10 min. The supernatants were stored at −20 °C until analyses.
2.3.2. Standard samples
Stock anisodamine solutions were prepared by dissolving anisodamine in methanol (1 mg/ml) and diluting to the desired concentration with methanol.
2.3.3. Urine extraction
2.3.3.1. Free fraction
An aliquot of 1 ml of mixed 0–24 h urine was loaded onto a C18 solid-phase extraction cartridge which was preconditioned with 2 ml of methanol and 1 ml of water. Then, the SPE cartridge was washed with 2 ml of water and the analytes were eluted with 1 ml of methanol. The elution solutions were filtered through 0.45 μm film and an aliquot of 10 μl was used for LC–MS/MS analyses.
2.3.3.2. Acidic hydrolysis
After optimizing the acidity and the heated time, 0.8 ml of 6 M HCl and 50 mg of cysteine were added to 1 ml of mixed 0–24 h urine samples. The mixture was heated at 100 °C for 60 min. After cooling to room temperature, it was neutralized to pH 8 with 6 M NaOH and extracted by SPE cartridge immediately, just like the procedure mentioned in Section 2.3.3.1.
2.3.3.3. Enzymatic hydrolysis
After optimizing the acidity, temperature, enzymatic content and the time of hydrolysis, 1 ml of mixed 0–24 h urine samples was adjusted to pH 5.0 with a few drops of glacial acetic acid. Then, 0.5 ml of acetate buffer (pH 5.0) and 0.2 ml of β-glucuronidase from E. coli (10,000 units/ml) were added to the solution prior to enzymatic hydrolyses. It took 5 h at 55 °C. After cooling, the solution was adjusted to pH 8 with 6 M NaOH and extracted by SPE cartridge immediately, just like the procedure mentioned in Section 2.3.3.1.
Free fraction was used for the comprehensive LC–MS/MS analyses of anisodamine and its metabolites. The target solutions after acidic and enzymatic hydrolyses were only used to assist in the investigation of phase II metabolites.
2.4. Chromatographic conditions
A reversed-phase column (AICHROM™ ReliAsil C18, 5 μm, 2 mm × 150 mm i.d., Agilent Technologies, Palo Alto, CA, USA) was connected with a guard column (cartridge 4.6 mm × 12.5 mm, 5 μm, Agilent Technologies) filled with the same packing material to separate the anisodamine and its metabolites in rat urine. After optimizing the column temperature and the acidity of the mobile phase using anisodamine standard, the temperature of the column was set at 40 °C. The mobile phase consisted of methanol and 0.01% triethylamine solution (adjusted to pH 3.5 with formic acid) (60:40, v/v), which was eluted at a flow rate of 0.2 ml/min during the whole run.
2.5. Mass spectrometry conditions
Mass spectral analyses were carried out using electrospray ionization (ESI) in positive ion detection mode, and only the structures of tropic acid and phase II metabolites were validated by LC–MS (MS/MS) in negative ion detection mode. Nitrogen was used as the sheath gas (40 arbitrary units). A typical source spray voltage of 5.0 kV, a capillary voltage of 45 V and a heated capillary temperature of 200 °C were obtained as optimal control conditions. The other parameters, including the voltages of octapole offset and tube lens offset, were optimised for maximum abundance of the ions of interest. The MS/MS product ion spectra were produced by collision induced dissociation (CID) of the protonated molecular ion [M + H]+ of analytes at their respective HPLC retention times utilizing helium in the ion trap, and the isolation width (m/z) was 1. The collision energy for each ion transition was optimized to produce the highest intensity of the selected ion peak. The optimized relative collision energy of 30% was used for all MSn works.
3. Results and discussion
3.1. LC–MS and LC–MS/MS analyses of standard
The first step of this work involved the characterization of the chromatographic and mass spectral properties of the parent drug. The chromatographic and mass spectrometry conditions were optimized using anisodamine standard. Full scan mass spectral analysis of anisodamine showed protonated molecular ion of m/z 306. The MS/MS product ion spectrum of the protonated molecular ion (m/z 306) and the predominant fragmentation patterns are shown in Fig. 1 . Fragmentation of protonated molecular ion of anisodamine in the ion trap leads to five main product ions at m/z: 288, 276, 140, 122 and 91. The product ions at m/z 288 and 276 were formed by the loss of H2O and HCHO from the parent ion at m/z 306, respectively. The most abundant product ion at m/z 140 was formed by the loss of tropic acid (C9H10O3, 166 Da). The m/z 122 ion was inferred to be produced by the loss of 184 Da (C9H10O3 + H2O). The product ion at m/z 91 was formed by the loss of NH2CH3 (31 Da) from the m/z 122 ion. It could be concluded that the ions at m/z 140, 122 and 91 were the characteristic product ions of anisodamine, and C9H10O3 (166 Da) and C9H12O4 (184 Da) were the characteristic neutral losses. These characteristic product ions and neutral losses were the key features that allowed identification of metabolites.
Fig. 1.
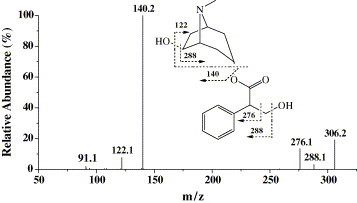
MS/MS product ion spectrum and the predominant fragmentation patterns of anisodamine.
The LC–MS2 chromatograms (parent ion at m/z 306) of blank urine and anisodamine (concentration at 50 ng/ml) were showed in Fig. 2 . In Fig. 2, the blank urine and anisodamine were analyzed in LC–MS2 mode in accordance with the detection mode of anisodamine and its metabolites in rat urine (Fig. 3 ). Anisodamine was eluted at 3.29 min under the experimental conditions (Fig. 2B). The specificity of the assay was evaluated by analyzing blank urine samples of rats, no impurity or endogenous interferences were found (Fig. 2A). The parent drug in blank urine and the extracted urine samples were stable for at least 2 months at 4 °C.
Fig. 2.
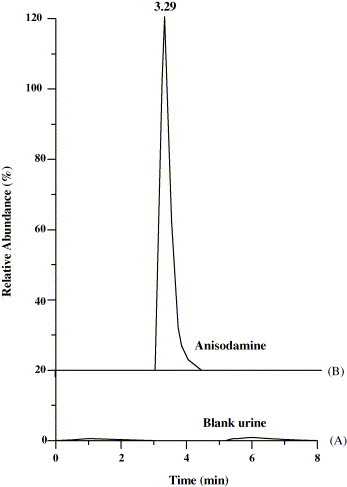
LC–MS2 chromatograms (parent ion at m/z 306) of (A) blank urine and (B) anisodamine standard (50 ng/ml).
Fig. 3.
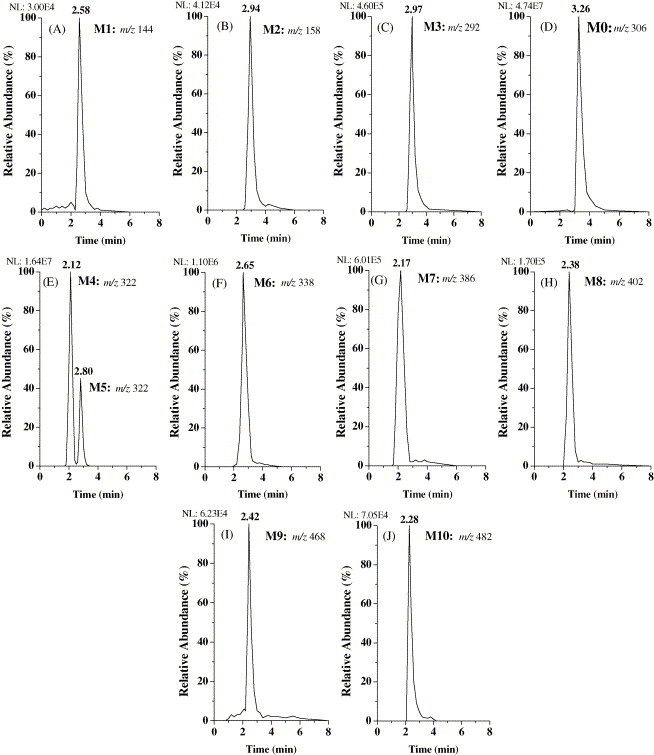
LC–MS2 chromatograms of anisodamine and its metabolites in rat urine.
In this work, the sensitivity of the method was determined using anisodamine standard, and its limit of detection (LOD) was lower than 6 ng/ml by LC–MS/MS. The mean recoveries (n = 5) were 76.7% at concentrations of 20 ng/ml.
3.2. LC–MS and LC–MS/MS analyses of metabolites
In order to identify the metabolites, the possible structures of metabolites have been speculated according to the metabolism rule of drugs firstly [24], [25]. The full scan mass spectrum of free fraction of rat urine after administration of anisodamine was compared with those of blank urine samples and anisodamine solution to find out the possible metabolites in rat urine. Then, these compounds were analyzed by LC–MS/MS. Their retention times, changes in observed mass (ΔM) and spectral patterns of product ions were compared with those of anisodamine standard to identify metabolites and elucidate their structures. Various extraction techniques (free fraction, enzyme hydrolyses and acid hydrolyses) and their comparison were carried out for investigation of the metabolism of anisodamine.
Based on the method mentioned above, the parent drug and its metabolites were found in rat urine after administration of anisodamine. Their molecular ions ([M + H]+) were at m/z 144, 158, 292, 306, 322, 338, 386, 402, 468 and 482, respectively. Their LC–MS2 chromatograms were presented in Fig. 3. The MS/MS product ion spectra of these analytes were shown in Fig. 4 . Among them, the retention time, the MS and MS/MS spectra of the molecular ion at m/z 306 (M0, Fig. 3, Fig. 4) were the same as those of anisodamine standard. Therefore, M0 can be confirmed as the unchanged parent drug.
Fig. 4.
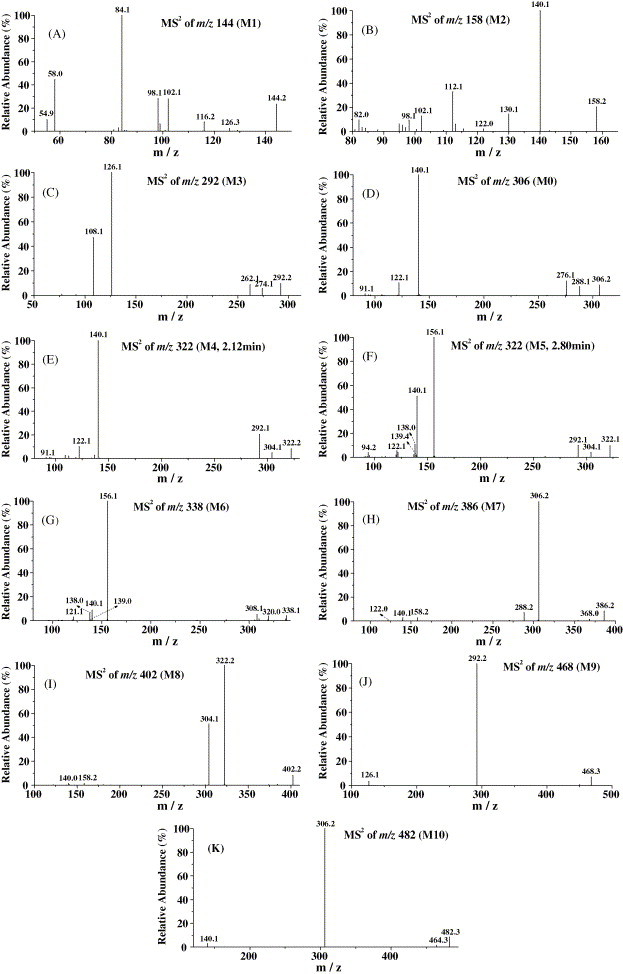
MS/MS product ion spectra of anisodamine and its metabolites in rat urine.
The characteristic product ions at m/z 140 and 122 of the parent drug appeared in the MS2 spectrum of m/z 158 (M2, Fig. 4B). The MS3 spectra of m/z 158 → 140 and m/z 158 → 122 was the same as those of m/z 306 → 140 and m/z 306 → 122 (anisodamine standard), respectively. So, M2 was identified as the hydrolysis product of anisodamine (6β-hydroxytropine).
The molecular ion at m/z 144 (M1) and its daughter ions at m/z 126, 116, 98 and 84 (Fig. 4A) were all 14 Da less than the molecular ion at m/z 158 (M2) and its daughter ions at m/z 140, 130, 112 and 98, respectively. These results indicated that M1 was the N-demethyl product of M2 (N-demethyl-6β-hydroxytropine).
The m/z 292 ion (M3, Fig. 4C) and its daughter ions at m/z 274, 262, 126 and 108 were all 14 Da less than the molecular ion of parent drug (m/z 306) and its daughter ions at m/z 288, 276, 140 and 122. Thus, M3 should be the N-demethyl product of anisodamine (N-demethylanisodamine).
The protonated molecular ion at m/z 322 was increased by 16 Da compared to that of the unchanged anisodamine. Two chromatographic peaks appeared in the LC–MS2 chromatogram of m/z 322 with retention times ca. 2.12 min (M4) and 2.80 min (M5), respectively (Fig. 3E). The appearances of the characteristic fragment ions at m/z 140, m/z 122 and characteristic neutral losses 182 Da (166 + 16) (m/z 322 → 140), 200 Da (184+16) (m/z 322 → 122) in the MS2 spectrum of molecular ion of M4 (Fig. 4E) indicated that M4 should be the hydroxylation product of anisodamine, and the localization of the hydroxyl group was at the tropic acid part. Reference [26] showed that in rat urine, the major metabolites of scopolamine, another kind of tropane alkaloids, were the three phenolic metabolites: p-hydroxy-, m-hydroxy-, and p-hydroxy-m-hydroxy-scopolamne, and no product of the benzylic carbon oxidation was found in rat urine. So, M4 was deduced as the hydroxyanisodamine hydroxylated at benzene ring.
The appearances of the predominant product ion at m/z 156 (140 + 16) and a pair of product ions at m/z 139 (156 − 17), m/z 138 (156 − 18) in the MS2 spectrum of the molecular ion of M5 (Fig. 4F) indicated that M5 should be the N-oxidation product of the parent drug (anisodamine N-oxide) because it is the cleavage feature of N-oxides to loss 17 and 18 Da. Cong [27] theoretically expounded the fragmentation feature of N-oxide: losing 17 and 18 Da from the parent molecule. The fragmentation feature has been validated by using oxymatrine in our experiment.
The characteristic product ions at m/z 140, 122 and 91 appeared in the MS2 spectrum of the molecular ion at m/z 338 (M6, Fig. 4G) which was increased by 32 Da compared to that of the parent drug. The product ion at m/z 156 (140 + 16) showed that the 6β-hydroxytropine part was oxidized, and the other oxidation should occur at tropic acid part. The m/z 139 (156 − 17) and 138 (156 − 18) ions showed the feature of N-oxide. So, M6 should be hydroxyscopolamine N-oxide. In Fig. 4F the fragment at m/z 140 should be produced from the ion at m/z 156 by the cleavage of N → O.
The product ions at m/z 306, 140 and 122 appeared in the MS2 spectrum of the molecular ion at m/z 386 (M7, Fig. 4H), and the MS3 spectrum of m/z 386 → 306 was the same as the MS2 spectrum of the protonated molecular ion of the parent drug (m/z 306). The product ion at m/z 306 was produced by neutral loss of 80 Da diagnostic of SO3 [28], [29]. Based on these data, M7 was identified as the sulfate conjugate of the parent drug. This deduction can be validated further by the fact that there was the parent ion at m/z 384 in the negative ion full scan LC–MS spectrum of the urine samples. The m/z 356 ([M + H–HCHO]+) ion was not found in the MS2 spectrum of m/z 386. So, from this fact, we inferred that the hydroxyl group at tropic acid was conjugated with sulfate in M7.
The protonated molecular ion at m/z 402 (M8) lost neutral fragment 80 Da (SO3) to produce the daughter ion at m/z 322 (Fig. 4I), and the MS3 spectrum of m/z 402 → 322 was the same as the MS2 spectrum of the protonated molecular ion of M4 (m/z 322, 2.12 min). There was the molecular ion at m/z 400 in the negative ion full scan MS spectrum of the urine samples. Consequently, M8 was identified as the sulfate conjugate of M4. Because phenolic hydroxyl has stronger affinity and higher speed than alcoholic hydroxyl in the sulfate esterifying reactions according to the rule of drug metabolism [24], M8 should be the sulfate conjugate of M4 esterified at its phenolic hydroxyl position. The selectivity of this conjugated reaction has been validated by many studies [24].
The MS2 spectrum of m/z 468 (M9) gave abundant daughter ion at m/z 292 (Fig. 4J), which was produced by neutral loss of 176 Da, and the MS3 spectrum of m/z 468 → 292 was the same as the MS2 spectrum of M3 (m/z 292). Besides, there was the molecular ion at m/z 466 in the negative ion full scan LC–MS spectrum of the urine samples, which gave the daughter ion at m/z 175 in its MS2 spectrum. Furthermore, the m/z 113 ion appeared in the MS3 spectrum of m/z 466 → 175. This fragmentation (m/z 466 → 175 → 113) is the cleavage feature of glucuronide conjugates [30], [31]. The m/z 438 ([M + H–HCHO]+) ion was not found in the MS2 spectrum of m/z 468. Thus, M9 was identified as the glucuronide conjugate of M3 conjugated with the hydroxyl group at tropic acid.
In the MS2 spectrum of m/z 482 (M10), the parent ion lost neutral fragment 176 Da to give its daughter ion at m/z 306 (Fig. 4K), and the MS3 spectrum of m/z 482 → 306 was the same as the MS2 spectrum of the protonated molecular ion of anisodamine (m/z 306). Besides, there was the m/z 480 ion in the negative ion full scan LC–MS spectrum of the urine samples, which gave the fragmentation of m/z 480 → 175 → 113 in its tandem MS spectra. The m/z 452 ([M + H–HCHO]+) ion was not found in the MS2 spectrum of m/z 482. So, M10 should be the glucuronide conjugate of anisodamine conjugated with the hydroxyl group at tropic acid.
The m/z 165 ion (M11) appeared in the negative ion full scan LC–MS spectrum of the urine samples. The appearances of the product ions at m/z 147 ([M − H–H2O]−) and 121 ([M − H–CO2]−) indicated that M11 was the hydrolysis product of anisodamine (tropic acid). No sulfate or glucuronide conjugate of M11 was found in rat urine.
Based on the above discussion, the proposed major metabolic pathway of anisodamine in rats was shown in Fig. 5 .
Fig. 5.
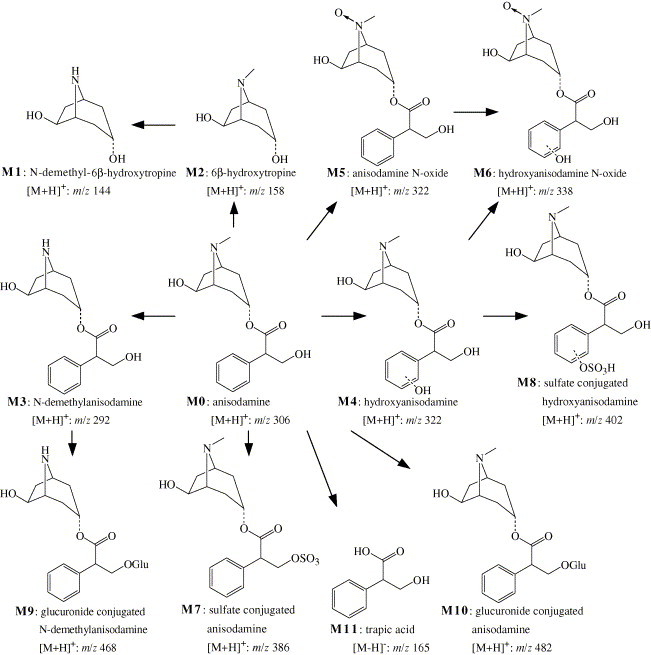
The proposed major metabolic pathway of anisodamine in rats.
These metabolites can be investigated further by comparing various extraction procedures (Table 1 ). Compared with free fraction, the peak areas of LC–MS2 chromatograms of M3, M0 and M4 increased markedly, and those of M7–M10 decreased after acidic hydrolyses. The peak areas of M3 and M0 increased, and those of M9 and M10 decreased after enzymatic hydrolysis. These results revealed that N-demethylanisodamine (M3), the unchanged anisodamine (M0) and hydroxyanisodamine (M4) excreted from rat urine as the free, sulfate conjugated or glucuronide conjugated forms.
Table 1.
Comparison between different extraction procedures
| Analyte | [M + H]+ | RT (min) | LC–MS2 chromatographic peak area (×106) |
||
|---|---|---|---|---|---|
| Free fraction | Acidic fraction | Enzymatic fraction | |||
| M1 | 144 | 2.58 | 0.58 | Trace | 0.61 |
| M2 | 158 | 2.94 | 1.7 | 1.9 | 1.7 |
| M3 | 292 | 2.97 | 21.2 | 26.6 | 25.0 |
| M0 | 306 | 3.26 | 3083.2 | 3121.7 | 3100.2 |
| M4 | 322 | 2.12 | 796.0 | 801.2 | 797.9 |
| M5 | 322 | 2.80 | 398.0 | 396.9 | 397.2 |
| M6 | 338 | 2.65 | 13.5 | 12.2 | 13.9 |
| M7 | 386 | 2.17 | 16.8 | Trace | 17.1 |
| M8 | 402 | 2.38 | 2.9 | NDa | 2.7 |
| M9 | 468 | 2.42 | 4.7 | ND | ND |
| M10 | 482 | 2.28 | 19.0 | Trace | Trace |
Not found.
The time of excretion of anisodamine and its metabolites in rat urine were detected by determining urine samples collected at different time-points (Fig. 6 ). The parent drug and its eleven metabolites were found in 0–1 h urine. The parent drug and its oxidation products (M4 and M5) could be detected for up 95 h using the tandem MS technique, and other metabolites could be detected for about 32–46 h.
Fig. 6.
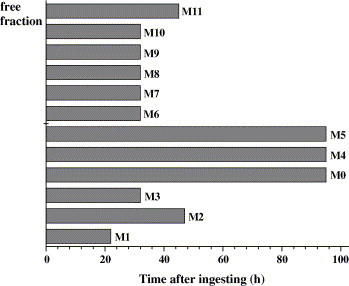
The excretion times of anisodamine and its metabolites in rat urine.
4. Conclusions
A liquid chromatography–electrospray ionization tandem mass spectrometry method was developed for the analysis of anisodamine and its major metabolites in rat urine. For the first time, eleven metabolites of anisodamine were identified through comparing their chromatographic retention times, changes in observed mass (ΔM) and tandem MS spectra with those of the parent drug. These metabolites included seven phase I metabolites (N-demethyl-6β-hydroxytropine, 6β-hydroxytropine, tropic acid, N-demethylanisodamine, hydroxyanisodamine, anisodamine N-oxide and hydroxyanisodamine N-oxide) and four phase II metabolites (glucuronide conjugated N-demethylanisodamine, sulfate conjugate and glucuronide conjugate of anisodamine, sulfate conjugated hydroxyanisodamine). Various extraction techniques and their comparison validated the presence of four phase II metabolites. The parent drug and its eleven metabolites were found in 0–1 h urine. The parent drug and its predominant metabolites (hydroxyanisodamine and anisodamine N-oxide) could be detected for up 95 h using the tandem MS technique.
The result showed that each metabolite has a specific “MSn Fingerprint” correlating to the MSn spectrum of parent drug. The structures of metabolites can be elucidated by comparing their MSn spectra to that of parent drug even without standards. Though the structures of metabolites cannot be determined conclusively by LC–MS/MS alone, the present method is still very valuable and dependable for the further study of the metabolism of anisodamine.
Acknowledgements
This paper was supported by the grant 020092325 of the Key Technology R&D Programme of China Hubei Provincial Science and Technology Department. The authors wish to thank their colleagues for their valuable technical assistance.
References
- 1.Hideko K., Ryoko A., Yoshikazu M., Junko K. J. Chromatogr. A. 2000;870:87. [Google Scholar]
- 2.Lam W., Ramanathan R. J. Am. Soc. Mass Spectrom. 2002;13:345. doi: 10.1016/S1044-0305(02)00346-X. [DOI] [PubMed] [Google Scholar]
- 3.Zhong D.F., Shi X.G., Sun L., Chen X.Y. J. Chromatogr. B. 2003;791:45. doi: 10.1016/s1570-0232(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z.H., Chen H.T., Chan K.K., Budd T., Ganapathi R.J. J. Chromatogr. B. 1999;728:85. doi: 10.1016/s0378-4347(99)00065-1. [DOI] [PubMed] [Google Scholar]
- 5.Spyridaki M.H., Lyris E., Georgoulakis I., Kouretas D., Konstantinidou M., Georgakopoulos C.G. J. Pharm. Biomed. Anal. 2004;35:107. doi: 10.1016/j.jpba.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Gu J.K., Zhong D.F., Chen X.Y., Zhou H., Shen J.C. Chem. J. Chin. Univ. 2000;21:690. [Google Scholar]
- 7.Hillis J., Morelli I., Neville D., Fox J., Leary A.C. Chromatographia. 2004;59:203. [Google Scholar]
- 8.Moller K., Crescenzi C., Nilsson U. Anal. Bioanal. Chem. 2004;378:197. doi: 10.1007/s00216-003-2267-5. [DOI] [PubMed] [Google Scholar]
- 9.Hsieh Y.S., Brisson J.M., Wang G.F., Ng K., Korfmacher W.A. J. Pharm. Biomed. Anal. 2003;33:251. doi: 10.1016/s0731-7085(03)00351-0. [DOI] [PubMed] [Google Scholar]
- 10.Zhong D.F., Chen X.Y., Gu J.K., Li X.Q., Guo J.F. Chin. Chim. Acta. 2001;313:147. doi: 10.1016/s0009-8981(01)00667-2. [DOI] [PubMed] [Google Scholar]
- 11.Siethoff C., Orth M., Ortling A., Brendel E., Wagner-Redeker W. J. Mass Spectrom. 2004;39:884. doi: 10.1002/jms.655. [DOI] [PubMed] [Google Scholar]
- 12.Cai Z.W., Qian T.X., Wong R.N.S., Jiang Z.H. Anal. Chim. Acta. 2003;492:283. [Google Scholar]
- 13.Yu X., Cui D.H., Davis M.R. J. Am. Soc. Mass Spectrom. 1999;10:175. doi: 10.1016/S1044-0305(98)00132-9. [DOI] [PubMed] [Google Scholar]
- 14.Appolpnova S.A., Shpak A.V., Semenov V.A. J. Chromatogr. B. 2004;800:281. doi: 10.1016/j.jchromb.2003.10.071. [DOI] [PubMed] [Google Scholar]
- 15.Molden E., Bøe G.H., Christensen H., Reubsaet L.J. J. Pharm. Biomed. Anal. 2003;33:275. doi: 10.1016/s0731-7085(03)00259-0. [DOI] [PubMed] [Google Scholar]
- 16.Gangl E., Utkin H., Gerber N., Vouros P. J. Chromatogr. A. 2002;974:91. doi: 10.1016/s0021-9673(02)01243-8. [DOI] [PubMed] [Google Scholar]
- 17.Ruan Q.R., Shong J.X., Deng Z.D. Chin. J. Pathol. 2000;29:212. [PubMed] [Google Scholar]
- 18.Chen J.Y. Clin. Med. Chin. 2000;16:649. [Google Scholar]
- 19.He X. J. Math. Med. 1998;11:250. [Google Scholar]
- 20.Zhan L.F., Zhang Y.Z., He M.S., Wang Y.Y., Chen Z.Y., He B.J. J. Chin. Med. Univ. 1989;18:271. [Google Scholar]
- 21.Ma P., Wu C.Y. Acta Pharm. Sin. 1992;27:763. [Google Scholar]
- 22.He X., Chen J. Acta Academiae Medicinae Xin Jiang. 1998;21:114. [Google Scholar]
- 23.Zhou H.B., Zhang X., He X. Chin. J. Chromatogr. 1996;14:60. [Google Scholar]
- 24.Zhang W.S., Li A.L. Higher Education Press; China: 1999. Medicinal Chemistry. pp. 43, 67–68. [Google Scholar]
- 25.Thomas G. Wiley; New York, NY: 2000. Medicinal Chemistry: A Introduction. [Google Scholar]
- 26.Wada S., Yoshimitsu T., Koga N., Yamada H., Oguri K., Yoshimura H. Xenobiotica. 1991;21:1289. doi: 10.3109/00498259109043204. [DOI] [PubMed] [Google Scholar]
- 27.Cong P.Z. Science Press; Beijing, China: 1997. The Application of Mass Spectroscopy in Natural Organic Chemistry. p. 406. [Google Scholar]
- 28.Stack R.F., Rudewicz P.J. J. Mass Spectrom. 1995;30:857. [Google Scholar]
- 29.Rudewicz P., Straub K.M. Anal. Chem. 1986;58:2928. doi: 10.1021/ac00127a008. [DOI] [PubMed] [Google Scholar]
- 30.Gu J.K., Zhong D.F., Chen X.Y. Fresenius J. Anal. Chem. 1993;65:553. [Google Scholar]
- 31.Chen X.Y., Zhong D.F., Hao J., Gu J.K. Acta Pharm. Sin. 1998;33:849. [Google Scholar]