

Abstract
The “exposome” is a term recently used to describe all environmental factors, both exogenous and endogenous, which we are exposed to in a lifetime. It represents an important tool in the study of autoimmunity, complementing classical immunological research tools and cutting-edge genome wide association studies (GWAS). Recently, environmental wide association studies (EWAS) investigated the effect of environment in the development of diseases. Environmental triggers are largely subdivided into infectious and non-infectious agents. In this review, we introduce the concept of the “infectome”, which is the part of the exposome referring to the collection of an individual's exposures to infectious agents. The infectome directly relates to geoepidemiological, serological and molecular evidence of the co-occurrence of several infectious agents associated with autoimmune diseases that may provide hints for the triggering factors responsible for the pathogenesis of autoimmunity. We discuss the implications that the investigation of the infectome may have for the understanding of microbial/host interactions in autoimmune diseases with long, pre-clinical phases. It may also contribute to the concept of the human body as a superorganism where the microbiome is part of the whole organism, as can be seen with mitochondria which existed as microbes prior to becoming organelles in eukaryotic cells of multicellular organisms over time. A similar argument can now be made in regard to normal intestinal flora, living in symbiosis within the host. We also provide practical examples as to how we can characterise and measure the totality of a disease-specific infectome, based on the experimental approaches employed from the “immunome” and “microbiome” projects.
Abbreviations: AMA, anti-mitochondrial antibody; ANA, anti-nuclear antibody; CMV, cytomegalovirus; CSF, cerebrospinal fluid; EBV, Epstein–Barr virus; EWAS, environmental-wide association study; FDR, first degree relatives; GWAS, genome wide association study; HHV6, human herpes virus 6; LC–MS/MS, liquid chromatography–tandem mass spectrometry (LC–MS/MS); MS, multiple sclerosis; IBS, irritable bowel syndrome; PBC, primary biliary cirrhosis; PCR, polymerase chain reaction; PDC, pyruvate dehydrogenase complex; SLE, systemic lupus erythematosus
Keywords: Autoantibodies, Autoimmunity, Autoimmune disease, Environment, Infection, Immunity, Microbiome
Highlights
► Genetics and environment are involved in the pathogenesis of autoimmunity. ► The study of individual infectious agents limits our understanding of the pathogenesis of autoimmunity. ► Exposome describes all environmental factors which we are exposed to. ► Infectome is the infectious component of the exposome. ► Investigation of the infectome is a holistic tool for the study of autoimmunity.
1. Introduction
It is widely accepted that the vast majority of diseases develop from the interaction between genes and the environment [1], [2], [3]. This concept has formed the basis for studying the pathogenesis of many diseases including autoimmune diseases [1], [2], [4]. There are now almost 100 categories of autoimmune diseases, both organ specific or systemic in nature. Although individual autoimmune diseases are relatively rare in any population that has been investigated so far, projected data estimate that approximately 5–20% of North Americans are affected by at least one autoimmune disease [5]. Some of the best known autoimmune diseases are type 1 diabetes, rheumatoid arthritis, multiple sclerosis, Grave's disease, Hashimoto's thyroiditis, myasthenia gravis, systemic lupus erythematosus, Sjögren's syndrome, scleroderma and autoimmune liver diseases such as autoimmune hepatitis, primary sclerosing cholangitis, and primary biliary cirrhosis (PBC) [5]. The observation that many autoimmune diseases may affect one individual, has led to the concept of the Mosaic of Autoimmunity [6], [7], [8], [9], [10], [11], [12], [13].
The study of genetic and epigenetic factors linking to autoimmunity is the focus of ongoing research [14]. Also, the exact signalling cascades that govern the perpetuation of inflammatory processes responsible for tissue destruction are poorly understood [15]. In recent years, genome wide association studies (GWAS) have identified numerous gene–disease associations, many of which include autoimmune diseases [2]. Although these associations are connected to genetic susceptibility, the genetic ‘dosage’ or number of associated genes required for disease development is not well defined [16]. Although GWAS have been instrumental in unlocking a pathogenetic starting point, exposure to environmental factors is also likely to contribute to the actual development of most diseases, and work with genetics in the induction of autoimmune disease [1], [2], [17]. For example, smoking has been indicated to increase the risk of developing MS in individuals with HLA-DRB1*1501 [18]. Epidemiological studies using toxicological, microbiological, biochemical, and immunological testing are important in order to identify these environmental agents, which include infectious organisms, xenobiotics, chemical compounds, heavy metals from prostheses and dental materials, radiation, vaccines, and foods to name but a few [1], [19], [20], [21], [22]. Heavy metals and vaccinations have also been implicated in the pathogenesis of autoimmune (auto-inflammatory) syndrome induced by adjuvants (ASIA) [23], [24], [25], [26], [27], [28], [29], [30], [31], [32]. In fact, siliconosis, Gulf war syndrome (GWS), macrophagic myofasciitis syndrome (MMF) and post-vaccination phenomena were linked with past exposure to adjuvants.
Since GWAS underlined the view that multiple genes are needed to induce autoimmunity [2], it is also likely that several environmental triggers either complement or substitute each other to provoke immune mediated processes which then lead to autoimmunity. The additive effects of these triggers, and their interaction with susceptible genes, remain poorly defined.
In recent years, the concept of an “exposome” has been introduced, as a means of collating, and possibly measuring the effects of environmental factors. The exposome takes into account all internal and external stimuli associated with a disease, and provides a potentially quantifiable way for the evaluation of environmental factors (Fig. 1 ). This review will examine the role of the exposome in relation to major exemplary autoimmune disease where genetics and environment most likely play key roles in pathogenesis. The role of the “infectome” as the infectious component of the microbiome/exposome that takes part (directly or indirectly) in the development of autoimmunity will also be introduced (Fig. 2 ).
Fig. 1.
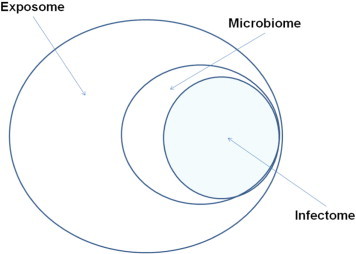
From exposome to infectome via microbiome.
“Exposome” describes all environmental factors which we are exposed to in a lifetime, both exogenous and endogenous, infectious and non-infectious. Environmental exposures are basically subdivided into infectious and non-infectious agents. The concept of “infectome” that we introduce, describes the part of the exposome which refers to the collection of an individual's exposures to infectious agents participating in the pathogenesis of autoimmune disease. The infectome can be considered a part of “microbiome”, the collection of the microbial products which the human body is exposed to at a given time.
Fig. 2.
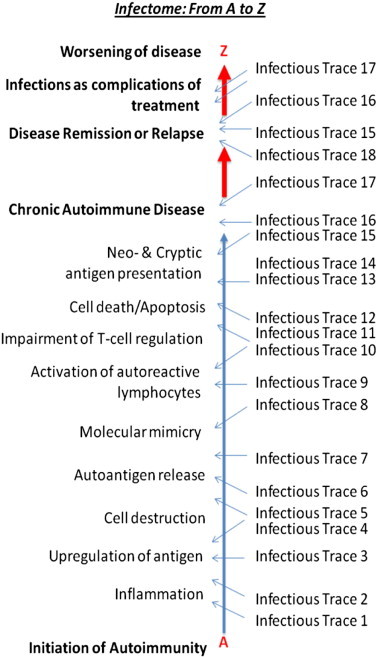
Tracing infectious triggers of autoimmunity: the infectome from A to Z. Infectious agents participating in a series of events critical for the initiation of autoimmunity and the development of autoimmune disease can be traced at various time points. The traces of these infectious agents may help us to understand the extent of their involvement in the loss of immunological breakdown and/or the maintenance of autoreactive immune responses leading to self destruction. Infections, at times different of those responsible for the chain reaction of events that led to autoimmune disease, may participate in the remission/relapse clinical patterns seen after the onset of the disease.
2. The exposome: what is it, and how do we measure it?
The exposome represents the totality of all environmental exposures, both exogenous and endogenous, which begins from conception, and extends throughout our lives [33], [34], [35], [36], [37]. This differs from previous epidemiological studies, which have largely concentrated on the external environment, and specifically to air, water, soil and food [35]. In contrast to approaches limited to external triggers, the study of the exposome needs to address endogenous factors directly or indirectly related to the environment [36], [37]. Endogenous sources include by-products of inflammation, lipid peroxidation, as well as oxidative stress [35]. Some of these components may act as nucleophiles or electrophiles, and as such, would be capable of DNA and protein modification [38]. Indeed, bacterial alterations in glycosylation patterns of immunoglobulins have been noted in several studies [39], [40], [41]. The collation of all these exposures may provide a fingerprint of a particular disease, which would likely complement data from GWAS [38].
The predominant problems in examining environmental exposures are the difficulties of quantification and normalisation as well as the determination of exposure sequences. This information is required, as alterations induced by one environmental agent may be necessary for a subsequent exposure to have its effect [2]. Moreover, environmental exposures within a population, as well as within an individual, are greatly varied. Given the myriad of causative possibilities in one individual, it may be useful clinically to look at causes within an individual in terms of exposure, sequence of exposure, hierarchy of exposure events, genetic and other risk factors that initiate or provoke exposure to various stimuli. Examined individually, these pieces may comprise the jigsaw puzzle that is loss of immunological tolerance, development of autoimmune disease and progression of clinically over disease and its complications. Although great variability may be seen from patient to patient, the overall collation of well-defined disease-related exposures individually may provide a picture of disease development in that patient. The exposome provides a potential platform by which these exposures may be measured. It has been suggested that their by-products in an individual, such as DNA and protein modification, can serve as biomarkers that are potentially measurable [38] in the blood or body fluids of patients [3], [36], [37], [38] via technologies such as liquid chromatography–tandem mass spectrometry (LC–MS/MS) [42], DNA adducts [43], functional measurements of antioxidant capacity, and breath analysis [34].
Rappaport suggests two methods in which the exposome may be measured: the ‘bottom-up’ method measures chemicals in air, water and the external environment, but is limited in that it does not take into account the actual uptake of those substances nor the internal environment of a subject [35]. As an alternative, the ‘top-down’ method measures biomarkers in the patient's blood, however, this method does not indicate a potential source of the toxicant. It is likely that both complementary methods, in conjunction with refined questionnaires, will define the exposome in individuals, and larger populations [35], [36], [37]. Frequent sampling could demonstrate changes of these markers over time, especially during critical phases in the development of a disease [35]. A study by Pleil and colleagues [34] indicates that breath analysis for particular biomarkers is capable of identifying whether one has been exposed, what the dosage was, how rapidly the body is eliminating the toxicant, as well as possibly identifying the short and long term effects [34]. Finally, a recent study by Patel and colleagues [44] has provided evidence that the exposome can indeed be measured and characterised. That study conducted an environmental-wide association study (EWAS) on type 2 diabetes mellitus, where epidemiological data was comprehensively interpreted in a manner similar to GWAS [44]. Associations were found with 37 environmental factors, including organochlorine, pesticides, nutrients/vitamins, polychlorinated biphenyls, and dioxins [44]. Other studies have shown a potential crossreactivity between antigens within the pancreatic islet cells and cow's milk casein in siblings of type 1 diabetics [45], and multiple sclerosis's myelin oligodendrocyte glycoprotein with milk butyrophilin [46], highlighting the potential role for food antigens as triggers of autoimmune disease. Similar approaches can be used in future studies on the role of the exposome in the development of autoimmune diseases, especially those which develop in genetically susceptible individuals several years or decades following the initial insult.
One major obstacle to the complete analysis of all these triggers is their heterogeneity. It is unlikely to cover all such components by homogeneous technology platforms, as all human biomarker measurements are subject to inter- and intra-subject variance. This includes the composition of the received data into one model, which appears to be a Sisyphean task even in the age of ultra-fast computing. Therefore, the break-down of this multiversity of components into easier accessible, homogeneous realms of markers seems to be a reasonable next step. Techniques that can be utilised to detect infectious agents like multi-parametric immunoassays for antibodies of various isotypes specific to bacterial or viral antigens, urine and stool cultures and polymerase chain reaction (PCR) can be considered to be robust and integrateable [36]. Routine screening to detect the presence of an infectious causative agent is used clinically in a small scale and for individual microbial agents on an everyday basis [36].
3. Exposome, infectome and autoimmunity
Infectious and non-infectious environmental agents have long been considered important for the development of autoimmunity [4], [47], [48], [49], [50], [51]. The list of non-infectious environmental factors which can trigger autoimmunity is vast, and includes tobacco smoke, pharmaceuticals, oestrogens, ultraviolet radiation, silica solvents, dietary components, heavy metals, dental materials, vaccines, and collagen/silicone implants (Table 1 ) [47], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [63], [64], [65], [66], [67], [68], [69], [70], [71], [72], [73], [74], [75], [76], [77], [78], [79], [80]. Infectious triggers implicated in autoimmunity are also numerous and include bacteria, viruses, parasites and fungi. An infection burden corresponding to geoepidemiological and serological evidence of the co-occurrence of anti-infectious agents has been observed and it is of interest that such an infection burden may differ from one autoimmune disease to another [4], [81], [82]. An analogous, autoantibody burden in non-autoimmune individuals during infection with various agents, further points towards the close relation of exposure to several infectious agents and the development of autoantibody reactivities [83]. From our point of view, it is helpful to define this subgroup of triggers as the “infectome” which is the part of the exposome referring to the collection of an individual's infectious exposures which are associated with disease, and in our case specific autoimmune diseases. It may also demonstrate the alteration in disease states, which are affected by alterations of the flora within the microbiome. Examples include treatment with antibiotics [84], [85], or exposure to toxic metals, silicone or other xenobiotics [30], [86], [87]. These exposures may very well alter the disease course or progression, by altering the flora within the microbiome, or by increasing the likelihood of infection with a disease causing infection. They may also increase the burden of oxidative stress. While the hygiene hypothesis underlines the protective role played by infections [88], [89], [90], [91], [92], clinical and experimental data in animal models implicate infections in the development of autoimmunity and autoimmune disease [4]. One of the best-studied examples of infection-induced autoimmunity is that of acute rheumatic fever presenting several weeks after infection with Streptococcus pyogenes. It is now well established that molecular mimicry between the bacterial M-protein and human lysoganglioside is responsible for the loss of immunological tolerance, and the development of autoimmunity in genetically susceptible individuals [93]. Other examples include the associations between Helicobacter pylori and autoimmune gastritis [94], as well as between Trypanosoma cruzi and Chagas' cardiomyopathy [95], and Mycoplasma with rheumatoid arthritis [96].
Table 1.
Non-infectious environmental agents associated with the development of autoimmunity. This table provides examples of several non-infectious agents from a variety of sources, which have been associated with the development of autoimmune disease. AIH, autoimmune hepatitis; AiLD, autoimmune liver disease; COPD, chronic obstructive pulmonary disease; DM, dermatomyositis; MG, myasthenia gravis; rheumatoid arthritis; SLE, systemic lupus erythematosus; SSc, systemic sclerosis.
| Non-infectious environmental triggers | Disease | Reference |
|---|---|---|
| Occupational exposures | ||
| Silica | RA, SLE, SSc, glomerulonephritis, small vessel vasculitis. | [52], [53], [54], [55], [56] |
| Solvents | SLE, AIH | [54], [55], [57], [58] |
| Pesticides | Autoimmune thyroid, RA, SLE, SSc | [59], [60], [61], [62], [63], [64], [65] |
| Ultraviolet radiation | SLE, RA, DM, PM, MS, type 1 diabetes mellitus | [54], [66], [67], [68], [69] |
| Drugs | ||
| Allopurinol | Immune haemolytic anaemia | [70] |
| Captopril | Autoimmune thrombocytopaenia | [71] |
| Chlorpromazine | Anti-phospholipid syndrome, haemolytic anaemia, SLE, AiLD | [72], [73], [74], [75], [76], [77], [78] |
| Estrogens | PBC, SLE, RA | [155], [156], [157], [158], [316], [317], [318], [319], [320] |
| Halothane | AIH | [77], [321], [322] |
| Iodine | Autoimmune thyroid | [62] |
| Penicillins | AiLD, immune haemolytic anaemia | [77], [323] |
| Rifampicin | AIH, autoimmune thyroid, immune haemolytic anaemia | [324], [325], [326] |
| Tetracyclines | AIH, DM, SLE | [79], [327], [328], [329], [330], [331], [332], [333], [334], [335], [336] |
| Miscellaneous | ||
| Vaccines | PBC, AIH, SLE, RA, MS, MG, DM, polyarteritis nodosa, | [279], [337], [338], [339], [340], [341], [342], [343], [344], [345], [346], [347], [348], [349] |
| Cigarette smoke | PBC, COPD, RA, autoimmune thyroid | [155], [156], [157], [158], [350], [351], [352], [353], [354], [355] |
| Collagen/silicone implants | SLE, Sjögren's, SSc | [353], [356], [357], [358], [359], [360], [361], [362] |
Although experimental models of autoimmune diseases and studies like those mentioned above provide data to support the role played by a single infectious trigger, the prevailing idea is that a multitude of infections from birth, in our term the infectome, contributes to the induction of autoimmunity [4]. In fact, Rolf Zinkernagel's hypothesis is that the increasing predisposition to autoimmune disease in the developing world may be due to the overall host–infection balance, beginning as early as the transfer of maternal antibodies via the placenta or via breast milk in the gastrointestinal tract. The question which then arises is how one can identify those specific infectious agents responsible for the initiation of an autoimmune disease. Studies in animals have provided some clues, but their resemblance to the human setting is poor in most cases.
Ideally, investigations have to be carried out in affected individuals. However, when work is performed on biological material obtained from patients with a given autoimmune disease, it is difficult to single out disease-triggering microbes. Undoubtedly, information from the collection of the microbial genes in a particular region (such as the gut or the oral cavity), also known as “microbiome”, can provide important hints for those potentially involved in the development of or protection against autoimmunity. However, this by itself cannot differentiate those with a pathogenic potential from the non-harmful ones, the latter representing the great majority. In fact, most of the isolated microbes have little to do with the initiation of immune-mediated inflammatory processes leading to a given disease. Moreover, induction of autoimmunity by viruses or bacteria is probably done by a ‘hit-and-run’ mechanism when the causative agent has been cleared from circulation by the time of diagnosis. Tracking down each individual's exposure to infectious agents as well as anti-microbial immune responses may be important for the establishment of a causative link between infection and autoimmunity.
Studies attempting to investigate the role of the “infectome” in the induction or remission/relapse state of autoimmune disease may need to focus on autoimmune diseases such as multiple sclerosis or rheumatoid arthritis, as patients with these diseases experience frequent remissions and relapses as well as fluctuations in disease activities that are poorly understood. Systemic lupus erythematosus, multiple sclerosis and rheumatoid arthritis are three of the best studied diseases so far for which specific viral agents have been considered to be important for the development of the disease and the breakdown of immunological tolerance [4]. As well, exposure to metals and various other chemical components have also been linked with these two conditions, and it would be interesting to see how these exposures alter the presence of infectious agents. It may be the case that exposure to certain xenobiotics increases susceptibility to disease causing microorganisms, or possibly eliminates organisms that are determined to be protective.
Arguably, the ideal clinical setting for the study of the role of the “infectome” in autoimmunity would involve typical autoimmune diseases with long prodromal stages [97]. Serial testing of biological material from individuals who progress from an asymptomatic sub-clinical phase to clinical disease would be optimal for the study of the “infectome”. Examples of this include systemic lupus erythematosus (SLE), multiple sclerosis (MS) and primary biliary cirrhosis (PBC) to name a few. All of those are characterised by highly specific antibodies that may appear years or even decades before the onset of symptoms [98], [99], [100]. These diseases also have long prodromal stages and their course highly varies among individuals. Relapse and remission states can be seen, but the reasons for this remain unknown. The characteristic features of these diseases and their reported pathogenic relationship with infectious agents make them ideal candidates for the study of the infectome.
4. SLE as an infectome model: known infections, stronger links
Systemic lupus erythematosus (SLE) is often characterised by a prodromal stage of antibody positivity (predominantly anti-Sm and anti-Ro) with no symptomatology [101], [102]. A recent study has also found that a significant proportion of first degree relatives of SLE patients are positive for several autoantibodies, namely ANA (mostly anti-dsDNA), anti-Ro/SSA, and many other specificities [103], [104], [105]. In fact, ANA titre ≥ 1:160, anti-dsDNA, anti-Ro/SSA and anti-chromatin may have a high predictive value for SLE diagnosis [103]. The reasons underlying the development of and/or the progression to clinical disease, as well as the mechanisms underlying disease flares, remain elusive [19]. However, several infectious agents have been implicated including EBV, cytomegalovirus (CMV) and parvovirus B19 [48], [101], [102], [104], [105], [106], [107], [108], [109], [110]. These features make SLE an ideal model for the infectome, given the strong evidence for an infectious trigger, in addition to its unpredictable clinical course.
EBV appears to have an association with SLE, with variations and susceptibility based on demographical and genetic characteristics [111]. Harley and colleagues [101] propose a progression of EBV infection followed by Epstein–Barr virus nuclear antigen 1 (EBNA-1) antibody production, which predisposes to the development of cross-reactive autoantibodies, with progression to clinical SLE. Harley and James [102] indicated that the first lupus specific antibodies are directed against EBNA-1, which interestingly also binds lupus specific antigens such as Sm and Ro. As with many implicated viruses, the exact mechanisms in which they induce autoimmunity are not yet clear. However, one group which has indicated molecular mimicry as a mechanism with regard to EBV and SLE, notes similarities between the EBV peptide PPPGRRP and the PPPGMRPP peptide of Sm [109]. That same group tested 196 ANA positive adult SLE patients, and two age, race, and sex matched controls per patient, for evidence of previous infection with EBV, CMV, herpes simplex virus-1 (HSV-1), HSV-2, and varicella zoster virus (VZV) by ELISA [109]. Among the SLE patients, 195 out of 196 had previous EBV infection compared to 370 controls [109]. No differences were found in regard to the rate of infection with other viruses [109]. A study by Wang et al. [112] found that the EBV-encoded latent membrane protein 2A induced a heightened sensitivity to toll-like receptor (TLR) ligand stimulation, which increased proliferation of anti-Sm B-cells, and/or increased antibody secreting cell differentiation.
Parvovirus B19 has also been found to be associated with SLE, although the evidence is not as clear cut as that of EBV. The group of Bengtsson [106] tested 99 SLE patients and 99 age and sex matched controls for parvovirus IgG antibodies. No evidence of parvovirus B19 was found in SLE compared to controls, and in one analysis, the controls had a higher positivity than the SLE group [106]. As the symptoms of SLE and parvovirus B19 infection may be similar, a prospective study involving 42 patients with acute parvovirus B19 infection attempted to determine whether the symptoms persisted, and whether infection contributed to the development of SLE [110]. Clinical and laboratory investigations were performed at 1, 2, 6, 12, and 24 months from initial infection. Arthralgias persisted for 2–6 months in three female patients, for greater than two years in one female, but resolved in the remaining cases within 2 weeks [110]. One female with persistent arthralgia over two years was ANA positive and had hypercomplementaemia, but did not fulfil the diagnostic criteria for SLE or RA [110]. Despite the lack of evidence linking parvovirus B19 to the induction of SLE, one case report of a 26 year old female with SLE, who had a disease flare-up following parvovirus B19 infection, suggests that parvovirus B19 causes disease flares in SLE patients [107].
As with parvovirus B19, the evidence linking CMV to SLE is scarce. Hrycek and colleagues found that 100% of female SLE patients have evidence of CMV infection, compared to 75% of controls [108]. As well, another study had found that the CMV pp65 antigen triggers humoral immunity in SLE patients and autoimmune prone mice [113]. The lack of evidence linking particular viruses to SLE does not infer non-involvement, but reflects the difficulty of detecting infectious organisms, which may be transient, in these patients. It is likely that diagnosis of SLE and/or SLE flares occurs after an infection. The infectome serves as a model in such cases. At-risk patients, such as first degree relatives of SLE patients, who are found to be positive for autoantibodies, may be screened at regular intervals. Likewise, SLE patients may be screened, and changes in clinical course, such as flares, may be correlated with infection. Several other diseases with a well defined connection such as that of H. pylori-induced idiopathic thrombocytopenic purpura could be explored. These methods are useful in that they not only indicate the who and when, but also provide a more narrowed-down list of organisms to evaluate in regard to the mechanism in which they induce autoimmunity.
5. Multiple sclerosis as an infectome model for relapse remittance clinical states
Multiple sclerosis (MS) serves as another example in which the infectome model may be applied. MS is a chronic autoimmune neurological syndrome characterised by chronic inflammation, demyelination and gliosis in the central nervous system (CNS) [114], [115]. It is characterised by periods of relapse and remission [114], [115], the reasons for which are not understood. Current evidence suggests that the risk for acquiring MS is spread over a long period of time, and is not limited to childhood or early adult life [116], [117], [118].
The most prevalent form of the syndrome is the so called relapsing–remitting MS, characterised by flares whereby pre-existing symptoms become more severe, or new symptoms develop [119]. These flares are followed by phases of complete or partial recovery and their duration is highly variably among affected individuals. A considerable proportion of these patients acquire progressive disability with or without disease relapses (secondary progressive MS). Another form of MS is characterised by a stable progression of the disease with worsening of the symptomatology over the course of the disease. This form is also known as primary progressive MS (PPMS). Two further forms of the disease are also found with totally contradictory outcomes. The benign form of MS usually presents with minimal or mild progression of disability, and is clinically characterised by full recovery of sporadic sensory episodes [119]. On the other hand, the Marburg variant of MS is characterised by rapidly progressive disease which leads to accumulating disability and eventually to death. The mechanisms responsible for the development of clinically distinct disease phenotypes are poorly understood [119].
The pathological hallmark of MS is inflammatory lesions (areas of demyelination) in the CNS composed of mononuclear cell infiltrates in the perivascular spaces that develop into plaques [114], [120]. These mononuclear cells are largely composed of T and B lymphocytes, plasma cells, macrophages and microglia [114], [120]. Positive staining for IgG is found in the peripheral regions of the plaques [114], [120]. Additionally, approximately 90% of MS patients show intrathecal IgG synthesis in the cerebrospinal fluid (CSF) [114].
Environmental and genetic components are clearly involved in the aetiology of MS, with geographical and twin data indicating a greater environmental component [114], [115], [121], [122]. However, identical twins are 100 times more likely to develop MS if their co-twin is affected, whereas non-twin siblings are 20 times more likely [123], [124]. Genetic and GWAS have implicated several alleles, with the strongest association being with HLA-DRB1*1501 [125]. Seven GWAS studies have been conducted, which included nearly 10,000 MS patients and 15,000 controls [115]. Implicated genes include IL7R, IL12RA, CLEC16, and CD226 [126], [127]. Nonetheless, implicated genes have only demonstrated an odds ratio of less than 1.3 [115], and MS twins only demonstrate a 30% concordance [114], indicating more of an environmental influence in the disease pathogenesis.
Non-infectious and infectious components have been implicated to be involved in the development of MS. Within the group of non-infectious agents, vitamin D is of particular interest, not only in MS but also in several other autoimmune diseases [115], [128], [129], [130], [131], [132], [133], [134], [135], [136], [137], [138], [139], [140]. Vitamin D appears to play a role in the modulation of pro-inflammatory pathways and T cell regulation [115]. Increased distance from the equator has been correlated with low vitamin D, and interestingly, MS rates increase as distance from the equator increases [114]. As well, populations with increased dietary vitamin D intake have lower rates of MS [115]. An Australian study found a decreased risk of a first demyelinating event in those with increased sun exposure, who also had increased levels of serum vitamin D [141]. This has also been found in other studies [142]. As well, the effect of month of birth on MS development was more apparent in familial MS groupings, suggesting an influence on prenatal vitamin D levels, as well as an interaction between genes and environment [115]. Like many conditions, smoking has also been associated with MS development, and a recent meta-analysis overwhelmingly associated smoking with MS [143]. Gene–environment interactions have been suggested in regard to smoking and the presence of HLA-DRB1*15 with the absence of HLA-A*02 [144]. Smokers with this genetic combination appeared to have an increased risk of developing MS [144]. Whether this is due to the effect of the nitrosamines or the heavy metals in cigarette smoke is not clear. There is limited evidence for the role of heavy metals in some patients with MS [145]. Infectious agents investigated in regard to MS have included bacteria and viruses (Table 3 ) [114], [115], [121], [122], [146], [147], and implicated viruses include EBV [114], [115], [121], [122] and human herpes virus 6 (HHV6). The relapse–remittance pattern of the human herpes virus 6 (HHV6) is similar to the clinical pattern of MS [115] and HHV6 reservoirs have been found in CNS tissue [148], [149], in addition to the CSF and serum of MS patients [150], [151]. Molecular mimicry has been indicated, as sequence homology has been found between myelin basic protein and HHV6 encoded U24, and cross-reactive T cells responding to both protein types are found to be elevated in MS patients [152]. Varicella zoster virus, torque teno virus, retroviruses, coronaviruses and JC virus have also been implicated, but with limited evidence [114], [115], [121], [122]. Reactivity to several viral peptides was found [153], which has led to the speculation that continual exposure to a variety of viruses can lead to T cell expansion reactive against highly conserved proteins, including self-peptides.
Table 3.
Examples of infectious agents implicated in primary biliary cirrhosis. This table provides several examples of infectious organisms which have been implicated in the pathogenesis of primary biliary cirrhosis (PBC). Strong evidence exists for some organisms such as Escherichia coli, while weaker evidence exists for others. This may be due to the lack of investigation into the prevalence of some organisms in PBC.
| Bacteria | Escherichia coli | [176], [178], [265], [267], [399], [400] |
| Chlamydia pneumoniae | [263], [401] | |
| Mycobacterium gordonae | [402], [403], [404] | |
| Novosphingobium aromaticivorans | [178], [399] | |
| Pseudomonas aeruginosa | [257], [405] | |
| Lactobacillus delbrueckii subsp bulgaricus | [259], [279] | |
| Yersinia enterocolitica | [260], [406] | |
| Salmonella typhimurium | [407] | |
| Salmonella minnesota | [408] | |
| Haemophilus influenzae | [257], [260] | |
| Streptococcus intermedius | [409] | |
| Paracoccus dentrificans | [410] | |
| Borrelia burgdorferi | [411] | |
| Propionibacterium acnes | [412] | |
| Mycoplasma pneumoniae | [413] | |
| Mycoplasma gallisepticum | [414] | |
| Viruses | Βetaretrovirus | [272], [276], [415], [416] |
| Cytomegalovirus | [257] | |
| Epstein Barr virus | [417] | |
| Parasites | Trypanosomes | [418], [419] |
| Ascaridia galli | [419] | |
| Other | Saccharomyces cerevisiae | [420] |
An infectome for the above conditions (and others) may clarify questions regarding aetiology, but may also identify the cause of certain disease characteristics, such as variable presentation and progression among PBC patients, disease flares in patients with SLE, or relapse–remittance among MS patients. As such, these conditions serve as models for an infectomal analysis.
6. Primary biliary cirrhosis as an infectome model disease
PBC can be used as a model disease to investigate the role of the exposome, and indeed of the infectome [154], as A) it is an autoimmune disease and it is not as rare as many other such diseases, B) it has a long silent phase in which well-established biomarkers can be determined in high-risk populations, C) it presents with subgroups of patients who have differing disease progressions and/or concomitant other autoimmune diseases, D) it is not treated with immunosuppessive drugs that might deteriorate the immunological assessment of the infectome, and E) there is growing evidence in support of genetic, environmental, and infectious factors involved in the pathogenesis of the disease such as recurrent urinary tract infection (UTI), cigarette smoke, and oestrogen deficiency [155], [156], [157], [158], [159], [160], [161], [162], [163]. A plethora of experimental and clinical data clearly indicate that the disease is indeed autoimmune in nature [98], [159], [164], [165], [166], [167], [168], [169], [170], [171], [172], [173], [174], [175], [176], [177], [178], [179], [180], [181], [182], [183], [184]. Infectious agents and xenobiotics mimicking or modifying the core epitopic region of PDC-E2, the major autoantigen in PBC, appear to induce immune-mediated destruction of the bile ducts. As PBC affects the liver, access to the affected organ for research purposes is possible at the time of diagnosis via liver biopsy, and over the course of the disease from early to advanced stages. This is not possible in diseases such as diabetes mellitus type I.
In line with the already discussed ideal situation of a long prodromal period, PBC has a long pre-clinical phase, characterised by an asymptomatic period with evidence of autoimmune markers in the form of autoantibodies, followed by biochemical evidence of liver damage and finally clinically-evident disease. The progression from asymptomatic to symptomatic PBC may take years or decades. Virtually, all patients with PBC have high-titre anti-mitochondrial antibodies (AMA) at the time of diagnosis [99], [175], [184], [185], [186], [187], [188], [189], [190], [191], [192], [193], [194], [195], [196], [197], [198], [199], [200], [201], [202], [203], [204], [205], [206], [207], [208], [209], [210]. Also, the presence of AMA is predictive of eventual disease development [211]. PBC patients with disease-specific anti-nuclear autoantibodies (ANA) appear to have more advanced disease and poorer prognosis [212], [213], [214], [215], [216], [217], [218], [219], [220], [221], [222], [223], [224], [225], [226], [227]. Individuals at the highest risk of developing PBC are family members of PBC patients. [228], [229], [230], [231], [232], [233], [234]. The vast majority of patients with PBC experience concomitant autoimmune diseases such as Sicca syndrome, autoimmune thyroiditis, rheumatoid arthritis, autoimmune hepatitis and systemic sclerosis. The reasons behind the fast pace of progression seen in some patients with PBC are unknown, and attempts to predefine those individuals which will progress faster than others have been largely unsuccessful.
PBC does not appear to respond to immunosuppressive treatment, making the disease perfect to study the involvement of infectious agents over the course of the disease without the need to consider the effects of immunosuppressive therapy on relapse/remission states. PBC patients are instead treated with ursodeoxycholic acid (UDCA) at adequate doses of 13–15 mg/kg/day [235], [236]. It is not clear as to whether UDCA has immunomodulatory properties or not.
A number of environmental factors have been implicated in PBC by ‘bottom-up’ approaches in various epidemiological studies examining associated risk factors [155], [156], [157], [158] including numerous xenobiotics and infectious agents [159], [160], [161], [162], [237]. Animal models have further supported this notion [159], [238], [239], [240], [241], [242]. The genetics of PBC appear to include HLA [243], [244], non-HLA [245], [246], [247], [248], [249], [250], [251] and sex-linked genes [252], [253], [254]. Particular allelic associations may be indirectly relevant to the pathogenesis of the disease, as they may influence the penetrance of infectious agents. For example HLA-DRB1*11 and HLA-DRB1*13 have a negative association with PBC, and are protective for several viruses that affect the liver, such as hepatitis B and C. The lack of HLA alleles protective for PBC, which are associated with resistance to specific pathogens, may lead to susceptibility to infection responsible for the induction of the disease [244], [255]. These findings underline the urgent need to investigate the infectome in parallel with GWAS. An infection burden involving EBV, CMV and Toxoplasma has been recently reported in patients with PBC.
However, the list of pathogens involved in PBC is vast, with some such as Escherichia coli, having a relatively strong evidence base (Table 2) [164], [182], [252], [256], [257], [258], [259], [260], [261], [262], [263], [264], [265], [266], [267], [268], [269], [270], [271], [272], [273], [274], [275], [276], [277], [278], [279]. Molecular mimicry has been considered a likely mechanism that could account for the initiation of liver autoimmune diseases, including PBC [164], [182], [256], [257], [258], [259], [260], [264], [274], [275], [279], [280], [281], [282], [283], [284], [285].
Table 2.
Examples of infectious agents implicated in multiple sclerosis. Several organisms have been implicated in the pathogenesis and clinical course of multiple sclerosis (MS), the vast majority of which are viruses.
| Viruses | Bacteria | ||
|---|---|---|---|
| Epstein Barr virus | [363], [364], [365], [366], [367], [368], [369], [370], [371], [372], [373], [374], [375], [376], [377], [378] | Chlamydia pneumoniae | [379] |
| Human herpes virus 6 | [148], [149], [150], [151], [152], [380], [381] | Borrelia burgdorferi | [382] |
| Varicella zoster virus | [383], [384], [385] | Mycobacterium tuberculosis | [386] |
| Human cytomegalovirus | [387] | ||
| Retroviruses | [388], [389] | ||
| Coronavirus | [390], [391] | ||
| Torque teno virus | [153] | ||
| JC virus | [392], [393] | ||
| Rubella virus | [394] | ||
| Parainfluenza virus I | [395] | ||
| Measles virus | [396], [397] | ||
| Mumps virus | [398] |
Much as the exposome reflects the collation of all exposures, an infectome may reflect all those bacterial, viral, or parasitic exposures which may contribute to the development of an autoimmune disease.
7. How to study the infectome
The study of the infectome needs to be customised taking into account the disease to be investigated. The type of the sample to be collected largely depends on the disease under investigation.
A generic, step-wise approach of infectome analysis at presentation would be the following:
-
1)
Determination of HLA class I and II is performed in all individuals under investigation. Ideally, this could be performed at birth, or at the baseline when the individual/patient presents for the first time in the clinic. This information is important in order to sub-group the individual into high or low risk.
-
2)
Collect urine, oro-nasal swabs, saliva, faecal material, and blood (for isolation of plasma, serum, and peripheral blood mononuclear cells — PBMCs).
-
3)
Regular follow-up (once yearly) with collection of samples;
-
4)
Meticulous recording of clinical data and collection of samples are needed when anti-infective treatment is applied for incidental/casual infections at the pre-clinical stages of the disease, as this may affect the final outcome of the underlying processes.
-
5)
Store samples until patient has laboratory and/or clinical features related to the disease;
-
6)
Analysis of collected samples for infectious agents with a known association with the disease, and “out of the box” analysis using multiplex technology for other infectious agents (see section “How to screen?” below). The analysis will provide information regarding the infection burdens at various-time points;
-
7)
Associations are analysed, providing evidence for known/unknown infectious agents in the development of symptomatic disease.
-
8)
Continuous analysis over the course of the disease, as it may reveal a close link between a specific agent and the clinical phenotypes of the disease.
The study of the role of infectious agents in the development of an autoimmune disease, whether RA, SLE, MS or PBC, may reveal which agents are involved in the development of the disease as well as other concomitant autoimmune diseases. It is likely that the combination of particular genes and particular infectious exposures is responsible for the development of an autoimmune disease, with or without a particular concomitant autoimmune disease. In other words, there may be several infectomes for SLE, some of which define SLE with a particular concomitant autoimmune disease. This of course applies to other autoimmune diseases.
8. Lessons that can be learned from the microbiome and immunome projects
The recently described microbiome concept can be separated from that of the infectome. The microbiome defines the collection of microbial genes in a particular region, such as the gut, inguinal crease, oral cavity, or virtually any body site [286], [287], [288], [289], [290], [291], [292], [293], [294], [295]. The microbiome may be reflective of the normal or pathological profile of organisms. For example, the microbiome of the gut has been analysed in healthy individuals [288], [289], [290], [291], [294], which identified three distinct profiles or clusters, known as enterotypes [287], [293]. Similar studies have been performed for the oral cavity as well [286], [295]. The microbiome may also be applied to a disease-affected body site, such as the gastrointestinal tract in children with irritable bowel syndrome (IBS) [292]. A study by Saulnier and colleagues [292] obtained 71 faecal samples from 22 children with IBS and 22 healthy children, all aged 7–12 years. These samples were analysed by 16S rRNA gene sequencing, which showed an elevated percentage of γ-proteobacteria in the gut flora of children with IBS, with Haemophilus parainfluenza being a prominent component [292].
So how does the infectome differ from this? First, the infectome relates to those infectious organisms which are associated with the disease in question, as opposed to a totality of all organisms, both pathological and non-pathological within the human microbiome. Second, the infectome reflects all sites of infection, as opposed to one body site usually represented by the microbiome. As well, microbiome studies to date have largely concentrated on bacterial species, whereas the infectome takes into account all pathological bacteria, viruses, parasites, and fungi. The infectome is not limited to the affected site or organ but includes biological fluids as well as sampling of various body-sites including the oral cavity. Some argue that the organisms of the human microbiome do not usually induce antigen specific systemic humoral and cellular immune responses provoking local or systemic inflammatory response, while others believe that these organisms may not be directly pathogenic, but create a dysbiosis of the gut flora and participate in the induction of autoimmunity. This is a key difference between the microbiome and the infectome as the former appreciates the direct or indirect effect of immune responses against infectious agents as pivotal for the initiation of autoaggression and immune-mediated, self-targeting pathology. For the infectome, monitoring of the microbial/host immunity is as important as the isolation of potentially harmful bacterial products, since the host/microbe immunological interaction is the likely cause of the self-destruction in the case of non-cytopathic viruses and microbes.
Although the infectome and microbiome are distinct entities, they may be used symbiotically to provide a micropathological profile (or profiles) of a particular disease. As well, the microbiome is essential to define what “normal” actually is. Recent microbiome studies have been able to provide an idea of what “normal” may be in the gut by performing metagenomic screens of bacterial populations in these regions [288], [289], [290], [291], [294]. For example, in the case of PBC it may be pertinent to understand the normal microbiome of the urinary bladder and vagina, as infections in these sites have been associated with PBC [155], [156], [157], [158], [296], [297]. Other body sites may also need investigation, as inhalation or consumption of potentially pathogenic organisms has also been implicated [275], [298]. All associated organisms located in all potential body-sites, would comprise the infectome. The establishment of what is normal as well as infectious may contribute to the understanding of the network of events or exposures which lead to disease development. It is possible that a series of exposures leads to increased susceptibility to further exposures, which could be defined by the infectomal model.
9. Who to screen?
Unlike GWAS and the microbiome, it is unlikely that population screening of infectomal components could be performed due to the lack of integrated analysis platforms. It may be more reasonable to screen particular groups of individuals who are at risk of developing autoimmune disease, like family members or individuals with an HLA profile conferring risk for a given disease. Further monitoring may reveal whether additional infections play a role in the progression of the disease to a symptomatic stage. Additionally, particular exposome/infectome profiles may be found among patients with rapidly progressive diseases (as in a subset of PBC patients), or in relapse remittance states in MS, and flares in SLE. This approach may delineate which infectious agents are responsible for disease development and/or progression, as well as identifying those that are associated with rapid versus slow progression, remittance, and flares.
10. How to screen?
The establishment of the infectome would have to rely on the detection of microbiological materials in patients, favourably in ease-to-sample material like blood, urine or saliva. There are several methods by which this may be done, and some of those have already been used for research purposes. Serological detection of IgM, IgA and IgG antibody responses to microbes, viruses, and fungi is likely to be the most common, fastest, and most cost effective method and independent of the actual presence of a respective infectious agent. Monitoring for seroconversion of antibody responses and isotype class switching from IgM to IgG is imperative for the identification of newly acquired infectious triggers over time. To this end, the part of the immunome which relates to infections is an integral part of the infectome and its primary goal to identify microbial triggers of autoimmunity. Immunoassays for the determination of antibodies against most of them, e.g. ELISA, are broadly available, have a high degree of robustness and standardisation and are comparably cheap. Furthermore in some diagnostic scenarios, multi-parametric immunoassays, e.g. lineblots for the detection of antibodies against tick-borne diseases exist that facilitate the determination of antibodies against several or even multiple entities at the same time with a limited amount of sample. The highest complexity up-to-date can be reached by using protein microarrays that allow for the screening of several thousand antibody entities at the same time [299], [300]. However, such platforms have so far only been used to screen for immune profiles of individuals, and small numbers of similar infectious agents and commercial solutions with a similar degree of standardisation as those containing human proteins are not available. A major breakthrough might result from the development of high-density peptidome libraries of relevant microbes and viruses similar to the concepts of peptidome libraries covering human proteins, e.g. T7-Pep library for the human peptidome [301].
A large German study used a PCR approach to detect a variety of organisms in archived liver tissues of PBC patients [302]. This approach has also been adopted in several other studies examining the role of mycobacteria in PBC [303], [304], [305], as well as in another group which used this method to detect beta-retroviral material in the liver and lymph node tissue from PBC patients [276], [277]. Similar studies have been conducted in other autoimmune diseases. Multi-parametric detection of viral and bacterial genetic material in tissues may be another promising approach. In recent years, DNA sequencing technology has undergone an outstanding upgrade, and the so-called ‘high-throughput DNA sequencers’ can determine hundreds of megabases of DNA sequences per run, enabling the analysis of a broad range of infectious agents [306], [307], [308], [309]. Massive, parallel sequencing might be the next step of this approach for that it is the most sensitive procedure available and allows for the detection of a multitude of infectious agents at the same time [310]. However, the considerable costs of this testing limit its wide use. Immunohistochemistry detecting several microbial agents in tissue samples can be applied [311], though it is likely that tissue based methods are not ideal in the establishment of the infectome, as they are time consuming, and tissue of the affected organ may not be readily available from all patients. The analysis of blood, faecal material, urine, or saliva is more plausible, and can be used for screening, reflecting the ‘top-down’ approach suggested by other researchers [35]. As mentioned, multiplex PCR is a useful tool for evaluating the presence of microorganisms from a variety of sample types. Microbiome studies have also highlighted the use of 16S/18S rRNA gene sequencing, which allows for the mass-analysis of samples [290], [292]. However, one drawback of this approach in comparison to immune profiles is the risk of missing the right time or site of sampling.
11. Where to screen?
The question also remains as to which body sites should be sampled. In addition to the affected organ, general or systemic infections should be an indication for sampling. This may include urine and stool samples in urinary and gastrointestinal tract infections respectively, or blood cultures in febrile patients. In addition to clinically overt infections, many patients with autoimmunity may have latent infections such as Lyme disease or Mycoplasma. Examples of this include hidden chronic low grade infection associated with dental treatments. In fact, the oral cavity is not commonly examined for infection in patients with autoimmune disease, and it would most likely be useful to examine and sample the oral cavity for such overt infections [312], [313], [314]. Accumulating evidence indicates that oral hygiene practices may induce alterations in the flora of the oral mucosa, which leads to a dysbiosis in the gut microbiome, and thereby contributes to the pathogenesis of IBD [312]. On the other hand, the increased frequency of dental problems in IBD patients may be due to alterations in oral flora. Complementing these findings, a study by the group of Helenius [314] indicates that patients with rheumatic conditions had various alterations in salivary flow and composition, and oral health. It remains to be seen whether the increased incidence of bacterial infections is common among all autoimmune diseases, and if so, which oral microorganisms are common among those conditions.
The proposed model of screening is not only useful in characterising the components which lead to autoimmune disease development/progression, but may also provide further evidence as to preventative measures. For example, if it is demonstrated that progression of an autoimmune disease is dependent upon infectious triggers, the proper use of antibiotic or even antiviral therapy may be initiated early on in the management of these patients. Antibiotic treatment for infectious agents associated with autoimmune disease may slow the progression of the disease in some individuals, and possibly prevent the development of the disease in individuals found to be at risk. If exposure to a particular microbe is proven to be critical in a particular autoimmune disease development, measures may be taken to reduce the exposure to this microbe, regardless of whether it is exogenous or endogenous. Such treatments can only be incorporated in the routine management of these diseases through consensus statements and authoritative position papers/guidelines; otherwise are potentially dangerous for the wellbeing of the patient [272], [276], [277], [315].
12. Conclusion
Genetic and environmental components are involved in the pathogenesis of most diseases. The GWAS have contributed greatly to the understanding of the genetic basis of disease. The theory of the exposome complementing GWAS is now evolving. The complexity of the exposome may require that it be broken down to several sub-components such as the infectome, which reflects the characterisation and measurement of all infectious organisms that we are exposed to. The relationship of the exposome/infectome to the development of a disease, in individuals with particular genetic characteristics, may also help us characterise disease initiation and progression.
Given the growing knowledge of the genetic basis of disease, it may one day be possible to screen newborns for HLA and genetic characteristics which infer susceptibility to autoimmune disease, which may be done as routinely as the current newborn screening. Infants with a particular susceptibility profile can then be monitored closely for exposure to environmental triggers that may contribute to the development of a particular autoimmune disease. It is therefore essential that we establish not only the genetic basis of the disease (via GWAS), but also the environmental component of the disease, which must be established through an exposomal model. The covalent binding of xenobiotics to genomic DNA has been extensively explored and should be re-visited in this context in the light of more recent findings. As there is a clear delineation between organic and inorganic materials, the establishment of the infectome will represent the infectious component, comprised of all potential infections that initiate the disease, or alter the disease course. It is important that the subgroups of the exposome are considered together alongside the detailed investigation of each component part. It is likely that the methods used to establish and analyse the infectome will be simple modifications of technology already in use. It remains to be seen how this approach may be applied to other potential subgroups of the exposome, such as xenobiotics.
Take-home messages
-
•
The infectious/host immunological interactions in autoimmunity are poorly understood.
-
•
A direct link between infection and autoimmunity is difficult to obtain.
-
•
Infectome refers to the collection of an individual's exposures to infectious agents.
-
•
Infectome can be characterised with approaches employed for the “immunome” and “microbiome” projects.
-
•
The study of the infectome can help us to delineate the triggers of autoimmunity.
Acknowledgements
We are indebted to Diego Vergani, Harold Baum, Lars Komorowski, and Andrew K. Burroughs for helpful comments and fruitful discussions leading to the initiation of the concept of infectome. We thank Timoklia Orfanidou and Maria G. Mytilinaiou for editing assistance.
References
- 1.Gourley M., Miller F.W. Mechanisms of disease: environmental factors in the pathogenesis of rheumatic disease. Nat Clin Pract Rheumatol. 2007;3:172–180. doi: 10.1038/ncprheum0435. [DOI] [PubMed] [Google Scholar]
- 2.Lettre G., Rioux J.D. Autoimmune diseases: insights from genome-wide association studies. Hum Mol Genet. 2008;17:R116–R121. doi: 10.1093/hmg/ddn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rappaport S.M., Smith M.T. Epidemiology. Environment and disease risks. Science. 2010;330:460–461. doi: 10.1126/science.1192603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kivity S., Agmon-Levin N., Blank M., Shoenfeld Y. Infections and autoimmunity—friends or foes? Trends Immunol. 2009;30:409–414. doi: 10.1016/j.it.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson D.L., Gange S.J., Rose N.R., Graham N.M. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–243. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 6.Blank M., Gershwin M.E. Autoimmunity: from the mosaic to the kaleidoscope. J Autoimmun. 2008;30:1–4. doi: 10.1016/j.jaut.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 7.Brickman C.M., Shoenfeld Y. The mosaic of autoimmunity. Scand J Clin Lab Invest Suppl. 2001;235:3–15. [PubMed] [Google Scholar]
- 8.Rahamim-Cohen D., Shoenfeld Y. The mosaic of autoimmunity. A classical case of inhalation of a polyclonal activating factor in a genetically and hormonally susceptible patient leading to multiple autoimmune diseases. Isr Med Assoc J. 2001;3:381–382. [PubMed] [Google Scholar]
- 9.Shepshelovich D., Shoenfeld Y. Prediction and prevention of autoimmune diseases: additional aspects of the mosaic of autoimmunity. Lupus. 2006;15:183–190. doi: 10.1191/0961203306lu2274rr. [DOI] [PubMed] [Google Scholar]
- 10.Shoenfeld Y., Blank M., Abu-Shakra M., Amital H., Barzilai O., Berkun Y. The mosaic of autoimmunity: prediction, autoantibodies, and therapy in autoimmune diseases—2008. Isr Med Assoc J. 2008;10:13–19. [PubMed] [Google Scholar]
- 11.Shoenfeld Y., Gilburd B., Abu-Shakra M., Amital H., Barzilai O., Berkun Y. The mosaic of autoimmunity: genetic factors involved in autoimmune diseases—2008. Isr Med Assoc J. 2008;10:3–7. [PubMed] [Google Scholar]
- 12.Asherson R.A., Gunter K., Daya D., Shoenfeld Y. Multiple autoimmune diseases in a young woman: tuberculosis and splenectomy as possible triggering factors? Another example of the “mosaic” of autoimmunity. J Rheumatol. 2008;35:1224–1226. [PubMed] [Google Scholar]
- 13.de Carvalho J.F., Pereira R.M., Shoenfeld Y. The mosaic of autoimmunity: the role of environmental factors. Front Biosci (Elite Ed) 2009;1:501–509. doi: 10.2741/e46. [DOI] [PubMed] [Google Scholar]
- 14.Costenbader K.H., Gay S., Alarcon-Riquelme M.E., Iaccarino L., Doria A. Genes, epigenetic regulation and environmental factors: which is the most relevant in developing autoimmune diseases? Autoimmun Rev. 2012;11:604–609. doi: 10.1016/j.autrev.2011.10.022. [DOI] [PubMed] [Google Scholar]
- 15.Mavropoulos A., Orfanidou T., Liaskos C., Smyk D.S., Billinis C., Blank M. p38 mitogen-activated protein kinase (p38 MAPK)-mediated autoimmunity: lessons to learn from ANCA vasculitis and pemphigus vulgaris. Autoimmun Rev. Nov 30 2012 doi: 10.1016/j.autrev.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Karlson E.W., Deane K. Environmental and gene–environment interactions and risk of rheumatoid arthritis. Rheum Dis Clin North Am. 2012;38:405–426. doi: 10.1016/j.rdc.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villeda S.A., Wyss-Coray T. The circulatory systemic environment as a modulator of neurogenesis and brain aging. Autoimmun Rev. Nov 29 2012 doi: 10.1016/j.autrev.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 18.Simon K.C., van der Mei I.A., Munger K.L., Ponsonby A., Dickinson J., Dwyer T. Combined effects of smoking, anti-EBNA antibodies, and HLA-DRB1*1501 on multiple sclerosis risk. Neurology. 2010;74:1365–1371. doi: 10.1212/WNL.0b013e3181dad57e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gatto M., Zen M., Ghirardello A., Bettio S., Bassi N., Iaccarino L. Emerging and critical issues in the pathogenesis of lupus. Autoimmun Rev. Sep 18 2012 doi: 10.1016/j.autrev.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Selmi C., Leung P.S., Sherr D.H., Diaz M., Nyland J.F., Monestier M. Mechanisms of environmental influence on human autoimmunity: a national institute of environmental health sciences expert panel workshop. J Autoimmun. 2012;39:272–284. doi: 10.1016/j.jaut.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Bogdanos D.P., Smith H., Ma Y., Baum H., Mieli-Vergani G., Vergani D. A study of molecular mimicry and immunological cross-reactivity between hepatitis B surface antigen and myelin mimics. Clin Dev Immunol. 2005;12:217–224. doi: 10.1080/17402520500285247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smyk D.S., Rigopoulou E.I., Muratori L., Burroughs A.K., Bogdanos D.P. Smoking as a risk factor for autoimmune liver disease: what we can learn from primary biliary cirrhosis. Ann Hepatol. 2012;11:7–14. [PubMed] [Google Scholar]
- 23.Israeli E., Agmon-Levin N., Blank M., Shoenfeld Y. Adjuvants and autoimmunity. Lupus. 2009;18:1217–1225. doi: 10.1177/0961203309345724. [DOI] [PubMed] [Google Scholar]
- 24.Nancy A.L., Shoenfeld Y. Chronic fatigue syndrome with autoantibodies—the result of an augmented adjuvant effect of hepatitis-B vaccine and silicone implant. Autoimmun Rev. 2008;8:52–55. doi: 10.1016/j.autrev.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 25.Perricone C., Agmon-Levin N., Valesini G., Shoenfeld Y. Vaccination in patients with chronic or autoimmune rheumatic diseases: the ego, the id and the superego. Joint Bone Spine. 2012;79:1–3. doi: 10.1016/j.jbspin.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Rosenblum H., Shoenfeld Y., Amital H. The common immunogenic etiology of chronic fatigue syndrome: from infections to vaccines via adjuvants to the ASIA syndrome. Infect Dis Clin North Am. 2011;25:851–863. doi: 10.1016/j.idc.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 27.Shoenfeld Y., Agmon-Levin N. ‘ASIA’ — autoimmune/inflammatory syndrome induced by adjuvants. J Autoimmun. 2011;36:4–8. doi: 10.1016/j.jaut.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Agmon-Levin N., Hughes G.R., Shoenfeld Y. The spectrum of ASIA: ‘Autoimmune (Auto-inflammatory) Syndrome induced by Adjuvants’. Lupus. 2012;21:118–120. doi: 10.1177/0961203311429316. [DOI] [PubMed] [Google Scholar]
- 29.Katzav A., Kivity S., Blank M., Shoenfeld Y., Chapman J. Adjuvant immunization induces high levels of pathogenic antiphospholipid antibodies in genetically prone mice: another facet of the ASIA syndrome. Lupus. 2012;21:210–216. doi: 10.1177/0961203311429550. [DOI] [PubMed] [Google Scholar]
- 30.Kivity S., Katz M., Langevitz P., Eshed I., Olchovski D., Barzilai A. Autoimmune syndrome induced by adjuvants (ASIA) in the Middle East: morphea following silicone implantation. Lupus. 2012;21:136–139. doi: 10.1177/0961203311429551. [DOI] [PubMed] [Google Scholar]
- 31.Zafrir Y., Agmon-Levin N., Paz Z., Shilton T., Shoenfeld Y. Autoimmunity following hepatitis B vaccine as part of the spectrum of ‘Autoimmune (Auto-inflammatory) Syndrome induced by Adjuvants’ (ASIA): analysis of 93 cases. Lupus. 2012;21:146–152. doi: 10.1177/0961203311429318. [DOI] [PubMed] [Google Scholar]
- 32.Blank M., Israeli E., Shoenfeld Y. When APS (Hughes syndrome) met the autoimmune/inflammatory syndrome induced by adjuvants (ASIA) Lupus. 2012;21:711–714. doi: 10.1177/0961203312438115. [DOI] [PubMed] [Google Scholar]
- 33.Arlt V.M., Schwerdtle T. UKEMS/Dutch EMS-sponsored workshop on biomarkers of exposure and oxidative DNA damage & 7th GUM-32P-postlabelling workshop, University of Munster, Munster, Germany, 28–29 March 2011. Mutagenesis. Sep 2011;26(5):679–685. doi: 10.1093/mutage/ger036. [DOI] [PubMed] [Google Scholar]
- 34.Pleil J.D., Stiegel M.A., Sobus J.R., Liu Q., Madden M.C. Observing the human exposome as reflected in breath biomarkers: heat map data interpretation for environmental and intelligence research. J Breath Res. 2011;5:037104. doi: 10.1088/1752-7155/5/3/037104. [DOI] [PubMed] [Google Scholar]
- 35.Rappaport S.M. Implications of the exposome for exposure science. J Expo Sci Environ Epidemiol. 2011;21:5–9. doi: 10.1038/jes.2010.50. [DOI] [PubMed] [Google Scholar]
- 36.Wild C.P. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev. 2005;14:1847–1850. doi: 10.1158/1055-9965.EPI-05-0456. [DOI] [PubMed] [Google Scholar]
- 37.Wild C.P. Environmental exposure measurement in cancer epidemiology. Mutagenesis. 2009;24:117–125. doi: 10.1093/mutage/gen061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith M.T., Zhang L., McHale C.M., Skibola C.F., Rappaport S.M. Benzene, the exposome and future investigations of leukemia etiology. Chem Biol Interact. 2011;192:155–159. doi: 10.1016/j.cbi.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collin M., Shannon O., Bjorck L. IgG glycan hydrolysis by a bacterial enzyme as a therapy against autoimmune conditions. Proc Natl Acad Sci U S A. 2008;105:4265–4270. doi: 10.1073/pnas.0711271105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allhorn M., Olin A.I., Nimmerjahn F., Collin M. Human IgG/Fc gamma R interactions are modulated by streptococcal IgG glycan hydrolysis. PLoS One. 2008;3:e1413. doi: 10.1371/journal.pone.0001413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCulloch J., Zhang Y.W., Dawson M., Harkiss G.D., Peterhans E., Vogt H.R. Glycosylation of IgG during potentially arthritogenic lentiviral infections. Rheumatol Int. 1995;14:243–248. doi: 10.1007/BF00262090. [DOI] [PubMed] [Google Scholar]
- 42.Polacco B.J., Purvine S.O., Zink E.M., Lavoie S.P., Lipton M.S., Summers A.O. Discovering mercury protein modifications in whole proteomes using natural isotope distributions observed in liquid chromatography–tandem mass spectrometry. Molecular & cellular proteomics. Mol Cell Proteomics. 2011 Aug;10(8) doi: 10.1074/mcp.M110.004853. [M110.004853] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLaren Howard J. The detection of DNA adducts (risk factors for DNA damage). A method for genomic DNA, the results and some effects of nutritional intervention. J Nutr Environ Med. 2002;12:19–31. [ΔΕΝ ΤΟ ΒΡΗΚΑ ΣΤΟ PUBMED] [Google Scholar]
- 44.Patel C.J., Bhattacharya J., Butte A.J. An Environment-Wide Association Study (EWAS) on type 2 diabetes mellitus. PLoS One. 2010;5:e10746. doi: 10.1371/journal.pone.0010746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karges W., Hammond-McKibben D., Cheung R.K., Visconti M., Shibuya N., Kemp D. Immunological aspects of nutritional diabetes prevention in NOD mice: a pilot study for the cow's milk-based IDDM prevention trial. Diabetes. 1997;46:557–564. doi: 10.2337/diab.46.4.557. [DOI] [PubMed] [Google Scholar]
- 46.Guggenmos J., Schubart A.S., Ogg S., Andersson M., Olsson T., Mather I.H. Antibody cross-reactivity between myelin oligodendrocyte glycoprotein and the milk protein butyrophilin in multiple sclerosis. J Immunol. 2004;172:661–668. doi: 10.4049/jimmunol.172.1.661. [DOI] [PubMed] [Google Scholar]
- 47.Molina V., Shoenfeld Y. Infection, vaccines and other environmental triggers of autoimmunity. Autoimmunity. 2005;38:235–245. doi: 10.1080/08916930500050277. [DOI] [PubMed] [Google Scholar]
- 48.Pordeus V., Szyper-Kravitz M., Levy R.A., Vaz N.M., Shoenfeld Y. Infections and autoimmunity: a panorama. Clin Rev Allergy Immunol. 2008;34:283–299. doi: 10.1007/s12016-007-8048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doria A., Sarzi-Puttini P., Shoenfeld Y. Infections, rheumatism and autoimmunity: the conflicting relationship between humans and their environment. Autoimmun Rev. 2008;8:1–4. doi: 10.1016/j.autrev.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 50.Tozzoli R., Barzilai O., Ram M., Villalta D., Bizzaro N., Sherer Y. Infections and autoimmune thyroid diseases: parallel detection of antibodies against pathogens with proteomic technology. Autoimmun Rev. 2008;8:112–115. doi: 10.1016/j.autrev.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 51.Corthesy B. Role of secretory IgA in infection and maintenance of homeostasis. Autoimmun Rev. Nov 29 2012 doi: 10.1016/j.autrev.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 52.Brown J.M., Pfau J.C., Pershouse M.A., Holian A. Silica, apoptosis, and autoimmunity. J Immunotoxicol. 2005;1:177–187. doi: 10.1080/15476910490911922. [DOI] [PubMed] [Google Scholar]
- 53.Otsuki T., Hayashi H., Nishimura Y., Hyodo F., Maeda M., Kumagai N. Dysregulation of autoimmunity caused by silica exposure and alteration of Fas-mediated apoptosis in T lymphocytes derived from silicosis patients. Int J Immunopathol Pharmacol. 2011;24:11S–16S. [PubMed] [Google Scholar]
- 54.Cooper G.S., Wither J., Bernatsky S., Claudio J.O., Clarke A., Rioux J.D. Occupational and environmental exposures and risk of systemic lupus erythematosus: silica, sunlight, solvents. Rheumatology. 2010;49:2172–2180. doi: 10.1093/rheumatology/keq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Finckh A., Cooper G.S., Chibnik L.B., Costenbader K.H., Watts J., Pankey H. Occupational silica and solvent exposures and risk of systemic lupus erythematosus in urban women. Arthritis Rheum. 2006;54:3648–3654. doi: 10.1002/art.22210. [DOI] [PubMed] [Google Scholar]
- 56.Parks C.G., Cooper G.S. Occupational exposures and risk of systemic lupus erythematosus: a review of the evidence and exposure assessment methods in population- and clinic-based studies. Lupus. 2006;15:728–736. doi: 10.1177/0961203306069346. [DOI] [PubMed] [Google Scholar]
- 57.Griffin J.M., Gilbert K.M., Lamps L.W., Pumford N.R. CD4(+) T-cell activation and induction of autoimmune hepatitis following trichloroethylene treatment in MRL +/+ mice. Toxicol Sci. 2000;57:345–352. doi: 10.1093/toxsci/57.2.345. [DOI] [PubMed] [Google Scholar]
- 58.Khan M.F., Kaphalia B.S., Prabhakar B.S., Kanz M.F., Ansari G.A. Trichloroethene-induced autoimmune response in female MRL +/+ mice. Toxicol Appl Pharmacol. 1995;134:155–160. doi: 10.1006/taap.1995.1179. [DOI] [PubMed] [Google Scholar]
- 59.Duntas L.H. Environmental factors and thyroid autoimmunity. Ann Endocrinol (Paris) 2011;72:108–113. doi: 10.1016/j.ando.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 60.Parks C.G., Walitt B.T., Pettinger M., Chen J.C., de Roos A.J., Hunt J. Insecticide use and risk of rheumatoid arthritis and systemic lupus erythematosus in the Women's Health Initiative Observational Study. Arthritis Care Res (Hoboken) 2011;63:184–194. doi: 10.1002/acr.20335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fortes C. Lupus erythematosus. Are residential insecticides exposure the missing link? Med Hypotheses. 2010;75:590–593. doi: 10.1016/j.mehy.2010.07.041. [DOI] [PubMed] [Google Scholar]
- 62.Burek C.L., Talor M.V. Environmental triggers of autoimmune thyroiditis. J Autoimmun. 2009;33:183–189. doi: 10.1016/j.jaut.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gold L.S., Ward M.H., Dosemeci M., De Roos A.J. Systemic autoimmune disease mortality and occupational exposures. Arthritis Rheum. 2007;56:3189–3201. doi: 10.1002/art.22880. [DOI] [PubMed] [Google Scholar]
- 64.Sobel E.S., Gianini J., Butfiloski E.J., Croker B.P., Schiffenbauer J., Roberts S.M. Acceleration of autoimmunity by organochlorine pesticides in (NZB × NZW)F1 mice. Environ Health Perspect. 2005;113:323–328. doi: 10.1289/ehp.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mayes M.D. Epidemiologic studies of environmental agents and systemic autoimmune diseases. Environ Health Perspect. 1999;107(Suppl. 5):743–748. doi: 10.1289/ehp.99107s5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Artukovic M., Ikic M., Kustelega J., Artukovic I.N., Kaliterna D.M. Influence of UV radiation on immunological system and occurrence of autoimmune diseases. Coll Antropol. 2010;34(Suppl. 2):175–178. [PubMed] [Google Scholar]
- 67.Handunnetthi L., Ramagopalan S.V. UV radiation, vitamin D, and multiple sclerosis. Proc Natl Acad Sci U S A. 2010;107:E130. doi: 10.1073/pnas.1004603107. [author reply E1] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prieto S., Grau J.M. The geoepidemiology of autoimmune muscle disease. Autoimmun Rev. 2010;9:A330–A334. doi: 10.1016/j.autrev.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 69.Kuhn A., Beissert S. Photosensitivity in lupus erythematosus. Autoimmunity. 2005;38:519–529. doi: 10.1080/08916930500285626. [DOI] [PubMed] [Google Scholar]
- 70.Tumgor G., Balkan C., Arikan C., Kavakli K., Aydogdu S. Immune haemolytic anaemia induced by allopurinol after liver transplantation. Acta Paediatr. 2006;95:762–763. doi: 10.1080/08035250500499465. [DOI] [PubMed] [Google Scholar]
- 71.Pujol M., Duran-Suarez J.R., Martin Vega C., Sanchez C., Tovar J.L., Valles M. Autoimmune thrombocytopenia in three patients treated with captopril. Vox Sang. 1989;57:218. doi: 10.1111/j.1423-0410.1989.tb00827.x. [DOI] [PubMed] [Google Scholar]
- 72.Gharavi A.E., Sammaritano L.R., Wen J., Miyawaki N., Morse J.H., Zarrabi M.H. Characteristics of human immunodeficiency virus and chlorpromazine induced antiphospholipid antibodies: effect of beta 2 glycoprotein I on binding to phospholipid. J Rheumatol. 1994;21:94–99. [PubMed] [Google Scholar]
- 73.Canoso R.T., de Oliveira R.M. Chlorpromazine-induced anticardiolipin antibodies and lupus anticoagulant: absence of thrombosis. Am J Hematol. 1988;27:272–275. doi: 10.1002/ajh.2830270408. [DOI] [PubMed] [Google Scholar]
- 74.Stein P.B., Inwood M.J. Hemolytic anemia associated with chlorpromazine therapy. Can J Psychiatry. 1980;25:659–661. doi: 10.1177/070674378002500810. [DOI] [PubMed] [Google Scholar]
- 75.Hadnagy C. Letter: Coombs-positive haemolytic anaemia provoked by chlorpromazine. Lancet. 1976;1:423. doi: 10.1016/s0140-6736(76)90251-8. [DOI] [PubMed] [Google Scholar]
- 76.Berglund S., Gottfries C.G., Gottfries I., Stormby K. Chlorpromazine-induced antinuclear factors. Acta Med Scand. 1970;187:67–74. doi: 10.1111/j.0954-6820.1970.tb02908.x. [DOI] [PubMed] [Google Scholar]
- 77.Liu Z.X., Kaplowitz N. Immune-mediated drug-induced liver disease. Clin Liver Dis. 2002;6:755–774. doi: 10.1016/s1089-3261(02)00025-9. [DOI] [PubMed] [Google Scholar]
- 78.Yung R.L., Richardson B.C. Drug-induced lupus. Rheum Dis Clin North Am. 1994;20:61–86. [PubMed] [Google Scholar]
- 79.Czaja A.J. Drug-induced autoimmune-like hepatitis. Dig Dis Sci. 2011;56:958–976. doi: 10.1007/s10620-011-1611-4. [DOI] [PubMed] [Google Scholar]
- 80.Chighizola C., Meroni P.L. The role of environmental estrogens and autoimmunity. Autoimmun Rev. 2012;11:A493–A501. doi: 10.1016/j.autrev.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 81.Shapira Y., Agmon-Levin N., Renaudineau Y., Porat-Katz B.S., Barzilai O., Ram M. Serum markers of infections in patients with primary biliary cirrhosis: evidence of infection burden. Exp Mol Pathol. 2012;93:386–390. doi: 10.1016/j.yexmp.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 82.Shapira Y., Agmon-Levin N., Shoenfeld Y. Defining and analyzing geoepidemiology and human autoimmunity. J Autoimmun. 2010;34:J168–J177. doi: 10.1016/j.jaut.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 83.Berlin T., Zandman-Goddard G., Blank M., Matthias T., Pfeiffer S., Weis I. Autoantibodies in nonautoimmune individuals during infections. Ann N Y Acad Sci. 2007;1108:584–593. doi: 10.1196/annals.1422.061. [DOI] [PubMed] [Google Scholar]
- 84.Saad R., Rizkallah M.R., Aziz R.K. Gut pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog. 2012;4:16. doi: 10.1186/1757-4749-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buccigrossi V., Nicastro E., Guarino A. Functions of intestinal microflora in children. Curr Opin Gastroenterol. 2013;29:31–38. doi: 10.1097/MOG.0b013e32835a3500. [DOI] [PubMed] [Google Scholar]
- 86.Gielda L.M., DiRita V.J. Zinc competition among the intestinal microbiota. MBio. 2012;3:e00171–e. doi: 10.1128/mBio.00171-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lidar M., Agmon-Levin N., Langevitz P., Shoenfeld Y. Silicone and scleroderma revisited. Lupus. 2012;21:121–127. doi: 10.1177/0961203311430703. [DOI] [PubMed] [Google Scholar]
- 88.Shoenfeld Y., Toubi E. Protective autoantibodies: role in homeostasis, clinical importance, and therapeutic potential. Arthritis Rheum. 2005;52:2599–2606. doi: 10.1002/art.21252. [DOI] [PubMed] [Google Scholar]
- 89.Toubi E., Shoenfeld Y. Predictive and protective autoimmunity in cardiovascular diseases: is vaccination therapy a reality? Lupus. 2005;14:665–669. doi: 10.1191/0961203305lu2196oa. [DOI] [PubMed] [Google Scholar]
- 90.Ram M., Anaya J.M., Barzilai O., Izhaky D., Porat Katz B.S., Blank M. The putative protective role of hepatitis B virus (HBV) infection from autoimmune disorders. Autoimmun Rev. 2008;7:621–625. doi: 10.1016/j.autrev.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 91.Meroni P.L., Shoenfeld Y. Predictive, protective, orphan autoantibodies: the example of anti-phospholipid antibodies. Autoimmun Rev. 2008;7:585–587. doi: 10.1016/j.autrev.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 92.Plot L., Amital H., Barzilai O., Ram M., Nicola B., Shoenfeld Y. Infections may have a protective role in the etiopathogenesis of celiac disease. Ann N Y Acad Sci. 2009;1173:670–674. doi: 10.1111/j.1749-6632.2009.04814.x. [DOI] [PubMed] [Google Scholar]
- 93.Guilherme L., Oshiro S.E., Fae K.C., Cunha-Neto E., Renesto G., Goldberg A.C. T-cell reactivity against streptococcal antigens in the periphery mirrors reactivity of heart-infiltrating T lymphocytes in rheumatic heart disease patients. Infect Immun. 2001;69:5345–5351. doi: 10.1128/IAI.69.9.5345-5351.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amedei A., Bergman M.P., Appelmelk B.J., Azzurri A., Benagiano M., Tamburini C. Molecular mimicry between Helicobacter pylori antigens and H +, K +-adenosine triphosphatase in human gastric autoimmunity. J Exp Med. 2003;198:1147–1156. doi: 10.1084/jem.20030530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cunha-Neto E., Coelho V., Guilherme L., Fiorelli A., Stolf N., Kalil J. Autoimmunity in Chagas' disease. Identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas' cardiomyopathy patient. J Clin Invest. 1996;98:1709–1712. doi: 10.1172/JCI118969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.da Rocha Sobrinho H.M., Jarach R., da Silva N.A., Shio M.T., Jancar S., Timenetsky J. Mycoplasmal lipid-associated membrane proteins and Mycoplasma arthritidis mitogen recognition by serum antibodies from patients with rheumatoid arthritis. Rheumatol Int. 2011;31:951–957. doi: 10.1007/s00296-010-1612-1. [DOI] [PubMed] [Google Scholar]
- 97.Tobon G.J., Pers J.O., Canas C.A., Rojas-Villarraga A., Youinou P., Anaya J.M. Are autoimmune diseases predictable? Autoimmun Rev. 2012;11:259–266. doi: 10.1016/j.autrev.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 98.Jones D.E. Pathogenesis of primary biliary cirrhosis. Gut. 2007;56:1615–1624. doi: 10.1136/gut.2007.122150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaplan M.M., Gershwin M.E. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–1273. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 100.Arbuckle M.R., McClain M.T., Rubertone M.V., Scofield R.H., Dennis G.J., James J.A. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 101.Harley J.B., Harley I.T., Guthridge J.M., James J.A. The curiously suspicious: a role for Epstein–Barr virus in lupus. Lupus. 2006;15:768–777. doi: 10.1177/0961203306070009. [DOI] [PubMed] [Google Scholar]
- 102.Harley J.B., James J.A. Epstein–Barr virus infection induces lupus autoimmunity. Bull NYU Hosp Jt Dis. 2006;64:45–50. [PubMed] [Google Scholar]
- 103.Navarra S.V., Ishimori M.I., Uy E.A., Hamijoyo L., Sama J., James J.A. Studies of Filipino patients with systemic lupus erythematosus: autoantibody profile of first-degree relatives. Lupus. 2011;20:537–543. doi: 10.1177/0961203310385164. [DOI] [PubMed] [Google Scholar]
- 104.Zandman-Goddard G., Berkun Y., Barzilai O., Boaz M., Blank M., Ram M. Exposure to Epstein–Barr virus infection is associated with mild systemic lupus erythematosus disease. Ann N Y Acad Sci. 2009;1173:658–663. doi: 10.1111/j.1749-6632.2009.04754.x. [DOI] [PubMed] [Google Scholar]
- 105.Barzilai O., Sherer Y., Ram M., Izhaky D., Anaya J.M., Shoenfeld Y. Epstein–Barr virus and cytomegalovirus in autoimmune diseases: are they truly notorious? A preliminary report. Ann N Y Acad Sci. 2007;1108:567–577. doi: 10.1196/annals.1422.059. [DOI] [PubMed] [Google Scholar]
- 106.Bengtsson A., Widell A., Elmstahl S., Sturfelt G. No serological indications that systemic lupus erythematosus is linked with exposure to human parvovirus B19. Ann Rheum Dis. 2000;59:64–66. doi: 10.1136/ard.59.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hemauer A., Beckenlehner K., Wolf H., Lang B., Modrow S. Acute parvovirus B19 infection in connection with a flare of systemic lupus erythematodes in a female patient. J Clin Virol. 1999;14:73–77. doi: 10.1016/s1386-6532(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 108.Hrycek A., Kusmierz D., Mazurek U., Wilczok T. Human cytomegalovirus in patients with systemic lupus erythematosus. Autoimmunity. 2005;38:487–491. doi: 10.1080/08916930500285667. [DOI] [PubMed] [Google Scholar]
- 109.James J.A., Neas B.R., Moser K.L., Hall T., Bruner G.R., Sestak A.L. Systemic lupus erythematosus in adults is associated with previous Epstein–Barr virus exposure. Arthritis Rheum. 2001;44:1122–1126. doi: 10.1002/1529-0131(200105)44:5<1122::AID-ANR193>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 110.Seishima M., Oyama Z., Yamamura M. Two-year follow-up study after human parvovirus B19 infection. Dermatology. 2003;206:192–196. doi: 10.1159/000068885. [DOI] [PubMed] [Google Scholar]
- 111.Parks C.G., Cooper G.S., Hudson L.L., Dooley M.A., Treadwell E.L., St Clair E.W. Association of Epstein–Barr virus with systemic lupus erythematosus: effect modification by race, age, and cytotoxic T lymphocyte-associated antigen 4 genotype. Arthritis Rheum. 2005;52:1148–1159. doi: 10.1002/art.20997. [DOI] [PubMed] [Google Scholar]
- 112.Wang H., Nicholas M.W., Conway K.L., Sen P., Diz R., Tisch R.M. EBV latent membrane protein 2A induces autoreactive B cell activation and TLR hypersensitivity. J Immunol. 2006;177:2793–2802. doi: 10.4049/jimmunol.177.5.2793. [DOI] [PubMed] [Google Scholar]
- 113.Chang M., Pan M.R., Chen D.Y., Lan J.L. Human cytomegalovirus pp 65 lower matrix protein: a humoral immunogen for systemic lupus erythematosus patients and autoantibody accelerator for NZB/W F1 mice. Clin Exp Immunol. 2006;143:167–179. doi: 10.1111/j.1365-2249.2005.02974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gilden D.H. Infectious causes of multiple sclerosis. Lancet Neurol. 2005;4:195–202. doi: 10.1016/S1474-4422(05)01017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kakalacheva K., Lunemann J.D. Environmental triggers of multiple sclerosis. FEBS Lett. 2011;23:3724–3729. doi: 10.1016/j.febslet.2011.04.006. [PMID: 21486562] [DOI] [PubMed] [Google Scholar]
- 116.Hammond S.R., English D.R., McLeod J.G. The age-range of risk of developing multiple sclerosis: evidence from a migrant population in Australia. Brain. 2000;123(Pt 5):968–974. doi: 10.1093/brain/123.5.968. [DOI] [PubMed] [Google Scholar]
- 117.Kurtzke J.F., Delasnerie-Laupretre N., Wallin M.T. Multiple sclerosis in North African migrants to France. Acta Neurol Scand. 1998;98:302–309. doi: 10.1111/j.1600-0404.1998.tb01738.x. [DOI] [PubMed] [Google Scholar]
- 118.Kurtzke J.F., Hyllested K. Multiple sclerosis in the Faroe Islands: I. Clinical and epidemiological features. Ann Neurol. 1979;5:6–21. doi: 10.1002/ana.410050104. [DOI] [PubMed] [Google Scholar]
- 119.Noseworthy J.H., Lucchinetti C., Rodriguez M., Weinshenker B.G. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 120.Frohman E.M., Racke M.K., Raine C.S. Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 121.Giovannoni G., Cutter G.R., Lunemann J., Martin R., Munz C., Sriram S. Infectious causes of multiple sclerosis. Lancet Neurol. 2006;5:887–894. doi: 10.1016/S1474-4422(06)70577-4. [DOI] [PubMed] [Google Scholar]
- 122.Giovannoni G., Ebers G. Multiple sclerosis: the environment and causation. Curr Opin Neurol. 2007;20:261–268. doi: 10.1097/WCO.0b013e32815610c2. [DOI] [PubMed] [Google Scholar]
- 123.Ebers G.C., Sadovnick A.D., Risch N.J. A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature. 1995;377:150–151. doi: 10.1038/377150a0. [DOI] [PubMed] [Google Scholar]
- 124.Sadovnick A.D., Ebers G.C., Dyment D.A., Risch N.J. Evidence for genetic basis of multiple sclerosis. The Canadian Collaborative Study Group. Lancet. 1996;347:1728–1730. doi: 10.1016/s0140-6736(96)90807-7. [DOI] [PubMed] [Google Scholar]
- 125.Barcellos L.F., Sawcer S., Ramsay P.P., Baranzini S.E., Thomson G., Briggs F. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet. 2006;15:2813–2824. doi: 10.1093/hmg/ddl223. [DOI] [PubMed] [Google Scholar]
- 126.Hafler D.A., Compston A., Sawcer S., Lander E.S., Daly M.J., De Jager P.L. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 127.Qu H.Q., Bradfield J.P., Belisle A., Grant S.F., Hakonarson H., Polychronakos C. The type I diabetes association of the IL2RA locus. Genes Immun. 2009;10(Suppl. 1):S42–S48. doi: 10.1038/gene.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Arnson Y., Amital H., Shoenfeld Y. Vitamin D and autoimmunity: new aetiological and therapeutic considerations. Ann Rheum Dis. 2007;66:1137–1142. doi: 10.1136/ard.2007.069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Carvalho J.F., Blank M., Kiss E., Tarr T., Amital H., Shoenfeld Y. Anti-vitamin D, vitamin D in SLE: preliminary results. Ann N Y Acad Sci. 2007;1109:550–557. doi: 10.1196/annals.1398.061. [DOI] [PubMed] [Google Scholar]
- 130.Shoenfeld N., Amital H., Shoenfeld Y. The effect of melanism and vitamin D synthesis on the incidence of autoimmune disease. Nat Clin Pract Rheumatol. 2009;5:99–105. doi: 10.1038/ncprheum0989. [DOI] [PubMed] [Google Scholar]
- 131.Shapira Y., Agmon-Levin N., Shoenfeld Y. Mycobacterium tuberculosis, autoimmunity, and vitamin D. Clin Rev Allergy Immunol. 2010;38:169–177. doi: 10.1007/s12016-009-8150-1. [DOI] [PubMed] [Google Scholar]
- 132.Amital H., Szekanecz Z., Szucs G., Danko K., Nagy E., Csepany T. Serum concentrations of 25-OH vitamin D in patients with systemic lupus erythematosus (SLE) are inversely related to disease activity: is it time to routinely supplement patients with SLE with vitamin D? Ann Rheum Dis. 2010;69:1155–1157. doi: 10.1136/ard.2009.120329. [DOI] [PubMed] [Google Scholar]
- 133.Toubi E., Shoenfeld Y. The role of vitamin D in regulating immune responses. Isr Med Assoc J. 2010;12:174–175. [PubMed] [Google Scholar]
- 134.Souberbielle J.C., Body J.J., Lappe J.M., Plebani M., Shoenfeld Y., Wang T.J. Vitamin D and musculoskeletal health, cardiovascular disease, autoimmunity and cancer: recommendations for clinical practice. Autoimmun Rev. 2010;9:709–715. doi: 10.1016/j.autrev.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 135.Agmon-Levin N., Blank M., Zandman-Goddard G., Orbach H., Meroni P.L., Tincani A. Vitamin D: an instrumental factor in the anti-phospholipid syndrome by inhibition of tissue factor expression. Ann Rheum Dis. 2011;70:145–150. doi: 10.1136/ard.2010.134817. [DOI] [PubMed] [Google Scholar]
- 136.Oren Y., Shapira Y., Agmon-Levin N., Kivity S., Zafrir Y., Altman A. Vitamin D insufficiency in a sunny environment: a demographic and seasonal analysis. Isr Med Assoc J. 2010;12:751–756. [PubMed] [Google Scholar]
- 137.Kivity S., Agmon-Levin N., Zisappl M., Shapira Y., Nagy E.V., Danko K. Vitamin D and autoimmune thyroid diseases. Cell Mol Immunol. 2011;8:243–247. doi: 10.1038/cmi.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lerner A., Shapira Y., Agmon-Levin N., Pacht A., Ben-Ami Shor D., Lopez H.M. The clinical significance of 25OH-Vitamin D status in celiac disease. Clin Rev Allergy Immunol. 2012;42:322–330. doi: 10.1007/s12016-010-8237-8. [DOI] [PubMed] [Google Scholar]
- 139.Hajas A., Sandor J., Csathy L., Csipo I., Barath S., Paragh G. Vitamin D insufficiency in a large MCTD population. Autoimmun Rev. 2011;10:317–324. doi: 10.1016/j.autrev.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 140.Cutolo M., Pizzorni C., Sulli A. Vitamin D endocrine system involvement in autoimmune rheumatic diseases. Autoimmun Rev. 2011;11:84–87. doi: 10.1016/j.autrev.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 141.Lucas R.M., Ponsonby A.L., Dear K., Valery P.C., Pender M.P., Taylor B.V. Sun exposure and vitamin D are independent risk factors for CNS demyelination. Neurology. 2011;76:540–548. doi: 10.1212/WNL.0b013e31820af93d. [DOI] [PubMed] [Google Scholar]
- 142.Munger K.L., Levin L.I., Hollis B.W., Howard N.S., Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296:2832–2838. doi: 10.1001/jama.296.23.2832. [DOI] [PubMed] [Google Scholar]
- 143.Handel A.E., Williamson A.J., Disanto G., Dobson R., Giovannoni G., Ramagopalan S.V. Smoking and multiple sclerosis: an updated meta-analysis. PLoS One. 2011;6:e16149. doi: 10.1371/journal.pone.0016149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hedstrom A.K., Sundqvist E., Baarnhielm M., Nordin N., Hillert J., Kockum I. Smoking and two human leukocyte antigen genes interact to increase the risk for multiple sclerosis. Brain. 2011;134:653–664. doi: 10.1093/brain/awq371. [DOI] [PubMed] [Google Scholar]
- 145.Stejskal V., Hudecek R., Stejskal J., Sterzl I. Diagnosis and treatment of metal-induced side-effects. Neuro Endocrinol Lett. 2006;27(Suppl. 1):7–16. [PubMed] [Google Scholar]
- 146.Fleming J.O. Helminths and multiple sclerosis: will old friends give us new treatments for MS? J Neuroimmunol. 2011;233:3–5. doi: 10.1016/j.jneuroim.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 147.Gaisford W., Cooke A. Can infections protect against autoimmunity? Curr Opin Rheumatol. 2009;21:391–396. doi: 10.1097/BOR.0b013e32832c2dee. [DOI] [PubMed] [Google Scholar]
- 148.Albright A.V., Lavi E., Black J.B., Goldberg S., O'Connor M.J., Gonzalez-Scarano F. The effect of human herpesvirus-6 (HHV-6) on cultured human neural cells: oligodendrocytes and microglia. J Neurovirol. 1998;4:486–494. doi: 10.3109/13550289809113493. [DOI] [PubMed] [Google Scholar]
- 149.Chan P.K., Ng H.K., Cheng A.F. Detection of human herpesviruses 6 and 7 genomic sequences in brain tumours. J Clin Pathol. 1999;52:620–623. doi: 10.1136/jcp.52.8.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kim J.S., Lee K.S., Park J.H., Kim M.Y., Shin W.S. Detection of human herpesvirus 6 variant A in peripheral blood mononuclear cells from multiple sclerosis patients. Eur Neurol. 2000;43:170–173. doi: 10.1159/000008158. [DOI] [PubMed] [Google Scholar]
- 151.Opsahl M.L., Kennedy P.G. Early and late HHV-6 gene transcripts in multiple sclerosis lesions and normal appearing white matter. Brain. 2005;128:516–527. doi: 10.1093/brain/awh390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mirandola P., Stefan A., Brambilla E., Campadelli-Fiume G., Grimaldi L.M. Absence of human herpesvirus 6 and 7 from spinal fluid and serum of multiple sclerosis patients. Neurology. 1999;53:1367–1368. doi: 10.1212/wnl.53.6.1367-a. [DOI] [PubMed] [Google Scholar]
- 153.Sospedra M., Zhao Y., zur Hausen H., Muraro P.A., Hamashin C., de Villiers E.M. Recognition of conserved amino acid motifs of common viruses and its role in autoimmunity. PLoS Pathog. 2005;1:e41. doi: 10.1371/journal.ppat.0010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bogdanos D.P., Gershwin M.E. What is new in primary biliary cirrhosis? Dig Dis. 2012;30(Suppl. 1):20–31. doi: 10.1159/000341118. [DOI] [PubMed] [Google Scholar]
- 155.Corpechot C., Chretien Y., Chazouilleres O., Poupon R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. J Hepatol. 2010;53:162–169. doi: 10.1016/j.jhep.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 156.Gershwin M.E., Selmi C., Worman H.J., Gold E.B., Watnik M., Utts J. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–1202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Parikh-Patel A., Gold E.B., Worman H., Krivy K.E., Gershwin M.E. Risk factors for primary biliary cirrhosis in a cohort of patients from the united states. Hepatology. 2001;33:16–21. doi: 10.1053/jhep.2001.21165. [DOI] [PubMed] [Google Scholar]
- 158.Prince M.I., Ducker S.J., James O.F. Case–control studies of risk factors for primary biliary cirrhosis in two United Kingdom populations. Gut. 2010;59:508–512. doi: 10.1136/gut.2009.184218. [DOI] [PubMed] [Google Scholar]
- 159.Invernizzi P., Selmi C., Gershwin M.E. Update on primary biliary cirrhosis. Dig Liver Dis. 2010;42:401–408. doi: 10.1016/j.dld.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Selmi C., Invernizzi P., Zuin M., Podda M., Gershwin M.E. Genetics and geoepidemiology of primary biliary cirrhosis: following the footprints to disease etiology. Semin Liver Dis. 2005;25:265–280. doi: 10.1055/s-2005-916319. [DOI] [PubMed] [Google Scholar]
- 161.Selmi C., Invernizzi P., Zuin M., Podda M., Seldin M.F., Gershwin M.E. Genes and (auto)immunity in primary biliary cirrhosis. Genes Immun. 2005;6:543–556. doi: 10.1038/sj.gene.6364248. [DOI] [PubMed] [Google Scholar]
- 162.Smyk D., Cholongitas E., Kriese S., Rigopoulou E.I., Bogdanos D.P. Primary biliary cirrhosis: family stories. Autoimmune Dis. 2011;2011:189585. doi: 10.4061/2011/189585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bogdanos D.P., Smyk D.S., Rigopoulou E.I., Mytilinaiou M.G., Heneghan M.A., Selmi C. Twin studies in autoimmune disease: genetics, gender and environment. J Autoimmun. 2012;38:J156–J169. doi: 10.1016/j.jaut.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 164.Gershwin M.E., Mackay I.R. The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology. 2008;47:737–745. doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]
- 165.Konikoff F., Pecht M., Theodor E., Shoenfeld Y. Primary biliary cirrhosis: lymphocyte subsets and function in a time frame. Hepatology. 1989;10:525–526. doi: 10.1002/hep.1840100427. [DOI] [PubMed] [Google Scholar]
- 166.Schlesinger M., Benbassat C., Shoenfeld Y. Complement profile in primary biliary cirrhosis. Immunol Res. 1992;11:98–103. doi: 10.1007/BF02918614. [DOI] [PubMed] [Google Scholar]
- 167.Shoenfeld Y. Primary biliary cirrhosis and autoimmune rheumatic diseases: prediction and prevention. Isr J Med Sci. 1992;28:113–116. [PubMed] [Google Scholar]
- 168.Weiss P., Shoenfeld Y. Primary biliary cirrhosis is a multisystem disorder. Ann Med Interne (Paris) 1991;142:283–287. [PubMed] [Google Scholar]
- 169.Weiss P., Shoenfeld Y. Primary biliary cirrhosis: increasing problem, approaching solution. Isr J Med Sci. 1992;28:726–728. [PubMed] [Google Scholar]
- 170.Benbassat C., Schlesinger M., Shoenfeld Y. The complement system and primary biliary cirrhosis. J Clin Lab Immunol. 1992;38:51–61. [PubMed] [Google Scholar]
- 171.Rotman P., Levy Y., Shoenfeld Y. Primary biliary cirrhosis—association or overlap with other autoimmune diseases. Isr J Med Sci. 1997;33:823–825. [PubMed] [Google Scholar]
- 172.Shapira S., Bar-Dayan Y., Gershwin M.E., Shoenfeld Y. Is it possible to predict and prevent primary biliary cirrhosis? Harefuah. 1999;137:39–41. [PubMed] [Google Scholar]
- 173.Aoki C.A., Roifman C.M., Lian Z.X., Bowlus C.L., Norman G.L., Shoenfeld Y. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun. 2006;27:50–53. doi: 10.1016/j.jaut.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 174.Barak V., Selmi C., Schlesinger M., Blank M., Agmon-Levin N., Kalickman I. Serum inflammatory cytokines, complement components, and soluble interleukin 2 receptor in primary biliary cirrhosis. J Autoimmun. 2009;33:178–182. doi: 10.1016/j.jaut.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 175.Bogdanos D.P., Baum H., Vergani D. Antimitochondrial and other autoantibodies. Clin Liver Dis. 2003;7:759–777. doi: 10.1016/s1089-3261(03)00104-1. [vi] [DOI] [PubMed] [Google Scholar]
- 176.Bogdanos D.P., Baum H., Vergani D., Burroughs A.K. The role of E. coli infection in the pathogenesis of primary biliary cirrhosis. Dis Markers. 2010;29:301–311. doi: 10.3233/DMA-2010-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bogdanos D.P., Vergani D. Origin of cross-reactive autoimmunity in primary biliary cirrhosis. Liver Int. 2006;26:633–635. doi: 10.1111/j.1478-3231.2006.01291.x. [DOI] [PubMed] [Google Scholar]
- 178.Bogdanos D.P., Vergani D. Bacteria and primary biliary cirrhosis. Clin Rev Allergy Immunol. 2009;36:30–39. doi: 10.1007/s12016-008-8087-9. [DOI] [PubMed] [Google Scholar]
- 179.Gershwin M.E., Mackay I.R. Primary biliary cirrhosis: paradigm or paradox for autoimmunity. Gastroenterology. 1991;100:822–833. doi: 10.1016/0016-5085(91)80033-6. [DOI] [PubMed] [Google Scholar]
- 180.Mackay I.R., Whittingham S., Fida S., Myers M., Ikuno N., Gershwin M.E. The peculiar autoimmunity of primary biliary cirrhosis. Immunol Rev. 2000;174:226–237. doi: 10.1034/j.1600-0528.2002.017410.x. [DOI] [PubMed] [Google Scholar]
- 181.Shimoda S., Nakamura M., Ishibashi H., Hayashida K., Niho Y. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune diseases. J Exp Med. 1995;181:1835–1845. doi: 10.1084/jem.181.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shimoda S., Nakamura M., Shigematsu H., Tanimoto H., Gushima T., Gershwin M.E. Mimicry peptides of human PDC-E2 163-176 peptide, the immunodominant T-cell epitope of primary biliary cirrhosis. Hepatology. 2000;31:1212–1216. doi: 10.1053/jhep.2000.8090. [DOI] [PubMed] [Google Scholar]
- 183.Shimoda S., Van de Water J., Ansari A., Nakamura M., Ishibashi H., Coppel R.L. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–1840. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Vergani D., Bogdanos D.P. Positive markers in AMA-negative PBC. Am J Gastroenterol. 2003;98:241–243. doi: 10.1111/j.1572-0241.2003.07270.x. [DOI] [PubMed] [Google Scholar]
- 185.Hohenester S., Oude-Elferink R.P., Beuers U. Primary biliary cirrhosis. Semin Immunopathol. 2009;31:283–307. doi: 10.1007/s00281-009-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Neuberger J. Primary biliary cirrhosis. Lancet. 1997;350:875–879. doi: 10.1016/S0140-6736(97)05419-6. [DOI] [PubMed] [Google Scholar]
- 187.Bogdanos D.P., Invernizzi P., Mackay I.R., Vergani D. Autoimmune liver serology: current diagnostic and clinical challenges. World J Gastroenterol. 2008;14:3374–3387. doi: 10.3748/wjg.14.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bogdanos D.P., Komorowski L. Disease-specific autoantibodies in primary biliary cirrhosis. Clin Chim Acta. 2011;412:502–512. doi: 10.1016/j.cca.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 189.Dahnrich C., Pares A., Caballeria L., Rosemann A., Schlumberger W., Probst C. New ELISA for detecting primary biliary cirrhosis-specific antimitochondrial antibodies. Clin Chem. 2009;55:978–985. doi: 10.1373/clinchem.2008.118299. [DOI] [PubMed] [Google Scholar]
- 190.Liu H., Norman G.L., Shums Z., Worman H.J., Krawitt E.L., Bizzaro N. PBC screen: an IgG/IgA dual isotype ELISA detecting multiple mitochondrial and nuclear autoantibodies specific for primary biliary cirrhosis. J Autoimmun. 2010;35:436–442. doi: 10.1016/j.jaut.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 191.Ma Y., Thomas M.G., Okamoto M., Bogdanos D.P., Nagl S., Kerkar N. Key residues of a major cytochrome P4502D6 epitope are located on the surface of the molecule. J Immunol. 2002;169:277–285. doi: 10.4049/jimmunol.169.1.277. [DOI] [PubMed] [Google Scholar]
- 192.Rigopoulou E.I., Davies E.T., Bogdanos D.P., Liaskos C., Mytilinaiou M., Koukoulis G.K. Antimitochondrial antibodies of immunoglobulin G3 subclass are associated with a more severe disease course in primary biliary cirrhosis. Liver Int. 2007;27:1226–1231. doi: 10.1111/j.1478-3231.2007.01586.x. [DOI] [PubMed] [Google Scholar]
- 193.Wen L., Ma Y., Bogdanos D.P., Wong F.S., Demaine A., Mieli-Vergani G. Pediatric autoimmune liver diseases: the molecular basis of humoral and cellular immunity. Curr Mol Med. 2001;1:379–389. doi: 10.2174/1566524013363672. [DOI] [PubMed] [Google Scholar]
- 194.Invernizzi P., Lleo A., Podda M. Interpreting serological tests in diagnosing autoimmune liver diseases. Semin Liver Dis. 2007;27:161–172. doi: 10.1055/s-2007-979469. [DOI] [PubMed] [Google Scholar]
- 195.Rigopoulou E.I., Bogdanos D.P., Liaskos C., Koutsoumpas A., Baum H., Vergani D. Anti-mitochondrial antibody immunofluorescent titres correlate with the number and intensity of immunoblot-detected mitochondrial bands in patients with primary biliary cirrhosis. Clin Chim Acta. 2007;380:118–121. doi: 10.1016/j.cca.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 196.Gilburd B., Ziporen L., Zharhary D., Blank M., Zurgil N., Scheinberg M.A. Antimitochondrial (pyruvate dehydrogenase) antibodies in leprosy. J Clin Immunol. 1994;14:14–19. doi: 10.1007/BF01541171. [DOI] [PubMed] [Google Scholar]
- 197.Konikoff F., Isenberg D.A., Barrison I., Theodor E., Shoenfeld Y. Antinuclear autoantibodies in chronic liver diseases. Hepatogastroenterology. 1989;36:341–345. [PubMed] [Google Scholar]
- 198.Konikoff F., Isenberg D.A., Kooperman O., Kennedy R.C., Rauch J., Theodor E. Common lupus anti-DNA antibody idiotypes in chronic liver diseases. Clin Immunol Immunopathol. 1987;43:265–272. doi: 10.1016/0090-1229(87)90134-6. [DOI] [PubMed] [Google Scholar]
- 199.Konikoff F., Shoenfeld Y., Isenberg D.A., Barrison I., Sobe T., Theodor E. Anti-Rnp antibodies in chronic liver diseases. Clin Exp Rheumatol. 1987;5:359–361. [PubMed] [Google Scholar]
- 200.Krause I., Hacham S., Gilburd B., Damianovitch M., Blank M., Shoenfeld Y. Absence of anti-idiotypic antibodies in IVIG preparations to autoantibodies of rare autoimmune diseases. Clin Immunol Immunopathol. 1995;77:229–235. doi: 10.1006/clin.1995.1148. [DOI] [PubMed] [Google Scholar]
- 201.Lorber M., Kra-Oz Z., Guilbrud B., Shoenfeld Y. Natural (antiphospholipid-PDH,-DNA) autoantibodies and their physiologic serum inhibitors. Isr J Med Sci. 1995;31:31–35. [PubMed] [Google Scholar]
- 202.Maran R., Dueymes M., Adler Y., Shoenfeld Y., Youinou P. Isotypic distribution of anti-pyruvate dehydrogenase antibodies in patients with primary biliary cirrhosis and their family members. J Clin Immunol. 1994;14:323–326. doi: 10.1007/BF01540986. [DOI] [PubMed] [Google Scholar]
- 203.Shoenfeld Y., Beresovski A., Zharhary D., Tomer Y., Swissa M., Sela E. Natural autoantibodies in sera of patients with Gaucher's disease. J Clin Immunol. 1995;15:363–372. doi: 10.1007/BF01541326. [DOI] [PubMed] [Google Scholar]
- 204.Zurgil N., Bakimer R., Kaplan M., Youinou P., Shoenfeld Y. Anti-pyruvate dehydrogenase autoantibodies in primary biliary cirrhosis. J Clin Immunol. 1991;11:239–245. doi: 10.1007/BF00918181. [DOI] [PubMed] [Google Scholar]
- 205.Zurgil N., Bakimer R., Moutsopoulos H.M., Tzioufas A.G., Youinou P., Isenberg D.A. Antimitochondrial (pyruvate dehydrogenase) autoantibodies in autoimmune rheumatic diseases. J Clin Immunol. 1992;12:201–209. doi: 10.1007/BF00918090. [DOI] [PubMed] [Google Scholar]
- 206.Zurgil N., Bakimer R., Slor H., Kaplan M., Moutsopoulos H., Shoenfeld Y. Pyruvate dehydrogenase as an antigen to detect antimitochondrial antibodies. Isr J Med Sci. 1990;26:682–685. [PubMed] [Google Scholar]
- 207.Zurgil N., Konikoff F., Bakimer R., Slor H., Shoenfeld Y. Detection of antimitochondrial antibodies: characterization by enzyme immunoassay and immunoblotting. Autoimmunity. 1989;4:289–297. doi: 10.3109/08916938909014705. [DOI] [PubMed] [Google Scholar]
- 208.Tishler M., Alosachie I., Barka N., Lin H.C., Gershwin M.E., Peter J.B. Primary Sjogren's syndrome and primary biliary cirrhosis: differences and similarities in the autoantibody profile. Clin Exp Rheumatol. 1995;13:497–500. [PubMed] [Google Scholar]
- 209.Bean P., Sutphin M.S., Liu Y., Anton R., Reynolds T.B., Shoenfeld Y. Carbohydrate-deficient transferrin and false-positive results for alcohol abuse in primary biliary cirrhosis: differential diagnosis by detection of mitochondrial autoantibodies. Clin Chem. 1995;41:858–861. [PubMed] [Google Scholar]
- 210.Agmon-Levin N., Shapira Y., Selmi C., Barzilai O., Ram M., Szyper-Kravitz M. A comprehensive evaluation of serum autoantibodies in primary biliary cirrhosis. J Autoimmun. 2010;34:55–58. doi: 10.1016/j.jaut.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 211.Metcalf J.V., Mitchison H.C., Palmer J.M., Jones D.E., Bassendine M.F., James O.F. Natural history of early primary biliary cirrhosis. Lancet. 1996;348:1399–1402. doi: 10.1016/S0140-6736(96)04410-8. [DOI] [PubMed] [Google Scholar]
- 212.Dubel L., Tanaka A., Leung P.S., Van de Water J., Coppel R., Roche T. Autoepitope mapping and reactivity of autoantibodies to the dihydrolipoamide dehydrogenase-binding protein (E3BP) and the glycine cleavage proteins in primary biliary cirrhosis. Hepatology. 1999;29:1013–1018. doi: 10.1002/hep.510290403. [DOI] [PubMed] [Google Scholar]
- 213.Leung P.S., Coppel R.L., Ansari A., Munoz S., Gershwin M.E. Antimitochondrial antibodies in primary biliary cirrhosis. Semin Liver Dis. 1997;17:61–69. doi: 10.1055/s-2007-1007183. [DOI] [PubMed] [Google Scholar]
- 214.Palmer J.M., Jones D.E., Quinn J., McHugh A., Yeaman S.J. Characterization of the autoantibody responses to recombinant E3 binding protein (protein X) of pyruvate dehydrogenase in primary biliary cirrhosis. Hepatology. 1999;30:21–26. doi: 10.1002/hep.510300106. [DOI] [PubMed] [Google Scholar]
- 215.Van de Water J., Fregeau D., Davis P., Ansari A., Danner D., Leung P. Autoantibodies of primary biliary cirrhosis recognize dihydrolipoamide acetyltransferase and inhibit enzyme function. J Immunol. 1988;141:2321–2324. [PubMed] [Google Scholar]
- 216.Bogdanos D.P., Liaskos C., Pares A., Norman G., Rigopoulou E.I., Caballeria L. Anti-gp210 antibody mirrors disease severity in primary biliary cirrhosis. Hepatology. 2007;45:1583. doi: 10.1002/hep.21678. [author reply -4] [DOI] [PubMed] [Google Scholar]
- 217.Invernizzi P., Podda M., Battezzati P.M., Crosignani A., Zuin M., Hitchman E. Autoantibodies against nuclear pore complexes are associated with more active and severe liver disease in primary biliary cirrhosis. J Hepatol. 2001;34:366–372. doi: 10.1016/s0168-8278(00)00040-4. [DOI] [PubMed] [Google Scholar]
- 218.Miyachi K., Hankins R.W., Matsushima H., Kikuchi F., Inomata T., Horigome T. Profile and clinical significance of anti-nuclear envelope antibodies found in patients with primary biliary cirrhosis: a multicenter study. J Autoimmun. 2003;20:247–254. doi: 10.1016/s0896-8411(03)00033-7. [DOI] [PubMed] [Google Scholar]
- 219.Nakamura M., Kondo H., Mori T., Komori A., Matsuyama M., Ito M. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis. Hepatology. 2007;45:118–127. doi: 10.1002/hep.21472. [DOI] [PubMed] [Google Scholar]
- 220.Rigopoulou E.I., Davies E.T., Pares A., Zachou K., Liaskos C., Bogdanos D.P. Prevalence and clinical significance of isotype specific antinuclear antibodies in primary biliary cirrhosis. Gut. 2005;54:528–532. doi: 10.1136/gut.2003.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Itoh S., Ichida T., Yoshida T., Hayakawa A., Uchida M., Tashiro-Itoh T. Autoantibodies against a 210 kDa glycoprotein of the nuclear pore complex as a prognostic marker in patients with primary biliary cirrhosis. J Gastroenterol Hepatol. 1998;13:257–265. doi: 10.1111/j.1440-1746.1998.01553.x. [DOI] [PubMed] [Google Scholar]
- 222.Lassoued K., Guilly M.N., Andre C., Paintrand M., Dhumeaux D., Danon F. Autoantibodies to 200 kD polypeptide(s) of the nuclear envelope: a new serologic marker of primary biliary cirrhosis. Clin Exp Immunol. 1988;74:283–288. [PMC free article] [PubMed] [Google Scholar]
- 223.Muratori P., Muratori L., Ferrari R., Cassani F., Bianchi G., Lenzi M. Characterization and clinical impact of antinuclear antibodies in primary biliary cirrhosis. Am J Gastroenterol. 2003;98:431–437. doi: 10.1111/j.1572-0241.2003.07257.x. [DOI] [PubMed] [Google Scholar]
- 224.Wesierska-Gadek J., Penner E., Battezzati P.M., Selmi C., Zuin M., Hitchman E. Correlation of initial autoantibody profile and clinical outcome in primary biliary cirrhosis. Hepatology. 2006;43:1135–1144. doi: 10.1002/hep.21172. [DOI] [PubMed] [Google Scholar]
- 225.Yang W.H., Yu J.H., Nakajima A., Neuberg D., Lindor K., Bloch D.B. Do antinuclear antibodies in primary biliary cirrhosis patients identify increased risk for liver failure? Clin Gastroenterol Hepatol. 2004;2:1116–1122. doi: 10.1016/s1542-3565(04)00465-3. [DOI] [PubMed] [Google Scholar]
- 226.Duarte-Rey C., Bogdanos D., Yang C.Y., Roberts K., Leung P.S., Anaya J.M. Primary biliary cirrhosis and the nuclear pore complex. Autoimmun Rev. 2012;11:898–902. doi: 10.1016/j.autrev.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Duarte-Rey C., Bogdanos D.P., Leung P.S., Anaya J.M., Gershwin M.E. IgM predominance in autoimmune disease: genetics and gender. Autoimmun Rev. 2012;11:A404–A412. doi: 10.1016/j.autrev.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 228.Bach N., Schaffner F. Familial primary biliary cirrhosis. J Hepatol. 1994;20:698–701. doi: 10.1016/s0168-8278(05)80137-0. [DOI] [PubMed] [Google Scholar]
- 229.Floreani A., Naccarato R., Chiaramonte M. Prevalence of familial disease in primary biliary cirrhosis in Italy. J Hepatol. 1997;26:737–738. doi: 10.1016/s0168-8278(97)80444-8. [DOI] [PubMed] [Google Scholar]
- 230.Tsuji K., Watanabe Y., Van De Water J., Nakanishi T., Kajiyama G., Parikh-Patel A. Familial primary biliary cirrhosis in Hiroshima. J Autoimmun. 1999;13:171–178. doi: 10.1006/jaut.1999.0299. [DOI] [PubMed] [Google Scholar]
- 231.Lazaridis K.N., Juran B.D., Boe G.M., Slusser J.P., de Andrade M., Homburger H.A. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46:785–792. doi: 10.1002/hep.21749. [DOI] [PubMed] [Google Scholar]
- 232.Douglas J.G., Finlayson N.D. Are increased individual susceptibility and environmental factors both necessary for the development of primary biliary cirrhosis? BMJ. 1979;2:419–420. doi: 10.1136/bmj.2.6187.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fagan E., Williams R., Cox S. Primary biliary cirrhosis in mother and daughter. BMJ. 1977;2:1195. doi: 10.1136/bmj.2.6096.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Tong M.J., Nies K.M., Reynolds T.B., Quismorio F.P. Immunological studies in familial primary biliary cirrhosis. Gastroenterology. 1976;71:305–307. [PubMed] [Google Scholar]
- 235.Poupon R. Primary biliary cirrhosis: a 2010 update. J Hepatol. 2010;52:745–758. doi: 10.1016/j.jhep.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 236.Corpechot C., Carrat F., Poupon R., Poupon R.E. Primary biliary cirrhosis: incidence and predictive factors of cirrhosis development in ursodiol-treated patients. Gastroenterology. 2002;122:652–658. doi: 10.1053/gast.2002.31880. [DOI] [PubMed] [Google Scholar]
- 237.Smyk D.S., Rigopoulou E.I., Bogdanos D.P. Potential roles for infectious agents in the pathophysiology of primary biliary cirrhosis: what's new? Curr Infect Dis Rep. Nov 29 2012 doi: 10.1007/s11908-012-0304-2. [Electronic publication ahead of print] [DOI] [PubMed] [Google Scholar]
- 238.Chuang Y.H., Ridgway W.M., Ueno Y., Gershwin M.E. Animal models of primary biliary cirrhosis. Clin Liver Dis. 2008;12:333–347. doi: 10.1016/j.cld.2008.02.011. [ix] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Hirschfield G.M., Invernizzi P. Progress in the genetics of primary biliary cirrhosis. Semin Liver Dis. 2011;31:147–156. doi: 10.1055/s-0031-1276644. [DOI] [PubMed] [Google Scholar]
- 240.Lleo A., Invernizzi P., Mackay I.R., Prince H., Zhong R.Q., Gershwin M.E. Etiopathogenesis of primary biliary cirrhosis. World J Gastroenterol. 2008;14:3328–3337. doi: 10.3748/wjg.14.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Selmi C., Zuin M., Gershwin M.E. The unfinished business of primary biliary cirrhosis. J Hepatol. 2008;49:451–460. doi: 10.1016/j.jhep.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wu S.J., Yang Y.H., Tsuneyama K., Leung P.S., Illarionov P., Gershwin M.E. Innate immunity and primary biliary cirrhosis: activated invariant natural killer T cells exacerbate murine autoimmune cholangitis and fibrosis. Hepatology. 2011;53:915–925. doi: 10.1002/hep.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Hirschfield G.M., Gershwin M.E. Primary biliary cirrhosis: one disease with many faces. Isr Med Assoc J. 2011;13:55–59. [PubMed] [Google Scholar]
- 244.Invernizzi P. Human leukocyte antigen in primary biliary cirrhosis: an old story now reviving. Hepatology. 2011;2:714–723. doi: 10.1002/hep.24414. [PMID: 21563204] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hemminki K., Li X., Sundquist K., Sundquist J. Shared familial aggregation of susceptibility to autoimmune diseases. Arthritis Rheum. 2009;60:2845–2847. doi: 10.1002/art.24749. [DOI] [PubMed] [Google Scholar]
- 246.Hirschfield G.M., Liu X., Han Y., Gorlov I.P., Lu Y., Xu C. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet. 2010;42:655–657. doi: 10.1038/ng.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hirschfield G.M., Liu X., Xu C., Lu Y., Xie G., Gu X. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. 2009;360:2544–2555. doi: 10.1056/NEJMoa0810440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Liu X., Invernizzi P., Lu Y., Kosoy R., Bianchi I., Podda M. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet. 2010;42:658–660. doi: 10.1038/ng.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Mells G.F., Floyd J.A., Morley K.I., Cordell H.J., Franklin C.S., Shin S.Y. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43:329–332. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Tanaka A., Invernizzi P., Ohira H., Kikuchi K., Nezu S., Kosoy R. Replicated association of 17q12-21 with susceptibility of primary biliary cirrhosis in a Japanese cohort. Tissue Antigens. 2011;78:65–68. doi: 10.1111/j.1399-0039.2011.01684.x. [DOI] [PubMed] [Google Scholar]
- 251.Tanaka A., Ohira H., Kikuchi K., Nezu S., Shibuya A., Bianchi I. Genetic association of Fc receptor-like 3 polymorphisms with susceptibility to primary biliary cirrhosis: ethnic comparative study in Japanese and Italian patients. Tissue Antigens. 2011;77:239–243. doi: 10.1111/j.1399-0039.2010.01600.x. [DOI] [PubMed] [Google Scholar]
- 252.Invernizzi P., Miozzo M., Battezzati P.M., Bianchi I., Grati F.R., Simoni G. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet. 2004;363:533–535. doi: 10.1016/S0140-6736(04)15541-4. [DOI] [PubMed] [Google Scholar]
- 253.Invernizzi P., Pasini S., Selmi C., Gershwin M.E., Podda M. Female predominance and X chromosome defects in autoimmune diseases. J Autoimmun. 2009;33:12–16. doi: 10.1016/j.jaut.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 254.Miozzo M., Selmi C., Gentilin B., Grati F.R., Sirchia S., Oertelt S. Preferential X chromosome loss but random inactivation characterize primary biliary cirrhosis. Hepatology. 2007;46:456–462. doi: 10.1002/hep.21696. [DOI] [PubMed] [Google Scholar]
- 255.Invernizzi P., Selmi C., Poli F., Frison S., Floreani A., Alvaro D. Human leukocyte antigen polymorphisms in Italian primary biliary cirrhosis: a multicenter study of 664 patients and 1992 healthy controls. Hepatology. 2008;48:1906–1912. doi: 10.1002/hep.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Bogdanos D.P., Baum H., Butler P., Rigopoulou E.I., Davies E.T., Ma Y. Association between the primary biliary cirrhosis specific anti-sp100 antibodies and recurrent urinary tract infection. Dig Liver Dis. 2003;35:801–805. doi: 10.1016/s1590-8658(03)00466-3. [DOI] [PubMed] [Google Scholar]
- 257.Bogdanos D.P., Baum H., Grasso A., Okamoto M., Butler P., Ma Y. Microbial mimics are major targets of crossreactivity with human pyruvate dehydrogenase in primary biliary cirrhosis. J Hepatol. 2004;40:31–39. doi: 10.1016/s0168-8278(03)00501-4. [DOI] [PubMed] [Google Scholar]
- 258.Bogdanos D.P., Pares A., Rodes J., Vergani D. Primary biliary cirrhosis specific antinuclear antibodies in patients from Spain. Am J Gastroenterol. 2004;99:763–764. doi: 10.1111/j.1572-0241.2004.04119.x. [author reply 5] [DOI] [PubMed] [Google Scholar]
- 259.Bogdanos D.P., Baum H., Okamoto M., Montalto P., Sharma U.C., Rigopoulou E.I. Primary biliary cirrhosis is characterized by IgG3 antibodies cross-reactive with the major mitochondrial autoepitope and its Lactobacillus mimic. Hepatology. 2005;42:458–465. doi: 10.1002/hep.20788. [DOI] [PubMed] [Google Scholar]
- 260.Bogdanos D.P., Baum H., Sharma U.C., Grasso A., Ma Y., Burroughs A.K. Antibodies against homologous microbial caseinolytic proteases P characterise primary biliary cirrhosis. J Hepatol. 2002;36:14–21. doi: 10.1016/s0168-8278(01)00252-5. [DOI] [PubMed] [Google Scholar]
- 261.Fussey S.P., Lindsay J.G., Fuller C., Perham R.N., Dale S., James O.F. Autoantibodies in primary biliary cirrhosis: analysis of reactivity against eukaryotic and prokaryotic 2-oxo acid dehydrogenase complexes. Hepatology. 1991;13:467–474. [PubMed] [Google Scholar]
- 262.Koutsoumpas A., Mytilinaiou M., Polymeros D., Dalekos G.N., Bogdanos D.P. Anti-Helicobacter pylori antibody responses specific for VacA do not trigger primary biliary cirrhosis-specific antimitochondrial antibodies. Eur J Gastroenterol Hepatol. 2009;21:1220. doi: 10.1097/MEG.0b013e32831a4807. [DOI] [PubMed] [Google Scholar]
- 263.Abdulkarim A.S., Petrovic L.M., Kim W.R., Angulo P., Lloyd R.V., Lindor K.D. Primary biliary cirrhosis: an infectious disease caused by Chlamydia pneumoniae? J Hepatol. 2004;40:380–384. doi: 10.1016/j.jhep.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 264.Burroughs A.K., Butler P., Sternberg M.J., Baum H. Molecular mimicry in liver disease. Nature. 1992;358:377–378. doi: 10.1038/358377a0. [DOI] [PubMed] [Google Scholar]
- 265.Burroughs A.K., Rosenstein I.J., Epstein O., Hamilton-Miller J.M., Brumfitt W., Sherlock S. Bacteriuria and primary biliary cirrhosis. Gut. 1984;25:133–137. doi: 10.1136/gut.25.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Butler P., Hamilton-Miller J., Baum H., Burroughs A.K. Detection of M2 antibodies in patients with recurrent urinary tract infection using an ELISA and purified PBC specific antigens. Evidence for a molecular mimicry mechanism in the pathogenesis of primary biliary cirrhosis? Biochem Mol Biol Int. 1995;35:473–485. [PubMed] [Google Scholar]
- 267.Butler P., Hamilton-Miller J.M., McIntyre N., Burroughs A.K. Natural history of bacteriuria in women with primary biliary cirrhosis and the effect of antimicrobial therapy in symptomatic and asymptomatic groups. Gut. 1995;36:931–934. doi: 10.1136/gut.36.6.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Butler P., Valle F., Hamilton-Miller J.M., Brumfitt W., Baum H., Burroughs A.K. M2 mitochondrial antibodies and urinary rough mutant bacteria in patients with primary biliary cirrhosis and in patients with recurrent bacteriuria. J Hepatol. 1993;17:408–414. doi: 10.1016/s0168-8278(05)80225-9. [DOI] [PubMed] [Google Scholar]
- 269.Donaldson P.T. Genetics of liver disease: immunogenetics and disease pathogenesis. Gut. 2004;53:599–608. doi: 10.1136/gut.2003.031732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Floreani A., Bassendine M.F., Mitchison H., Freeman R., James O.F. No specific association between primary biliary cirrhosis and bacteriuria? J Hepatol. 1989;8:201–207. doi: 10.1016/0168-8278(89)90008-1. [DOI] [PubMed] [Google Scholar]
- 271.Leung P.S., Park O., Matsumura S., Ansari A.A., Coppel R.L., Gershwin M.E. Is there a relation between Chlamydia infection and primary biliary cirrhosis? Clin Dev Immunol. 2003;10:227–233. doi: 10.1080/10446670310001642429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Mason A., Xu L., Shen Z., Fodera B., Joplin R., Neuberger J. Patients with primary biliary cirrhosis make anti-viral and anti-mitochondrial antibodies to mouse mammary tumor virus. Gastroenterology. 2004;127:1863–1864. doi: 10.1053/j.gastro.2004.10.024. [author reply 4–5] [DOI] [PubMed] [Google Scholar]
- 273.McNally R.J., Ducker S., James O.F. Are transient environmental agents involved in the cause of primary biliary cirrhosis? Evidence from space–time clustering analysis. Hepatology. 2009;50:1169–1174. doi: 10.1002/hep.23139. [DOI] [PubMed] [Google Scholar]
- 274.Selmi C., Balkwill D.L., Invernizzi P., Ansari A.A., Coppel R.L., Podda M. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology. 2003;38:1250–1257. doi: 10.1053/jhep.2003.50446. [DOI] [PubMed] [Google Scholar]
- 275.Smyk D., Mytilinaiou M.G., Rigopoulou E.I., Bogdanos D.P. PBC triggers in water reservoirs, coal mining areas and waste disposal sites: from Newcastle to New York. Dis Markers. 2010;29:337–344. doi: 10.3233/DMA-2010-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Xu L., Sakalian M., Shen Z., Loss G., Neuberger J., Mason A. Cloning the human betaretrovirus proviral genome from patients with primary biliary cirrhosis. Hepatology. 2004;39:151–156. doi: 10.1002/hep.20024. [DOI] [PubMed] [Google Scholar]
- 277.Xu L., Shen Z., Guo L., Fodera B., Keogh A., Joplin R. Does a betaretrovirus infection trigger primary biliary cirrhosis? Proc Natl Acad Sci U S A. 2003;100:8454–8459. doi: 10.1073/pnas.1433063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Selmi C., De Santis M., Cavaciocchi F., Gershwin M.E. Infectious agents and xenobiotics in the etiology of primary biliary cirrhosis. Dis Markers. 2010;29:287–299. doi: 10.3233/DMA-2010-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Bogdanos D., Pusl T., Rust C., Vergani D., Beuers U. Primary biliary cirrhosis following Lactobacillus vaccination for recurrent vaginitis. J Hepatol. 2008;49:466–473. doi: 10.1016/j.jhep.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 280.Oldstone M.B. Molecular mimicry and autoimmune disease. Cell. 1987;50:819–820. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- 281.Bogdanos D.P., Choudhuri K., Vergani D. Molecular mimicry and autoimmune liver disease: virtuous intentions, malign consequences. Liver. 2001;21:225–232. doi: 10.1034/j.1600-0676.2001.021004225.x. [DOI] [PubMed] [Google Scholar]
- 282.Vergani D., Bogdanos D.P., Baum H. Unusual suspects in primary biliary cirrhosis. Hepatology. 2004;39:38–41. doi: 10.1002/hep.20028. [DOI] [PubMed] [Google Scholar]
- 283.Rigopoulou E.I., Smyk D.S., Matthews C.E., Billinis C., Burroughs A.K., Lenzi M. Epstein–Barr virus as a trigger of autoimmune liver diseases. Adv Virol. 2012;2012:987471. doi: 10.1155/2012/987471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Vergani D., Choudhuri K., Bogdanos D.P., Mieli-Vergani G. Pathogenesis of autoimmune hepatitis. Clin Liver Dis. 2002;6:727–737. doi: 10.1016/s1089-3261(02)00018-1. [DOI] [PubMed] [Google Scholar]
- 285.Vergani D., Longhi M.S., Bogdanos D.P., Ma Y., Mieli-Vergani G. Autoimmune hepatitis. Semin Immunopathol. 2009;31:421–435. doi: 10.1007/s00281-009-0170-7. [DOI] [PubMed] [Google Scholar]
- 286.Ahn J., Yang L., Paster B.J., Ganly I., Morris L., Pei Z. Oral microbiome profiles: 16S rRNA pyrosequencing and microarray assay comparison. PLoS One. 2011;6:e22788. doi: 10.1371/journal.pone.0022788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Arumugam M., Raes J., Pelletier E., Le Paslier D., Yamada T., Mende D.R. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Gill S.R., Pop M., Deboy R.T., Eckburg P.B., Turnbaugh P.J., Samuel B.S. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kurokawa K., Itoh T., Kuwahara T., Oshima K., Toh H., Toyoda A. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Marchesi J.R. Human distal gut microbiome. Environ Microbiol. 2011 doi: 10.1111/j.1462-2920.2011.02574.x. [DOI] [PubMed] [Google Scholar]
- 291.Qin J., Li R., Raes J., Arumugam M., Burgdorf K.S., Manichanh C. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Saulnier D.M., Riehle K., Mistretta T.A., Diaz M.A., Mandal D., Raza S. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Siezen R.J., Kleerebezem M. The human gut microbiome: are we our enterotypes? Microb Biotechnol. 2011;4:550–553. doi: 10.1111/j.1751-7915.2011.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Turnbaugh P.J., Hamady M., Yatsunenko T., Cantarel B.L., Duncan A., Ley R.E. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Nasidze I., Li J., Schroeder R., Creasey J.L., Li M., Stoneking M. High diversity of the saliva microbiome in Batwa pygmies. PLoS One. 2011;6:e23352. doi: 10.1371/journal.pone.0023352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Smyk D.S., Bogdanos D.P., Kriese S., Billinis C., Burroughs A.K., Rigopoulou E.I. Urinary tract infection as a risk factor for autoimmune liver disease: from bench to bedside. Clin Res Hepatol Gastroenterol. 2011 doi: 10.1016/j.clinre.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 297.Varyani F.K., West J., Card T.R. An increased risk of urinary tract infection precedes development of primary biliary cirrhosis. BMC Gastroenterol. 2011;11:95. doi: 10.1186/1471-230X-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Smyk D., Rigopoulou E.I., Baum H., Burroughs A.K., Vergani D., Bogdanos D.P. Autoimmunity and environment: am I at risk? Clin Rev Allergy Immunol. 2011 doi: 10.1007/s12016-011-8259-x. [DOI] [PubMed] [Google Scholar]
- 299.Natesan M., Ulrich R.G. Protein microarrays and biomarkers of infectious disease. Int J Mol Sci. 2010;11:5165–5183. doi: 10.3390/ijms11125165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Quintana F.J., Hagedorn P.H., Elizur G., Merbl Y., Domany E., Cohen I.R. Functional immunomics: microarray analysis of IgG autoantibody repertoires predicts the future response of mice to induced diabetes. Proc Natl Acad Sci U S A. 2004;101(Suppl. 2):14615–14621. doi: 10.1073/pnas.0404848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Larman H.B., Zhao Z., Laserson U., Li M.Z., Ciccia A., Gakidis M.A. Autoantigen discovery with a synthetic human peptidome. Nat Biotechnol. 2011;29:535–541. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Drebber U., Kasper H.U., Ratering J., Wedemeyer I., Schirmacher P., Dienes H.P. Hepatic granulomas: histological and molecular pathological approach to differential diagnosis—a study of 442 cases. Liver Int. 2008;28:828–834. doi: 10.1111/j.1478-3231.2008.01695.x. [DOI] [PubMed] [Google Scholar]
- 303.O'Donohue J., Fidler H., Garcia-Barcelo M., Nouri-Aria K., Williams R., McFadden J. Mycobacterial DNA not detected in liver sections from patients with primary biliary cirrhosis. J Hepatol. 1998;28:433–438. doi: 10.1016/s0168-8278(98)80317-6. [DOI] [PubMed] [Google Scholar]
- 304.Tanaka A., Prindiville T.P., Gish R., Solnick J.V., Coppel R.L., Keeffe E.B. Are infectious agents involved in primary biliary cirrhosis? A PCR approach. J Hepatol. 1999;31:664–671. doi: 10.1016/s0168-8278(99)80346-8. [DOI] [PubMed] [Google Scholar]
- 305.Vilagut L., Pares A., Rodes J., Vila J., Vinas O., Gines A. Mycobacteria—related to the aetiopathogenesis of primary biliary cirrhosis? J Hepatol. 1996;24:125. doi: 10.1016/s0168-8278(96)80198-x. [DOI] [PubMed] [Google Scholar]
- 306.Chen E.C., Miller S.A., DeRisi J.L., Chiu C.Y. Using a pan-viral microarray assay (Virochip) to screen clinical samples for viral pathogens. J Vis Exp. 2011 doi: 10.3791/2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kistler A., Avila P.C., Rouskin S., Wang D., Ward T., Yagi S. Pan-viral screening of respiratory tract infections in adults with and without asthma reveals unexpected human coronavirus and human rhinovirus diversity. J Infect Dis. 2007;196:817–825. doi: 10.1086/520816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Nakamura S., Nakaya T., Iida T. Metagenomic analysis of bacterial infections by means of high-throughput DNA sequencing. Exp Biol Med (Maywood) 2011;236:968–971. doi: 10.1258/ebm.2011.010378. [DOI] [PubMed] [Google Scholar]
- 309.Tang P., Chiu C. Metagenomics for the discovery of novel human viruses. Future Microbiol. 2010;5:177–189. doi: 10.2217/fmb.09.120. [DOI] [PubMed] [Google Scholar]
- 310.Moore R.A., Warren R.L., Freeman J.D., Gustavsen J.A., Chenard C., Friedman J.M. The sensitivity of massively parallel sequencing for detecting candidate infectious agents associated with human tissue. PLoS One. 2011;6:e19838. doi: 10.1371/journal.pone.0019838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Broome U., Scheynius A., Hultcrantz R. Induced expression of heat-shock protein on biliary epithelium in patients with primary sclerosing cholangitis and primary biliary cirrhosis. Hepatology. 1993;18:298–303. doi: 10.1002/hep.1840180212. [DOI] [PubMed] [Google Scholar]
- 312.Singhal S., Dian D., Keshavarzian A., Fogg L., Fields J.Z., Farhadi A. The role of oral hygiene in inflammatory bowel disease. Dig Dis Sci. 2011;56:170–175. doi: 10.1007/s10620-010-1263-9. [DOI] [PubMed] [Google Scholar]
- 313.Ochoa-Reparaz J., Mielcarz D.W., Ditrio L.E., Burroughs A.R., Foureau D.M., Haque-Begum S. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009;183:6041–6050. doi: 10.4049/jimmunol.0900747. [DOI] [PubMed] [Google Scholar]
- 314.Helenius L.M., Meurman J.H., Helenius I., Kari K., Hietanen J., Suuronen R. Oral and salivary parameters in patients with rheumatic diseases. Acta Odontol Scand. 2005;63:284–293. doi: 10.1080/00016350510020043. [DOI] [PubMed] [Google Scholar]
- 315.Mason A.L., Xu L., Guo L., Munoz S., Jaspan J.B., Bryer-Ash M. Detection of retroviral antibodies in primary biliary cirrhosis and other idiopathic biliary disorders. Lancet. 1998;351:1620–1624. doi: 10.1016/S0140-6736(97)10290-2. [DOI] [PubMed] [Google Scholar]
- 316.Alvaro D., Alpini G., Onori P., Franchitto A., Glaser S.S., Le Sage G. Alfa and beta estrogen receptors and the biliary tree. Mol Cell Endocrinol. 2002;193:105–108. doi: 10.1016/s0303-7207(02)00103-x. [DOI] [PubMed] [Google Scholar]
- 317.Cunningham M., Gilkeson G. Estrogen receptors in immunity and autoimmunity. Clin Rev Allergy Immunol. 2011;40:66–73. doi: 10.1007/s12016-010-8203-5. [DOI] [PubMed] [Google Scholar]
- 318.Duvic M., Steinberg A.D., Klassen L.W. Effect of the anti-estrogen, Nafoxidine, on NZB/W autoimmune disease. Arthritis Rheum. 1978;21:414–417. doi: 10.1002/art.1780210403. [DOI] [PubMed] [Google Scholar]
- 319.Holmdahl R. Estrogen exaggerates lupus but suppresses T-cell-dependent autoimmune disease. J Autoimmun. 1989;2:651–656. doi: 10.1016/s0896-8411(89)80004-6. [DOI] [PubMed] [Google Scholar]
- 320.Walker S.E. Estrogen and autoimmune disease. Clin Rev Allergy Immunol. 2011;40:60–65. doi: 10.1007/s12016-010-8199-x. [DOI] [PubMed] [Google Scholar]
- 321.Mizutani T., Shinoda M., Tanaka Y., Kuno T., Hattori A., Usui T. Autoantibodies against CYP2D6 and other drug-metabolizing enzymes in autoimmune hepatitis type 2. Drug Metab Rev. 2005;37:235–252. doi: 10.1081/dmr-200028798. [DOI] [PubMed] [Google Scholar]
- 322.Obermayer-Straub P., Strassburg C.P., Manns M.P. Autoimmune hepatitis. J Hepatol. 2000;32:181–197. doi: 10.1016/s0168-8278(00)80425-0. [DOI] [PubMed] [Google Scholar]
- 323.Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009:73–79. doi: 10.1182/asheducation-2009.1.73. [DOI] [PubMed] [Google Scholar]
- 324.Khokhar O., Gange C., Clement S., Lewis J. Autoimmune hepatitis and thyroiditis associated with rifampin and pyrazinamide prophylaxis: an unusual reaction. Dig Dis Sci. 2005;50:207–211. doi: 10.1007/s10620-005-1302-0. [DOI] [PubMed] [Google Scholar]
- 325.Takasu N., Takara M., Komiya I. Rifampin-induced hypothyroidism in patients with Hashimoto's thyroiditis. N Engl J Med. 2005;352:518–519. doi: 10.1056/NEJM200502033520524. [DOI] [PubMed] [Google Scholar]
- 326.Ahrens N., Genth R., Salama A. Belated diagnosis in three patients with rifampicin-induced immune haemolytic anaemia. Br J Haematol. 2002;117:441–443. doi: 10.1046/j.1365-2141.2002.03416.x. [DOI] [PubMed] [Google Scholar]
- 327.Heurgue-Berlot A., Bernard-Chabert B., Diebold M.D., Thiefin G. Drug-induced autoimmune-like hepatitis: a case of chronic course after drug withdrawal. Dig Dis Sci. 2011;56:2504–2505. doi: 10.1007/s10620-011-1786-8. [author reply 5] [DOI] [PubMed] [Google Scholar]
- 328.Bjornsson E., Talwalkar J., Treeprasertsuk S., Kamath P.S., Takahashi N., Sanderson S. Drug-induced autoimmune hepatitis: clinical characteristics and prognosis. Hepatology. 2010;51:2040–2048. doi: 10.1002/hep.23588. [DOI] [PubMed] [Google Scholar]
- 329.Geddes M.R., Sinnreich M., Chalk C. Minocycline-induced dermatomyositis. Muscle Nerve. 2010;41:547–549. doi: 10.1002/mus.21487. [DOI] [PubMed] [Google Scholar]
- 330.Angulo J.M., Sigal L.H., Espinoza L.R. Minocycline induced lupus and autoimmune hepatitis. J Rheumatol. 1999;26:1420–1421. [PubMed] [Google Scholar]
- 331.Bachmeyer C., Cadranel J.F. Minocycline-induced lupus and autoimmune hepatitis: family autoimmune disorders as possible risk factors. Dermatology. 2002;205:185–186. doi: 10.1159/000063890. [DOI] [PubMed] [Google Scholar]
- 332.Bhat G., Jordan J., Jr., Sokalski S., Bajaj V., Marshall R., Berkelhammer C. Minocycline-induced hepatitis with autoimmune features and neutropenia. J Clin Gastroenterol. 1998;27:74–75. doi: 10.1097/00004836-199807000-00016. [DOI] [PubMed] [Google Scholar]
- 333.Chamberlain M.C., Schwarzenberg S.J., Akin E.U., Kurth M.H. Minocycline-induced autoimmune hepatitis with subsequent cirrhosis. J Pediatr Gastroenterol Nutr. 2006;42:232–235. doi: 10.1097/01.mpg.0000184923.47507.ae. [DOI] [PubMed] [Google Scholar]
- 334.Colmegna I., Perandones C.E., Chaves J.G. Minocycline induced lupus and autoimmune hepatitis. J Rheumatol. 2000;27:1567–1568. [PubMed] [Google Scholar]
- 335.Gough A., Chapman S., Wagstaff K., Emery P., Elias E. Minocycline induced autoimmune hepatitis and systemic lupus erythematosus-like syndrome. BMJ. 1996;312:169–172. doi: 10.1136/bmj.312.7024.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Healy J., Alexander B., Eapen C., Roberts-Thomson I.C. Minocycline-induced autoimmune hepatitis. Intern Med J. 2009;39:487–488. doi: 10.1111/j.1445-5994.2009.01971.x. [DOI] [PubMed] [Google Scholar]
- 337.Della Corte C., Carlucci A., Francalanci P., Alisi A., Nobili V. Autoimmune hepatitis type 2 following anti-papillomavirus vaccination in a 11-year-old girl. Vaccine. 2011;29:4654–4656. doi: 10.1016/j.vaccine.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 338.Karali Z., Basaranoglu S.T., Karali Y., Oral B., Kilic S.S. Autoimmunity and hepatitis A vaccine in children. J Investig Allergol Clin Immunol. 2011;21:389–393. [PubMed] [Google Scholar]
- 339.Stubgen J.P. Neuromuscular disorders associated with hepatitis B vaccination. J Neurol Sci. 2010;292:1–4. doi: 10.1016/j.jns.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 340.Cacoub P., Terrier B. Hepatitis B-related autoimmune manifestations. Rheum Dis Clin North Am. 2009;35:125–137. doi: 10.1016/j.rdc.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 341.Aron-Maor A., Shoenfeld Y. Vaccination and systemic lupus erythematosus: the bidirectional dilemmas. Lupus. 2001;10:237–240. doi: 10.1191/096120301673085478. [DOI] [PubMed] [Google Scholar]
- 342.Borchers A.T., Keen C.L., Shoenfeld Y., Silva J., Jr., Gershwin M.E. Vaccines, viruses, and voodoo. J Investig Allergol Clin Immunol. 2002;12:155–168. [PubMed] [Google Scholar]
- 343.Chen R.T., Pless R., Destefano F. Epidemiology of autoimmune reactions induced by vaccination. J Autoimmun. 2001;16:309–318. doi: 10.1006/jaut.2000.0491. [DOI] [PubMed] [Google Scholar]
- 344.Cohen A.D., Shoenfeld Y. Vaccine-induced autoimmunity. J Autoimmun. 1996;9:699–703. doi: 10.1006/jaut.1996.0091. [DOI] [PubMed] [Google Scholar]
- 345.Nadler J.P. Multiple sclerosis and hepatitis B vaccination. Clin Infect Dis. 1993;17:928–929. doi: 10.1093/clinids/17.5.928-b. [DOI] [PubMed] [Google Scholar]
- 346.Ravel G., Christ M., Horand F., Descotes J. Autoimmunity, environmental exposure and vaccination: is there a link? Toxicology. 2004;196:211–216. doi: 10.1016/j.tox.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 347.Shoenfeld Y., Aharon-Maor A., Sherer Y. Vaccination as an additional player in the mosaic of autoimmunity. Clin Exp Rheumatol. 2000;18:181–184. [PubMed] [Google Scholar]
- 348.Shoenfeld Y., Aron-Maor A. Vaccination and autoimmunity-‘vaccinosis’: a dangerous liaison? J Autoimmun. 2000;14:1–10. doi: 10.1006/jaut.1999.0346. [DOI] [PubMed] [Google Scholar]
- 349.Shoenfeld Y., Aron-Maor A., Tanai A., Ehrenfeld M. Bcg and autoimmunity: another two-edged sword. J Autoimmun. 2001;16:235–240. doi: 10.1006/jaut.2000.0494. [DOI] [PubMed] [Google Scholar]
- 350.Costenbader K.H., Gay S., Riquelme M.E., Iaccarino L., Doria A. Genes, epigenetic regulation and environmental factors: which is the most relevant in developing autoimmune diseases? Autoimmun Rev. 2011 doi: 10.1016/j.autrev.2011.10.022. [DOI] [PubMed] [Google Scholar]
- 351.Arnson Y., Shoenfeld Y., Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun. 2010;34:J258–J265. doi: 10.1016/j.jaut.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 352.Brusselle G.G., Joos G.F., Bracke K.R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378:1015–1026. doi: 10.1016/S0140-6736(11)60988-4. [DOI] [PubMed] [Google Scholar]
- 353.Simard J.F., Costenbader K.H. What can epidemiology tell us about systemic lupus erythematosus? Int J Clin Pract. 2007;61:1170–1180. doi: 10.1111/j.1742-1241.2007.01434.x. [DOI] [PubMed] [Google Scholar]
- 354.Costenbader K.H., Karlson E.W. Cigarette smoking and autoimmune disease: what can we learn from epidemiology? Lupus. 2006;15:737–745. doi: 10.1177/0961203306069344. [DOI] [PubMed] [Google Scholar]
- 355.Rubin R.L., Hermanson T.M., Bedrick E.J., McDonald J.D., Burchiel S.W., Reed M.D. Effect of cigarette smoke on autoimmunity in murine and human systemic lupus erythematosus. Toxicol Sci. 2005;87:86–96. doi: 10.1093/toxsci/kfi217. [DOI] [PubMed] [Google Scholar]
- 356.Asherson R.A., Shoenfeld Y., Jacobs P., Bosman C. An unusually complicated case of primary Sjogren's syndrome: development of transient “lupus-type” autoantibodies following silicone implant rejection. J Rheumatol. 2004;31:196–197. [PubMed] [Google Scholar]
- 357.Bar-Meir E., Eherenfeld M., Shoenfeld Y. Silicone gel breast implants and connective tissue disease—a comprehensive review. Autoimmunity. 2003;36:193–197. doi: 10.1080/08916931000148818. [DOI] [PubMed] [Google Scholar]
- 358.Bar-Meir E., Teuber S.S., Lin H.C., Alosacie I., Goddard G., Terybery J. Multiple autoantibodies in patients with silicone breast implants. J Autoimmun. 1995;8:267–277. doi: 10.1006/jaut.1995.0020. [DOI] [PubMed] [Google Scholar]
- 359.Vasey F.B., Zarabadi S.A., Seleznick M., Ricca L. Where there's smoke there's fire: the silicone breast implant controversy continues to flicker: a new disease that needs to be defined. J Rheumatol. 2003;30:2092–2094. [PubMed] [Google Scholar]
- 360.Vermeulen R.C., Scholte H.R. Rupture of silicone gel breast implants and symptoms of pain and fatigue. J Rheumatol. 2003;30:2263–2267. [PubMed] [Google Scholar]
- 361.Zandman-Goddard G., Blank M., Ehrenfeld M., Gilburd B., Peter J., Shoenfeld Y. A comparison of autoantibody production in asymptomatic and symptomatic women with silicone breast implants. J Rheumatol. 1999;26:73–77. [PubMed] [Google Scholar]
- 362.Hajdu S.D., Agmon-Levin N., Shoenfeld Y. Silicone and autoimmunity. Eur J Clin Invest. 2011;41:203–211. doi: 10.1111/j.1365-2362.2010.02389.x. [DOI] [PubMed] [Google Scholar]
- 363.Alotaibi S., Kennedy J., Tellier R., Stephens D., Banwell B. Epstein–Barr virus in pediatric multiple sclerosis. JAMA. 2004;291:1875–1879. doi: 10.1001/jama.291.15.1875. [DOI] [PubMed] [Google Scholar]
- 364.DeLorenze G.N., Munger K.L., Lennette E.T., Orentreich N., Vogelman J.H., Ascherio A. Epstein–Barr virus and multiple sclerosis: evidence of association from a prospective study with long-term follow-up. Arch Neurol. 2006;63:839–844. doi: 10.1001/archneur.63.6.noc50328. [DOI] [PubMed] [Google Scholar]
- 365.Goodin D.S. The causal cascade to multiple sclerosis: a model for MS pathogenesis. PLoS One. 2009;4:e4565. doi: 10.1371/journal.pone.0004565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Levin L.I., Munger K.L., O'Reilly E.J., Falk K.I., Ascherio A. Primary infection with the Epstein–Barr virus and risk of multiple sclerosis. Ann Neurol. 2010;67:824–830. doi: 10.1002/ana.21978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Levin L.I., Munger K.L., Rubertone M.V., Peck C.A., Lennette E.T., Spiegelman D. Temporal relationship between elevation of Epstein–Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA. 2005;293:2496–2500. doi: 10.1001/jama.293.20.2496. [DOI] [PubMed] [Google Scholar]
- 368.Lindberg C., Andersen O., Vahlne A., Dalton M., Runmarker B. Epidemiological investigation of the association between infectious mononucleosis and multiple sclerosis. Neuroepidemiology. 1991;10:62–65. doi: 10.1159/000110248. [DOI] [PubMed] [Google Scholar]
- 369.Lunemann J.D., Edwards N., Muraro P.A., Hayashi S., Cohen J.I., Munz C. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain. 2006;129:1493–1506. doi: 10.1093/brain/awl067. [DOI] [PubMed] [Google Scholar]
- 370.Lunemann J.D., Huppke P., Roberts S., Bruck W., Gartner J., Munz C. Broadened and elevated humoral immune response to EBNA1 in pediatric multiple sclerosis. Neurology. 2008;71:1033–1035. doi: 10.1212/01.wnl.0000326576.91097.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Lunemann J.D., Jelcic I., Roberts S., Lutterotti A., Tackenberg B., Martin R. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med. 2008;205:1763–1773. doi: 10.1084/jem.20072397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Operskalski E.A., Visscher B.R., Malmgren R.M., Detels R. A case–control study of multiple sclerosis. Neurology. 1989;39:825–829. doi: 10.1212/wnl.39.6.825. [DOI] [PubMed] [Google Scholar]
- 373.Peferoen L.A., Lamers F., Lodder L.N., Gerritsen W.H., Huitinga I., Melief J. Epstein Barr virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain. 2010;133:e137. doi: 10.1093/brain/awp296. [DOI] [PubMed] [Google Scholar]
- 374.Sargsyan S.A., Shearer A.J., Ritchie A.M., Burgoon M.P., Anderson S., Hemmer B. Absence of Epstein–Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology. 2010;74:1127–1135. doi: 10.1212/WNL.0b013e3181d865a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Serafini B., Rosicarelli B., Franciotta D., Magliozzi R., Reynolds R., Cinque P. Dysregulated Epstein–Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899–2912. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Thorley-Lawson D.A. Epstein–Barr virus: exploiting the immune system. Nat Rev Immunol. 2001;1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 377.Warner H.B., Carp R.I. Multiple sclerosis and Epstein–Barr virus. Lancet. 1981;2:1290. doi: 10.1016/s0140-6736(81)91527-0. [DOI] [PubMed] [Google Scholar]
- 378.Willis S.N., Stadelmann C., Rodig S.J., Caron T., Gattenloehner S., Mallozzi S.S. Epstein–Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain. 2009;132:3318–3328. doi: 10.1093/brain/awp200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Sriram S., Stratton C.W., Yao S., Tharp A., Ding L., Bannan J.D. Chlamydia pneumoniae infection of the central nervous system in multiple sclerosis. Ann Neurol. 1999;46:6–14. [PubMed] [Google Scholar]
- 380.Challoner P.B., Smith K.T., Parker J.D., MacLeod D.L., Coulter S.N., Rose T.M. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A. 1995;92:7440–7444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Dockrell D.H., Smith T.F., Paya C.V. Human herpesvirus 6. Mayo Clin Proc. 1999;74:163–170. doi: 10.4065/74.2.163. [DOI] [PubMed] [Google Scholar]
- 382.Chmielewska-Badora J., Cisak E., Dutkiewicz J. Lyme borreliosis and multiple sclerosis: any connection? A seroepidemic study. Ann Agric Environ Med. 2000;7:141–143. [PubMed] [Google Scholar]
- 383.Mancuso R., Delbue S., Borghi E., Pagani E., Calvo M.G., Caputo D. Increased prevalence of varicella zoster virus DNA in cerebrospinal fluid from patients with multiple sclerosis. J Med Virol. 2007;79:192–199. doi: 10.1002/jmv.20777. [DOI] [PubMed] [Google Scholar]
- 384.Ordonez G., Pineda B., Garcia-Navarrete R., Sotelo J. Brief presence of varicella-zoster viral DNA in mononuclear cells during relapses of multiple sclerosis. Arch Neurol. 2004;61:529–532. doi: 10.1001/archneur.61.4.529. [DOI] [PubMed] [Google Scholar]
- 385.Sotelo J., Martinez-Palomo A., Ordonez G., Pineda B. Varicella-zoster virus in cerebrospinal fluid at relapses of multiple sclerosis. Ann Neurol. 2008;63:303–311. doi: 10.1002/ana.21316. [DOI] [PubMed] [Google Scholar]
- 386.Birnbaum G., Kotilinek L., Albrecht L. Spinal fluid lymphocytes from a subgroup of multiple sclerosis patients respond to mycobacterial antigens. Ann Neurol. 1993;34:18–24. doi: 10.1002/ana.410340106. [DOI] [PubMed] [Google Scholar]
- 387.Wroblewska Z., Gilden D., Devlin M., Huang E.S., Rorke L.B., Hamada T. Cytomegalovirus isolation from a chimpanzee with acute demyelinating disease after inoculation of multiple sclerosis brain cells. Infect Immun. 1979;25:1008–1015. doi: 10.1128/iai.25.3.1008-1015.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Perron H., Garson J.A., Bedin F., Beseme F., Paranhos-Baccala G., Komurian-Pradel F. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc Natl Acad Sci U S A. 1997;94:7583–7588. doi: 10.1073/pnas.94.14.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Rasmussen H.B., Geny C., Deforges L., Perron H., Tourtelotte W., Heltberg A. Expression of endogenous retroviruses in blood mononuclear cells and brain tissue from multiple sclerosis patients. Acta Neurol Scand Suppl. 1997;169:38–44. doi: 10.1111/j.1600-0404.1997.tb08148.x. [DOI] [PubMed] [Google Scholar]
- 390.Murray R.S., Brown B., Brian D., Cabirac G.F. Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann Neurol. 1992;31:525–533. doi: 10.1002/ana.410310511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Stewart J.N., Mounir S., Talbot P.J. Human coronavirus gene expression in the brains of multiple sclerosis patients. Virology. 1992;191:502–505. doi: 10.1016/0042-6822(92)90220-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Ferrante P., Omodeo-Zorini E., Caldarelli-Stefano R., Mediati M., Fainardi E., Granieri E. Detection of JC virus DNA in cerebrospinal fluid from multiple sclerosis patients. Mult Scler. 1998;4:49–54. doi: 10.1177/135245859800400202. [DOI] [PubMed] [Google Scholar]
- 393.Stoner G.L., Agostini H.T., Ryschkewitsch C.F., Baumhefner R.W., Tourtellotte W.W. Characterization of JC virus DNA amplified from urine of chronic progressive multiple sclerosis patients. Mult Scler. 1996;1:193–199. [PubMed] [Google Scholar]
- 394.Forghani B., Cremer N.E., Johnson K.P., Ginsberg A.H., Likosky W.H. Viral antibodies in cerebrospinal fluid of multiple sclerosis and control patients: comparison between radioimmunoassay and conventional techniques. J Clin Microbiol. 1978;7:63–69. doi: 10.1128/jcm.7.1.63-69.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.ter Meulen V., Koprowski H., Iwasaki Y., Kackell Y.M., Muller D. Fusion of cultured multiple-sclerosis brain cells with indicator cells: presence of nucleocapsids and virions and isolation of parainfluenza-type virus. Lancet. 1972;2:1–5. doi: 10.1016/s0140-6736(72)91273-1. [DOI] [PubMed] [Google Scholar]
- 396.Haase A.T., Ventura P., Gibbs C.J., Jr., Tourtellotte W.W. Measles virus nucleotide sequences: detection by hybridization in situ. Science. 1981;212:672–675. doi: 10.1126/science.7221554. [DOI] [PubMed] [Google Scholar]
- 397.Jacobson S., Flerlage M.L., McFarland H.F. Impaired measles virus-specific cytotoxic T cell responses in multiple sclerosis. J Exp Med. 1985;162:839–850. doi: 10.1084/jem.162.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Alperovitch A., Berr C., Cambon-Thomsen A., Puel J., Dugoujon J.M., Ruidavets J.B. Viral antibody titers, immunogenetic markers, and their interrelations in multiple sclerosis patients and controls. Hum Immunol. 1991;31:94–99. doi: 10.1016/0198-8859(91)90011-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Ortega-Hernandez O.D., Levin N.A., Altman A., Shoenfeld Y. Infectious agents in the pathogenesis of primary biliary cirrhosis. Dis Markers. 2010;29:277–286. doi: 10.3233/DMA-2010-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Shigematsu H., Shimoda S., Nakamura M., Matsushita S., Nishimura Y., Sakamoto N. Fine specificity of T cells reactive to human PDC-E2 163-176 peptide, the immunodominant autoantigen in primary biliary cirrhosis: implications for molecular mimicry and cross-recognition among mitochondrial autoantigens. Hepatology. 2000;32:901–909. doi: 10.1053/jhep.2000.18714. [DOI] [PubMed] [Google Scholar]
- 401.Liu H.Y., Deng A.M., Zhang J., Zhou Y., Yao D.K., Tu X.Q. Correlation of Chlamydia pneumoniae infection with primary biliary cirrhosis. World J Gastroenterol. Jul 14, 2005;11(26):4108–4110. doi: 10.3748/wjg.v11.i26.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Bogdanos D.P., Pares A., Baum H., Caballeria L., Rigopoulou E.I., Ma Y. Disease-specific cross-reactivity between mimicking peptides of heat shock protein of Mycobacterium gordonae and dominant epitope of E2 subunit of pyruvate dehydrogenase is common in Spanish but not British patients with primary biliary cirrhosis. J Autoimmun. 2004;22:353–362. doi: 10.1016/j.jaut.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 403.Vilagut L., Pares A., Vinas O., Vila J., Jimenez de Anta M.T., Rodes J. Antibodies to mycobacterial 65-kD heat shock protein cross-react with the main mitochondrial antigens in patients with primary biliary cirrhosis. Eur J Clin Invest. 1997;27:667–672. doi: 10.1046/j.1365-2362.1997.1690724.x. [DOI] [PubMed] [Google Scholar]
- 404.Vilagut L., Vila J., Vinas O., Pares A., Gines A., Jimenez de Anta M.T. Cross-reactivity of anti-Mycobacterium gordonae antibodies with the major mitochondrial autoantigens in primary biliary cirrhosis. J Hepatol. 1994;21:673–677. doi: 10.1016/s0168-8278(94)80117-7. [DOI] [PubMed] [Google Scholar]
- 405.Kita H., Matsumura S., He X.S., Ansari A.A., Lian Z.X., Van de Water J. Analysis of TCR antagonism and molecular mimicry of an HLA-A0201-restricted CTL epitope in primary biliary cirrhosis. Hepatology. 2002;36:918–926. doi: 10.1053/jhep.2002.35616. [DOI] [PubMed] [Google Scholar]
- 406.Yamaguchi H., Miura H., Ohsumi K., Ishimi N., Taguchi H., Ishiyama N. Detection and characterization of antibodies to bacterial heat-shock protein 60 in sera of patients with primary biliary cirrhosis. Microbiol Immunol. 1994;38:483–487. doi: 10.1111/j.1348-0421.1994.tb01813.x. [DOI] [PubMed] [Google Scholar]
- 407.Nickowitz R.E., Worman H.J. Autoantibodies from patients with primary biliary cirrhosis recognize a restricted region within the cytoplasmic tail of nuclear pore membrane glycoprotein Gp210. J Exp Med. 1993;178:2237–2242. doi: 10.1084/jem.178.6.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Stemerowicz R., Hopf U., Moller B., Wittenbrink C., Rodloff A., Reinhardt R. Are antimitochondrial antibodies in primary biliary cirrhosis induced by R(rough)-mutants of Enterobacteriaceae? Lancet. 1988;2:1166–1170. doi: 10.1016/s0140-6736(88)90235-8. [DOI] [PubMed] [Google Scholar]
- 409.Haruta I., Kikuchi K., Hashimoto E., Kato H., Hirota K., Kobayashi M. A possible role of histone-like DNA-binding protein of Streptococcus intermedius in the pathogenesis of bile duct damage in primary biliary cirrhosis. Clin Immunol. 2008;127:245–251. doi: 10.1016/j.clim.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 410.Sayers T.J., Baum H. Possible cross-reactivity of human anti-mitochondrial antibodies with membrane vesicles of Paracoccus denitrificans. Biochem Soc Trans. 1976;4:138–139. doi: 10.1042/bst0040138. [DOI] [PubMed] [Google Scholar]
- 411.Bogdanos D.P., Koutsoumpas A., Baum H., Vergani D. Borrelia burgdorferi: a new self-mimicking trigger in primary biliary cirrhosis. Dig Liver Dis. 2006;38:781–782. doi: 10.1016/j.dld.2006.05.010. [author reply 2–3] [DOI] [PubMed] [Google Scholar]
- 412.Harada K., Tsuneyama K., Sudo Y., Masuda S., Nakanuma Y. Molecular identification of bacterial 16S ribosomal RNA gene in liver tissue of primary biliary cirrhosis: is Propionibacterium acnes involved in granuloma formation? Hepatology. 2001;33:530–536. doi: 10.1053/jhep.2001.22653. [DOI] [PubMed] [Google Scholar]
- 413.Berg C.P., Kannan T.R., Klein R., Gregor M., Baseman J.B., Wesselborg S. Mycoplasma antigens as a possible trigger for the induction of antimitochondrial antibodies in primary biliary cirrhosis. Liver Int. 2009;29:797–809. doi: 10.1111/j.1478-3231.2008.01942.x. [DOI] [PubMed] [Google Scholar]
- 414.Jan G., Le Henaff M., Fontenelle C., Wroblewski H. Biochemical and antigenic characterisation of Mycoplasma gallisepticum membrane proteins P52 and P67 (pMGA) Arch Microbiol. 2001;177:81–90. doi: 10.1007/s00203-001-0364-4. [DOI] [PubMed] [Google Scholar]
- 415.Ninomiya M., Ueno Y., Shimosegawa T. PBC: animal models of cholangiopathies and possible endogenous viral infections. Int J Hepatol. 2012;2012 doi: 10.1155/2012/649290. (649290) (Electronic publication ahead of print 2011 Aug 8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Zhang G., Chen M., Graham D., Subsin B., McDougall C., Gilady S. Mouse mammary tumor virus in anti-mitochondrial antibody producing mouse models. J Hepatol. 2011;55:876–884. doi: 10.1016/j.jhep.2011.01.037. [DOI] [PubMed] [Google Scholar]
- 417.Morshed S.A., Nishioka M., Saito I., Komiyama K., Moro I. Increased expression of Epstein–Barr virus in primary biliary cirrhosis patients. Gastroenterol Jpn. 1992;27:751–758. doi: 10.1007/BF02806528. [DOI] [PubMed] [Google Scholar]
- 418.Uzoegwu P., Baum H., Williamson J. Correlation of oligomycin-sensitive ATPase activity in trypanosomes with their content of an antigen to primary biliary cirrhosis. Cell Biol Int Rep. 1984;8:981–986. doi: 10.1016/0309-1651(84)90196-6. [DOI] [PubMed] [Google Scholar]
- 419.Uzoegwu P.N., Baum H., Williamson J. The occurrence and localization in trypanosomes and other endo-parasites of an antigen cross-reacting with mitochondrial antibodies of primary biliary cirrhosis. Comp Biochem Physiol B. 1987;88:1181–1189. doi: 10.1016/0305-0491(87)90022-8. [DOI] [PubMed] [Google Scholar]
- 420.Sakly W., Jeddi M., Ghedira I. Anti-Saccharomyces cerevisiae antibodies in primary biliary cirrhosis. Dig Dis Sci. 2008;53:1983–1987. doi: 10.1007/s10620-007-0092-y. [DOI] [PubMed] [Google Scholar]