

Abstract
Disturbances in sleep/wake cycle are a common complaint of individuals with Huntington’s disease (HD) and are displayed by HD mouse models. The underlying mechanisms, including the possible role of the circadian timing system, have been the topic of a number of recent studies. The (z)Q175 mouse is a knock-in model in which the human exon 1 sequence of the huntingtin gene is inserted into the mouse DNA with approximately 190 CAG repeats. Among the numerous models available, the heterozygous Q175 offers strong construct validity with a single copy of the mutation, genetic precision of the insertion and control of mutation copy number. In this review, we will summarize the evidence that this model exhibits disrupted diurnal and circadian rhythms in locomotor activity. We found overwhelming evidence for autonomic dysfunction including blunted daily rhythms in heart rate and core body temperature, reduced heart rate variability, and almost a complete failure of the sympathetic arm of the autonomic nervous system to function during the baroreceptor reflex. Mechanistically, the Q175 mouse model exhibits deficits in the neural output of the central circadian clock, the suprachiasmatic nucleus (SCN) along with an enhancement of at least one type of potassium current in these neurons. Finally, we report a novel network analysis examining the phase coherence between activity, core body temperature, and cardiovascular measures. Such analyses found that even young Q175 mutants (heterozygous or homozygous) show coherence degradation, and suggests that loss of phase coherence is a variable that should be considered as a possible biomarker for HD.
Keywords: autonomic nervous system, cardiovascular function, circadian rhythms, Huntington’s disease, phase coherence, Q175, Suprachiasmatic nucleus
Introduction
Huntington’s disease is a genetically-determined neurodegenerative disease
Huntington’s disease (HD) patients suffer from a progressive neurodegenerative disorder that inflicts cognitive, psychiatric and motor impairments (Bates et al., 2015; McColgan and Tabrizi, 2018). HD is caused by a CAG repeat expansion within the first exon of the huntingtin (HTT) gene, which produces a polyglutamine repeat leading to protein misfolding, soluble aggregates and inclusion bodies (Saft et al., 2005; Ciammola et al., 2006). The normal function of the protein (Huntingtin) is unknown; however, the mutated form leads to dysfunction of a broad range of cellular processes including cytoskeletal organization, metabolism and transcriptional activities. Based on the broad distribution of the HTT, the mutation would be expected to produce symptoms throughout the body. Indeed, recent work suggests HD is a systemic illness affecting the entire body and data from animal studies imply that core features of the disease can be modified by treatments targeting other tissues besides the central nervous system (Carroll et al., 2015).
Sleep and circadian dysfunction are an integral component of Huntington’s disease pathophysiology
Sleep disorders are prevalent in HD patients and have detrimental effects on the daily functioning and quality of life of patients and their caregivers (Morton et al., 2005; Goodman et al., 2011). The most common symptoms include a delay in sleep onset, fragmented sleep during the night and daytime sleepiness. Importantly, these disruptions in the sleep/wake cycle occur early in the disease progression and so could serve as a biomarker for HD as well as a target for interventions. The sleep/wake cycle is conceptualized as being driven by two anatomically-distinct processes: a homeostatic sleep mechanism (process S) as well as by the circadian timing system (process C). To date, there is little evidence that HD impacts sleep homeostasis in patients, but there is growing evidence for HD-driven disruption in circadian timing. Still, it is difficult to determine whether the disease alters the circadian timing system in humans and animal models provide critical insights.
To further preclinical research, a large number of animal models of HD have been developed, each with strengths and weaknesses (Rangel-Barajas and Rebec, 2018). These models display a rapid and progressive breakdown of the circadian rest/activity cycle with close resemblance to the human conditions, e.g. loss of consolidated sleep, increased activity during the rest phase and sleepiness during wake (Table 1). Importantly, these disruptions are present whether the animals are held under the regular light/dark (LD) cycle or in constant darkness (DD). The latter is critical to identify a compromised circadian system. Disorganized circadian timing alters the function of key organ systems throughout the body (Colwell, 2015). Collectively this prior research supports the hypothesis that circadian dysfunction should be recognized as an integral component of HD pathophysiology. Here, we summarize our progress using Q175 line of mice to explore the role of circadian system in HD and describe findings obtained by carrying out a computational/network analysis of our published data.
Table 1:
Here we summarize the described circadian phenotypes from HD models.
| Phenotype | Models | Citations |
|---|---|---|
| Low amplitude activity rhythms | R6/2, BACHD, Q175, HD rats, HD sheep | Morton et al., 2005; Bode et al., 2009; Kudo et al., 2011; Oakeshott et al., 2011; Balci et al., 2013; Loh et al., 2013; Morton et al., 2014; Kuljis et al., 2016 |
| Hypoactivity during active phase | BACHD, Q175, | Morton et al., 2005; Kudo et al., 2011; Loh et al., 2013; Kuljis et al., 2016; |
| Hyperactivity during rest phase | BACHD, Q175 | Kudo et al., 2011; Loh et al., 2013; Kuljis et al., 2016; |
| Increase in cycle to cycle variation | BACHD, Q175 | Kudo et al., 2011; Loh et al., 2013; Kuljis et al., 2016; |
| Disrupted rhythms in sleep, sleep fragmentation | R6/2, R6/1, Q175 | Fisher et al., 2013; Kantor et al., 2013; Loh et al., 2013; Lebreton et al., 2015; Fisher et al., 2016 |
| Low amplitude rhythms in autonomic driven rhythms in heart rate, core body temperature. | R6/1, R6/2, BACHD, Q175 | Kudo et al., 2011; Kiriazis et al., 2012; Mielcarek et al., 2014; Schroeder et al., 2016; Cutler et al., 2017 |
| Disrupted rhythms in hormones | R6/2 | Dufour and McBride, 2016; Rudenko et al., 2019. |
| Loss of melanopsin expressing retinal ganglia cells that carry light information to the central circadian clock (SCN) | R6/2, N171-82Q | Ouk et 2016; Lin et al., 2019 |
| Disrupted rhythms in electrical activity in the SCN | BACHD, Q175 | Kuljis et al., 2016; Kuljis et al., 2018 |
| Disrupted gene expression rhythms in central clock | R6/2 | Morton et al., 2005 |
| Disrupted gene expression rhythms outside SCN | R6/2 | Morton et al., 2005; Maywood et al., 2010 |
| Pathology in central clock | R6/2, BACHD | Fahrenkrug et al., 2007; Kuljis et al., 2016; |
| Circadian based treatments improving function | R6/2, BACHD, Q175 | Pallier et al., 2007; Maywood et al., 2010; Cuesta et al., 2014; Skillings et al., 2014; Wang et al., 2017; Whittaker et al., 2017; Wang et al., 2018; |
Methods
The methods and raw data used in this study have been previously published (Loh et al., 2013; Cutler et al., 2015; Kuljis et al., 2018). In the present study, we re-analyzed this prior work using new standards of statistical analysis and graphical representation of the data.
Animals
All the experimental protocols used to collect the data used in the present report were approved by the UCLA Animal Research Committee, and followed the guidelines and recommendations for animal use and welfare set by the UCLA Division of Laboratory Animal Medicine and National Institutes of Health. Data were collected using homozygous (Hom) and heterozygous (Het) for the Q175 allele mice and wild types (WT) from 3 to 12 months of age. The Q175 mice arose from a spontaneous expansion of the CAG repeat in the CAG140 transgenic knock-in line (Menalled et al., 2012) and have around 190 CAG repeats. Mutant mice were obtained from the Jackson Laboratory (Bar Harbor, Maine) from a colony managed by the CHDI Foundation. Only male mice were used in this study as female mice exhibit a delay in the expression of the symptoms (Kuljis et al., 2016). Genotyping was performed at 15 days of age by tail snips, and after weaning, littermates were group housed by sex. All animals were housed in sound proof, humidity controlled chambers with controlled lighting conditions, using a 12 hr light, 12 hr dark cycle (12:12 LD, intensity 300 lux) for at least two-weeks prior to experimentation.
Modeling
Telemetry data was collected from the same mice when they were young (3–6 months-old, mo) and middle age (9–12 mo) under constant dark (DD) conditions. The analysis examined 5 variables in each recording, all binned to means of 20 second intervals: heart rate (HR), heart rate variability (HRV), R-R interval (RR), core body temperature (CBT), and locomotor activity (Act). Missing data were impugned using a linear interpolation. Data were then transformed into Z-scores for each individual in each age group.
Frequency characteristics of each variable within each individual were generated using wavelet transformations (Leise, 2013; Smarr et al., 2017) which were then compared using wavelet coherence (Chang and Glover, 2010). Briefly, wavelet transforms provide frequency-by-time information for non-stationary signals (those with inconstant frequency); this is as opposed to a Fourier transform, which lacks temporal resolution. Here we used a morlet wavelet, which is appropriate because it has an established history of use in distinguishing circadian and ultradian changes in rodents and humans (Leise, 2013; Smarr et al., 2016; 2017). Wavelet coherence is a method to compare wavelet transforms of two different variables, providing a measure of how much change in frequency strength at a given time in one corresponds to similar change in the other – how coherent are the two signals, as measured by frequency-by-time. Wavelet transforms were run for all individuals for all variables, and ultradian bands were extracted by taking the timepoint-by-timepoint median power across all frequencies between 2–5h, creating an average power of ultradian frequency per time. Ultradian bands of each pair of variables were then compared by wavelet coherence, and the circadian band of coherence was then extracted by taking the timepoint-by-timepoint median power across all frequencies between 23–25h, creating an average power of circadian frequency per time. This in summary generated a quantitative measure of the extent to which ultradian rhythms for each pair of variables within an individual are being similarly modulated by the time of day.
Networks were constructed of nodes connected by edges. Each variable was defined as a node, or intersection point on the network. Edges were then defined by connecting each unique variable pair with an edge of weight equal to the sum over time of power of the pair’s circadian coherence band. Network graphs were created for each individual at each age, and group representative networks were constructed using the medians of these edge weights. Edge weights were then normalized across all 6 graphs (3 genotypes x 2 ages) so that colors could be assigned by relative edge weight to allow for visual comparison across graphs.
Statistical Analysis
Statistical significance for the behavioral and physiological experiments was determined by two-way analysis of variance (ANOVA) with genotype (WT, Het Q175, Hom Q175) and time as factors. If the data did not exhibit a normal distribution, a two-way ANOVA on ranks was used. Post-hoc t-test was applied to determine significant differences when appropriate. If the measurements were obtained at only one age, we used one-way ANOVA followed by Bonferroni’s multiple comparison test or one-way ANOVA on ranks. Statistical analyses were performed using the SigmaStat statistical software (San Jose, CA). Values are reported as the mean ± standard deviation (SD).
For network analyses, comparisons of average graph edge weights by condition, as well as within-individual comparisons of specific edge-weight change with age, were accomplished using Kruskal Wallis non-parametric test. Post-hoc non-parametric rank-sum tests were adjusted for multiple comparison by the Bonferroni method. Non-parametric tests were chosen to avoid assumptions of normality. For the specific edge-weight change with age of RR-CBT, a centroid was calculated as the center of weight of a k-means cluster defined by the WT distribution, and geometric distance from that centroid was then calculated for each individual.
Results & Discussion
When looking for possible circadian dysfunction in mouse models, the first analysis is commonly measurement of locomotor activity rhythms when mice are held in constant darkness using 24/7 automated monitoring. These rhythms are most commonly recorded under conditions in which the mouse has the opportunity to run on a wheel and thus represent voluntary motor activity. We commonly measure period, phase, fragmentation, and amplitude of the rhythms. For circadian biologists seeking to understand the mechanisms underlying the molecular clock, the measurement of circadian period has proven to be the most reliable phenotype (Takahashi et al., 2008). However, disease processes do not seem to target the core molecular clock mechanisms but instead impact the parameters of phase, fragmentation (or coherence), and amplitude. These parameters can be influenced by processes downstream of the core circadian clock and are more vulnerable to disruption.
Decline in circadian rhythms of wheel running activity
In previous work, we used wheel-running activity to characterize the impact of the Q175 mutation on circadian locomotor activity from 3 to 12 months of age (Loh et al., 2013). Measured parameters from young (3–6 mo) homozygous (Hom) and heterozygous (Het) Q175 mutant mice were not significantly different from their WT counterparts, showing equally consolidated and precise rhythms in activity that were nocturnal in nature under 12:12 light dark (LD) or constant dark (DD) conditions (Loh et al., 2013). However, as the mice aged, the strength of the rhythm (power) declined sharply in the Q175 Hom mice, and by middle age (9–12 mo), circadian dysfunction was apparent (Fig. 1A). Quantification of the wheel-running activity measured in DD at 12 mo of age (one-way AVOVA on Ranks, Fig. 1B–E) found significant differences in the activity levels (H = 18.354, P < 0.001), power (H = 15.839, P < 0.001) and cycle-to-cycle variation in the activity onset (H = 10.978, P = 0.04). The Q175 Het exhibited a much less impacted age-related decline in activity levels and power, suggesting an effect of gene dosage (Loh et al., 2013; Fig. 1). A variety of studies has now demonstrated that four distinct HD models exhibit a progressive and rapid breakdown of the circadian rest/activity cycle (Bode et al., 2009; Kudo et al., 2011; Loh et al., 2013; Morton et al., 2005; Oakeshott et al., 2011). These assays have the advantage that the mice are undisturbed in their home cages during the test and circadian disruption occurs early in disease progression. While these changes were concomitant with a decline in general locomotor activity, it is critical to note that precision of the daily onset of activity also deteriorated at the same time, suggesting that the circadian disruption is not simply due to motor dysfunction. To our way of thinking, the decline in precision and power of rhythms is indicative of an intrinsically weaker circadian oscillator whereas a reduction in the amount of activity is more likely driven by a weaker motor system. The cycle-length (tau) of the activity rhythms was not affected by the mutation implying that the core molecular feedback loop driving circadian oscillations is still functional. Nevertheless, we feel that these data indicate that circadian disruption of locomotor activity provides a sensitive marker of disease progression.
Fig. 1.

Circadian deficits in locomotor activity in Q175 mutant mice. (A) Representative double plotted actograms of wheel running activity from age-matched WT (left), Het Q175 (middle) and Hom Q175 Hom (right) mice under 12:12 light:dark (LD) and constant darkness (DD) are shown. The white/black bars on top indicate the LD cycle, and gray shading indicates darkness. Successive days of activity are plotted from top to bottom. An analysis of the data the data collected from 12 month old mice in DD found significant alterations in the wheel running activity (B), power (C), and cycle-to-cycle variation (D) in activity onset. The free-running period did not vary with genotype although the Hom Q175 exhibit higher variation (E). Individual data points are shown with WT (black circle, n=8), Het Q175 (grey triangle, n=7) and Hom Q175 (red diamonds, n=8). The bar graphs show means and standard deviation from the mean. The asterisk indicates significant difference (P < 0.05) from WT. Data from Loh et al., 2013.
In order to function adaptively, the circadian timing system must to be synchronized to the environment and timing of light is particularly important for this entrainment. The Q175 line did show some deficits in the light-response by middle age (Loh et al., 2013). For example, the Hom Q175 were more activity in the light than is typical for a nocturnal organism and; in an LD cycle, the mutant mice exhibited high levels of cycle-to-cycle variability whereas WT mice exhibit precise activity onset. Intrinsically photosensitive retinal ganglion cells (ipRGCs), which express the photopigment melanopsin, are photosensitive neurons in the retina and are essential for circadian synchronization (e.g. Schmidt et al., 2011). Five subtypes of ipRGCs (M1-M5) have been identified in mice and recent work suggests that the M1 subtype is reduced in other HD models (R6/2 and N171–82Q) prior to the onset of motor deficits (Ouk et al., 2016; Lin et al., 2019). The reduced innervation of M1 ipRGCs is likely to contribute to a diminished light-induction of c-fos in the central clock, the suprachiasmatic nucleus (SCN), which may explain the impaired circadian light response in these HD mice. Based on these data, we expect that we would ultimately detect loss of the M1 cell population in the retina of older Q175 mice.
Another important signaling pathway that may be involved in the reduction of the photic regulation of the circadian system is brain-derived neurotrophic factor (BDNF). Deficits in BDNF expression, trafficking, and signaling have been found in tissue from HD patients and HD mouse models (e.g. Zuccato and Cattaneo, 2007; Smith-Dijak et al., 2019). BDNF also appears to be an important regulator of synaptic input into the SCN. The transcriptional repressor methyl-CpG-binding protein 2 (MECP2) is expressed in the SCN and this Bdnf regulator is phosphorylated in response to light (Zhou et al., 2006). Both BDNF and its high-affinity tropomyosin-related receptor kinase (TrkB) receptor are expressed in the SCN (Liang et al., 1998; Allen & Earnest, 2005; Baba et al., 2008). Physiological data has demonstrated that BDNF and neurotrophin receptors can enhance glutamatergic synaptic transmission within a subset of SCN neurons (Kim et al., 2006) and potentiate glutamate-induced phase shifts of the circadian rhythm of neural activity in the SCN (Michel et al., 2006). Functionally, both BDNF- and TrkB-deficient mice exhibit a reduction in the behavioral effects of light on the circadian system (Liang et al., 2000; Allen et al., 2005). Therefore, an HD-driven reduction in BDNF would be expected to disrupt photic regulation of the circadian system.
Deficits in temporal patterning of sleep
The circadian system is intimately involved in the control of the temporal patterning of sleep although apparently not in the homeostatic regulation of sleep (e.g. Eban-Rothschild et al., 2017). Next, we used video recording to measure sleep as defined by time spent immobile, in combination with automated mouse tracking analysis software (Loh et al., 2013). The 6-hr bins of hourly immobility-defined sleep in mice held in DD clearly showed a decline in the amount of total sleep per cycle in the middle age Q175 Hom mice (Fig. 2A) with a significant effect of both genotype (F2,91 = 15.375, P < 0.001) and time (F3,91 = 63.072, P < 0.001). In particular, the Hom Q175 mutants spent less time sleeping throughout the circadian cycle (Fig. 2B; one-way ANOVA, F2,21 = 10.174, P < 0.001) and specifically slept less during the subjective day (CT 0–5 & 6–11, Fig. 2C; one-way ANOVA, F2,21 = 13.351, P = 0.001), and no significant changes were observed during the subjective night (data not shown). To examine sleep quality in the Q175 model, fragmentation analysis was performed to determine the number of sleep bouts and the average duration during both the (subjective) day and night. At 9–12mo, increased fragmentation became apparent with more sleep bouts of a shorter average duration in the Hom Q175 mutant (Loh et al., 2013). Hence, Q175 mutation appears to selectively impact daytime sleep in the Hom mutants, with a specific reduction of the amount of time spent asleep, as well as increased fragmentation of an already reduced sleep.
Fig. 2:

Sleep behavior is altered in the Hom Q175. (A) The amount of sleep (6 hr bins) measured from 12 month old mice of each genotype is plotted. All of the mice slept more in the subjective day than in the subjective night. By convention, CT 12 indicates the onset of daily activity in nocturnal animals held in constant darkness. The Hom Q175 sleep less at CT 0–5 and 6–11 but no significant differences were observed in the subjective night. The Hom Q175 mice exhibited less sleep through the circadian cycle (B) and during the subjective day (C). Individual data points are shown with WT (black circle, n=8), Het Q175 (grey triangle, n=7) and Hom Q175 (red diamonds, n=7). Data from Loh et al., 2013.
The video-based assay that is independent of the animal’s ability to turn a wheel, we found that daytime sleep was selectively affected by the Q175 mutation. While video analysis of sleep does not allow us to draw conclusions regarding the depth of sleep nor the relative progression from one state of sleep to another (e.g. NREM vs REM), such measurements have been shown to accurately measure sleep and to highly correlate with simultaneously recorded electroencephalogram (EEG)-defined sleep (Fisher et al., 2012). Prior work has carefully characterized age-related changes in the EEG of both Hom and Het Q175 (Fisher et al., 2016). The Q175 mice exhibited disrupted EEG starting at 4 mo of age providing clear evidence that the EEG can serve as an early biomarker in these models. However, there was no effect of genotype on the high amplitude delta waves (0.5–4 Hz) associated with slow-wave sleep (SWS) or in sleep time in response to sleep deprivation. Therefore, the circadian behavior and physiological rhythms were disrupted, while the sleep homeostatic mechanisms remain intact. In the R6/2 model, EEG-defined sleep showed strong fragmentation of the sleep/wake cycle and significant changes in the neural oscillatory patterns that can be seen on an EEG (Kantor et al., 2013; Fisher et al., 2013). In response to a homeostatic challenge, the R6/2 line did show deficits in recovery sleep (Fisher et al., 2013). The increase in sleep fragmentation is commonly associated with the sleep disturbances observed in HD patients (e.g. Goodman et al., 2011) and there is a growing appreciation that quantitative EEG may serve as a biomarker for HD. (Leuchter et al., 2017; Odish et al., 2018).
Deficits in generalized activity as measured by telemetry
We examined the circadian rhythms in general activity in young and middle aged Q175 mice by continuously monitoring the animals with a telemetry system (Cutler et al., 2017). Telemetry measurements of activity would record all movements as compared to running wheels that only capture voluntary motor activity. This sensitive method detected significant effects of both age and gene dosage, and most importantly, deficits in nocturnal activity even in the Het and Hom young mice in DD (Fig. 3A). Analysis of the activity with 2-way ANOVA found significant effects of genotype (F2,99 = 20.412, P < 0.001) and time (F3,99 = 50.875, P < 0.001). These deficits were dramatically accentuated in the middle aged mutants (Fig. 3B) with significant effects of both genotype (F2,91 = 20.772, P < 0.001) and time (F3,99 = 35.704, P < 0.001). At both ages, the most significant changes were observed during the active phase (subjective night, CT 12–17 and 18–23).
Fig 3:
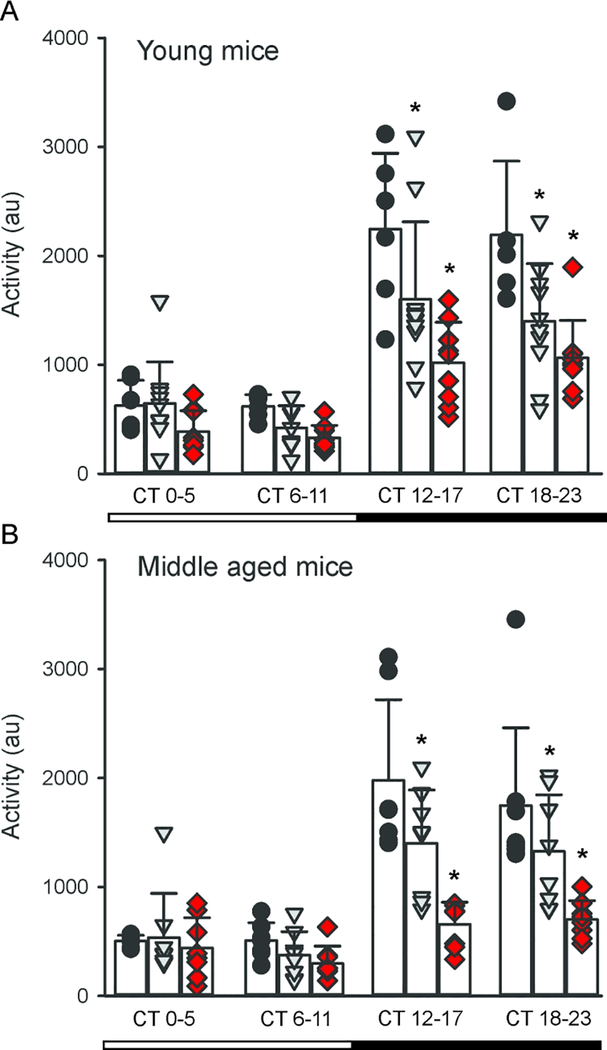
Q175 mice exhibited reduced amplitude circadian rhythms of the activity as measured by telemetry. Whereas wheel-running represents voluntary motor activity, telemetry captures all movement. All of the mice exhibited significant subjective day-night differences in activity. (A) The activity of young mice (3–4 months) was unaltered during the subjective day but significantly reduced throughout the subjective night in the Het and Hom Q175 compared to WT. (B) At middle age (9–12 mo), a very similar pattern is observed. Individual data points are shown with WT (black circle, n=6), Het Q175 (grey triangle, n=10) and Hom Q175 (red diamonds, n=9). Data from Cutler et al., 2017.
Difficulty in maintaining CBT
The circadian clock in the SCN regulates daily rhythms in core body temperature (CBT) and these rhythms offer a fairly direct measure of SCN output (Szymusiak, 2018). The circadian rhythms in CBT are independent of locomotor activity but dependent on an intact SCN (Stephan and Nunez, 1977; Ruby et al., 2002; Scheer et al., 2005). The SCN projects to the dorsal subparaventricular zone (SPZ) (Leak and Moore, 2001), which is necessary for driving circadian rhythms of body temperature (Lu et al., 2001). We used telemetry to examine circadian rhythms in CBT in young and middle aged mice Q175 mice (Cutler et al., 2017). Under DD conditions, the measurements of CBT obtained by telemetry showed the expected rhythms with higher body temperature during the active phase in the young Q175 mutant mice and their WT counterparts (Fig. 4A), with a significant effect of time (F3,99 = 248.655, P < 0.001) but not of genotype (F2,99 = 0.268, P = 0.765). By middle age, both genotype (F2,91 = 17.236, P < 0.001) and time (F3,91 = 65.568, P < 0.001) (Fig. 4B) had a significant effect on CBT rhythm. In particular, the Hom Q175 exhibited difficulty in maintaining the body temperature in the first half of the day (subjective day, CT 0–5). The HD mutants presented with hypothermic episodes during which they failed to maintain their body temperature. Overall, the Q175 seemed to lack the capability to regulate their CBT and this problem emerged early in the Hom Q175 and was more pronounced by middle age.
Fig. 4:

Hom Q175 mice exhibited disrupted ability to maintain core body temperature (CBT) by middle age. All of the mice exhibited significant subjective day-night differences in CBT. (A) There were no differences between the genotypes the rhythms in CBT in the young adult mice (3–6 mo). (B) By middle age (9–12 mo), the Hom Q175 exhibited reduced CBT in the early subjective day. The mutant mice exhibited episodes of hypothermia that were most severe in the Hom Q175. Individual data points are shown with WT (black circle, n=6), Het Q175 (grey triangle, n=10) and Hom Q175 (red diamonds, n=9). Data from Cutler et al., 2017.
The circadian timing system is intimately linked to metabolism at a cellular, molecular and system level (Bass and Takashahi, 2010; Panda, 2016; Reinke and Asher, 2019). One of the most dramatic daily rhythms in the body is the feeding/fasting cycle in which an organism has a number of hours with abundant glucose followed by hours without. The circadian system regulates ingestive behaviors, the metabolic systems by which the food is processed, as well as the sleep/wake cycle. Thus, the circadian clock coordinates appropriate metabolic responses within peripheral tissues with the LD cycle. For example, the liver clock promotes gluconeogenesis and glycogenolysis during the sleep/fasting period, while fostering glycogen and cholesterol synthesis during the wake/feeding period. The CBT measures suggest that the circadian regulation of metabolism is compromised in the Q175 line and fits with prior work that demonstrated that HD patients as well as the Q175 mice have deficits in the tricarboxylic acid cycle (Naseri et al., 2015) responsible for the production of ATP. The diurnal variation in CBT was also attenuated in the R6/2 (Fisher et al., 2013) and BACHD mice (Kudo et al., 2011). Like the Q175 mutants, the R6/2 mice also exhibited episodes of hypothermia, particularly evident in the dark period. Overall, there is strong evidence for mitochondrial dysfunction in HD as well as in other neurodegenerative diseases (Kuhl et al., 1982; Naseri et al., 2015; Dubinsky, 2017; Carmo et al., 2018; Franco-Iborra et al., 2018). Unintended weight loss is a hallmark of HD patients and a recent study found that a high body mass index (BMI) is associated with a significantly slower rate of functional, motor and cognitive deterioration (van der Burg et al., 2017). Altogether, these findings justify further analysis of the links between circadian rhythms and metabolism in the context of HD.
Deficits in rhythms in heart rate as well as in heart rate variability
In nocturnal mice, diurnal and circadian of cardiovascular output peak in the night when the mice are active. While influenced by activity levels, the rhythms occur even when measured in windows of time when the mice were inactive. We recorded HR when the animals were physically inactive, i.e. the resting HR (Cutler et al., 2017). The WT and young Q175 mutant mice again showed similar rhythms in DD (Fig. 5A), again with no effect of genotype (F2,99 = 2.429, P = 0.094) but a significant effect of time (F3,99 = 307.788, P < 0.001). By middle age (Fig. 5B), we detected lower resting HR in the Hom Q175 kept in DD with significant effects of both genotype (F2,91 = 7.001, P = 0.002) and time (F3,91 = 25.035, P < 0.001). To further explore a possible autonomic dysfunction in the Q175 line, we measured HRV throughout the 24-hr cycle. HRV measures the variability of the time between individual heartbeats and reflects the balance of sympathovagal signals to the heart. High HRV is associated with cardiovascular health, while low HRV is a sign of poor cardiovascular function. We observed the normal high HRV during the day and lower at night (Fig. 6) in WT mice. Conversely, the HRV was abnormally low in the HD mutants (Hom & Het) and this damping was largest during the sleep phases (CT 0–11). HRV measures were separated into low frequency (LF, 0.2–1.5 Hz) and high frequency (HF, 1.5–4.0 Hz) bands, which are commonly used to quantify parasympathetic and sympathetic regulation, respectively (Campen et al., 2005). The power of the HF band was highly disrupted in the middle-aged Hom Q175 (Fig. 6A) exhibiting significant effects of both genotype (F2,91 = 9.396, P < 0.001) and time (F3,91 = 21.818, P < 0.001). In contrast, there were no differences in the power of the LF band (Fig. 6B) with genotype (F2,91 = 0.186, P = 0.831) but there were significant differences with time (F3,91 = 5.575, P = 0.002). These results clearly demonstrate that the temporal patterning as well as the overall level of autonomic regulation of the cardiovascular system is compromised in the Q175 mice.
Fig. 5:

Q175 mice exhibited altered circadian rhythms in resting HR by middle age. (A) There were no differences in the rhythms in HR between the genotypes in the young adult mice (3–6 mo). (B) By middle age (9–12 mo), the Hom Q175 show low resting HR in the early subjective day. At other phases the HR of the Hom mutants trended lower but these effects were not significant. Individual data points are shown with WT (black circle, n=6), Het Q175 (grey triangle, n=10) and Hom Q175 (red diamonds, n=9). Data from Cutler et al., 2017.
Fig. 6:

Q175 mice exhibited low HRV in the high frequency band. HRV measures were separated into high frequency (HF, 0.15–0.4 Hz) and low frequency (LF, 0.04–0.15 Hz) and bands, which are commonly used to quantify sympathetic and parasympathetic regulation, respectively. All genotypes exhibited subjective day/night differences in both frequency bands, The power of the HF band was disrupted with the middle-aged Hom Q175 (A) whereas the LF domain was unaltered (B). Individual data points are shown with WT (black circle, n=6), Het Q175 (grey triangle, n=10) and Hom Q175 (red diamonds, n=9).
Diurnal and circadian rhythms in HR are also disrupted in the BACHD, R6/1, R6/2 lines of mice (Mihm et al., 2007; Kudo et al., 2011, Wood et al., 2012; Schroeder et al., 2016). In agreement, we saw evidence in the BACHD line of the circadian system failing to lower blood pressure (BP) during sleep (Schroeder et al., 2011). The HR rhythms in R6/1 mice are disrupted with the mutants showing a higher HR than WT littermates as young adults but ultimately exhibiting low HR later in life (Kiriazis et al., 2012). In two other HD models (R6/2 and HdhQ150 lines), the HR was significantly reduced in symptomatic mice (Mielcarek et al., 2014). Altogether, data from several studies using different mouse models all found overlapping evidence for disrupted diurnal or circadian rhythms of HR. As far as we know, the question of whether the rhythms in HR and BP are disrupted have not been examined in HD patients. The preclinical data suggest that the disruptions are most likely to occur at the beginning of sleep and can be difficult to diagnose. Office visits are unlikely to coincide with the expression of hypertension. This type of “masked” or nocturnal hypertension is associated with poor clinical outcomes as the symptoms are likely to be untreated and patients frequently develop organ damage prior to transitioning to sustained hypertension which can be detected (Franklin et al., 2017; Bowles et al., 2018). Based on these data, HD patients should be strong candidates for 24-hr BP monitoring, which is the best method for uncovering this type of hypertension.
A decrease in HRV has also been reported during the pre-symptomatic and early stages of HD progression (Andrich et al., 2002; Kobal et al., 2004; Bär et al., 2008; Kobal et al., 2010). Prior studies reported HRV deficits during the Valsalva maneuver, hand-grip and head up tilt tests in HD patients (Aminoff and Gross, 1974; Den Heijer et al., 1988; Sharma et al., 1999; Bär et al., 2008). As far as we know, the spectral power analysis has not been carried out on data from patients. In human subjects, HRV changes with daily cycle and with sleep state (Boudreau et al., 2013) but we do not have this data from the HD patients. Thus, both the preclinical and clinical work is consistent with a disruption in the sympathetic branch early in disease progression that is reflected as a decreased HRV.
Deficits in neural activity in the suprachiasmatic nucleus
The central circadian clock is the SCN of the hypothalamus, whose rhythmic output drives rhythms in locomotor activity, sleep and the autonomic nervous system (Kalsbeek et al., 2006; Mohawk et al., 2012). Rhythmic spontaneous firing rates (SFR) are an intrinsic property of individual SCN neurons (Webb et al., 2009) and are always high during the day in both diurnal as well as nocturnal species. These SFR are driven by the rhythmic expression of ionic currents that depolarize the neuronal membrane potential toward the action potential threshold during the day and hyperpolarize the membrane potential to silence neurons at night (Allen et al., 2017). Normally, dSCN neurons exhibit high SFRs during the day and low SFRs during the night, but Het and Hom (6–7 mo) Q175 dSCN neurons displayed low SFRs both during both the day and the night (Kuljis et al., 2018; Fig. 7A). This dysfunction was significantly affected by both genotype (F2,117 = 6.309, P = 0.003) and time (F1,117 = 6.116, P = 0.015). One of the key potassium (K+) currents regulating AP frequency as well as hyperpolarizing the membrane at night is the large-conductance calcium activated potassium (BK) currents (Whitt et al., 2016). We measured the BK current in the Het Q175 in the daytime and found that the SCN neurons exhibited significantly greater BK current at some voltage steps (Fig. 7B). An analysis of this data found effects of genotype (F1,399 = 8.912, P = 0.003) and voltage (F9,399 = 51.786, P < 0.001). In particular, significant effects were seen when the membrane potential was stepped to 60 and 80mV. Thus, the Q175 HD model exhibits low daytime electrical activity with enhanced BK currents in the SCN.
Fig. 7:

Q175 mice exhibited reduced spontaneous firing rate in SCN during the day. (A) Using the current-clamp recording technique in the whole-cell configuration, the SFR in dSCN neurons was measured during the day and night. The Q175 dSCN neurons did not exhibit the day/night difference in SFR seen in WT neurons. Firing rate was measured in the day (WT, n=12 neurons, Het Q175, n=21, Hom Q175, n=22) and night (WT, n=23 neurons, Het Q175, n=17, Hom Q175, n=23). (B) BK currents measured in Het Q175 dSCN neurons were significantly larger than those in WT during the day (ZT 5–7). Two-way ANOVA was used to identify significant effects of genotype and/or voltage step magnitude on evoked currents. There were significant effects of genotype on the BK current at the 60 and 80 mV steps. BK current were measured in WT (n=11 neurons) and Het Q175 (grey triangle, n=9). Data from Kuljis et al., 2018.
These findings demonstrate that the SCN neurons in the Q175 mouse models exhibit malfunctioning likely triggered, at least in part, by dysregulation of BK current. Prior work has also found some evidence of altered transcription of genes coding for BK channels (Langfelder et al., 2016) in the Q175 line. This study found that mutants had altered expression of Kcnmb2 in the hypothalamus and of Kcnmb4 in cortical tissue. HD associated transcription dysregulation, including increased CRE-mediated transcription, has previously been shown (Obrietan and Hoyt, 2004; Sugars and Rubinsztein, 2003; Langfelder et al., 2016). CREB is a known positive regulator of BK expression (Wang, Ghezzi, Yin, & Atkinson, 2009) and we have shown alterations in Kcnma1 transcript levels in the BACHD SCN (Kuljis et al., 2018).
A disruption in the neural output from the SCN provides a plausible explanation for the disrupted behavioral and physiological rhythms seen in HD models but, of course, is not yet conclusive. Furthermore, we have shown that the SCN neurons display significantly reduced daytime SFR also in the BACHD line (Kuljis et al., 2018); however, work in the R6/2 model did not find similar firing rate deficits (Pallier et al., 2007; Williams et al., 2011). Instead, it was reported that the R6/2 mice exhibit a reduction in the neural activity in orexin neurons. This cell population in the lateral hypothalamus receives input from the SCN and is critical regulator of arousal state (e.g. Arrigoni et al., 2018). Therefore, it is not yet clear whether a reduction in daytime firing rate in the SCN is a common feature across these mouse models. Future work should use in vivo recording techniques to measure the neural activity rhythms within the intact SCN look at different phases within the daily cycle. Since the mutant mice are behaviorally rhythmic until the late stages of disease progression, we speculate that we will still see a rhythm of neural activity in the mutants, just at a lower amplitude than what we measure in WT mice.
In other brain regions, these is evidence for disrupted potassium currents in neurons and astrocytes in HD models (Zhang et al., 2018). For example, striatal neurons in the R6/2 and Q175 models exhibit increased excitability already before the onset of motor deficits (Sebastianutto et al., 2017). These changes are associated with decreased function of inward rectifier and voltage-gated potassium channels. To provide another example, Surmeier and co-workers (2019) recently found that the dendrites of the neurons in the striatal indirect pathway are hypoexcitable. This low activity was due to an increased association of dendritic Kv4 potassium channels with auxiliary KChIP subunits and could be reversed by knocking down the Kv4 channels or by just lowering the mutant-Htt (mHtt) levels with a zinc finger protein (Carrillo-Reid et al., 2019). These broader data support our inference that malfunctioning potassium channels in the SCN are responsible for the weakened SCN output.
Aged SCN also exhibits reduced amplitude of SFR rhythms (Nakamura et al., 2011; Farajnia et al., 2012) reminiscent of the phenotype observed in Q175 SCN. Prior work indicates a selective loss of circadian modulation of the fast-delayed rectifier and A-type potassium currents in the aging SCN (Farajnia et al., 2012), as well as loss of circadian modulation of BK channel activity resulting in a reduction of nighttime BK currents (Farajnia et al., 2015). Loss of nighttime BK current in aged SCN neurons was associated with diminished AP afterhyperpolarization, depolarized resting membrane potential, widened AP and increased intracellular calcium levels. While there are clearly differences between the findings in aged WT and Q175 SCN pathophysiology, these findings suggest that alterations in BK currents may be a common path to generate inappropriate neuronal activity in the SCN.
Network analysis: a different re-analysis of old data
The telemetry recordings allowed the simultaneous measurement of activity, CBT, HR, R-R interval, and HRV from individual mice for 5 days at age 3 months (young), and for another 5 days at age 9 months (aged). Due to the findings of sleep and circadian degradation, we sought to investigate whether degradation might be related to internal desynchrony, or loss of organizational coherence from one physio-behavioral variable to another. We Z-transformed the data to highlight relative timing of change across variables. Visual inspection revealed that each variable was composed of within-a-day ultradian episodes of rapid change, and that these were organized by time-of-day (Fig. 8A). Comparison confirmed that in healthy animals, the timing of these ultradian episodes appeared coordinated across variables (Fig. 8B). To quantify each individual’s internal synchrony, we generated a feature of temporal organization based on the strength of circadian coherence across each pair of variables’ ultradian rhythms (see Methods).
Fig. 8:

Example data from one healthy individual showing phase relationships in circadian and ultradian rhythms (less than 24 hr, UR). Records of averages per 20 seconds (A, top to bottom) of the heart’s R-R interval, heart rate variability (HRV), average heart rate (HR), core body temperature (CBT) and locomotor activity (Act). Aligning these reveals that some variables show phase alignment at both circadian and ultradian time scales (e.g. HR and CBT, B), while others display anti-phase relationships at both frequencies (e.g. RR and Act, C). In all cases, ultradian rhythm amplitude is modulated by time of day, so that the circadian-ultradian interaction must be taken into account when comparing phase alignment synchrony across these different physio-behavioral outputs. Data from Cutler et al., 2017.
We then used these features to compare internal coordination across each pair of variables, constructing a graph network for each group which captured the average strength of coordination for each pair of variables when young and also when aged, for each genotype (Fig. 9). The graphs show coherence as edges (lines, blue is high coherence, red is low) between two nodes (variables at vertices). Each edge is normalized so that edge colors are comparable across all 6 networks shown. We found a significant difference in overall network coherence (median edge weight) across conditions (χ2 = 23.11, P = 0.0003). Post hoc comparisons revealed that there had been significant decreases in the Het and Hom groups with age, but not in the WT groups (Fig. 9B). These effects are visually clear from the networks, but it is also clear that different edges respond differently to condition and age.
Fig. 9:

Network visualizations allow identification of specific relationships for deeper future explorations, as compared to pan-network group averages. (A) Networks constructed of all possible pairs of circadian coherence of ultradian rhythms between RR, HR, HRV, CBT, and Act allow visual and quantitative assessment of the overall physio-behavioral synchrony within each genotype (from left to right: WT, Het Q175, and Hom Q175). Blue indicates higher relative coherence, red indicates lower relative coherence. (B) Scatter plots compare the average edge strengths across time and genotype (WT: black circle, n=10; Het: grey triangle, n=9; Hom: red diamond, n=8). Edges show significant decreases between young and aged Het and Hom, but not WT networks (*), suggesting they can act as biomarkers for detection of deterioration of network coherence with age in Q175 mice. Data from Cutler et al., 2017.
Because the network model presents all relationships simultaneously, such network graphs might be useful in guiding future investigations into particular relationships. For example, in the edge RR-HR, there is a significant (and visually clear; χ2 = 6.1, P = 0.048) loss of coherence with age in a Q175 dose-dependent manner (Fig. 10A). By contrast, in the edge RR-CBT there is not a significant loss of coherence, but there is an increase in dissimilarity (scattering away from the healthy average) across individuals within each genotype (Fig. 10B; χ2 = 10.07, P = 0.0065). These edge-wise comparisons suggest that different mechanisms may be implicated in different forms of progressive physio-behavioral disorganization that arise in HD.
Fig. 10:

Specific edges can be examined for their role in identifying changes by age, genotype, or interactions. Edge-wise comparisons within individuals’ network graphs finds that different edges contain different kinds of information. RR-HR coherence significantly declines with age in a gene-dose dependent manner (A,B), seen by an increase in average distance from the line x=y (dotted, A; * B). By comparison, RR-CBT coherence shows no effect of age, but there is a gene-dose dependent increase in the population variance of coherence values (C, D), seen as distance from the WT centroid (pale grey dot), so that topographically the coherence values form a target shape, with a tightly clustered WT (black circles) bull’s eye, and wider rings of heterozygote (grey triangles) and homozygote (red diamonds). These findings demonstrate that coherence relationships across variables will provide a rich feature set for future exploration of potentially useful biomarkers. Data from Cutler et al., 2017.
In summary, these data support a coupled oscillator network model of health, in which temporal coordination across systems creates a stable, healthy network, while disorganization leads to loss of wellness. Importantly, these data demonstrate that disorganization also generates a detectable deviation in oscillating signals that can be detected to follow disease progression in groups and in individuals. Additionally, these finding support the idea that information from one measured parameter carries information about other parameters. There may be shared outputs, including HRv, that provide high temporal-resolution information about the entire network through noninvasive means. This type of analysis may provide useful biomarkers for looking at the impact of interventions on HD progression. With the rapid development of wearable technology, we can speculate that we may be able to generate predictive measures of outcome with noninvasive, biologically relevant data gathered continuously within individuals from broad populations.
Future work
In summary, disturbances in the timing of the sleep/wake cycle are a well-established symptom of HD and commonly present in other neurodegenerative diseases. These behavioral symptoms raise questions about the underlying mechanisms and the possible involvement of the circadian system. Using mouse models (Table 1), we have demonstrated behavioral and physiological circadian disruptions early in disease progression. Notably, these findings do not preclude the involvement of other well-known structures involved in HD, such as the basal ganglia, in eliciting a disrupted circadian output. The dysfunction found in the circadian system of multiple HD models does however raise the question of whether it is possible to ameliorate such symptoms and, in the end, to “treat” HD and other neurodegenerative diseases using environmental manipulations designed to improve circadian rhythms and enhance the sleep/wake cycle by increasing the excitatory drive onto the SCN (Morton, 2013; Schroeder and Colwell, 2013). Prior work using the R6/2 model found that a combination of bright-light therapy and scheduled voluntary exercise was beneficial in delaying disease symptoms (Cuesta et al., 2014). We have reported that exposing Q175 mice to a blue-wavelength enhanced lighting (Wang et al., 2017) and time-restricted feeding (Wang et al., 2018) was similarly beneficial in delaying symptom progression. Since behavioral changes may be difficult to implement, thus, it is also important to develop pharmacological treatments. In the Q175 model, timed administration of a blocker of histamine receptor type 3 blocker (Whittaker et al., 2017) has proven to be beneficial. Hence, preclinical HD models are clearly advantageous for exploring disease mechanisms as well as the impact of circadian treatments and therapies on disease progression.
Significance.
Here we summarize evidence that several of the phenotypes of the Q175 line were found to vary with a diurnal and circadian phase dependence. Thus, the time of measurement and the circadian cycle should be considered a critical factor in the expression and assessment of HD symptoms.
The Q175 mouse model of HD exhibits cardiovascular symptoms similar to those seen in HD patients with a prominent sympathetic dysfunction during the resting phase. This line of mice provides a suitable model for preclinical studies of therapeutics designed to prolong health-span in HD.
A network analysis found evidence for disrupted phase coherence between activity, core body temperature, and cardiovascular measures even in young Q175 mutants. This result suggests that loss of phase coherence is a variable that should be considered as a possible biomarker for HD.
Acknowledgments
We would like to acknowledge the excellent training and support from Dr. M. Levine. Besides making many seminal contributions to research into basal ganglia function, Dr. Levine has been an important contributor to the vitality of the UCLA neuroscience community.
REFERENCES
- Allen CN, Nitabach MN, Colwell CS. 2017. Membrane Currents, Gene Expression, and Circadian Clocks. Cold Spring Harb Perspect Biol. 9(5). pii: a027714. doi: 10.1101/cshperspect.a027714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GC, Earnest DJ. 2005. Overlap in the distribution of TrkB immunoreactivity and retinohypothalamic tract innvervation of the rat suprachiasmatic nucleus. Neurosci. Lett, 376: 200–204. [DOI] [PubMed] [Google Scholar]
- Allen GC, Qu X, Earnest DJ. 2005. TrkB-deficient mice show diminished phase shifts to the circadian activity rhythm in response to light. Neurosci. Lett, 378: 150–155. [DOI] [PubMed] [Google Scholar]
- Aminoff MJ, Gross M. 1974. Vasoregulatory activity in patients with Huntington’s chorea. J Neurol Sci. 21: 33–38. [DOI] [PubMed] [Google Scholar]
- Andrich J, Schmitz T, Saft C, Postert T, Kraus P, Epplen JT, et al. 2002. Autonomic nervous system function in Huntington’s disease. J Neurol Neurosurg Psychiatr. 72: 726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni E, Chee MJS, Fuller PM. 2018. To eat or to sleep: That is a lateral hypothalamic question. Neuropharmacology. pii: S0028–3908(18)30853–0. doi: 10.1016/j.neuropharm.2018.11.017. [DOI] [PubMed] [Google Scholar]
- Baba K, Ono D, Honma S, Honma K. 2008. A TTX-sensitive local circuit is involved in the expression of PK2 and BDNF circadian rhythms in the mouse suprachiasmatic nucleus. Eur J Neurosci. 27(4):909–16. [DOI] [PubMed] [Google Scholar]
- Balci F, Oakeshott S, Shamy JL, El-Khodor BF, Filippov I, Mushlin R, Port R, Connor D, Paintdakhi A, Menalled L, Ramboz S, Howland D, Kwak S, Brunner D. 2013. High-Throughput Automated Phenotyping of Two Genetic Mouse Models of Huntington’s Disease. PLoS Curr. 5 pii: ecurrents.hd.124aa0d16753f88215776fba102ceb29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bär KJ, Boettger MK, Andrich J, Epplen JT, Fischer F, Cordes J, et al. 2008. Cardiovagal modulation upon postural change is altered in Huntington’s disease. Eur J Neurol. 15: 869–871. doi: 10.1111/j.1468-1331.2008.02173.x. [DOI] [PubMed] [Google Scholar]
- Bass J, Takahashi JS. 2010. Circadian integration of metabolism and energetics. Science 330(6009):1349–54. 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. 2015. Huntington disease. Nat Rev Dis Primers. 1: 15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- Bode FJ, Stephan M, Wiehager S, Nguyen HP, Björkqvist M, von Hörsten S, Bauer A, and Petersén A 2009. Increased numbers of motor activity peaks during light cycle are associated with reductions in adrenergic alpha(2)-receptor levels in a transgenic Huntington’s disease rat model. Behav. Brain Res 205: 175–182. [DOI] [PubMed] [Google Scholar]
- Boudreau P, Yeh W-H, Dumont GA, Boivin DB. 2013. Circadian variation of heart rate variability across sleep stages. Sleep. 36: 1919–1928. doi: 10.5665/sleep.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles NP, Thosar SS, Herzig MX, Shea SA. 2018. Chronotherapy for Hypertension. Curr Hypertens Rep. 20: 97. doi: 10.1007/s11906-018-0897-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campen MJ, Tagaito Y, Jenkins TP, Balbir A, and O’Donnell CP 2005. Heart rate variability responses to hypoxic and hypercapnic exposures in different mouse strains. J. Appl. Physiol 99:807–813. [DOI] [PubMed] [Google Scholar]
- Carmo C, Naia L, Lopes C, Rego AC. 2018. Mitochondrial Dysfunction in Huntington’s Disease. Adv Exp Med Biol. 1049:59–83. doi: 10.1007/978-3-319-71779-1_3. [DOI] [PubMed] [Google Scholar]
- Carrillo-Reid L, Day M, Xie Z, Melendez AE, Kondapalli J, Plotkin JL, Wokosin DL, Chen Y, Kress GJ, Kaplitt M, Ilijic E, Guzman JN, Chan SS, Surmeier DJ. 2019. Mutant huntingtin enhances activation of dendritic Kv4 K+ channels in striatal spiny projection neurons. Elife. 8 pii: e40818. doi: 10.7554/eLife.40818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JB, Bates GP, Steffan J, Saft C, Tabrizi SJ. 2015. Treating the whole body in Huntington’s disease. Lancet Neurol. 14(11):1135–42. doi: 10.1016/S1474-4422(15)00177-5. [DOI] [PubMed] [Google Scholar]
- Ciammola A, Sassone J, Alberti L, Meola G, Mancinelli E, Russo MA, et al. 2006. Increased apoptosis, huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington’s disease subjects. Cell Death and Differentiation. 13: 2068–2078. doi: 10.1038/sj.cdd.4401967. [DOI] [PubMed] [Google Scholar]
- Circadian Medicine. Edited by CS Colwell Wiley; 2015, ISBN 978-1-118-46778-7. [Google Scholar]
- Cuesta M, Aungier J, Morton AJ. 2014. Behavioral therapy reverses circadian deficits in a transgenic mouse model of Huntington’s disease. Neurobiol Dis 63:85–91. doi: 10.1016/j.nbd.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Cutler TS, Park S, Loh DH, Jordan MC, Yokota T, Roos KP, et al. 2017. Neurocardiovascular deficits in the Q175 mouse model of Huntington’s disease. Physiol Rep. 5. doi: 10.14814/phy2.13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Heijer JC, Bollen WL, Reulen JP, van Dijk JG, Kramer CG, Roos RA, et al. 1988. Autonomic nervous function in Huntington’s disease. Arch Neurol. 45: 309–312. [DOI] [PubMed] [Google Scholar]
- Dubinsky JM. 2017. Towards an Understanding of Energy Impairment in Huntington’s Disease Brain. J Huntingtons Dis. 6(4):267–302. doi: 10.3233/JHD-170264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour BD, McBride JL. 2016. Corticosterone dysregulation exacerbates disease progression in the R6/2 transgenic mouse model of Huntington’s disease. Exp Neurol. 283(Pt A):308–17. doi: 10.1016/j.expneurol.2016.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eban-Rothschild A, Giardino WJ, de Lecea L. 2017. To sleep or not to sleep: neuronal and ecological insights. Curr Opin Neurobiol. 44:132–138. doi: 10.1016/j.conb.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenkrug J, Popovic N, Georg B, Brundin P, Hannibal J. 2007. Decreased VIP and VPAC2 receptor expression in the biological clock of the R6/2 Huntington’s disease mouse. J Mol Neurosci. 31(2):139–48. [DOI] [PubMed] [Google Scholar]
- Farajnia S, Michel S, Deboer T, Vanderleest HT, Houben T, Rohling JHT, Ramkisoensing A, Yasenkov R, Meijer JH. 2012. Evidence for neuronal desynchrony in the aged suprachiasmatic nucleus clock. J Neurosci 32:5891–5899. doi: 10.1523/JNEUROSCI.0469-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farajnia S, Meijer JH, Michel S. 2015. Age-related changes in large-conductance calcium-activated potassium channels in mammalian circadian clock neurons. Neurobiol Aging 36(6):2176–83. doi: 10.1016/j.neurobiolaging.2014.12.040. [DOI] [PubMed] [Google Scholar]
- Fisher SP, Black SW, Schwartz MD, Wilk AJ, Chen TM, Lincoln WU, Liu HW, Kilduff TS, Morairty SR. 2013. Longitudinal analysis of the electroencephalogram and sleep phenotype in the R6/2 mouse model of Huntington’s disease. Brain. 136(Pt 7):2159–72. [DOI] [PubMed] [Google Scholar]
- Fisher SP, Godinho SIH, Pothecary CA, Hankins MW, Foster RG, et al. 2012. Rapid assessment of sleep-wake behavior in mice. J Biol Rhythms 27: 48–58. doi: 10.1177/0748730411431550.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SP, Schwartz MD, Wurts-Black S, Thomas AM, Chen TM, Miller MA, Palmerston JB, Kilduff TS, Morairty SR. 2016. Quantitative Electroencephalographic Analysis Provides an Early-Stage Indicator of Disease Onset and Progression in the zQ175 Knock-In Mouse Model of Huntington’s Disease. Sleep. 39(2):379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Iborra S, Vila M, Perier C. 2018. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front Neurosci. 12:342. doi: 10.3389/fnins.2018.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AOG, Rogers L, Pilsworth S, McAllister CJ, Shneerson JM, et al. 2011. Asymptomatic sleep abnormalities are a common early feature in patients with Huntington’s disease. Curr Neurol Neurosci Rep 11: 211–217. doi: 10.1007/s11910-010-0163-x. [DOI] [PubMed] [Google Scholar]
- Kantor S, Szabo L, Varga J, Cuesta M, Morton AJ. 2013. Progressive sleep and electroencephalogram changes in mice carrying the Huntington’s disease mutation. Brain. 136(Pt 7):2147–58. [DOI] [PubMed] [Google Scholar]
- Kim YI, Choi HJ, Colwell CS. 2006. Brain-derived neurotrophic factor regulation of N-methyl-D-aspartate receptor-mediated synaptic currents in suprachiasmatic nucleus neurons. J Neurosci Res. 84(7):1512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriazis H, Jennings NL, Davern P, Lambert G, Su Y, Pang T, et al. 2012. Neurocardiac dysregulation and neurogenic arrhythmias in a transgenic mouse model of Huntington’s disease: Neurocardiac phenotype in Huntington’s disease mice. The Journal of Physiology. 590: 5845–5860. doi: 10.1113/jphysiol.2012.238113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobal J, Meglic B, Mesec A, Peterlin B. 2004. Early sympathetic hyperactivity in Huntington’s disease. European Journal of Neurology. 11: 842–848. doi: 10.1111/j.1468-1331.2004.00894.x [DOI] [PubMed] [Google Scholar]
- Kobal J, Melik Z, Cankar K, Bajrovic FF, Meglic B, Peterlin B, et al. 2010. Autonomic dysfunction in presymptomatic and early symptomatic Huntington’s disease. Acta Neurol Scand. 121: 392–399. doi: 10.1111/j.1600-0404.2009.01251.x [DOI] [PubMed] [Google Scholar]
- Kuhl DE, Phelps ME, Markham CH, Metter EJ, Riege WH, Winter J. 1982. Cerebral metabolism and atrophy in huntington’s disease determined by 18FDG and computed tomographic scan. Ann Neurol. 12:425–434. [DOI] [PubMed] [Google Scholar]
- Kuljis DA, Gad L, Loh DH, MacDowell Kaswan Z, Hitchcock ON, Ghiani CA, et al. 2016. Sex Differences in Circadian Dysfunction in the BACHD Mouse Model of Huntington’s Disease. PLoS ONE. 11: e0147583. doi: 10.1371/journal.pone.0147583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuljis D, Kudo T, Tahara Y, Ghiani CA, Colwell CS. 2018. Pathophysiology in the suprachiasmatic nucleus in mouse models of Huntington’s disease. Journal of Neuroscience Research. 96: 1862–1875. doi: 10.1002/jnr.24320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, et al. 2011. Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Experimental Neurology. 228: 80–90. doi: 10.1016/j.expneurol.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, Deverasetty S, Tebbe A, Schaab C, Lavery DJ, Howland D, Kwak S, Botas J, Aaronson JS, Rosinski J, Coppola G, Horvath S, Yang XW. 2016. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. 19(4):623–33. doi: 10.1038/nn.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leak RK, Moore RY (2001) Topographic organization of suprachiasmatic nucleus projection neurons. J Comp Neurol. 433(3):312–34. [DOI] [PubMed] [Google Scholar]
- Lebreton F, Cayzac S, Pietropaolo S, Jeantet Y, Cho YH. 2015. Sleep Physiology Alterations Precede Plethoric Phenotypic Changes in R6/1 Huntington’s Disease Mice. PLoS One. 10(5):e0126972. doi: 10.1371/journal.pone.0126972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leise TL (2013) Wavelet analysis of circadian and ultradian behavioral rhythms. J. Circadian Rhythms. 11:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuchter MK, Donzis EJ, Cepeda C, Hunter AM, Estrada-Sánchez AM, Cook IA, Levine MS, Leuchter AF. 2017. Quantitative Electroencephalographic Biomarkers in Preclinical and Human Studies of Huntington’s Disease: Are They Fit-for-Purpose for Treatment Development? Front Neurol. 8:91. doi: 10.3389/fneur.2017.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang FQ, Allen G, Earnest D. 2000. Role of brain-derived neurotrophic factor in the circadian regulation of the suprachiasmatic pacemaker by light. J. Neurosci, 20: 2978–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MS, Liao PY, Chen HM, Chang CP, Chen SK, Chern Y. 2019. Degeneration of ipRGCs in Mouse Models of Huntington’s Disease Disrupts Non-Image-Forming Behaviors Before Motor Impairment. J Neurosci. 39(8):1505–1524. doi: 10.1523/JNEUROSCI.0571-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh DH, Kudo T, Truong D, Wu Y, Colwell CS. 2013. The Q175 mouse model of Huntington’s disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PLoS ONE. 8: e69993. doi: 10.1371/journal.pone.0069993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Zhang YH, Chou TC, Gaus SE, Elmquist JK, Shiromani P, Saper CB. 2001. Contrasting effects of ibotenate lesions of the paraventricular nucleus and subparaventricular zone on sleep-wake cycle and temperature regulation. J Neurosci. 21(13):4864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, Fraenkel E, McAllister CJ, Wood N, Reddy AB, Hastings MH, Morton AJ. 2010. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J Neurosci. 30(30):10199–204. doi: 10.1523/JNEUROSCI.1694-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColgan P, Tabrizi SJ. 2018. Huntington’s disease: a clinical review. Eur J Neurol. 25(1):24–34. doi: 10.1111/ene.13413. [DOI] [PubMed] [Google Scholar]
- Michel S, Clark JP, Ding JM, Colwell CS. 2006. Brain-derived neurotrophic factor and neurotrophin receptors modulate glutamate-induced phase shifts of the suprachiasmatic nucleus. Eur J Neurosci. 24(4):1109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Inuabasi L, Bondulich MK, Muller T, Osborne GF, Franklin SA, et al. 2014. Dysfunction of the CNS-heart axis in mouse models of Huntington’s disease. PLoS Genet. 10: e1004550. doi: 10.1371/journal.pgen.1004550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihm MJ, Amann DM, Schanbacher BL, Altschuld RA, Bauer JA, Hoyt KR. 2007. Cardiac dysfunction in the R6/2 mouse model of Huntington’s disease. Neurobiology of Disease. 25: 297–308. doi: 10.1016/j.nbd.2006.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, Takahashi JS. 2012. Central and Peripheral Circadian Clocks in Mammals. Annual Review of Neuroscience. 35: 445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ. Circadian and sleep disorder in Huntington’s disease. 2013. Exp Neurol. 243: 34–44. doi: 10.1016/j.expneurol.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Rudiger SR, Wood NI, Sawiak SJ, Brown GC, Mclaughlan CJ, Kuchel TR, Snell RG, Faull RL, Bawden CS. 2014. Early and progressive circadian abnormalities in Huntington’s disease sheep are unmasked by social environment. Hum Mol Genet. 23(13):3375–83. doi: 10.1093/hmg/ddu047. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. 2005. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci. 25: 157–163. doi: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TJ, Nakamura W, Yamazaki S, Kudo T, Cutler T, Colwell CS, Block GD. 2011. Age-related decline in circadian output. J Neurosci 31:10201–10205 doi: 10.1523/JNEUROSCI.0451-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseri NN, Xu H, Bonica J, Vonsattel JP, Cortes EP, Park LC, Arjomand J, Gibson GE. 2015. Abnormalities in the tricarboxylic Acid cycle in Huntington disease and in a Huntington disease mouse model. J Neuropathol Exp Neurol. 74(6):527–37. doi: 10.1097/NEN.0000000000000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakeshott S, Balci F, Filippov I, Murphy C, Port R, Connor D, Paintdakhi A, Lesauter J, Menalled L, Ramboz S, et al. 2011. Circadian Abnormalities in Motor Activity in a BAC Transgenic Mouse Model of Huntington’s Disease. PLoS Curr. 3: RRN1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrietan K, Hoyt KR 2004. CRE-mediated transcription is increased in Huntington’s disease transgenic mice. Journal of Neuroscience 24(4): 791–796. 10.1523/JNEUROSCI.3493-03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odish OFF, Johnsen K, van Someren P, Roos RAC, van Dijk JG. 2018. EEG may serve as a biomarker in Huntington’s disease using machine learning automatic classification. Sci Rep. 8(1):16090. doi: 10.1038/s41598-018-34269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouk K, Hughes S, Pothecary CA, Peirson SN, Morton AJ. 2016. Attenuated pupillary light responses and downregulation of opsin expression parallel decline in circadian disruption in two different mouse models of Huntington’s disease. Hum Mol Genet. 25(24): pii: ddw359. doi: 10.1093/hmg/ddw359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallier PN, Maywood ES, Zheng Z, Chesham JE, Inyushkin AN, Dyball R, Hastings MH, Morton AJ. 2007. Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J Neurosci 27:7869–7878. doi: 10.1523/JNEUROSCI.0649-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S 2016. Circadian physiology of metabolism. Science 354(6315):1008–1015. 10.1126/science.aah4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Barajas C Rebec GV. 2018. Overview of Huntington’s Disease Models: Neuropathological, Molecular, and Behavioral Differences. Curr Protoc Neurosci. 83(1): e47. doi: 10.1002/cpns.47. [DOI] [PubMed] [Google Scholar]
- Reinke H, Asher G (2019) Crosstalk between metabolism and circadian clocks. Nat Rev Mol Cell Biol. 20(4):227–241. [DOI] [PubMed] [Google Scholar]
- Ruby NF, Dark J, Burns DE, Heller HC, Zucker I (2002) The suprachiasmatic nucleus is essential for circadian body temperature rhythms in hibernating ground squirrels. J Neurosci. 22(1):357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko O, Springer C, Skov LJ, Madsen AN, Hasholt L, Nørremølle A, Holst B. 2019. Ghrelin-mediated improvements in the metabolic phenotype in the R6/2 mouse model of Huntington’s disease. J Neuroendocrinol. e12699. doi: 10.1111/jne.12699. [DOI] [PubMed] [Google Scholar]
- Saft C, Zange J, Andrich J, Müller K, Lindenberg K, Landwehrmeyer B, et al. 2005. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov Disord. 20: 674–679. doi: 10.1002/mds.20373. [DOI] [PubMed] [Google Scholar]
- Scheer FA, Pirovano C, Van Someren EJ, Buijs RM. 2005. Environmental light and suprachiasmatic nucleus interact in the regulation of body temperature. Neurosci. 132(2):465–77. [DOI] [PubMed] [Google Scholar]
- Schmidt TM, Chen SK, Hattar S. 2011. Intrinsically photosensitive retinal ganglion cells: many subtypes, diverse functions. Trends Neurosci. 34(11):572–80. doi: 10.1016/j.tins.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder AM, Colwell CS. 2013. How to fix a broken clock. Trends Pharmacol Sci 34(11): 605–19. doi: 10.1016/j.tips.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder AM, Loh DH, Jordan MC, Roos KP, Colwell CS. 2011. Baroreceptor reflex dysfunction in the BACHD mouse model of Huntington’s disease. PLoS Currents 3: RRN1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder AM, Wang HB, Park S, Jordan MC, Gao F, Coppola G, Fishbein MC, Roos KP, Ghiani CA, Colwell CS. 2016. Cardiac Dysfunction in the BACHD Mouse Model of Huntington’s Disease. PLOS ONE 11: e0147269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastianutto I, Cenci MA, Fieblinger T. 2017. Alterations of striatal indirect pathway neurons precede motor deficits in two mouse models of Huntington’s disease. Neurobiol Dis. 105:117–131. doi: 10.1016/j.nbd.2017.05.011. [DOI] [PubMed] [Google Scholar]
- Sharma KR, Romano JG, Ayyar DR, Rotta FT, Facca A, Sanchez-Ramos J. 1999. Sympathetic skin response and heart rate variability in patients with Huntington disease. Arch Neurol. 56: 1248–1252. [DOI] [PubMed] [Google Scholar]
- Skillings EA, Wood NI, Morton AJ. 2014. Beneficial effects of environmental enrichment and food entrainment in the R6/2 mouse model of Huntington’s disease. Brain Behav. 4(5):675–86. doi: 10.1002/brb3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smarr BL, Grant AD, Zucker I, Prendergast BJ, Kriegsfeld LJ. 2017. Sex differences in variability across timescales in BALB/c mice. Biol. Sex Differ. 8: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smarr BL, Zucker I and Kriegsfeld LJ. 2016. Detection of Successful and Unsuccessful Pregnancies in Mice within Hours of Pairing through Frequency Analysis of High Temporal Resolution Core Body Temperature Data. PLOS ONE 11: e0160127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Dijak AI, Sepers MD, Raymond LA. 2019. Alterations in synaptic function and plasticity in Huntington disease. J Neurochem. doi: 10.1111/jnc.14723. [DOI] [PubMed] [Google Scholar]
- Stephan FK, Nunez AA. 1977. Elimination of circadian rhythms in drinking, activity, sleep, and temperature by isolation of the suprachiasmatic nuclei. Behav Biol. 20(1):1–61. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. 2003. Transcriptional abnormalities in Huntington disease. Trends in Genetics, 19(5), 233–238. 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Szymusiak R 2018. Body temperature and sleep. Handb Clin Neurol. 156: 341–351. doi: 10.1016/B978-0-444-63912-7.00020-5. [DOI] [PubMed] [Google Scholar]
- Takahashi JS, Shimomura K, Kumar V. 2008. Searching for genes underlying behavior: lessons from circadian rhythms. Science. 322(5903): 909–12. doi: 10.1126/science.1158822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg JMM, Gardiner SL, Ludolph AC, Landwehrmeyer GB, Roos RAC, Aziz NA. 2017. Body weight is a robust predictor of clinical progression in Huntington disease. Ann Neurol. 82(3):479–483. doi: 10.1002/ana.25007. [DOI] [PubMed] [Google Scholar]
- Van Wamelen DJ, Aziz NA, Anink JJ, Van Steenhoven R, Angeloni D, Fraschini F, et al. 2013. Suprachiasmatic Nucleus Neuropeptide Expression in Patients with Huntington’s Disease. Sleep. 36 (1):117–25. doi: 10.5665/sleep.2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Loh DH, Whittaker DS, Cutler T, Howland D, Colwell CS. 2018. Time-Restricted Feeding Improves Circadian Dysfunction as well as Motor Symptoms in the Q175 Mouse Model of Huntington’s Disease. eNeuro 3: 5(1). doi: 10.1523/ENEURO.0431-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Whittaker DS, Truong D, Mulji AK, Ghiani CA, Loh DH, Colwell CS. 2017. Blue light therapy improves circadian dysfunction as well as motor symptoms in two mouse models of Huntington’s disease. Neurobiol Sleep Circadian Rhythms 2:39–52. doi: 10.1016/j.nbscr.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker DS, Wang H-B, Loh DH, Cachope R, Colwell CS. 2017. Possible use of a H3R antagonist for the management of non-motor symptoms in the Q175 mouse model of Huntington’s disease. Pharma Res Per e00344. doi: 10.1002/prp2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RH, Morton AJ, Burdakov D. 2011. Paradoxical function of orexin/hypocretin circuits in a mouse model of Huntington’s disease. Neurobiol Dis. 42(3):438–45. doi: 10.1016/j.nbd.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AB, Angelo N, Huettner JE, Herzog ED. 2009. Intrinsic, nondeterministic circadian rhythm generation in identified mammalian neurons. Proc Natl Acad Sci USA. 106: 16493–16498. doi: 10.1073/pnas.0902768106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood NI, Sawiak SJ, Buonincontri G, Niu Y, Kane AD, Carpenter TA, et al. 2012. Direct evidence of progressive cardiac dysfunction in a transgenic mouse model of Huntington’s disease. J Huntingtons Dis. 1: 57–64. doi: 10.3233/JHD-2012-120004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wan JQ, Tong XP. 2018. Potassium channel dysfunction in neurons and astrocytes in Huntington’s disease. CNS Neurosci Ther. 24(4):311–318. doi: 10.1111/cns.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. 2006. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 52(2):255–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. 2007. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol 81(5–6):294–330. [DOI] [PubMed] [Google Scholar]