

Abstract
Disturbances in sleep/wake cycle are a common complaint of individuals with Huntington’s disease (HD) and are displayed by HD mouse models. The underlying mechanisms, including the possible role of the circadian timing system, are not well established. The BACHD mouse model of HD exhibits disrupted behavioral and physiological rhythms, including decreased electrical activity in the central circadian clock (suprachiasmatic nucleus, SCN). In this study, electrophysiological techniques were used to explore the ionic underpinning of the reduced spontaneous neural activity in male mice. We found that SCN neural activity rhythms were lost early in the disease progression and was accompanied by loss of the normal daily variation in resting membrane potential in the mutant SCN neurons. The low neural activity could be transiently reversed by direct current injection or application of exogenous NMDA thus demonstrating that the neurons have the capacity to discharge at WT levels. Exploring the potassium currents known to regulate the electrical activity of SCN neurons, our most striking finding was that these cells in the mutants exhibited an enhancement in the large-conductance calcium activated K+ (BK) currents. The expression of the pore forming subunit (Kcnma1) of the BK channel was higher in the mutant SCN. We found a similar decrease in daytime electrical activity and enhancement in the magnitude of the BK currents early in disease in another HD mouse model (Q175). These findings suggest that SCN neurons of both HD models exhibit early pathophysiology and that dysregulation of BK current may be responsible.
Keywords: BACHD, BK current, circadian rhythms, Huntington’s disease, Q175, Suprachiasmatic nucleus
Graphical Abstract
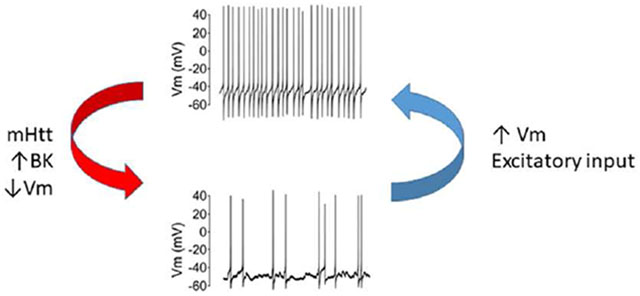
We found decreased daytime electrical activity in the central circadian clock (SCN) in two mouse models of HD. The mutant neurons have the capacity to generate action potentials at a higher frequency in response to direct current injection or application of exogenous NMDA. We also found that the BK current was enhanced in the mutant SCN providing a possible mechanistic explanation for the low daytime electrical activity.
Introduction
Huntington’s disease (HD) is caused by an expanded trinucleotide repeat in the gene encoding the huntingtin protein (HTT) that results in degeneration within cortical and striatal tissues causing cognitive and motor deficits (McDonald et al., 1993). Specific patterns of cell loss and neuronal circuit alterations within hypothalamic tissues are thought to underlie non-motor symptoms such as sleep and mood disruptions (Halliday et al., 1998; Petersén et al., 2005; Paulsen et al., 2006; Petersén and Björkqvist, 2006; Wamelen et al., 2013). Although motor symptom onset is requisite for the HD diagnosis (Ross and Tabrizi, 2011), non-motor symptoms often appear earlier and are a prominent feature of the disease (Paulsen and Conybeare, 2005; Pallier et al., 2007; Aziz et al., 2010; Morton, 2013; Wamelen et al., 2013). Disruptions in the sleep/wake cycle as well as temporal changes to melatonin secretion rhythms implicate circadian system dysfunction in HD, but the mechanism is not well understood (Morton et al., 2005; Aziz et al., 2009; Goodman et al., 2011; Morton, 2013; Kalliolia et al., 2014).
The circadian system is composed of cell autonomous clock gene expression rhythms that are synchronized and adaptively phase aligned in tissues throughout the body by rhythmic output from the central circadian clock located in the suprachiasmatic nucleus (SCN) of the hypothalamus (Kalsbeek et al., 2006; Mohawk et al., 2012).The SCN’s unique circuit properties allow it to generate robust and self-sustaining rhythms in clock gene expression and electrical activity using synaptic and peptidergic signaling mechanisms (Welsh et al., 2010; Herzog et al., 2017). Individual SCN neurons express rhythms in spontaneous firing rate (SFR), with high firing rates observed during the day and low rates at night (Webb et al., 2009). These SFR rhythms are driven by rhythmic expression of ionic currents that depolarize neuronal membrane potential toward action potential (AP) threshold during the day and hyperpolarize membrane potential to silence neurons at night (Allen et al., 2017). Additionally, two key potassium (K+) currents that regulate AP frequency include the fast delayed rectifier (FDR) (Itri et al., 2005; Kudo et al., 2011a), and big-conductance Ca2+ activated K+ (BK) currents (Cloues and Sather, 2003; Meredith et al., 2006; Pitts et al., 2006; Kent and Meredith, 2008; Whitt et al., 2016). These currents are critically involved in driving the circadian rhythm in AP frequency that defines SCN physiology.
Like other mouse models of HD (Morton et al., 2005; Menalled et al., 2012; Loh et al., 2013), the bacterial artificial chromosome transgenic mouse model of HD (BACHD) exhibits disrupted rhythms in sleep, activity, and physiology (Kudo et al., 2011; Kuljis et al., 2016; Schroeder et al., 2016). Early in the disease progression (3 month of age), mutant dorsal SCN (SCN) neurons lose their daily rhythms in electrical activity (Kudo et al., 2011b; Kuljis et al., 2016), suggesting circadian behavioral deficits are caused by pathophysiology of the SCN. In this study, we first sought to determine whether the SCN firing rate deficits continue when GABA-mediated synaptic transmission is blocked, and whether the daily rhythms in resting membrane potential (RMP) and conductance are disrupted. Second, we tested whether mutant SCN neurons have the capacity of firing at a higher rate when depolarized with direct current injection or in response to exogenous NMDA treatment. Thirdly, to identify mechanisms underlying aberrant electrical activity we assessed whether the mutation impacted the two main K+ currents known to regulate SCN firing rate – the FDR, and BK currents, and measured gene expression levels of the BK channel subunits. Finally, we measured SCN firing rate and BK currents in a second HD model (the Q175 mouse) to determine whether our key pathophysiology findings could be observed across multiple HD mouse models.
Materials and Methods
Animals and housing
The UCLA Animal Research Committee approved all experimental protocols used in this study, which followed guidelines and recommendations for animal use and welfare set by the UCLA Division of Laboratory Animal Medicine and National Institutes of Health. The BACHD model that we used in this study expresses the full length human mutant Htt gene encoding 97 glutamine repeats under the control of endogenous regulatory machinery (BACHD) (Gray et al., 2008). Female BACHD dams backcrossed on a C57BL/6J background (minimum 12 generations) were bred with C57BL/6J (WT) males from The Jackson Laboratory (Bar Harbor, Maine) in our own breeding facility to obtain male and female offspring, either WT or heterozygous for the BACHD transgene. The Q175 mice arose from a spontaneous expansion of the CAG repeat in the CAG140 transgenic knock-in line (Menalled et al., 2012). The Q175 mice have previously been shown to have around 175 CAG repeats and we used mice heterozygous (Het) and homozygous (Hom) for the Q175 allele. Mutant mice were obtained from the Jackson Laboratory (Bar Harbor, Maine) from a colony managed by the CHDI Foundation. Only male mice were used in this study. Genotyping was performed at 15 days of age by tail snips, and after weaning, littermates were group housed by sex, until otherwise noted. All animals were housed in sound proof, humidity controlled chambers with controlled lighting conditions, using a 12 hr light, 12 hr dark cycle (12:12 LD, intensity 300 lux) for at least two-weeks prior to experimentation. For all experiments, a light meter (BK precision, Yorba Linda, CA) was used to measure light-intensity (lux).
We used male mice for these studies as we have previously demonstrated that there are sex differences in the circadian phenotype and SCN pathophysiology in the BACHD line (Kuljis et al., 2016).
Acute slice preparation
Methods for electrophysiology recordings were similar to those previously reported (Kuljis et al., 2013; 2016). All animals were between 2.5 and 3.5 mo of age, unless otherwise noted. Zeitgeber time (ZT) 0 corresponds to the time of lights-on when nocturnal animals are held in an LD cycle. For daytime (ZT4-6) and nighttime (ZT14-16) recordings, animals were briefly placed into a charged isofluorane chamber before decapitation and brain removal at ZT2 and ZT11.5 respectively. Brains were chilled in ice-cold slice solution (in mM: 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 125 NaCl, 3 KCl, 5 MgCl2, 1 CaCl2; all chemicals from Sigma-Aldrich, St. Louis, MO) before trimming and slicing using a Leica VT1200S vibratome (Nussloch, Germany). Two to three coronal slices (250 μm) containing the SCN were transferred into artificial cerebrospinal fluid (ACSF) solution (in mM: 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 125 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2) and incubated at 32°C for 30 mins before a room temperature incubation for one hour. All solutions were adjusted for pH (7.20 - 7.40), osmolarity (290 – 310), and aerated continually with 95% O2/ 5% CO2 for at least 15 mins before use. Following room-temperature incubation, slices were placed into a recording chamber (PH-1, Warner Instruments, Hamden, CT) attached to the stage of a fixed upright differential interference contrast (DIC) microscope (Olympus, Tokyo, Japan) and superfused continuously (2 ml/min) with room temperature ACSF. The mid-SCN slice was identified based on third-ventricle and optic chiasm morphology, as well as SCN cell density and area as visualized under magnification (40x objective). Each cell was determined to be within the dorsal mid-SCN by directly visualizing its location with reference to anatomical markers. Dorsal SCN (dSCN) neurons were classified by their location just dorsal to the tip of third ventricle. Neurons in this region are known to exhibit robust rhythms in electrical activity and were the focus of the present study.
Whole-cell patch-clamp electrophysiology
Whole-cell patch-clamp recordings used electrode micropipettes (3-7 MΩ) pulled from borosilicate glass capillaries (WPI, Sarasota, FL) using a multistage puller (Sutter P-97, Novato, CA) and were filled with internal solution (in mM: 112.5 K-gluconate, 4 NaCl, 17.5 KCl, 0.5 CaCl2, 1 MgCl2, 5 MgATP, 1 EGTA, 10 HEPES, 1 GTP, 0.1 Leupeptin, 10 Phosphocreatine; pH adjusted to 7.2 using KOH and osmolarity adjusted to 290 using sucrose; all chemicals from Sigma-Aldrich), unless otherwise specified. Single-cell recordings were made using the Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) and monitored on-line with pCLAMP (Ver. 10, Molecular Devices). The amplifier’s voltage-offset was used to cancel the junction potential between the pipette’s internal solution and the extracellular solution (ACSF). While in voltage-clamp mode (0 mV holding), cells were approached by electrode micropipettes with slight positive-pressure to clear extracellular matrix. By switching to negative-pressure and gradually lowering the holding potential to −70 mV, a high resistance seal (2-10 GΩ) was formed. A second pulse of negative pressure was used to break the membrane and enter whole cell mode. Membrane holding current, cell capacitance, and access resistance were tested using a 5mV step applied at 5 Hz from the −70 mV holding potential. Cells with holding currents greater than 0 or less than −20 pA, capacitances greater than 20 pF, or access resistances greater than 60 MΩ (typically 10 to 40 MΩ) were excluded from analysis. These membrane parameters were monitored during the experiment, and if they changed significantly, or if a cell was unable to decrease firing rate or re-hyperpolarize membrane potential following excitatory treatment, that cell’s data was discarded. Pipette-resistance and series-resistance compensation (80%) was performed for all voltage-clamp recordings unless otherwise noted. All recordings were collected using 10kHz sampling rate and 5 kHz lowpass Bessel filter.
Action potential frequency
Following the formation of a high-resistance seal and going whole-cell, the amplifier mode was switched from voltage- to current-clamp, and AP frequency, here defined as spontaneous firing rate (SFR), was recorded during the subsequent minute without any current injection. SFR was calculated using the total number of APs recorded over 60 secs, unless otherwise noted. Drug treatments were performed by dissolving gabazine (10 μM; Tocris Bioscience, Minneapolis, MN) or N-methyl-d-aspartate (NMDA, 25 μM; Tocris) in ACSF, and were delivered to the slice bath using a rapid gravity feed system. Slices were treated with gabazine alone for at least 3-4 mins, or until synaptic activity was abolished as determined with on-line monitoring of membrane activity. SFR was examined over the subsequent minute of drug treatment. AP properties were analyzed using Clampfit Software (Ver 10.4, Molecular Devices) and MiniAnalysis (Ver. 6.0.3, Synaptosoft, Inc., Fort Lee, NJ).
Resting membrane properties
Previous studies have shown that Per1::GFP SCN neurons show daily rhythms in multiple resting membrane properties (Kuhlman and McMahon, 2004). A similar protocol was used to examine the intrinsic excitability of SCN neurons in this study. Resting membrane properties of dSCN neurons were examined during the daytime and nighttime in slices treated for at least 2 min with tetrodotoxin (TTX, 1 μM; Tocris Bioscience, Minneapolis, MN) and GABAA receptor antagonist gabazine (10 μM; Tocris Bioscience) to block APs and GABAergic synaptic potentials. After forming a high resistance seal, going whole-cell, and obtaining membrane parameters for the selected neuron, the amplifier was switched from voltage-clamp to current-clamp mode. Cells typically reached a stable Vm within the first 10 secs of the switch. Resting Vm was recorded over the subsequent 1 to 3 mins and calculated as the average Vm over 50 seconds during that time. Next, increasingly hyperpolarizing current steps (500 msec, −5 to −25 pA in 5 pA steps) were applied to each neuron at least 3 times. Membrane voltage response traces were filtered for electrical interference before analysis (harmonics 1:1, 119 cycles to average, auto-reference frequency). Peak hyperpolarization (using 5 smoothing points) was identified for each current injection step, then averaged across replications. For all other voltage response metrics (hyperpolarization peak time, area, and slope of the response between 10 and 90% of its value), the average trace of the three replications was used. Cells which did not maintain a steady Vm between current injection treatments were excluded from analysis.
K+ currents
K+ current magnitudes were examined in BACHD and WT littermate SCN neurons during the daytime (ZT 4-6) in voltage-clamp mode. After forming a high resistance seal, going whole-cell, and obtaining membrane parameters for the selected neuron, cells were held at −70 mV before treatment with a specific voltage-step protocol to activate the FDRor BK K+ currents (protocol details below) twice per min. After baseline, current readings became stable (generally within 2 min), the current specific protocol was applied. All current magnitudes were normalized to cell capacitance.
FDR current was examined using a similar protocol as published previously (Itri et al., 2005; Farajnia et al., 2012). FDR current was elicited by applying an inactivating −100 mV step (100 msec), followed by voltage-steps from −50 to 60 mV (10mV step, 400 msec duration), after which Vm was returned to −100 mV (20 msec.). Control recording medium contained Cd++ (25 μM), TTX (1 μM), and gabazine (25 μM) in ACSF to block voltage-gated Ca2+ channels, voltage-gated sodium channels, and GABAA receptors. Control currents typically stabilized within 5 mins. Cells were then treated for 4 mins with tetra-ethyl ammonium (TEA; 1mM) to block the FDR current and were then subjected to the same voltage-step protocol. TEA current was subtracted from control current to calculate FDR current. Current magnitude was measured as the mean current during the last 200 msec of voltage step. Traces used in figures were filtered for noise using Bessel (8-pole, 60 Hz) and Boxcar (99-smoothing points) filtering.
BK currents were tested using methods similar to those previously published (Pitts et al., 2006). Recording conditions were the same as those described above, except that internal solution contained EGTA (0.5 mM) so as not to interfere with calcium activation of the BK current. In addition, the BK current is sensitive to both voltage and intracellular Ca2+ levels so these experiments were not performed in TTX which reduce Ca2+ levels. In whole-cell mode, cells were voltage-clamped at −60 mV for 3 to 5 min. Holding potential was then clamped at −70 mV to fully inactivate voltage-gated channels (100 msec.) before Vm was moved from −100 to +80 mV (20mV steps, 180 msec) to activate BK-currents. At the end of each current step Vm was returned back to −70 mV (200 msec). Currents were measured during the last 20 to 30 msec of each voltage step. Baseline currents were measured twice per min until stable readings were obtained for three consecutive trials. Cells were then treated with iberiotoxin (IbTX, 100 nM; Sigma) for 5 to 8 min to block BK currents. Currents were then measured every min and IbTX treatment was terminated when no further reduction of currents was detected. Cells were then washed for 10 to 30 min to test for partial or full restoration of currents back to baseline levels.
Quantitative Real-time RT–PCR
The SCN from 3mo WT (n=6) and BACHD (n=7) were rapidly dissected and frozen in the daytime (ZT 4-6). Total RNA (200ng) was extracted using TRIzol® reagent (Life Technologies; Carlsbad, CA) following the manufacturer’s protocol, and further purified by treatment with Ambion® TURBO DNA-free™ (Life Technologies, Waltham, MA), followed by a second extraction with phenol/chloroform. Sample concentrations and purity were assessed using a NanoDrop One Microvolume UV-Vis Spectrophotometer (ThermoScientific; Canoga Park, CA). RNA was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA), and then analyzed for the transcript expressions of three Bk channel subunits (Kcnma1, Kncmb2, and Kcnmb4) on a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories). Reactions were set up using iQ SYBR Green Supermix (Bio-Rad Laboratories), QuantiTect Primer Assay (Qiagen, Valencia, CA), and performed in duplicate as previously described (Ghiani et al., 2011; Schroeder et al., 2016). Negative controls (samples in which the reverse transcriptase was omitted) were amplified individually using the same primer sets to ensure the absence of genomic DNA contamination. Amplification specificity was assessed by melting curve, while standard curves made from serial dilutions of control RNA were used for quantification. Expression standardization was done using the geometric mean of the housekeeping genes: Ppia (peptidylprolyl isomerase A or Cyclophilin A; Gene ID 268373; Mm_Ppia_1_SG QuantiTect Primer Assay, Cat. no. QT00247709) and Rplp0 (PO) (ribosomal protein, large, P0; Gene ID 11837; Mm_Rplp0_1_SG QuantiTect Primer Assay, Cat. No. QT00249375). Data are shown as the mean ± SEM of the specific ratio gene of interest/housekeeping genes geometric mean for WT (n=6) and BACHD (n=7). When normalized one to the other, the expression levels of Ppia (WT: 0.8440 ± 0.05567; BACHD: 0.8178 ± 0.02205) or Rplp0 (WT: 1.208 ± 0.07056; BACHD: 1.228 ± 0.03333) did not differ between genotypes (t-test, P > 0.05).
Statistics:
Mice were randomly assigned to different experimental and control groups. Scientists masked as to the experimental condition carried out the analysis of the experimental data. Sample size was initially determined by power analysis using Sigmaplot (SYSTAT Software, San Jose, CA) with power set at 0.8 and alpha at 0.05. Data was excluded if the neuron did not exhibit properties of a live cell including exhibiting a resting membrane potential and the ability to generate an action potential. Outliers are defined as values which were twice the standard deviation or more from the mean and are removed before analysis.
The data sets were analyzed by test for equal variance and normal distribution to help select the appropriate test. For the analysis of day/night variation in SFR, Vm and conductance, 2-way analysis of variance (ANOVA) was used to evaluate significance with genotype and time as factors. A t-test or Mann-Whitney Rank sum test was used for the analysis of AP parameters recorded in the two genotypes during the day. The analysis of the impact of current injection and NMDA treatment was performed by 2-way ANOVA with genotype and treatment as factors. Significant variation in K+ currents were analyzed by 2-way ANOVA with genotype and voltage as factors. When significant effects were detected, post-hoc multiple pairwise comparison testing using Holm-Sidak method was used to identify significant group differences. Genotypic differences in BK subunit expression levels by t-test. SigmaPlot (SYSTAT Software, San Jose, CA) or GraphPad Prism 7 (GraphPad Software; La Jolla, CA) was used to run all statistical analysis. Significance levels for all statistical procedures were set at P < 0.05. All reported values are means ± standard error of the means (SEMs).
RESULTS
Reduced daytime electrical activity in SCN neurons from BACHD
Previously we have reported that BACHD SCN neurons lose their rhythm in electrical activity due to depression of daytime spontaneous firing rates (SFR) (Kudo et al., 2011b). Here, we re-examined and extend our understanding of this finding. As expected at baseline (Fig. 1A, C), WT dSCN neurons displayed high SFRs during the day (5.7 ± 0.4 Hz) and low SFRs during the night (3.0 ± 0.4 Hz), while BACHD dSCN neurons have low SFRs both during the day (1.9 ± 0.3 Hz.) and night (2.4 ± 0.4 Hz). 2-way ANOVA analysis showed significant effects of genotype (F(1,67) = 33.39, n=68, P < 0.001) and time (F(1,67) = 6.97, n=68, P = 0.01) as well as a significant interaction (F(1,67) = 15.86, P < 0.001). Post-hoc multiple comparisons found that BACHD neurons lacked the day/night difference in firing rate (t(29) = 0.99, P = 0.32) typical of WT neurons (t(37) = 4.49, P < 0.001). The SCN is a GABAergic neuronal network, and alterations in this circuit’s properties may cause altered inhibitory tone. Thus, we tested whether the observed depressed SFRs were a property of altered GABAergic signaling in the BACHD line by examining SFR rhythms in the presence of gabazine (10 μM), a GABAA-R antagonist (Fig. 1B, C). Significant effects of genotype (F(1,61) = 55.91, n=62, P < 0.001) and time (F(1,61) = 12.01, n=62, P = 0.001) with a significant interaction (F(1,61) = 18.92, P < 0.001) on GABA-independent SFR were detected by 2-way ANOVA. The same genotypic differences persisted in the presence of gabazine. WT dSCN neurons showed high SFRs during the day (7.1 ± 0.46 Hz) and low SFRs at night (3.8 ± 0.5 Hz; t(27) = 5.44, P < 0.001), and the BACHD neurons still had low SFRs during both the day (1.7 ± 0.3 Hz) and night (2.0 ± 1.0 Hz, t(32) = 0.64, P = 0.53). Finally, we analyzed the waveforms of the APs recorded in the presence of gabazine in dSCN neurons in the daytime (Table 1). The APs recorded from the BACHD neurons exhibited significantly larger after spike hyperpolarization (AHP) area compared to controls although the peak amplitude of the AHP did not vary between genotypes. The measurements associated with AP amplitude or rise were not altered (Table 1). Thus, early in the disease progression, BACHD SCN neurons exhibit clear deficits in neural activity.
Fig. 1:
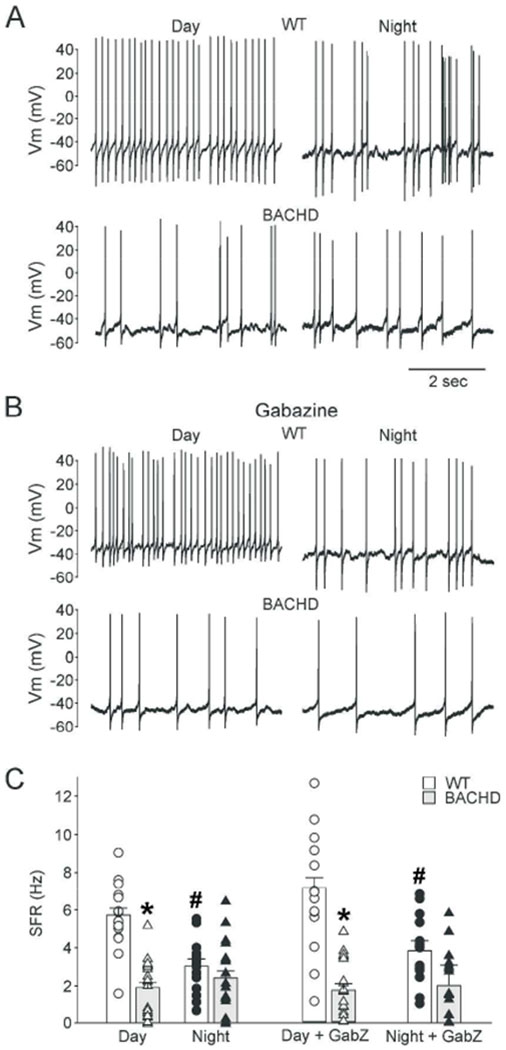
Depressed daytime spontaneous firing rates (SFR) in BACHD dSCN is not GABA-mediated. (A) Representative traces of dSCN neuron electrical activity recorded during the day (left) or night (right) from young WT (top) and BACHD (bottom) mouse brain slices. (B) Representative trace recorded in presence of the GABAA-R antagonist gabazine (10 μM) with the same organization as (A). (C) Summary of WT (white) and BACHD (grey) dSCN neuron SFR with or without GABAzine. Data are shown as the means ± SEM for each group (bars), the scatter plots show the individual values: WT day (white circles, n = 16), WT night (black circles, n = 14), BACHD day (white triangles, n = 21), BACHD night (black triangles, n = 17). 2-way ANOVA with genotype and time as factors. * = significant difference between genotypes; # = significant difference between day and night.
Table 1:
The AP waveforms of dSCN neurons of each genotype were analyzed in the presence of the gabazine. All data was collected during the day (ZT 4-6). A t-test or Mann-Whitney Rank-sum test were used to evaluate possible genotypic differences. Afterhyperpolarization, AHP.
| WT | BACHD | t or U | P | |
|---|---|---|---|---|
| Peak Amplitude (mV) | 88 ± 6 | 100.7 ± 5 | 45.0 | 0.207 |
| Half-Amplitude (mV) | 44 ± 3 | 50 ± 3 | −0.98 | 0.336 |
| Area (mVxmsec) | 205 ± 6 | 231 ± 8* | −2.52 | 0.019 |
| Half-Width (msec) | 2.4 ± 0.2 | 3.6 ± 0.2 | 0.175 | 0.863 |
| Rise Tau (msec) | 2.7 ± 0.6 | 3.6 ± 0.7 | 50.0 | 0.340 |
| Time to Peak (msec) | 1.6 ± 0.1 | 1.8 ± 0.1 | −1.16 | 0.260 |
| Decay Tau (msec) | 1.1 ± 0.1 | 2.9 ± 0.6* | 19.0 | 0.004 |
| AHP Area (mVxmsec) | −7776 ± 650 | −10225 ± 299* | 12.0 | 0.001 |
| AHP Peak (mV) | −31.8 ± 4 | −32 ± 3 | 0.04 | 0.971 |
Prior work indicates circadian SFR rhythm is driven by rhythmic closure of potassium channels and depolarization of membrane potential during the daytime (Kuhlman and McMahon, 2004). We measured input resistance and resting membrane potential (RMP) of dSCN neurons in the presence of GABAzine and TTX (Fig. 2). WT dSCN neurons had a relatively depolarized RMP during the day (−48.5 ± 1.2 mV) and a hyperpolarized RMP during the night (−57.7 ± 1.8 mV), while BACHD dSCN neurons displayed a similar RMP during the day (−53.1 ± 1.5 mV) and the night (−54.6 ± 1.7 mV) (Fig. 2A). 2-way ANOVA analysis identified significant main effect of time (F(1,128) = 11.217, n=129, P = 0.001) and an interaction of genotype and time on RMP (F(1,128) = 5.95, n=129, P = 0.016), but no main effect of genotype (F(1,128) = 0.217, P = 0.642). Post-hoc multiple comparison testing determined BACHD neurons lacked the day/night difference in RMP (t(60) = 0.63, P = 0.53) seen for WT SCN neurons (t(68) = 4.21, P = 0.001). The RMP was significantly hyperpolarized in the BACHD compared to WT neurons in the day (t(80) = 2.30, P = 0.023) but not in the night (t(46) = 1.27, P = 0.206). Next we measured peak voltage response to negative current step injection (−5 to −25pA, Δ-5pA; Fig. 2B, C). Effects of genotype and time on peak responses were tested using Two-Way ANOVA, which identified main effect of time (F(1,118) = 5.867, P = 0.017) but not genotype (F(1,118) = 1.3497, P = 0.248). For WT dSCN neurons, there was a larger deflection in the day than the night (Mann-Whitney Rank Sum, U(60) = 288, P = 0.010). This rhythm was lost in the mutants (t-test, t(55) = −0.381, P = 0.705). Similarly, in WT dSCN neuronal input resistance was rhythmic (Day = 3.8 ± 0.2 GΩ, Night = 2.9 ± 0.1 GΩ, U(60) = 282.0, P = 0.008), but the rhythm was absent in the mutants (Day = 3.2 ± 0.2 GΩ, Night = 3.0 ± 0.2 GΩ, t(55) = 0.381, P = 0.705). These findings clearly suggest a loss of the day/night rhythms in Vm and conductance that underlie the neural activity rhythms in the BACHD SCN neurons.
Fig. 2:
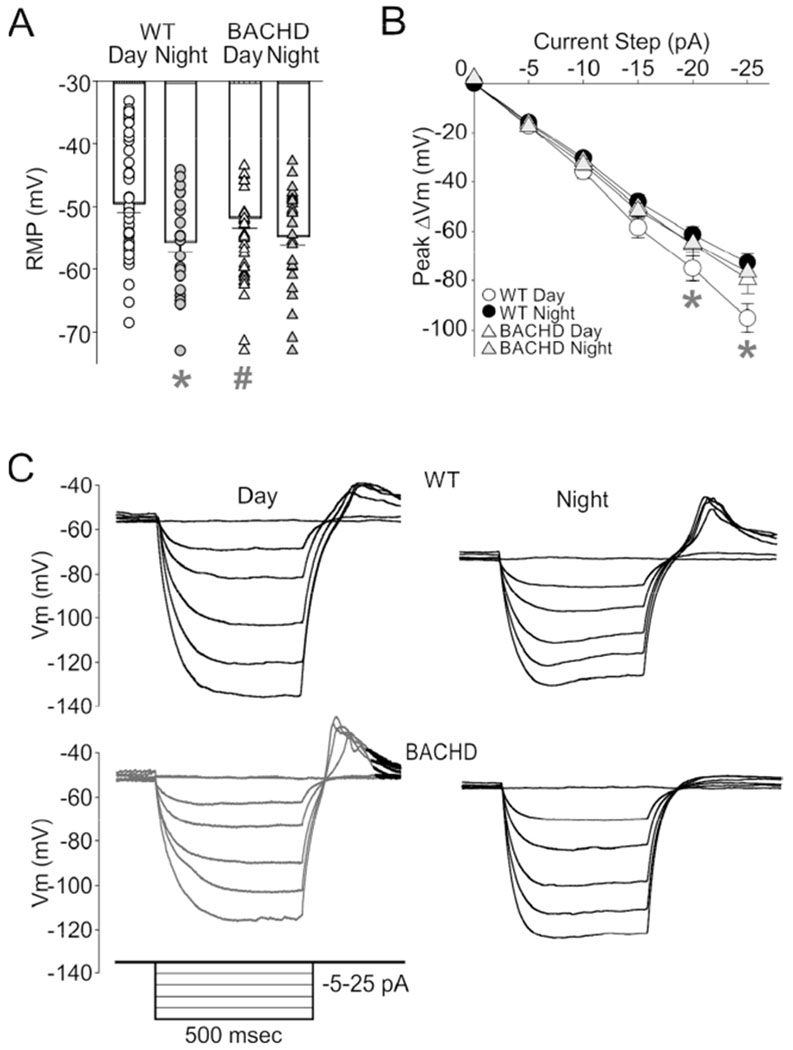
BACHD dSCN neurons lose day/night rhythms in resting membrane potential (RMP) and input resistance. (A) Resting Vm of WT (white bars) and BACHD (gray bars) cells recorded in TTX and gabazine. Bar is means ± SEM, individual neuron values overlaid. WT day (white circles, n = 48), WT night (black circles, n = 21), BACHD day (white triangles, n = 33), BACHD night (black triangles, n = 27). (B) Current-voltage response curve for negative current injection steps. WT day (white circles, n = 36), WT night (black circles, n = 26), BACHD day (white triangles, n = 29), and BACHD night (grey triangles, n = 28). (C) Representative trace examples of dSCN neurons responses to negative current injection during the day (left) and night (right). The insert shows the current injection protocol used to evoked the voltage responses. The magnitude of post-inhibitory rebound potentials did not vary between the genotypes. Two-way ANOVA with genotype and time as factors. * = significant difference between day and night; # = significant difference between genotype.
Current injection restored SFR to BACHD dSCN neurons.
During the day in an active slice dSCN BACHD neurons exhibited a slightly hyperpolarized Vm with reduced electrical activity during the day. It is possible that the membrane hyperpolarization is responsible for the reduced electrical activity and, thus, we examined the possibility that direct current injection could rescue the firing rate. Positive current was directly injected into WT and BACHD neurons (Fig. 3A, B). For all WT and BACHD neurons examined, progressively larger current injection steps (0, 5, and 10 pA steps, 1 min duration) caused significant increases in inter-spike Vm relative to the previous current step. For example the 10pA injection depolarized the WT RMP by 5.2 ± 0.7 mV (paired t-test, t(6) = , n = 7, P = 0.00004) and the BACHD RMP by 5.5 ± 0.7 mV (t(10) = −4.810, n=11, P = 0.0007). In WT neurons, the current injection and resultant membrane depolarization only increased firing less than half of the neurons examined (3/7) and overall there was no significant change in SFR (paired t-test, t(6) = 1.747, n =7, P = 0.131). In contrast, in the mutant neurons, the current injection increased firing in all neurons examined (11/11) and significantly increased SFR (paired t-test, t(10) = −5.287, n=11, P = 0.0003; Fig. 3B). Next, we sought to determine whether activation of the NMDA class of glutamate receptors could also rescue firing rate. Exogenous NMDA (25 μM, 5 min) was applied in the bath during the day to WT and BACHD dSCN neurons (Fig. 3C, D), and increased the SFR of both BACHD and WT neurons to a similar degree (WT: +2.7 ± 0.9 Hz; BACHD: +3.0 ± 1.0 Hz). SFR was affected by genotype (F(1,54) = 34.6, n=55, P < 0.001) and treatment (F(1,54) = 15.5, n=55, P = 0.001), but not by their interaction (F(1,54) = 1.7, P = 0.19). The average firing rate of BACHD neurons treated with NMDA (5.4 ± 1.4 Hz) was still below WT control levels (6.7 ± 0.8 Hz) but was no longer significantly different (t(25) = 1.22, P = 0.234, Fig. 3E). Thus, both direct current injection and treatment with NMDA successfully restored SFRs of BACHD neurons to WT levels and demonstrated that the mutant neurons still have the capacity to generate APs at a higher rate.
Fig. 3:
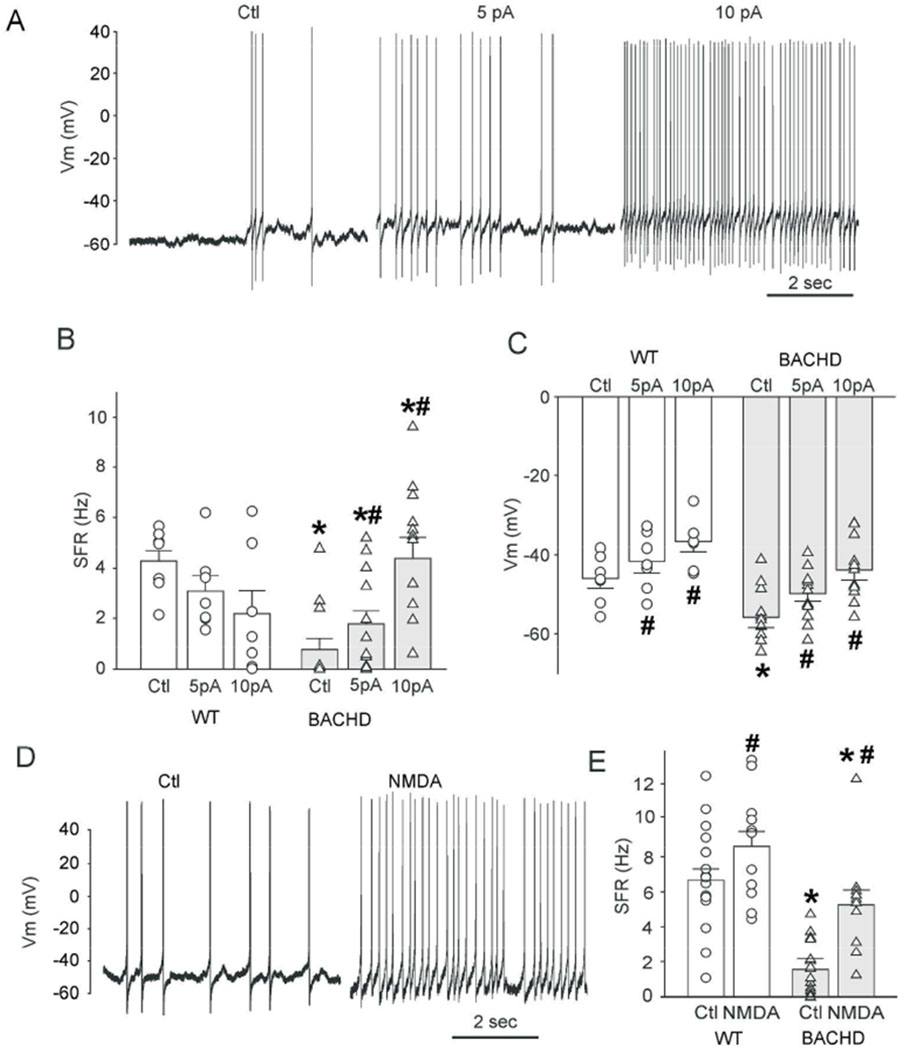
Direct current injection or NMDA treatment rescues depressed daytime SFR of BACHD dSCN neurons. (A) Traces illustrating that positive current injection (5 or 10pA) increases the SFR of BACHD dSCN neurons during the day. (B) Summary of SFR and (C) Vm responses to current injection for WT (white bars and circles, n = 7) and BACHD (grey bars and white triangles, n = 12) neurons. (D) Example trace of BACHD dSCN neuron response to NMDA treatment (25 μM, 5 min). (E) Summary of NMDA SFR responses for WT (white circles, n = 11) and BACHD cells (white triangles, day, n =11). Bars are mean ± SEM. Two-way ANOVA with genotype and treatment as factors. Data were analyzed using a two-way ANOVA with genotype and treatment as factors. * = significant difference between genotypes; # = significant difference between treated and control.
K+ currents altered during the daytime in BACHD dSCN
Two K+ currents that play an important role in mediating fast daytime firing rates in SCN neurons include the FDR and BK currents (Fig. 4). For the TEA-sensitive FDR current, a main effect of voltage (F(11, 754) = 104.669, P < 0.001), but not of genotype (F(1, 754) = 0.4, P = 0.076) was observed (Fig. 4A, C). Peak FDR currents very similar in WT (67 ± 12 pA/pF, n=28) and BACHD neurons (63 ± 17 pA/pF, n=34). Second, we tested the IbTX-sensitive BK currents (Fig. 4B, D), 2- way ANOVA identified significant effect of genotype (F(1, 399) = 8.9, P = 0.003), voltage (F(9, 399)=52, P < 0.001), and their interaction (F(9, 399) = 2.1, P = 0.03). BACHD neurons exhibited significantly greater BK current at depolarized voltage steps (VC+60mV: WT = 44.8 ± 5.1 pA/pF; BACHD = 65.7 ± 9.8 pA/pF, t(38) = 3.1, n=40, P = 0.002; VC+80mV: WT = 50.3 ± 5.3 pA/pF, BACHD = 75.1 ± 11.9 pA/pF, t(38) = 3.7, n=40 P < 0.001). Since the BK current has been shown to play an important role in regulating firing rate and Vm in SCN neurons (Whitt et al., 2016), the enhancement of the BK current provides a plausible explanation for the decreased spontaneous activity found in the mutant SCN neurons.
Fig. 4:
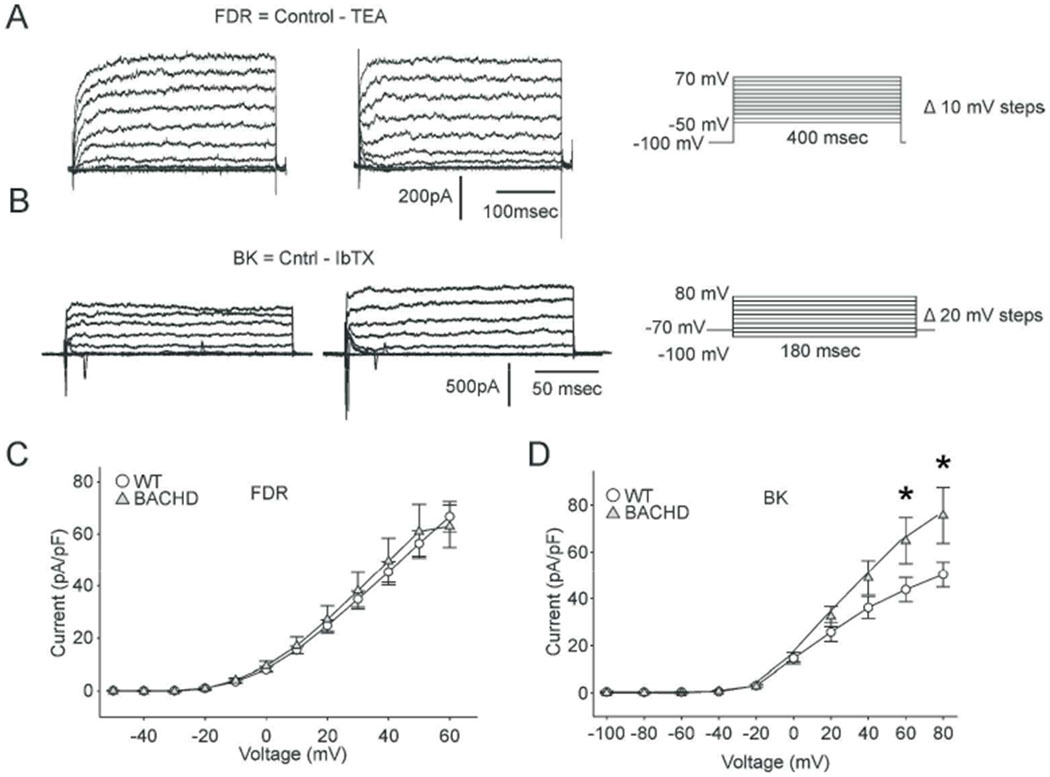
The BK potassium (K+) current is enhanced in BACHD dSCN neurons. (A) FDR, and (B) BK current trace examples recorded from WT (left) and BACHD (right) dSCN neurons during the daytime. Voltage step protocol shown to right. Current-voltage response curve summary for (C) FDR (n = 35), and (D) BK (n = 22) currents measured from WT (circles) and BACHD (grey triangles) neurons. Symbols and error bars represent means ± SEM. Two-way ANOVA was used to identify significant effects of genotype and/or voltage step magnitude on evoked currents (P ± 0.05). * = significant difference between genotypes.
An increased expression of the pore forming subunit was observed in the BACHD mutants.
BK channels are composed of alpha (Kcnma1) and regulatory beta (Kcnmb1, 2, 4) subunits, and changes in subunit expression levels or channel subunit composition may underline the observed alteration in BK currents in BACHD. We measured the expression levels of the genes encoding alpha and beta-2 and −4 subunits in 3 month old WT and BACHD SCN (Fig. 5A–C). While we did not see any change in the expression of Kcnmb2 (t(11) = −0.369, P = 0.719) or Kcnmb4 (t(11) = −0.043, P = 0.966), we did see a modest (15%) but significant (t(11) = −2.863, P = 0.015) increase in Kcnma1 transcript levels. The observed potentiation in Kcnma1 transcript levels provides a probable explanation for the increased magnitude of the BK current observed in BACHD SCN neurons.
Fig. 5:
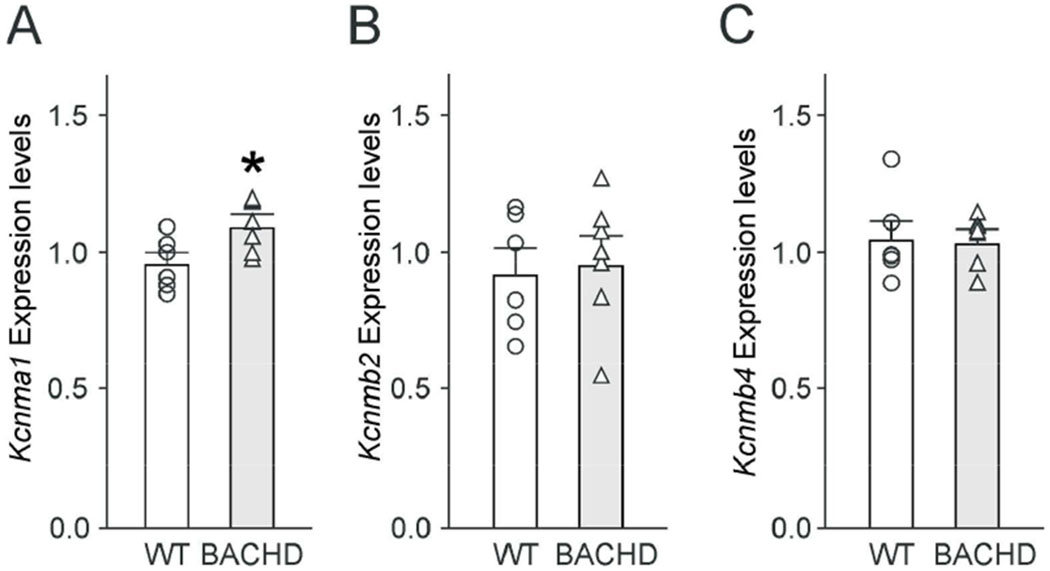
BACHD SCN displayed increased expression of the alpha BK subunit. Kcnma1 expression was modestly higher in the mutant SCN. Gene expression levels of the BK subunits Kcnma1 (A), Kcnmb2 (B), Kcnmb4 (C) were analyzed in WT and BACHD SCN tissue by QPCR, normalized to the geometric mean of the housekeeping genes: Ppia and Rplp0, and are reported as the mean ± SEM of WT (n=6) and BACHD (n=7). When normalized one to the other, the expression levels of Ppia (WT: 0.8440 ± 0.05567; BACHD: 0.8178 ± 0.02205) or Rplp0 (WT: 1.208 ± 0.07056; BACHD: 1.228 ± 0.03333) did not differ between genotypes. * = significant difference between genotypes, t-tests.
Reduced daytime electrical activity and enhanced magnitude of BK currents are found in SCN neurons from the Q175 HD model.
In the final set of experiments, we wanted to determine if our key findings obtained in the BACHD SCN neurons could be proven in another HD model (Q175 mice). Q175 Het and Hom (6-7 mo) dSCN neurons displayed low SFRs both during the day (Het = 2.0 ± 0.4 Hz, Hom = 1.9 ± 0.4 Hz) and night (Het = 1.8 ± 0.5 Hz, Hom = 1.7 ± 0.5 Hz), whereas WT littermate dSCN neurons had high SFRs during the day (5.2 ± 0.8 Hz) and low SFRs during the night (1.7 ± 0.5 Hz) (Fig. 6A). 2-way ANOVA analysis indicated significant effects of genotype (F(2, 115) = 6.419, P < 0.002) and time on SFR (F(1, 115) = 6.521, P = 0.012), and interaction (F(2, 115) = 4.546, P = 0.013). Like the BACHD, Het and Hom Q175 neurons lacked the day/night difference in firing rate (Het: t(36) = 0.120, n = 38, P = 0.090; Hom: t(43) = 0.237, n = 45, P = 0.905), whereas, the WT neurons displayed a significant difference (t(31) = 3.635, n = 33, P < 0.001; Post-hoc multiple comparisons). Additionally, Het and Hom Q175 neurons displayed significantly lower daytime SFRs than WT neurons (Het: t(29) = 3.341, n = 31, P <0.001; Hom: t(30) = 3.28, n = 32, P < 0.001).
Fig. 6:
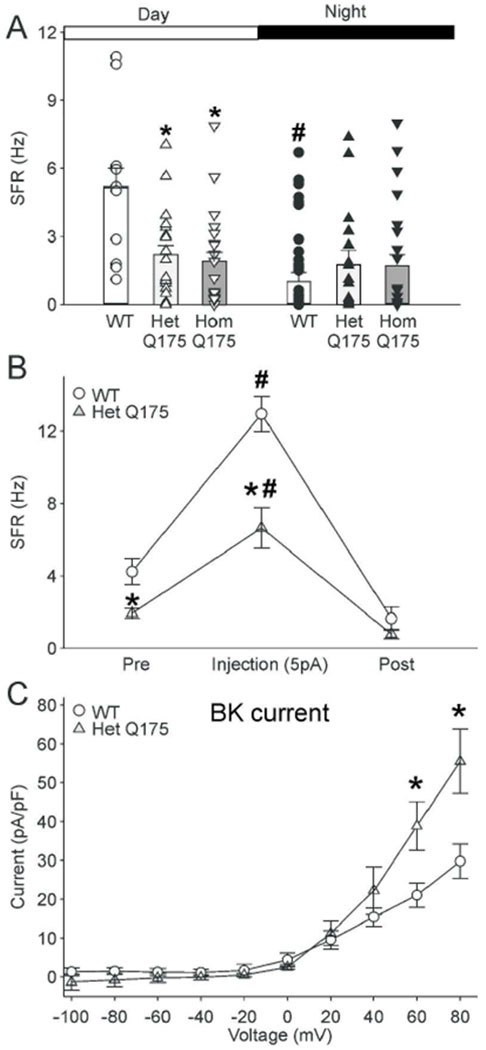
Key findings with the BACHD model were confirmed in the Q175 mouse model. (A) The daily peak of SFR in SCN neurons is reduced in Het and Hom Q175 mice. Using the current-clamp recording technique in the whole-cell configuration, we measured the SFR in dSCN neurons during the day (ZT 5-7) and night (ZT 17-19). Bar graphs plot the mean and SEM for each of the groups. Scatter plots show values for the individual dSCN neurons: WT (white circles, day, n = 10; black circles, night, n = 23); Het Q175 (white upward triangles, day, n = 21; black upward triangles, night, n = 17); Hom Q175 (white downward triangles, day, n = 22; black downward triangles, night, n = 23). The data was analyzed using two-way ANOVA with genotype and time as factors. The Q175 dSCN neurons did not exhibit the day/night difference in SFR seen in WT neurons. * = significant difference between genotypes; # = significant difference between day and night. (B) Current injection (5pA) increased the firing rate in the SCN in WT and Het Q175 mice (n=14 per genotype), but the increase seen in WT is significantly larger than that measured in the Het Q175. Symbols show mean ± SEM of the firing rate before, during, and after current injection. Two-way ANOVA was used to identify significant effects of genotype and treatment on firing rate. * = significant difference between genotypes; # = significant difference between control and treated. (C) BK currents measured in Het Q175 dSCN neurons (n = 9) were significantly larger than those in WT (n = 6) during the day (ZT 5-7). Symbols and error bars represent means ± SEM. Two-way ANOVA was used to identify significant effects of genotype and/or voltage step magnitude on evoked currents. There were significant effects of genotype on the BK current at the 60 and 80 mV steps. * = significant difference between genotypes.
Next, we sought to determine if direct current injection into the SCN neurons could overcome the decreased SFR in the Het Q175 mice. Using current-clamp recording in the whole cell patch configuration, we injected a current of 5 pA in dSCN neurons. Current injection increased SFR in both WT (Pre: 3.9 ± 0.7 Hz, Injection: 12.9 ± 1.0 Hz; paired t-test, t(13) = −12.142, n = 14, P < 0.001) and Het Q175 mice (Pre: 1.9 ± 0.3 Hz, Injection: 6.6 ± 1.1 Hz, t(13) = 5.811, n = 14, P < 0.001) (Fig. 6B). These results demonstrated that SCN neurons from Het Q175 mice still had the capacity to generate APs at the normal frequency.
Finally, we measured the BK current in the Het Q175 in the daytime (Fig. 6C), and found significant effects of genotype (F(1, 149) = 9.401, n = 15, P = 0.003), voltage (F(9, 149)= 49.730, n = 15, P < 0.001), and interaction effect of genotype and voltage (F(9, 149)= 5.374, n = 15, P < 0.001). Post-hoc multiple pairwise comparison testing determined that Q175 neurons exhibited significantly greater BK current at 60 mV (WT = 21 ± 3 pA/pF; Q175 = 39 ± 6 pA/pF, t(13) = 4.181, P < 0.001) and 80 mV (WT = 30 ± 4 pA/pF; Q175 = 55 ± 8 pA/pF, t(13) = 6.035, P < 0.001) steps. Thus, the Q175 HD model exhibits low daytime electrical activity with enhanced BK currents as described in the BACHD model.
Discussion
The characteristic spontaneous rhythmic neural activity of central circadian clock located in the SCN is compromised in the BACHD mouse model (Kudo et al., 2011b; Kuljis et al., 2016). In the present study, we sought to identify the ionic mechanisms underlying depressed daytime SFR and the loss of electrical activity rhythms in BACHD dSCN neurons. Our first hypothesis was that enhanced GABA signaling within the GABAergic SCN circuit inhibited electrical activity of BACHD SCN neurons, but treatment with the GABAA-R antagonist gabazine failed to restore BACHD dSCN SFR (Fig. 1). At the level of the individual cell, the interplay of multiple ionic currents regulates daily rhythms in SFR. APs recorded from the BACHD neurons in the day had significantly larger AP AHP areas compared to WT controls (Table 1), suggesting K+ channels may be involved.
Previous studies have shown that night-time hyperpolarization of Per1 expressing SCN neurons is associated with reduced input resistance and increased K+ conductance (Kuhlman and McMahon, 2004). The model suggested by this study along with earlier work in Bulla (reviewed in Block and Colwell, 2014) is that the daily increase in electrical activity is driven by the closing of K+ channels and the resulting depolarization of the membrane potential. Consistent with this model, in WT littermate dSCN neurons, we found day/night differences in resting Vm and input resistance. During the day, neuronal membrane potential was depolarized and input resistance high, and during the night membrane potential was hyperpolarized and input resistance low. Importantly, these fundamental rhythms in resting membrane properties are lost in the dSCN neurons from the BACHD model (Fig. 2), again pointing to dysfunction in K+ currents in the BACHDSCN.
In the BACHD mouse, SCN neurons’ daytime SFR is depressed, which may have been accounted for either by an inability to generate APs or membrane hyperpolarization. To distinguish between these two possibilities, we examined the impact of direct current injection and pharmacological excitation with NMDA (Fig. 3). Both treatments were effective in depolarizing Vm and increasing BACHD SFRs to WT levels. At least for younger animals, WT littermate SCN neurons fire close to their maximal rates during the daytime. Direct current injection did not result in increased discharge in the SCN neurons from younger animals (3 months of age) likely due to “depolarization block”. It is interesting, although unexplained, that direct current injection did increase firing rate in older WT controls for the Q175 line (7 months of age). NMDA treatment did increase the SFR of WT neurons of both ages. NMDA-R are calcium permeable, and activation of calcium-sensitive K+ channels might account for different WT responses between the two excitation methods. Overall, BACHD SCN neurons displayed the ability to generate APs at WT levels with both excitatory treatments, suggesting interventions that increase excitatory drive to the SCN or changes in intrinsic excitability during the day may be helpful for improving circadian system function.
K+ currents critical for determining spiking rates of SCN neurons include the FDR and the BK currents (Colwell, 2011; Allen et al., 2017). During the daytime in BACHD SCN, when the greatest effects of excitation on SFR were observed, we found the FDR current was unaltered (Fig. 4A, C), while the BK current was enlarged at positive Vm’s (Fig. 4B, D). The later finding is consistent with AP-driven enhancement of K+ conductances. Prior work by Meredith and colleagues indicate that the BK current is a critical regulator of both daytime firing rate and Vm (Meredith et al., 2006; Montgomery and Meredith, 2012; Whitt et al., 2016), with currents typically active during AP repolarization and AP afterhyperpolarization. The larger AP afterhyperpolarization area (Table 1), RMP hyperpolarization (Fig. 2), enhanced BK currents (Fig. 4D), and similar (15-20%) degree of Kcnma1 gene expression potentiation (Fig. 5), all implicate aberrant BK channel regulation in SCN of the HD mouse.
BK regulation of neural excitability depends on the channel’s subunit composition, which is highly regulated in the nervous system. Multiple splice isoforms of the pore-forming alpha subunit exist and can be differentially associated with multiple accessory beta subunits to yield diverse BK currents with different activation/inactivation kinetics and calcium-modulated voltage activations (see review Contet et al., 2016). Fast-activating/inactivating BK currents, such as those associated with the β2-accessory subunit, are thought to predominantly contribute to the AP repolarization phase to mediate fast spike rates (Martinez-Espinosa et al., 2014; Whitt et al. 2016). This is in contrast to slowly-activating/inactivating BK currents, such as those associated with the β4 accessory subunit, which are thought to contribute predominantly to the AP afterhyperpolarization phase to decrease spike rates (Brenner et al., 2005; Petrik et al., 2011). Rhythmic transcription of BK channel subunits is an essential mechanism underlying rhythmic BK current regulation in SCN (Montgomery et al., 2013). HD associated transcription dysregulation, including increased CRE-mediated transcription, has previously been shown (Sugars and Rubinsztein, 2003; Obrietan and Hoyt, 2004), and considering CREB is a known positive regulator of BK expression (Wang et al., 2009), HD-related transcriptional dysregulation may account for the increased Kcnma1 transcript levels we observed in BACHD SCN. Without potentiation of the inactivating β2-subunit’s transcription, higher daytime BK alpha subunit expression may enhance BK contribution to AP afterhyperpolarization and lengthen interspike intervals. Evidence of HD-driven channelopathies have been observed in other brain regions (Ariano et al., 2005; Dallerac et al., 2015; Sebastianutto et al., 2017), but these studies generally found a reduction in K+ currents that resulted in membrane depolarization and did not explore BK currents specifically. More work is required to determine whether alterations to BK currents are a common feature of neural circuits affected by the HD causing mutation and future studies utilizing genetic manipulation of the levels of the BK subunits would be very helpful to clarify these issues. Our findings are consistent with the possibility that the larger BK current seen in SCN of BACHD and Q175 mice is a critical piece of the circadian pathology observed in these mutants.
Aged mice show reduced amplitude activity and physiological rhythms as well as increases in behavioral circadian rhythm period and fragmentation similar to those exhibited by young presymptomatic BACHD mice (Valentinuzzi et al., 1997; Nakamura et al., 2011; Farajnia et al., 2014). Aged SCN also exhibits reduced amplitude of SFR rhythms (Nakamura et al., 2011; Farajnia et al., 2012), reminiscent of the phenotype observed in BACHD SCN. Prior work indicates that in aged SCN, BK currents are reduced during the nighttime, and this effect is associated with diminished AP afterhyperpolarization, depolarized RMP, widened AP, and increased intracellular Ca2+ levels (Farajnia et al., 2015). While there are clearly differences between the findings in aged (>23 months) and BACHD (~3 months) SCN pathophysiology, these findings suggest alterations of BK currents may elicit inappropriate neuronal activity in the SCN.
Among the mouse models of HD, the heterozygote (Het) Q175 offers advantages of single copy of the mutation, genetic precision of the insertion and control of mutation copy number (Pouladi et al., 2013). Prior work with this model has demonstrated that the Q175 line exhibits age-dependent disruptions in motor performance (Heikkinen et al., 2012; Menalled et al., 2012) as well as circadian behavior (Loh et al., 2013; Cutler et al., 2017) when compared to WT. In the present study, we sought to confirm our principle findings in the BACHD model with the Q175 model (Fig. 6). The fact that these key observations were exhibited by two unrelated mouse models strengthens our argument that SCN pathophysiology is a fundamental symptom of HD.
In summary, disturbances in the timing of the sleep/wake cycle are a well-established symptom of HD and other neurodegenerative diseases. These behavioral symptoms raise questions about the mechanisms underlying these disturbances and the possible involvement of the circadian system. Using mouse models, we have been able to demonstrate that the neurons within the central circadian clock (SCN) exhibit pathophysiology early in disease progression (Kudo et al., 2011; Kuljis et al., 2016; present study). Notably, these findings do not preclude the involvement of other well-known structures involved in HD, such as the basal ganglia, in eliciting a disrupted circadian output. The dysfunction found in the circadian system of multiple models does however raise the question of whether it is possible to ameliorate the symptoms and, in the end, to “treat” HD and other neurodegenerative diseases using environmental manipulations designed to improve circadian rhythms and enhance the sleep/wake cycle by increasing excitatory drive onto SCN (Morton, 2013; Schroeder and Colwell, 2013). Prior work using the R6/2 HD model found that a combination of bright-light therapy and scheduled voluntary exercise was beneficial in delaying disease symptoms (Cuesta et al., 2014), and more recently that exposing the Q175 model to blue-wavelength enhanced lighting (Wang et al., 2017) and time-restricted feeding (Wang et al., 2018) was similarly beneficial in delaying symptom progression. Since behavior changes may be difficult to implement, it is also important to develop pharmacological treatments. In HD models, timed administration of GABAA-R agonists (Pallier et al., 2007) as well as a blocker of histamine receptor type 3 blocker (Whittaker et al., 2017) have proven to be beneficial. Thus, preclinical HD models are proving to be advantageous for exploring disease mechanisms and the impact of circadian treatments and therapies on disease progression.
Significance:
We found decreased daytime electrical activity in the central circadian clock (SCN) in two mouse models of HD. The mutant neurons have the capacity to generate action potentials at a higher frequency in response to direct current injection or application of exogenous NMDA. Thus, the SCN neurons are still early in the degenerative process and rescue is still possible by treatments that alter intrinsic excitability or the balance of excitatory/inhibitory synaptic transmission. Finally, we found that the BK current was enhanced in the mutant SCN providing a possible mechanistic explanation for the low daytime electrical activity.
Acknowledgements:
Dr. Dika Kuljis: 159C Mellon Institute, Dept. of Biological Sciences, Carnegie Mellon University 4400 Fifth Avenue, Pittsburgh, PA 15213. dika.kuljis@gmail.com.
Dr. Takashi Kudo: Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, Kunigami-gun, Okinawa, Japan 904-0495. tkudo3@gmail.com.
Funding was provided by CHDI Foundation grant #A-7293, DAK was supported by a training grant from the Laboratory of Neuroendocrinology (T32 HD 07228-26).
Footnotes
Conflict of Interest Statement:
The authors have no conflict of interest to declare.
References
- Allen CN, Nitabach MN, Colwell CS. 2017. Membrane Currents, Gene Expression, and Circadian Clocks. Cold Spring Harb Perspect Biol 9(5). pii: a027714. doi: 10.1101/cshperspect.a027714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariano MA, Cepeda C, Calvert CR, Flores-Hernández J, Hernández-Echeagaray E, Klapstein GJ, Chandler SH, Aronin N, DiFiglia M, Levine MS. 2005. MSN in R6/2 reductions in both inwardly and outwardly rectifying K+ currents. J Neurophysiol 93(5):2565–74. doi: 10.1152/jn.00791.2004. [DOI] [PubMed] [Google Scholar]
- Aziz NA, Anguelova GV, Marinus J, Lammers GJ, Roos RAC. 2010. Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism Relat Disord 16:345–350. doi: 10.1016/j.parkreldis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Aziz NA, Pijl H, Frölich M, Schröder-van der Elst JP, van der Bent C, Roelfsema F, Roos RAC. 2009. Delayed onset of the diurnal melatonin rise in patients with Huntington’s disease. J Neurol 256:1961–1965. doi: 10.1007/s00415-009-5196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block GD and Colwell CS. 2014. Molluskan Ocular Pacemakers: Lessons Learned pp 213–232. In: The Retina and Circadian Rhythms, Edited by Tosini G, luvone PM, McMahon DG, Collin SP. Springer-Verlag; New York City. doi: 10.1007/978-1-4614-9613-7 [DOI] [Google Scholar]
- Bouskila Y, Dudek FE. 1995. A rapidly activating type of outward rectifier K+ current and A-current in rat suprachiasmatic nucleus neurones. J Physiol 488(Pt 2):339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. 2005. BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci 8(12):1752–9. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, Besing RC, Menaker M, Gewirtz AT, Davidson AJ. 2010. Dysregulation of inflammatory responses by chronic circadian disruption. J Immunol 185:5796–5805. doi: 10.4049/jimmunol.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloues RK, Sather WA. 2003. Afterhyperpolarization regulates firing rate in neurons of the suprachiasmatic nucleus. J Neurosci 23:1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS. 2011. Linking neural activity and molecular oscillations in the SCN. Nat Rev Neurosci 12:553–569. doi: 10.1038/nrn3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contet C, Goulding SP, Kuljis DA, Barth AL. 2016. BK Channels in the Central Nervous System. Int Rev Neurobiol 128:281–342. doi: 10.1016/bs.irn.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta M, Aungier J, Morton AJ. 2014. Behavioral therapy reverses circadian deficits in a transgenic mouse model of Huntington’s disease. Neurobiol Dis 63:85–91. doi: 10.1016/j.nbd.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Cutler TS, Park S, Loh DH, Jordan MC, Yokota T, Roos KP, Ghiani CA, Colwell CS. 2017. Neurocardiovascular deficits in the Q175 mouse model of Huntington’s disease. Physiol Rep 5(11). pii: e13289. doi: 10.14814/phy2.13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallérac GM, Levasseur G, Vatsavayai SC, Milnerwood AJ, Cummings DM, Kraev I, Huetz C, Evans KA, Walters SW, Rezaie P, Cho Y, Hirst MC, Murphy KP. 2015. Dysfunctional Dopaminergic Neurones in Mouse Models of Huntington’s Disease: A Role for SK3 Channels. Neurodegener Dis 15(2):93–108. doi: 10.1159/000375126. [DOI] [PubMed] [Google Scholar]
- Farajnia S, Deboer T, Rohling JH, Meijer JH, Michel S. 2014. Aging of the suprachiasmatic clock. Neuroscientist 20(1):44–55. doi: 10.1177/1073858413498936. [DOI] [PubMed] [Google Scholar]
- Farajnia S, Michel S, Deboer T, Vanderleest HT, Houben T, Rohling JHT, Ramkisoensing A, Yasenkov R, Meijer JH. 2012. Evidence for neuronal desynchrony in the aged suprachiasmatic nucleus clock. J Neurosci 32:5891–5899. doi: 10.1523/JNEUROSCI.0469-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farajnia S, Meijer JH, Michel S. 2015. Age-related changes in large-conductance calcium-activated potassium channels in mammalian circadian clock neurons. Neurobiol Aging 36(6):2176–83. doi: 10.1016/j.neurobiolaging.2014.12.040. [DOI] [PubMed] [Google Scholar]
- Foster RG, Wulff K. 2005. The rhythm of rest and excess. Nat Rev Neurosci 6:407–414. doi: 10.1038/nrn1670. [DOI] [PubMed] [Google Scholar]
- Goodman AOG, Morton AJ, Barker RA. 2010. Identifying sleep disturbances in Huntington ‘ s disease using a simple disease-focused questionnaire. PLoS Curr HD 2:RRN1189. doi: 10.1371/currents.RRN1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AOG, Rogers L, Pilsworth S, McAllister CJ, Shneerson JM, Morton AJ, Barker RA. 2011. Asymptomatic sleep abnormalities are a common early feature in patients with Huntington’s disease. Curr Neurol Neurosci Rep 11:211–217. doi: 10.1007/s11910-010-0163-x. [DOI] [PubMed] [Google Scholar]
- Granados-Fuentes D, Hermanstyne TO, Carrasquillo Y, Nerbonne JM, Herzog ED. 2015. IA Channels Encoded by Kv1.4 and Kv4.2 Regulate Circadian Period of PER2 Expression in the Suprachiasmatic Nucleus. J Biol Rhythms. 30(5):396–407. doi: 10.1177/0748730415593377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granados-Fuentes D, Norris AJ, Carrasquillo Y, Nerbonne JM, Herzog ED. 2012. I(A) channels encoded by Kv1.4 and Kv4.2 regulate neuronal firing in the suprachiasmatic nucleus and circadian rhythms in locomotor activity. J Neurosci 32(29):10045–52. doi: 10.1523/JNEUROSCI.0174-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, Andre VM, Wilburn B, Lu X-H, Tao J, Yamazaki I, Li S-H, Sun YE, Li X-J, Levine MS, Yang XW. 2008. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM, McRitchie DA, Macdonald V, Double KL, Trent RJ, McCusker E. 1998. Regional specificity of brain atrophy in Huntington’s disease. Exp Neurol 154:663–672. DOI: 10.1006/exnr.1998.6919. [DOI] [PubMed] [Google Scholar]
- Heikkinen T, Lehtimäki K, Vartiainen N, Puoliväli J, Hendricks SJ, Glaser JR, Bradaia A, Wadel K, Touller C, Kontkanen O, Yrjänheikki JM, Buisson B, Howland D, Beaumont V, Munoz-Sanjuan I, Park LC. 2012. Characterization of Neurophysiological and Behavioral Changes, MRI Brain Volumetry and 1H MRS in Q175 Knock-In Mouse Model of Huntington’s Disease. PLoS ONE 7: e50717. doi: 10.1371/journal.pone.0050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanstyne TO, Granados-Fuentes D, Mellor RL, Herzog ED, Nerbonne JM. 2017. Acute Knockdown of Kv4.1 Regulates Repetitive Firing Rates and Clock Gene Expression in the Suprachiasmatic Nucleus and Daily Rhythms in Locomotor Behavior. eNeuro 4(3). pii: ENEURO.0377-16.2017. doi: 10.1523/ENEURO.0377-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog ED, Hermanstyne T, Smyllie NJ, Hastings MH. 2017. Regulating the Suprachiasmatic Nucleus (SCN) Circadian Clockwork: Interplay between Cell-Autonomous and Circuit-Level Mechanisms. Cold Spring Harb Perspect Biol 9(1). pii: a027706. doi: 10.1101/cshperspect.a027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itri JN, Vosko AM, Schroeder A, Dragich JM, Michel S, Colwell CS. 2010. Circadian regulation of A-type potassium currents in the suprachiasmatic nucleus. J Neurophysiol 103:632–640. doi: 10.1152/jn.00670.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itri JN, Michel S, Vansteensel MJ, Meijer JH, Colwell CS. 2005. Fast delayed rectifier potassium current is required for circadian neural activity. Nat Neurosci 8:650–656. doi: 10.1038/nn1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliolia E, Silajdžić E, Nambron R, Hill NR, Doshi A, Frost C, Watt H, Hindmarsh P, Björkqvist M, Warner TT. 2014. Plasma melatonin is reduced in Huntington’s disease. Mov Disord 29:1511–1515. doi: 10.1002/mds.26003. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, Palm IF, La Fleur SE, Scheer FAJL, Perreau-Lenz S, Ruiter M, Kreier F, Cailotto C, Buijs RM. 2006. SCN outputs and the hypothalamic balance of life. J Biol Rhythms 21:458–469. DOI: 10.1177/0748730406293854. [DOI] [PubMed] [Google Scholar]
- Kent J, Meredith AL. 2008. BK channels regulate spontaneous action potential rhythmicity in the suprachiasmatic nucleus. PLoS One 3:e3884 10.1371/journal.pone.0003884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Loh DH, Kuljis D, Constance C, Colwell CS. 2011a. Fast delayed rectifier potassium current: critical for input and output of the circadian system. J Neurosci 31:2746–2755. 10.1523/JNEUROSCI.5792-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, Colwell CS. 2011b. Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Exp Neurol 228:80–90. doi: 10.1016/j.expneurol.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman SJ, McMahon DG. 2004. Rhythmic regulation of membrane potential and potassium current persists in SCN neurons in the absence of environmental input. Eur J Neurosci 20:1113–1117. doi: 10.1111/j.1460-9568.2004.03555.x [DOI] [PubMed] [Google Scholar]
- Kuljis DA, Gad L, Loh DH, MacDowell Kaswan Z, Hitchcock ON, Ghiani CA, Colwell CS. 2016. Sex Differences in Circadian Dysfunction in the BACHD Mouse Model of Huntington’s Disease. PLoS One. 11(2):e0147583. doi: 10.1371/journal.pone.0147583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuljis DA, Loh DH, Truong D, Vosko AM, Ong ML, McClusky R, Arnold AP, Colwell CS. 2013. Gonadal- and sex-chromosome-dependent sex differences in the circadian system. Endocrinology 154:1501–1512. DOI: 10.1210/en.2012-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuljis D, Schroeder AM, Kudo T, Loh DH, Willison DL, Colwell CS. 2012. Sleep and circadian dysfunction in neurodegenerative disorders: insights from a mouse model of Huntington’s disease. Minerva Pneumol 51:93–106. [PMC free article] [PubMed] [Google Scholar]
- Kyle BD, Braun AP. 2014. The regulation of BK channel activity by pre- and post-translational modifications. Front Physiol 5:1–10 doi: 10.3389/fphys.2014.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh DH, Kudo T, Truong D, Wu Y, Colwell CS. 2013. The Q175 mouse model of Huntington’s disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PLoS One 8:e69993. doi: 10.1371/journal.pone.0069993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Espinosa PL, Yang C, Gonzalez-Perez V, Xia XM, Lingle CJ. 2014. Knockout of the BK β2 subunit abolishes inactivation of BK currents in mouse adrenal chromaffin cells and results in slow-wave burst activity. J Gen Physiol. 144(4):275–95. doi: 10.1085/jgp.201411253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, Fraenkel E, McAllister CJ, Wood N, Reddy AB, Hastings MH, Morton AJ. 2010. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J Neurosci 30:10199–10204. DOI: 10.1523/JNEUROSCI.1694-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C et al. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, Connor D, Murphy C, Oakeshott S, Kwan M, Beltran J, Ghavami A, Brunner D, Park LC, Ramboz S, Howland D. 2012. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS One 7(12): e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith AL, Wiler SW, Miller BH, Takahashi JS, Fodor AA, Ruby NF, Aldrich RW. 2006. BK calcium-activated potassium channels regulate circadian behavioral rhythms and pacemaker output. Nat Neurosci 9:1041–1049. doi: 10.1038/nn1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, Takahashi JS. 2012. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci 35:445–62. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JR, Meredith AL. 2012. Genetic activation of BK currents in vivo generates bidirectional effects on neuronal excitability. Proc Natl Acad Sci USA 109:18997–19002. doi: 10.1073/pnas.1205573109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JR, Whitt JP, Wright BN, Lai MH, Meredith AL. 2013. Mis-expression of the BK K(+) channel disrupts suprachiasmatic nucleus circuit rhythmicity and alters clock-controlled behavior. Am J Physiol Cell Physiol. 304(4):C299–311. doi: 10.1152/ajpcell.00302.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ. 2013. Circadian and sleep disorder in Huntington’s disease. Exp Neurol 243:34–44. doi: 10.1016/j.expneurol.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. 2005. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci 25:157–163. DOI: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TJ, Nakamura W, Yamazaki S, Kudo T, Cutler T, Colwell CS, Block GD. 2011. Age-related decline in circadian output. J Neurosci 31:10201–10205 doi: 10.1523/JNEUROSCI.0451-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrietan K and Hoyt KR. 2004. CRE-Mediated Transcription Is Increased in Huntington’s Disease Transgenic Mice. J. Neurosci 24(4) 791–796. DOI: 10.1523/JNEUROSCI.3493-03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallier PN, Maywood ES, Zheng Z, Chesham JE, Inyushkin AN, Dyball R, Hastings MH, Morton AJ. 2007. Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J Neurosci 27:7869–7878. doi: 10.1523/JNEUROSCI.0649-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen J, Conybeare R. 2005. Cognitive changes in Huntington’s disease. Adv Neurol 96:209–225. [PubMed] [Google Scholar]
- Paulsen JS, Magnotta VA, Mikos AE, Paulson HL, Penziner E, Andreasen NC, Nopoulos PC. 2006. Brain structure in preclinical Huntington’s disease. Biol Psychiatry 59:57–63. DOI: 10.1016/j.biopsych.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Petersén A, Björkqvist M. 2006. Hypothalamic-endocrine aspects in Huntington’s disease. Eur J Neurosci 24:961–967. DOI: 10.1111/j.1460-9568.2006.04985.x [DOI] [PubMed] [Google Scholar]
- Petersén A, Gil J, Maat-Schieman MLC, Björkqvist M, Tanila H, Araújo IM, Smith R, Popovic N, Wierup N, Norlén P, Li J-Y, Roos RAC, Sundler F, Mulder H, Brundin P. 2005. Orexin loss in Huntington’s disease. Hum Mol Genet 14:39–47 DOI: 10.1093/hmg/ddi004. [DOI] [PubMed] [Google Scholar]
- Petrik D, Wang B, Brenner R. Modulation by the BK accessory β4 subunit of phosphorylation-dependent changes in excitability of dentate gyrus granule neurons. Eur J Neurosci. (September, 2011). 34(5):695–704. doi: 10.1111/j.1460-9568.2011.07799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts GR, Ohta H, McMahon DG. 2006. Daily rhythmicity of large-conductance Ca2+-activated K+ currents in suprachiasmatic nucleus neurons. Brain Res 1071:54–62. doi: 10.1016/j.brainres.2005.11.078. [DOI] [PubMed] [Google Scholar]
- Pouladi MA, Morton AJ, and Hayden MR. 2013. Choosing an animal model for the study of Huntington’s disease. Nat. Rev. Neurosci 14:708–721. doi: 10.1038/nrn3570. [DOI] [PubMed] [Google Scholar]
- Reddy AB, O’Neill JS. 2010. Healthy clocks, healthy body, healthy mind. Trends Cell Biol 20:36–44. doi: 10.1016/j.tcb.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. 2011. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- Rüger M, Scheer FAJL. 2009. Effects of circadian disruption on the cardiometabolic system. Rev Endocr Metab Disord 10:245–260. doi: 10.1007/s11154-009-9122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder AM, Colwell CS. 2013. How to fix a broken clock. Trends PharmacolSci 34(11):605–19. doi: 10.1016/j.tips.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder AM, Wang HB, Park S, Jordan MC, Gao F, Coppola G, Fishbein MC, Roos KP, Ghiani CA, Colwell CS. 2016. Cardiac Dysfunction in the BACHD Mouse Model of Huntington’s Disease. PLoS One 11(1):e0147269. doi: 10.1371/journal.pone.0147269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastianutto I, Cenci MA, Fieblinger T. 2017. Alterations of striatal indirect pathway neurons precede motor deficits in two mouse models of Huntington’s disease. Neurobiol Dis. 105:117–131. doi: 10.1016/j.nbd.2017.05.011. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. 2003. Transcriptional abnormalities in Huntington disease. Trends Genet 19(5):233–8. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Valentinuzzi VS, Scarbrough K, Takahashi JS, Turek FW. 1997. Effects of aging on the circadian rhythm of wheel-running activity in C57BL/6 mice. Am J Physiol 273:R1957–64. PMID: 9435649. [DOI] [PubMed] [Google Scholar]
- Videnovic A, Noble C, Reid KJ, Peng J, Turek FW, Marconi A, Rademaker AW, Simuni T, Zadikoff C, Zee PC. 2014. Circadian Melatonin Rhythm and Excessive Daytime Sleepiness in Parkinson Disease. JAMA Neurol 02114:1–7. doi: 10.1001/jamaneurol.2013.6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wamelen DJ, Aziz NA, Anink JJ, Steenhoven R Van, Angeloni D, Fraschini F, Jockers R, Roos RAC, Swaab DF. 2013. Suprachiasmatic Nucleus Neuropeptide Expression in Patients with Huntington’s Disease. Sleep 36:117–125. doi: 10.5665/sleep.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Loh DH, Whittaker DS, Cutler T, Howland D, Colwell CS. 2018. Time-Restricted Feeding Improves Circadian Dysfunction as well as Motor Symptoms in the Q175 Mouse Model of Huntington’s Disease. eNeuro 3: 5(1). doi: 10.1523/ENEURO.0431-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-B, Whittaker DS, Truong D, Mulji AK, Ghiani CA, Loh DH, Colwell CS. 2017. Blue light therapy improves circadian dysfunction as well as motor symptoms in two mouse models of Huntington’s disease. Neurobiol Sleep Circadian Rhythms 2:39–52. doi: 10.1016/j.nbscr.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ghezzi A, Yin JCP, Atkinson NS. 2009. CREB regulation of BK channel gene expression underlies rapid drug tolerance. Genes Brain Behav 8(4):369–376. doi: 10.1111/j.1601-183X.2009.00479.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AB, Angelo N, Huettner JE, Herzog ED. 2009. Intrinsic, nondeterministic circadian rhythm generation in identified mammalian neurons. Proc Natl Acad Sci USA 106:16493–16498. doi: 10.1073/pnas.0902768106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DK, Takahashi JS, Kay SA. 2010. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol 72:551–577. doi: 10.1146/annurev-physiol-021909-135919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitt JP, Montgomery JR, Meredith AL. 2016. BK channel inactivation gates daytime excitability in the circadian clock. Nat Commun 7:10837. doi: 10.1038/ncomms10837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker DS, Wang H-B, Loh DH, Cachope R, Colwell CS. 2017. Possible use of a H3R antagonist for the management of non-motor symptoms in the Q175 mouse model of Huntington’s disease. Pharma Res Per e00344. doi: 10.1002/prp2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]