

Abstract
Like in many tumor types, immunotherapy is currently under investigation to assess its potential efficacy in glioblastoma patients. Trials are under way to assess the efficacy of new immune checkpoint inhibitors including anti-PD-1 or CTLA4. We here investigate the expression and efficacy of a novel immune-checkpoint inhibitor, called LAG-3. We show that LAG-3 is expressed in human glioblastoma samples and in a mouse glioblastoma model we show that knock out or LAG-3 inhibition with a blocking antibody is efficacious against glioblastoma and can be used in combination with other immune checkpoint inhibitors toward complete eradication of the model glioblastoma tumors. From a mechanistic standpoint we show that LAG-3 expression is an early marker of T cell exhaustion and therefore early treatment with LAG-3 blocking antibody is more efficacious than later treatment. These data provide insight and support the design of trials that incorporate LAG-3 in the treatment of glioblastoma.
Keywords: glioblastoma, anti-LAG-3, anti-PD-1, T cell exhaustion, IFN-γ
Background
Glioblastoma is the most frequent and lethal primary brain tumor.1 Despite significant advances in understanding of the pathophysiology and genetics of the disease no significant breakthrough has been made in treatment of this disease. This is in part related to the fact that glioblastoma has developed multiple mechanisms to evade detection and destruction by the immune system including the secretion of immunosuppressive cytokines and downregulation of major histocompatibility complex (MHC) molecules.2 Furthermore, the use of cytotoxic therapy leads to further immune dysfunction.3,4 Our prior pre-clinical studies indicate that reversal of the immune suppressive milieu of glioblastoma is possible in the murine model, GL261, by combining first generation immune checkpoint blockade [anti-PD-1 (programmed death 1)], anti-CTLA4 (cytotoxic T lymphocyte associated protein 4)] in combination with radiation or chemotherapy.3,5–7 These agents have established significant efficacy in other cancer types and have specifically achieved FDA approval for many end stage cancers.8–10 Due to the efficacy of these first generation immune checkpoint inhibitors a set of second generation of checkpoint inhibitors, including anti-LAG-3 (lymphokine activation gene 3) antibodies (Abs) is under preclinical testing.
LAG-3 (also known as CD223) is a cell surface molecule expressed on a variety of T cells (CD4 primarily but also CD8) and is binding primarily with the MHCII molecule in antigen presenting cells (APCs) promoting apoptosis, decreasing proliferation and increasing T cell tolerance.11,12 The use of anti-LAG-3 Abs in a melanoma tumor model led to increased CD8+ IFNγ producing cells and decreased tumor growth compared to non-treated mice. The combination of anti-LAG-3 and anti-PD-1 in a variety of tumor models has led to synergistic antitumor efficacy.13,14. We are building upon prior published work of our co-author (CGD) that has shown that LAG-3 inhibition skews CD4 cells away from Treg phenotype and combination of anti-PD-1 and anti-LAG-3 is synergistic in noncentral nervous system tumors.14,15
In this manuscript our group is investigating the expression of LAG-3 in glioblastoma, the efficacy of anti-LAG-3 Ab alone or in combination with anti-PD-1 and we are exploring the mechanism of efficacy of anti-LAG-3 Ab. By utilizing LAG-3 knockout mice we provide mechanistic evidence and further support on the importance of inhibiting the LAG-3 pathway as a way to control glioblastoma growth. A clinical trial involving the use of anti-LAG-3 in combination with anti-PD-1 for the treatment of glioblastoma is currently ongoing (Clinical Trial identifier: NCT02658981) that will shed light on the clinical significance of LAG-3 in glioblastoma.
Methods
Human glioblastoma samples
Archival formal fixed paraffin embedded tumor tissue from 10 human glioblastoma patients operated by a single surgeon from 2016 to 2017 was used to stain for the LAG-3 epitope.
Immunohistochemistry (IHC) in human glioblastoma samples w human LAG-3 Ab
IHC for LAG-3 was performed using a primary mouse anti-human mAb (clone 17B4, Lifespan Bioscience) at a concentration of 0.1 μg/mL followed by an antigen retrieval of 10 min in citrate buffer, pH 6.0 at 120°C. A secondary anti-mouse IgG1-biotin antibody was used at 1.0 μg/ml. Amplification was performed using the CSA kit (DAKO Cat.K1500), and was visualized using streptavidin-HRP (DAKO LSAB2 Cat. K0675) and DAB (Sigma Cat.D0426). Human tonsil was used as positive control for this study.
Cell line
GL-261 luciferase positive cells (GL-261 LUC) were purchased from Caliper Life Sciences (Hopkinton, MA). Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies, Grand Island, NY) with 10% Fetal Bovine Serum (FBS, Gemini Bio-Products, West Sacramento, CA) plus 1% Penicillin/Streptomycin (Life Technologies, Grand Island, NY) and 100 μg/mL of G418 (Invitrogen, San Diego, CA) in an incubator maintained at 37°C with 5% CO2.
Tumor model
Female C57BL/6J mice (The Jackson Laboratories, Bar Harbor, ME), 6–8 weeks old, were implanted (Day 0) with GL-261 LUC cells to establish intracranial gliomas, as previously described.16 Briefly, mice were anesthetized with ketamine/xylazine (100 mg/kg ketamine, 10 mg/kg xylazine). A small midline incision was made to the head and a burr hole was then drilled directly over the striatum and 130,000 GL-261 LUC cells were implanted at a depth of 3 mm from the cortical surface. The tumor take rate was 100%. Day 7 post implantation, mice were imaged to assess the progress of tumor growth using an IVIS platform (In Vivo Imaging System, Caliper Life Sciences, Hopkinton, MA). Mice were then stratified into experimental treatment groups based on luminescence. Each treatment group had 5–15 mice in the survival experiments. The treatment groups were as follows: Control (PBS administered intra-peritoneally with isotype control), anti-PD-1, anti-LAG-3, anti-PD-1+ anti-LAG-3, control LAG-3−/− mice (KO) anti-PD-1 mAb in LAG-3−/− mice. All experiments were repeated at least in triplicate unless otherwise stated. The experimental schedules are explained in Figure 1.
Figure 1.
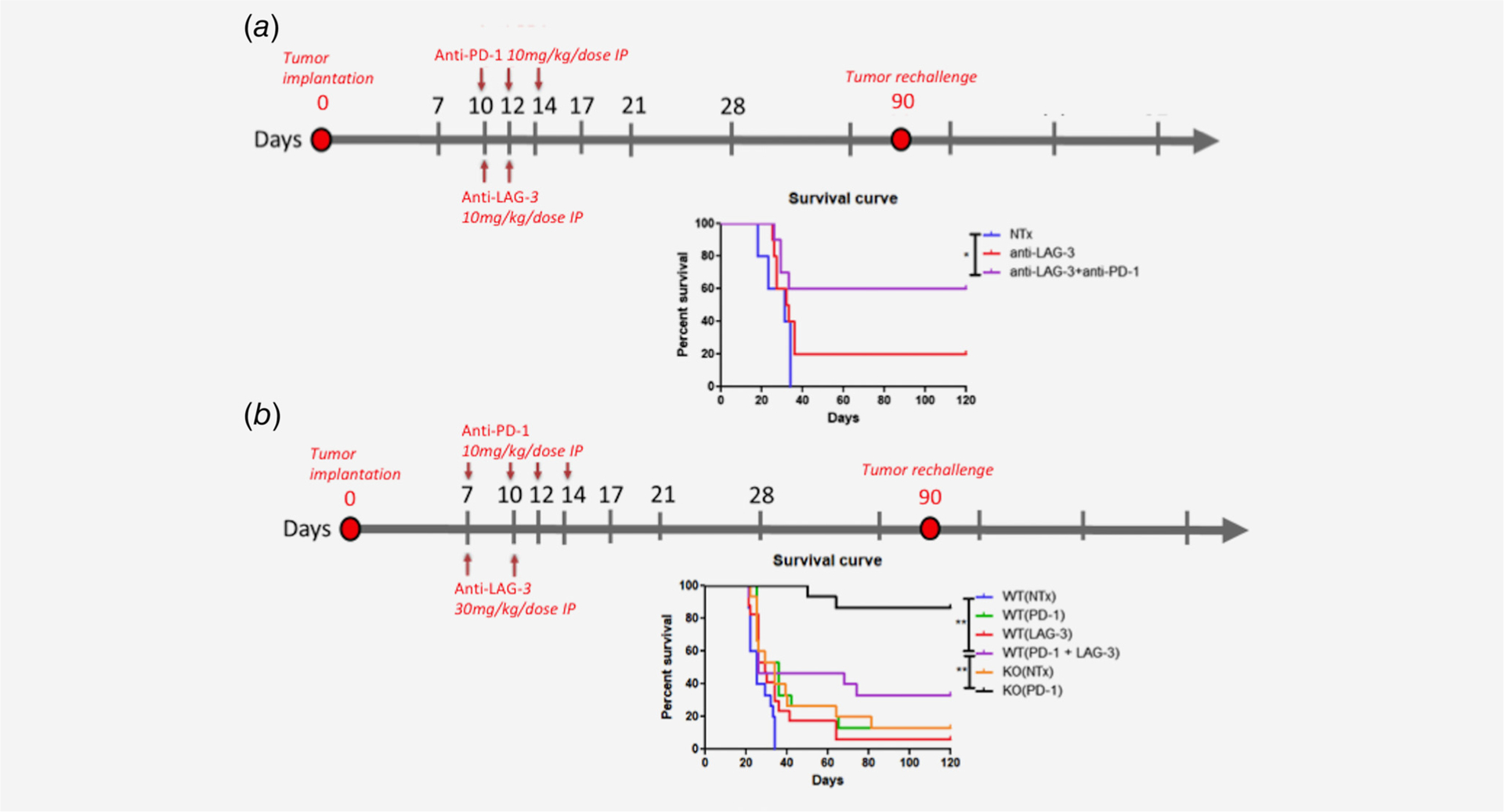
(a) Late treatment with anti-PD-1 and/or anti-LAG3 (starting day 10) led to moderate survival benefit compared to NTx. Anti-LAG3 alone showed moderate survival benefit compared to NTx (p = 0.15). Anti-LAG-3 + anti-PD-1 showed moderate survival benefit compared to anti-LAG-3 alone (p = 0.1). However, anti-PD-1 + anti-LAG-3 showed statistically improved survival benefit compared to NTx mice (p = 0.03). N = 10 mice. (b) Early treatment with anti-PD-1 and/or anti-LAG3 (starting day 7) led to significant survival benefit of anti-LAG3 alone (p = 0.04) or anti-PD-1 (p = 0.002) compared to NTx. Anti-PD1 + anti-LAG3 did not show statistically different survival differences compared to anti-PD-1 or anti-LAG3. Striking survival benefit was obtained in LAG3 KO mice treated with anti-PD-1 (0.002) compared to ant-PD1 + anti-LAG3. N = 15 mice for every treatment group
Rechallenge experiment
Mice from all the treatment groups involved in this study were observed for 90 days at which point they were imaged to assure no residual tumor signal via IVIS imaging. Mice were re-implanted with flank tumors as a way of tumor re-challenge. Matrigel (BD Biosciences) and cell solution (106 cells) were mixed in a 1:1 ratio in a total volume of 100 μl and were then immediately injected in the flank of all the long term surviving mice. Mice were followed for tumor recurrence.
Anti-PD-1 and anti-LAG-3 monoclonal antibodies
Hamster anti-murine PD-1 monoclonal antibody-producing hybridoma (G4) was used to produce antibody as previously described.17 Anti-LAG-3 mAb (C9B7W, IgG1) was produced as previously described.18 About 200 μg of anti-PD-1 and 200 μg of anti-LAG-3 were used for each dose. Hamster immunoglobulin isotype (Rockland Immunochemicals Inc., Gilbertsville, PA) antibody was administered to animals receiving no treatment.
Flow cytometry
At day 21 post tumor implantation, mice were sacrificed using a lethal dose of ketamine/xylazine cocktail. The brain and cervical lymph nodes (LN) were harvested and passed through a 40-μm strainer. A 30–37–60% Percoll gradient (GE Healthcare, Buck-inghamshire, UK) was used to isolate immune cell populations from brain tumors and the draining lymph nodes (LNs). After centrifugation, the 37–60% interface contained lymphocytes, monocytes and microglia in the case of brain tumors, and contained lymphocytes and monocytes in the case of draining LNs.
For flow cytometric analysis, lymphocytes were stained with CD8 PerCp-Cy5.5 Clone: 53–6.7 (eBioscience), CD3 FITC Clone: 17A2 (eBioscience), CD4 APCH7 Clone: GK1.5 (BD Biosciences), FoxP3 PE Clone: NNRF-30 (eBioscience), IFNγ BV421 Clone: XMG 1.2 (Biolegend), LAG-3 APC Clone: C9B7W, PD-1 PE-Cy7 Clone: J43 and fixable aqua L/D stain (Life Technologies). Appropriate isotype controls were used.
All flow cytometry experiments were performed on a LSRII (BD Biosciences) and analysis was performed using FlowJo software (TreeStar, Ashland, OR).
IVIS imaging
The progression of tumor burden in vivo was tracked by IVIS imaging at days 0, 7, 14 and 21. All animals were anesthetized with isoflurane-oxygen mix before they were inserted in the IVIS imaging platform (Perkins Elmer) provided in our animal facility. Mice were injected with 200 μl of firefly D-luciferin solution. Images were obtained and values of bioluminescence intensity were used to quantify tumor volume.
Knockout mice
LAG-3−/− mice have been evaluated in previous publication14 by one of our authors group (CGD).
Statistics
Survival was plotted using Kaplan–Meier curves, and curves were analyzed with the log-rank Mantel–Cox test using GraphPad Prism software (GraphPad Software, La Jolla, CA). For comparison of cell numbers and percentages between treatment groups in flow cytometry experiments, a two-tailed unpaired t test was used. The p values ≤0.05 were considered significant.
Results
Survival experiments
Two different treatment schedules were used to assess efficacy of anti-LAG-3 with or without anti-PD-1. The first treatment schedule we used was in accordance with prior timeline we have used in our lab in previous experiments with immune checkpoint inhibitors3 where treatment with anti-LAG-3 and/or anti-PD-1 starts on day 10. Mice were treated with anti-PD-1 at days 10, 12, 14 and anti-LAG-3 at days 10 and 12 (Fig. 1a). This treatment schedule showed modest results in terms of efficacy of anti-LAG-3 compared to NTx (No treatment) mice (p = 0.15). Combination of anti-PD-1 and anti-LAG-3 showed statistically significant survival benefit compared to NTx mice (p = 0.03) but no statistical difference between anti-LAG-3 and anti-PD-1+ anti-LAG-3 groups (p = 0.1).
A cohort of mice was implanted with GL-261 tumors and was treated with anti-PD-1 (Days 7, 10, 12 and 14) and/or anti-LAG-3 Abs (Days 7 and 10) (Fig. 1b). Mice treated with anti-PD-1 had a significantly different survival compared to NTx group (p = 0.002). Mice treated with anti-LAG-3 had a significantly different survival compared to NTx group (p = 0.04). The combination of anti-PD-1 with anti-LAG-3 was significantly different than NTx group (p = 0.0096) but was not significantly different than each treatment alone (p = 0.128 for anti-LAG-3 and 0.355 for anti-PD-1). NTx LAG-3−/− mice had a higher survival than the NTx wild type (WT) mice but this difference did not reach statistical significance (p = 0.07). Interestingly enough LAG-3−/− mice treated with anti-PD-1 had a significantly improved survival compared to WT mice treated with anti-PD-1 or with anti-PD-1 and anti-LAG-3 (p = 0.0001 and p = 0.002) indicating that knocking out LAG-3 does improve the efficacy of anti-PD-1 significantly unlike anti-LAG-3 blockade. Detailed statistics of the Kaplan–Meir survival analysis can be found in Table 1.
Table 1.
Treatment group comparison of overall survival for survival superiority for the experimental groups in Figure 1b
| Comparison | Superior OS | p-value |
|---|---|---|
| KO(PD-1) vs. KO(NTx) | KO(PD-1) | 0.0001 |
| KO(PD-1) vs. WT(PD-1) | KO(PD-1) | 0.0001 |
| KO(PD-1) vs. WT(LAG-3) | KO(PD-1) | 0.0001 |
| KO(PD-1) vs. WT(PD-1 + LAG-3) | KO(PD-1) | 0.002 |
| KO(PD-1) vs. WT(NTx) | KO(PD-1) | 0.0001 |
| KO(NTx) vs. WT(NTx) | KO(NTx) | 0.007 |
| WT(PD-1) vs. WT(NTx) | WT(PD-1) | 0.002 |
| WT(LAG-3) vs. WT(NTx) | WT(LAG-3) | 0.04 |
| WT(PD-1+ LAG-3) vs. WT(NTx) | WT(PD-1+ LAG-3) | 0.0096 |
| WT(PD-1+ LAG-3) vs. WT(LAG-3) | WT(LAG-3) | 0.128 |
| WT(PD-1+ LAG-3) vs. WT(PD-1) | WT(PD-1) | 0.348 |
| WT(PD-1) vs. WT(LAG-3) | WT(PD-1) | 0.355 |
| KO(NTx) vs. WT(LAG-3) | *KO(NTx) | 0.484 |
| KO(NTx) vs. WT(PD-1) | /Wt(pd-i) | 0.888 |
All comparisons were done by the Log-rank test, two-sided.
Flow cytometry
GL-261-luc implanted mice treated in the first cohort presented above (Fig. 1a), were sacrificed on day 21 and tumor infiltrating lymphocytes as well as lymphocytes from the draining lymph nodes were immunophenotypically analyzed.
The percent of Tregs was only statistically different in the anti-PD-1 treated group compared to NTx group (p = 0.05) (Figs. 2a and 2b). However, although anti-PD-1 and anti-LAG-3 group showed a trend toward decreased percent of Tregs the result was not statistically significant (p = 0.25). Similar to this result, the percent of CD8 or CD4 IFNγ producing cells (T effector cells) was not significantly different among groups although the combination of anti-PD-1+ and anti-LAG-3 trended toward higher percentage of effector cells compared to anti-PD-1 or anti-LAG-3 alone.
Figure 2.
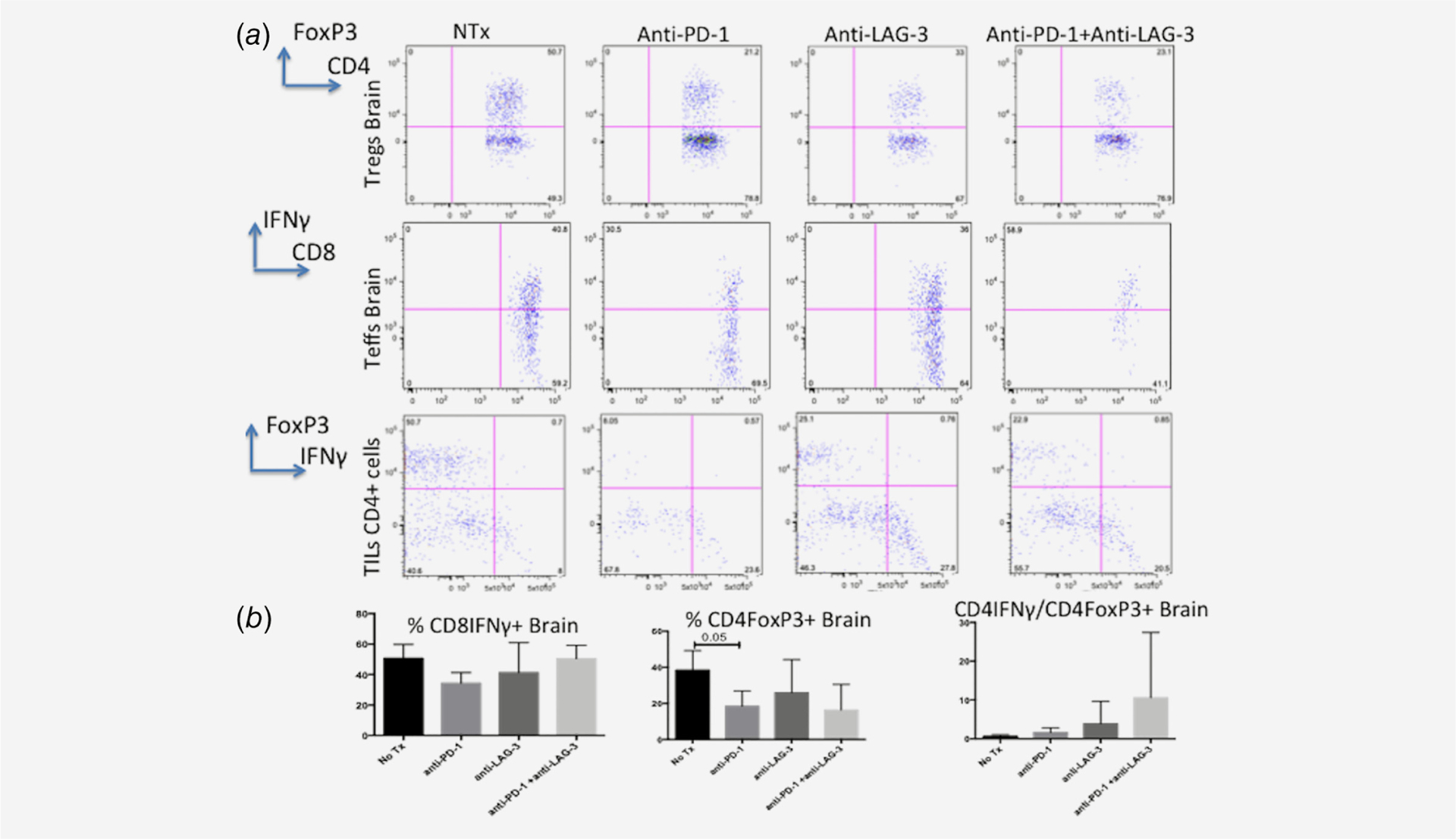
(a, b) Flow cytometric analysis of mice treated with the late anti-PD-1 and anti-LAG-3 schedule and sacrificed on experimental day 21. The only statistically significant difference noticed on the analysis was reduction in the percent of Tregs in the anti-PD-1 group compared to NTx group (p = 0.05). A trend to higher CD4 effector:Tregs was observed in the combination group (anti-PD-1 + anti-LAG-3) compared to all the other groups but the result was not statistically significant due to high variation.
We were able to further characterize the functional capacity (production of IFNγ) of CD4 and CD8 cells based on expression of surface PD-1 and/or LAG-3 (Fig. 3). In both CD4 and CD8 PD-1–/LAG-3+ T cells the production of IFNγ was minimal (4% of cells producing IFNγ). PD-1+/LAG-3+ cells had higer percent of IFNγ producing cells (10%). PD-1–/LAG-3- cells had even higher production of IFNγ(36%) whereas the highest percent of IFNγ production was present in the PD-1+/LAG-3-cells.
Figure 3.
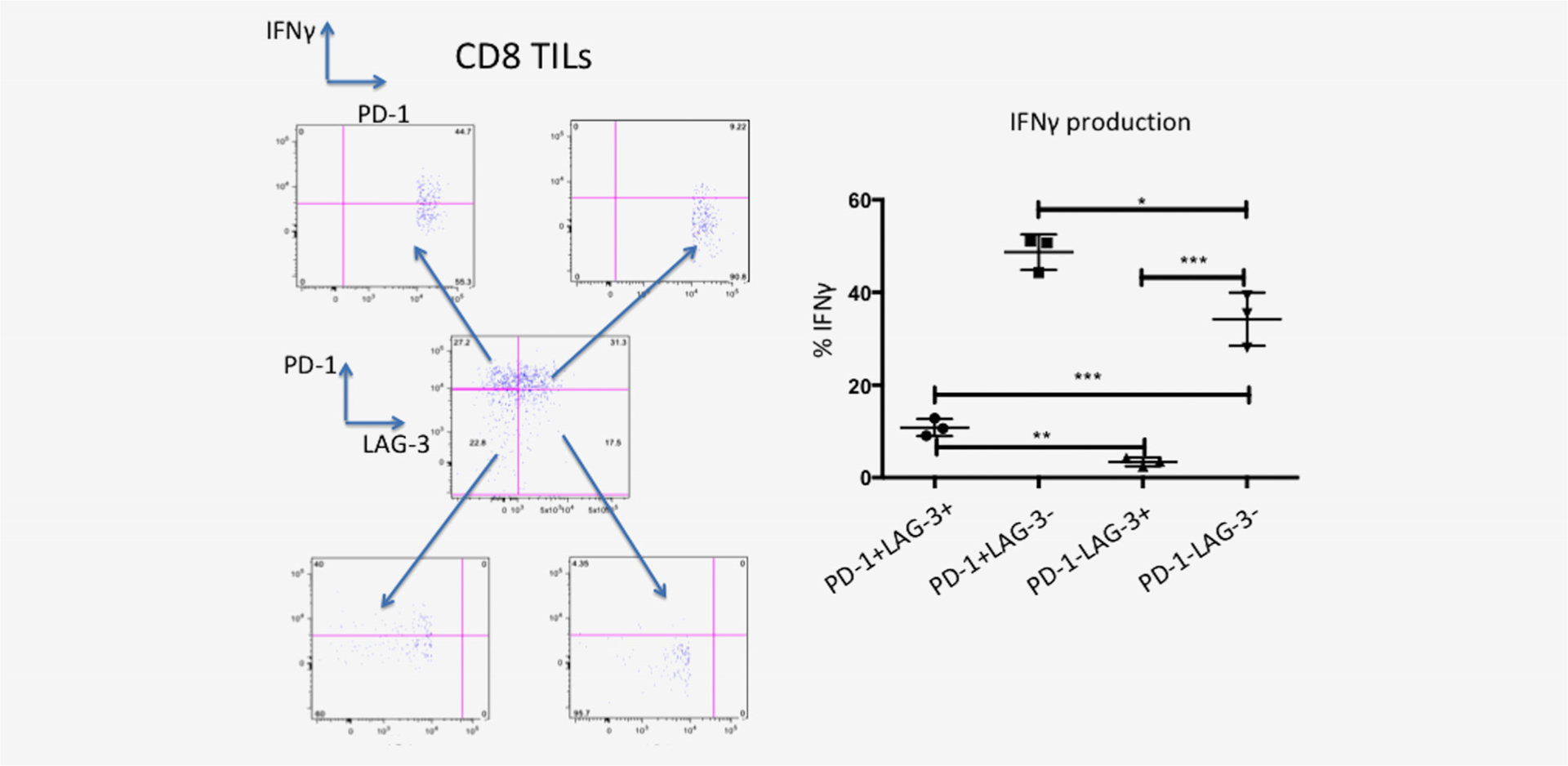
Functional analysis of CD8+ cells after PMA/Ionomyocin activation shows an exhaustion phenotype (decreased IFN-γ production) for LAG-3+ PD-1-cells. PD-1-LAG-3+ cells show minimal IFNγ production compared to PD-1+ LAG-3+ cells (p = 0.002). PD-1+ LAG-3+ cells had a fourfold reduction of IFNγ production compared to double negative cells(PD-1-LAG-3-) [p < 0.001] and a fivefold reduction of PD-1+ LAG-3-cells (p < 0.001). This indicates that LAG-3 when expressed on T cells shows an exhaustion phenotype. When PD-1 is expressed on T cells this indicates a state of immune activation where PD-1 is overexpressed to control.
Analysis of DLN lymphocytes showed no significant difference in the immunophenotypic profile of these cells among different treatment groups.
Combination anti-PD-1 and anti-LAG3 Abs allows for immune memory response upon tumor re-challenge
All mice surviving greater than 90 days post implantation were considered “long-term survivors” and re-challenged with flank tumors to evaluate for a host anti-tumor memory response. Tumors were implanted subcutaneously and monitored for growth. 100% of mice from the combination therapy group rejected tumor whereas 100% of naïve mice developed rapidly enlarging flank tumors.
IHC in human glioblastoma samples
About 10 patients with histologically confirmed (by a senior board certified pathologist, PCB) glioblastoma, IDH1, 2 wild type were stained with human anti-LAG-3 antibody. One sample was excluded from the analysis as there was not enough tissue to be stained after processing. About 6/9 samples (66%) were positively stained with the LAG-3 Ab. LAG-3 was present on tumor infiltrating lymphocytes (TILs) as well as lymphocytes of the perivascular niche of the tumor (Fig. 4). LAG-3 was not expressed on tumor cells or macrophages or other myeloid lineage cells. The clinical characteristics and the staining pattern are summarized in Table 2. Notably there was variable expression of LAG-3 on glioblastoma samples with all the positive samples having less than 1% LAG3 positive cells/high power field (HPF).
Figure 4.
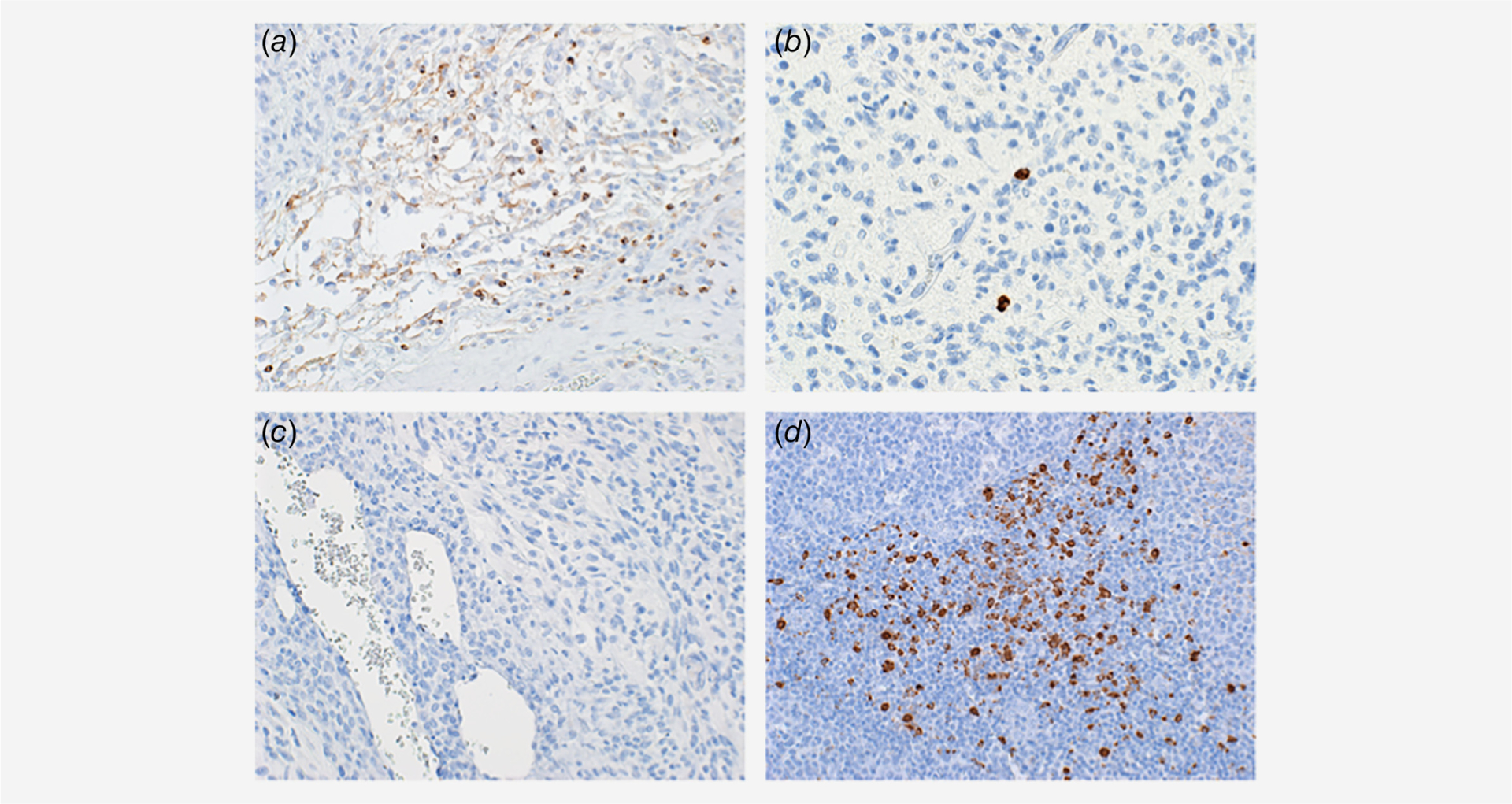
Representative photomicrographs of human glioblastoma tissue demonstrating the LAG-3 expression in the glioblastoma microenvironment. (a) LAG-3 expression is avidly expressed in perivascular lymphocytes. (b) LAG-3 expression was present on tumor infiltrating lymphocytes. (c) Internal tissue control showing no non-specific binding of LAG-3 Ab. (d) Positive control for LAG-3 expression in human tonsil showing avid expression of LAG-3 in a subset of lymphocytes.
Table 2.
Clinical characteristics of the glioblastoma patients analyzed for presence of LAG-3 by IHC in this cohort
| Sample name | Histological diagnosis-genetics | Age | Gender | Primary vs recurrent tumor | Treatment | LAG-3 staining | LAG-3 pattern |
|---|---|---|---|---|---|---|---|
| 1 | Glioblastoma IDH WT | 54 | M | Primary | Postop XRT/temodar | Positive | Scattered-intratumoral and perivascular < 0.1% p HPF |
| 2 | Glioblastoma | 57 | F | Primary | Postop XRT/temodar | Positive | Scattered-intratumoral and perivascular <1% p HPF |
| 3 | Glioblastoma IDH WT, sarcomatoid features | 57 | M | Recurrent | Prior surgery XRT/temodar | Positive | Scattered-intratumoral and perivascular <0.1% p HPF |
| 4 | Glioblastoma IDH WT | 61 | F | Primary | Postop XRT/temodar | Positive | Scattered-intratumoral and perivascular <1% p HPF |
| 5 | Glioblastoma IDH WT, spindle cell | 56 | M | Recurrent | Prior surgery (x2), temodar, avastin | Positive | Scattered-intratumoral and perivascular <0.1% p HPF |
| 6 | Glioblastoma IDH WT | 67 | M | Primary | Postop XRT/temodar | Positive | Scattered-intratumoral and perivascular <0.1% p HPF |
| 7 | Glioblastoma IDH WT, unmethylated MGMT | 51 | F | Recurrent | Prior surgery XRT/temodar | Negative | N/A |
| 8 | Glioblastoma IDH WT | 65 | M | Primary | Postop XRT | Negative | N/A |
| 9 | Glioblastoma IDH WT | 57 | M | Primary | - | Negative | N/A |
The percent of LAG-3 positive cells is calculated based on number of LAG-3 positive cells over the total number of cells per high power field.
Discussion
In this study inhibition of LAG-3 either by knock down or anti-LAG-3 Ab improves survival in a preclinical glioblastoma model and considerably improves the efficacy of anti-PD-1 treatment. LAG-3 expression on T cells has been considered as a marker of T cell exhaustion. We show that the expression of LAG-3 on CD4 or CD8 cells significantly decreases their ability to produce IFNγ. Expression of PD-1 on T cells is a marker of activating T cells, as confirmed by our flow analysis and high expression of IFNγ on PD-1 high T cells. These data support the strategy of using LAG-3 inhibition early on in treatment rather than in late stages as disease progression and high levels of LAG-3 leads to T cells exhaustion and tolerance. These data further support the idea of combining immune checkpoint inhibitors to achieve better treatment outcomes. Furthermore, data from our human glioblastoma samples show expression of LAG-3 on infiltrating immune cells as well as immune cells in the perivascular niche in 66% of the human samples (however, the percentage of LAG-3 positive cells/HPF in the samples are <1%), providing rationale for initiation of human clinical trials of anti-LAG-3 Ab in glioblastoma.
The mechanism of immunological function of LAG-3 has been studied in pathogen-associated immunity more so than in cancer. LAG-3 has been shown to be upregulated on exhausted T cells compared to effector or memory T cells in LCMV infection. In the context of cancer, LAG-3 is upregulated on TILs and blockade of LAG-3 can enhance anti-tumor T cell responses.11–14 In addition, dual blockade of the PD-1 pathway and LAG-3 has been shown in mice and humans to be more effective for anti-tumor immunity than blocking either molecule alone in fibrosarcoma and MC38 model.14 However, the role of LAG-3 in glioblastoma is yet to be determined. In this study we show that LAG-3 is expressed on T cells in a mouse glioblastoma model. We found that LAG-3 is co-expressed with PD-1 on T cells. High expression of LAG-3 correlated with significantly less IFNγ release upon activation, corroborating with the findings of other studies indicating LAG-3 as a marker of T cell exhaustion. Furthermore, the blockade of LAG-3 shows significant improvement in survival and tumor eradication. Knocking out LAG-3 was more effective in tumor eradication compared to anti-LAG-3 mAb. These results could be explained either by incomplete blockade of LAG-3 epitope by the mAb or by the fact that LAG-3 expression early on in tumorigenesis causes a significant immunosuppressive effect that can only be partially reversed in later stages of tumor progression by monoclonal Ab blockade. Additionally, the combination of LAG-3 blockade and PD-1 blockade showed synergistic improvement of survival in mice with glioblastoma. Specifically 87% of LAG-3−/− mice treated with anti-PD-1 were long-term survivors showing no evidence of disease 90 days post-implantation and upon re-challenged they were able to prevent tumor formation.
Conclusively, LAG-3 is expressed on TILs and tumor associated perivascular lymphocytes of human glioblastoma samples. The data from our preclinical model indicates that LAG-3 alone or in combination with anti-PD-1 is very effective at eradicating glioblastoma mouse tumors. This combination is more efficacious when LAG-3 is given at an early time point. Additionally from a mechanistic standpoint our data indicate that LAG-3 is an early marker of exhaustion of T cell effector function. The above data provide preclinical basis and direct human evidence for launching clinical trials to establish safety and efficacy of anti-LAG-3 with or without anti-PD-1 treatment in combination with the current standard of care in the primary tumor setting as well as at the setting of tumor recurrence.
Novelty and Impact.
We show LAG-3 blockage alone or in combination with anti-PD-1 leads to eradication of glioblastoma tumors in mice. This is supported by evidence LAG-3 is an early marker of exhaustion of effector T cells and thus its early blockade can release an antitumor immune response. Additionally, we show LAG-3 is expressed in human glioblastoma. These data can guide the design of clinical trials that are already under way for the treatment of glioblastoma.
What’s new?
Glioblastoma derives some of its lethality from its ability to escape destruction by the immune system. Researchers have begun investigating immune checkpoint inhibitors as a tool to combat glioblastoma. Here, the authors report on a novel immune-checkpoint inhibitor, LAG-3. In a mouse model of glioblastoma, they successfully improved survival by eliminating LAG-3, either by genetic knockout or using antibodies against it. They show that TILs from human glioblastoma samples express LAG-3, and that high LAG-3 expression correlates with reduced interferon release. The authors propose that anti-LAG-3, alone or in combination with other anti-PD-1 treatment, could improve glioblastoma treatment.
Abbreviations:
- APC
antigen presenting cell
- CTLA-4
cytotoxic T lymphocyte associated protein 4
- HPF
high power field
- IHC
immunohistochemistry
- KO
knockout
- LAG-3
lymphokine activation gene 3
- LN
lymph nodes
- mAbs
monoclonal antibodies
- MHC
major histocompatibility complex
- NTx
no treatment group
- PD-1
programmed death 1
- TILs
tumor infiltrating lymphocytes
- WT
wild type
Footnotes
Conflicts of Interest: JMT is a consultant for Bristol Meyers Squibb (BMS), Merck and AstraZeneca and receives research support from BMS. MS, CT and AK are employees of BMS and MS and CT own BMS stocks. CGD is a consultant for Agenus, Dendreon, Janssen, Lilly, Merck, MedImmune, Pierre Fabre, Roche/Genentech; receives research support from Aduro Biotech, BMS, Janssen; has ownership interests in Compugen, Harpoon, Kleo, Potenza, Tizona; and holds patents with BMS, AstraZeneca, MedImmune and Janssen. ML is a consultant for Agenus, BMS, Regeneron, Oncorus, Boston Biomedical, Tocagen, SQZ Biotechnologies, Stryker and Baxter and receives research support from Arbor, Agenus, Altor, BMS, Immunocellular, Celldex, Accuray and DNAtrix. All other authors declare no relevant disclosures.
References
- 1.Ostrom QT, Gittleman H, Xu J, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro Oncol 2016;18: v1–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nduom EK, Weller M, Heimberg AB. Immuno-suppressive mechanisms in glioblastoma. Neuro Oncol 2015;17(Suppl 7):vii9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathios D, Kim JE, Mangraviti A, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med 2016;8:370ra180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campian JL, Piotrowski AF, Ye X, et al. Serial changes in lymphocyte subsets in patients with newly diagnosed high grade astrocytomas treated with standard radiation and temozolomide. J Neurooncol 2017;135:343–51. [DOI] [PubMed] [Google Scholar]
- 5.Kim JE, Patel MA, Mangraviti A, et al. Combination therapy with anti-pd-1, anti-tim-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res 2017;23:124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel MA, Kim JE, Theodros D, et al. Agonist anti-GITR monoclonal antibody and stereotactic radiation induce immune-mediated survival advantage in murine intracranial glioma. J Immunother Cancer 2016;4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathios D, Park CK, Marcus WD, et al. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int J Cancer 2016; 138:187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18: 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538–12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:2461–71. [DOI] [PubMed] [Google Scholar]
- 10.Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol 2016;17:1558–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andrews LP, Marciscano AE, Drake CG, et al. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev 2017;276:80–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat Rev Immunol 2015;15:45–56. [DOI] [PubMed] [Google Scholar]
- 13.Huang RY, Eppolito C, Lele S, et al. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015;6:27359–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 2012;72:917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durham NM, Nirschl CJ, Jackson CM, et al. Lymphocyte activation gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS One 2014;9: e109080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys 2013;86: 343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirano F, Kaneko K, Tamura H, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 2005;65:1089–96. [PubMed] [Google Scholar]
- 18.Workman CJ, Rice DS, Dugger KJ, et al. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur J Immunol 2002;32:2255–63. [DOI] [PubMed] [Google Scholar]