

Summary Sections
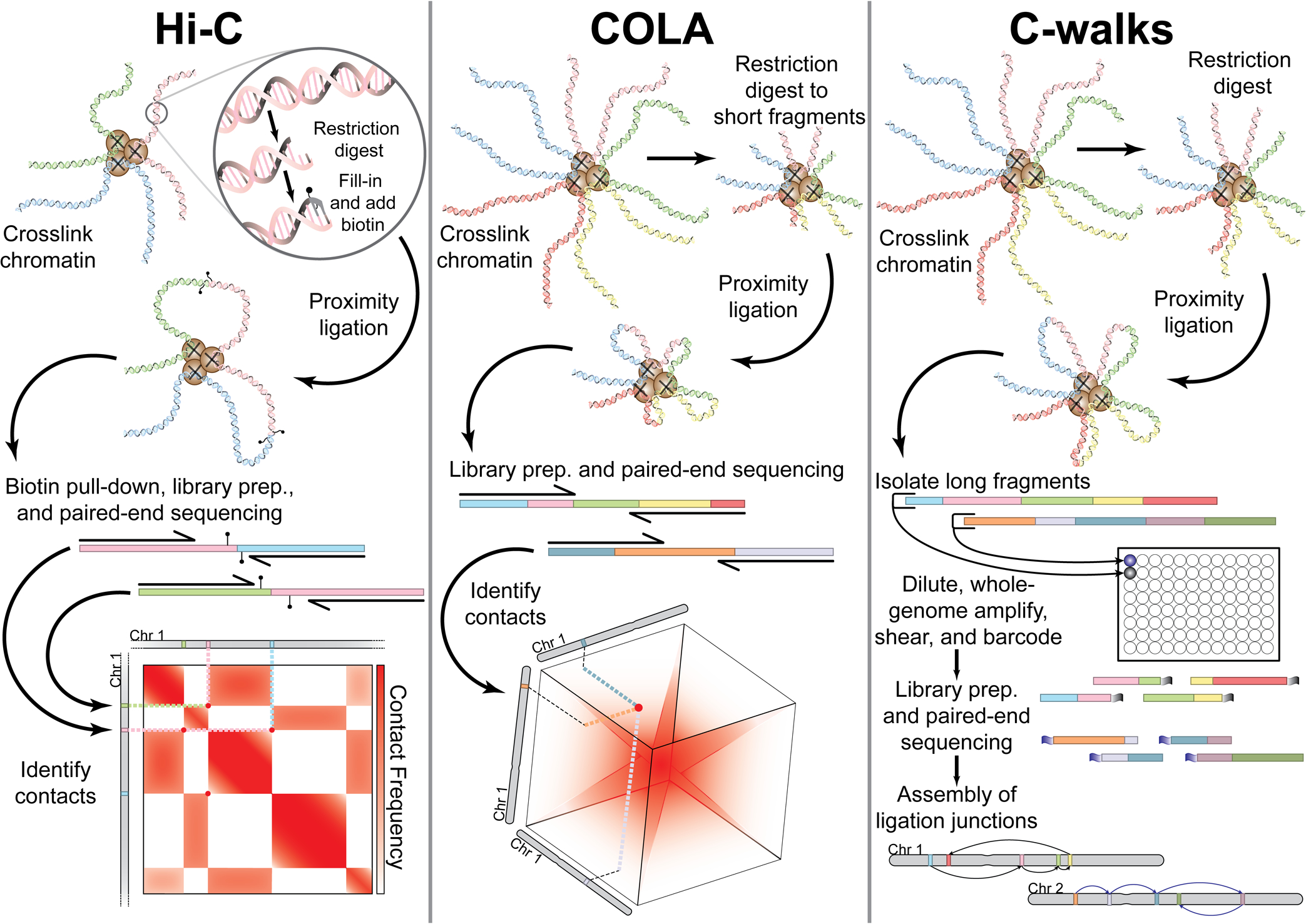
Nuclear architecture has remained mysterious due to inadequate methods for observing DNA folding. New technologies aim to probe chromosome organization by mapping chromatin contacts. Chromosome conformation capture (3C) originally relied on chemical crosslinking of chromatin followed by proximity ligation to recover spatial information of neighboring genomic loci. Ligation-based methods have progressed from measuring pairwise contacts (Hi-C), to a handful (COLA, Tri-C, MC-4C) and dozens (C-walks) of interactions genome-wide.
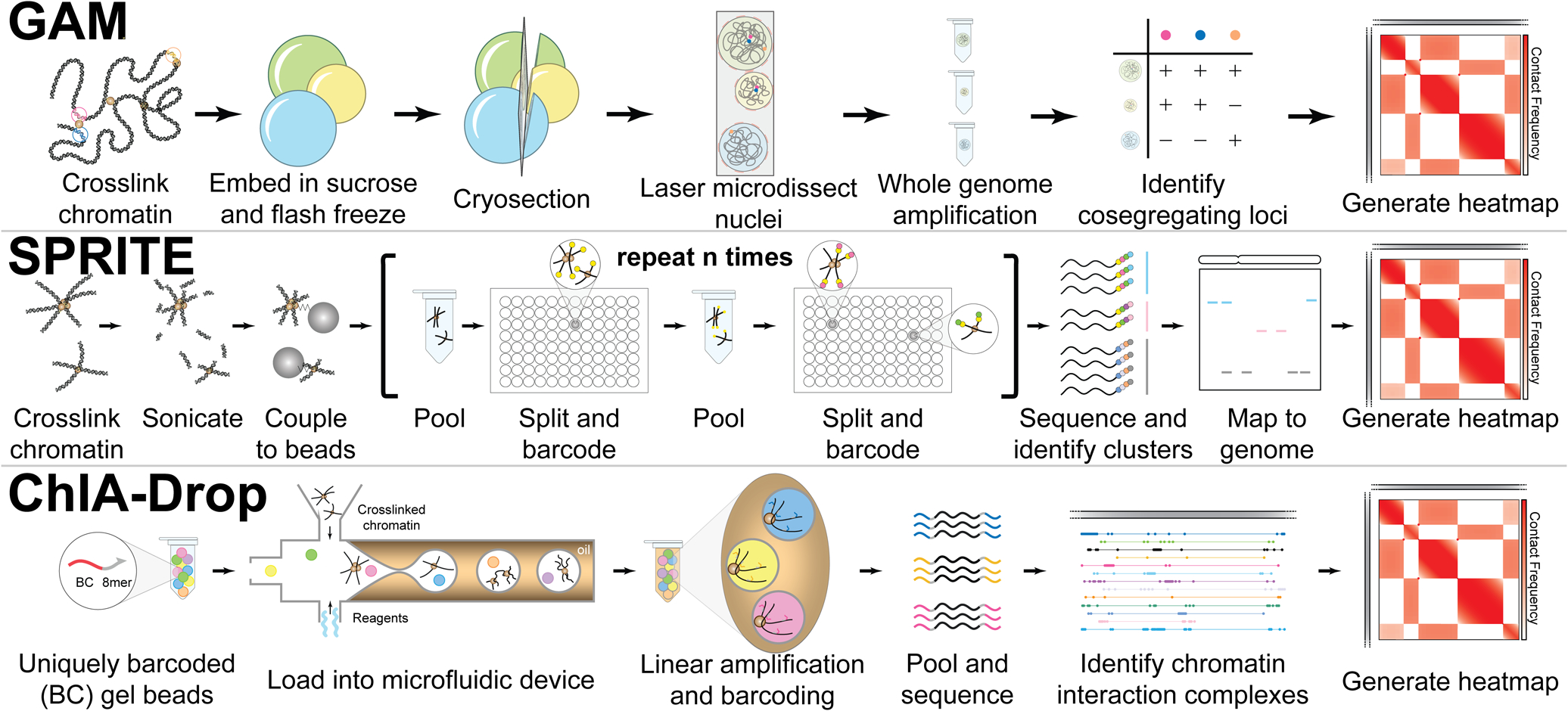
Technologies based on proximity ligation inefficiently detect multiple interactions per locus. Ligation-free methods enhance the identification of higher-order interactions among distinct chromosomal regions as well as those within nuclear compartments. These novel methods provide unbiased spatial information from low cell inputs (Genome Architecture Mapping [GAM]), identify simultaneous interactions between multiple genomic loci (Split-pool Recognition of Interactions by Tag Extension [SPRITE]), and determine precise interactions at the single molecule level (ChIA-Drop).
Advantages
- Hi-C
- High-resolution (~1 kb) analysis possible
- Reduced noise by enriching for ligation junctions
- COLA
- Shorter cross-linked fragments can recover more concatemers
- C-Walks
- Isolation of longer DNA concatamers can reveal hubs of interaction
- COLA & C-walks
- Can detect multiway interactions, beyond pairwise contacts
- GAM
- Captures multivalent interactions using only a small number of cells
- Uniquely isolates nuclear slices versus processing of cross-linked chromatin
- SPRITE
- Maps both multiple DNA-DNA contacts as well as DNA-RNA contacts
- Does not require encapsulation nor extensive whole genome amplification
- ChIA-Drop
- Single-molecule precision
- Requires only a few thousand cells
Challenges
- Hi-C, COLA, & C-Walks
- Proximity ligation-based approach
- Bulk, cell population data
- Hi-C
- Lower probability of capturing multiway interactions
- COLA & C-Walks
- Sparse data, particularly as the valency of interactions increases
- GAM
- Time-consuming
- Requires special equipment and training
- SPRITE
- Relies on extensive fixation
- ChIA-Drop
- Background noise (singleton DNA fragments)
- Comprehensiveness limited by number of droplet-containing chromatin complexes
- Low resolution for mammalian systems
All methods infer, rather than directly observe, chromatin folding
Acknowledgements
We are grateful to Haneen Ammouri and Qi Yu for helpful reading of this manuscript and apologize to those whose work could not be cited due to space limitations. Research in the Eagen Lab is supported by an NIH Director’s Early Independence Award DP5OD024587.
References
- 1.Allahyar A et al. (2018) Enhancer hubs and loop collisions identified from single-allele topologies. Nat Genet 50, 1151–1160 [DOI] [PubMed] [Google Scholar]
- 2.Beagrie RA et al. (2017) Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darrow EM et al. (2016) Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proc National Acad Sci 113, E4504–E4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dekker J et al. (2002) Capturing chromosome conformation. Science 295, 1306–1311 [DOI] [PubMed] [Google Scholar]
- 5.Lieberman-Aiden E et al. (2009) Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 326, 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olivares-Chauvet P et al. (2016) Capturing pairwise and multi-way chromosomal conformations using chromosomal walks. Nature 540, 296. [DOI] [PubMed] [Google Scholar]
- 7.Oudelaar MA et al. (2018) Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nat Genet 50, 1744–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinodoz SA et al. (2018) Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao S et al. (2014) A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng M et al. (2019) Multiplex chromatin interactions with single-molecule precision. Nature 566, 558–562 [DOI] [PMC free article] [PubMed] [Google Scholar]