

Abstract
The metalloprotease ADAM17 (a disintegrin and metalloprotease 17) is a key regulator of tumor necrosis factor α (TNFα), interleukin 6 receptor (IL-6R), and epidermal growth factor receptor (EGFR) signaling. ADAM17 maturation and function depend on the seven-membrane–spanning inactive rhomboid-like proteins 1 and 2 (iRhom1/2 or Rhbdf1/2). Most studies to date have focused on overexpressed iRhom1 and -2, so only little is known about the properties of the endogenous proteins. Here, we show that endogenous iRhom1 and -2 can be cell surface–biotinylated on mouse embryonic fibroblasts (mEFs), revealing that endogenous iRhom1 and -2 proteins are present on the cell surface and that iRhom2 also is present on the surface of lipopolysaccharide-stimulated primary bone marrow–derived macrophages. Interestingly, very little, if any, iRhom2 was detectable in mEFs or bone marrow–derived macrophages lacking ADAM17, suggesting that iRhom2 is stabilized by ADAM17. By contrast, the levels of iRhom1 were slightly increased in the absence of ADAM17 in mEFs, indicating that its stability does not depend on ADAM17. These findings support a model in which iRhom2 and ADAM17 are obligate binding partners and indicate that iRhom2 stability requires the presence of ADAM17, whereas iRhom1 is stable in the absence of ADAM17.
Keywords: ADAM, transmembrane domain, protein stability, myeloid cell, metalloprotease, ADAM17, Rhbdf1, inactive Rhomboid 1, iRhom1, Rhbdf2, inactive Rhomboid 2, iRhom2, STING, stimulator of interferon genes, cell-surface enzyme, membrane protein
Introduction
A disintegrin and metalloprotease 17 (ADAM17)2 is a cell-surface metalloprotease that is required for the proteolytic processing of tumor necrosis factor α (TNFα) and is therefore also referred to as TACE (TNFα convertase). In addition, ADAM17 has a crucial role in the proteolytic release and activation of several ligands of the epidermal growth factor receptor (EGFR) as well as of the IL-6 receptor (IL-6R) and other membrane proteins (1–7). Major functions of ADAM17 include the regulation of the EGFR signaling pathway during development (4, 8–10) and protection of the skin and intestinal barrier in adults (11–14). Moreover, ADAM17 can contribute to cancers that involve inappropriate EGFR signaling (15, 16) and to pathologies involving dysregulated TNFα and IL-6R pathways, including autoimmune diseases such as rheumatoid arthritis (17, 18).
ADAM17 can be rapidly and post-translationally activated by a number of different signaling pathways (19–23) and requires its transmembrane domain, but not its cytoplasmic domain, for this rapid posttranslational activation (19). The seven-membrane–spanning protein iRhom2 (inactive Rhomboid 2, also referred to as Rhbdf2, Rhomboid 5 homolog 2) was identified as a crucial regulator of the maturation of ADAM17 in bone marrow-derived macrophages (BMDM) (24, 25). Additional insight into the relationship of ADAM17 and iRhom2 was provided by a point mutation in the first transmembrane domain (TMD) of iRhom2, termed sinecure, which results in a strong reduction of ADAM17-dependent TNFα release from BMDM (26). Mice that are homozygous for the iRhom2 sinecure mutation and also lack the related iRhom1 resemble previously described iRhom1/2−/− double knockout mice (27, 28), demonstrating that the sinecure point mutation in the first TMD of iRhom2 results in a strongly hypomorphic phenotype. Moreover, the substrate selectivity of ADAM17 is differentially regulated by iRhom1 and -2 (29), and point mutations in the TMD of ADAM17 that were predicted to affect the interaction with iRhom2 strongly reduced iRhom2/ADAM17-dependent shedding events, without affecting iRhom1/ADAM17-dependent shedding (28). The different substrate selectivity of iRhom2/ADAM17 and iRhom1/ADAM17-dependent shedding (29), the effects of the sinecure mutation on ADAM17, and the effects of point mutations in the TMD of ADAM17 on ADAM17/iRhom2-dependent shedding (28) suggested that iRhom2 and ADAM17 form a heteromeric complex. Presumably, this complex associates in the endoplasmic reticulum (ER) and remains together to regulate iRhom2/ADAM17-dependent shedding on the cell surface or in the late secretory pathway. This model is further supported by recent studies demonstrating that mutations in cytoplasmic phosphorylation sites of iRhom2 affect the activation of ADAM17 (30, 31). In addition, a newly discovered iRhom2-interacting protein, termed Frmd8 (FERM domain–containing 8) or iTAP (iRhom tail–associated protein), was found to regulate endocytosis of iRhom2/ADAM17 and degradation in the lysosome (32, 33).
Most studies on iRhom1 and -2 to date have focused on the overexpressed proteins (29–34), so there is a paucity of information on the cell biological properties of endogenous iRhom1 and only limited information on iRhom2 (25). The main goal of the current study was to perform a biochemical characterization of endogenous iRhom2 using primary mouse macrophage cultures and mouse embryonic fibroblasts. In light of the crucial role of iRhom2 in regulating the maturation and function of ADAM17 (27), we were also interested in whether ADAM17 reciprocally affects the stability of iRhom2 and the related iRhom1 and their transport to the cell surface. Moreover, because iRhom2 has been shown to interact with the multimembrane-spanning protein stimulator of interferon genes (STING) (35), this raised questions about the role of STING in the stability and maturation of iRhom2 and ADAM17.
Results
Characterization of murine iRhom2 in primary bone marrow–derived macrophages
To characterize endogenous mouse iRhom2, we raised rabbit polyclonal antibodies (pAbs) against a portion of the N-terminal cytoplasmic domain of murine iRhom2 (amino acid residues 1–376; see “Experimental procedures” for details). These anti-iRhom2 pAbs were tested on lysates of primary BMDM isolated from iRhom2−/− mice or WT controls. Because the expression of iRhom2 is up-regulated by treatment with LPS (36), we compared untreated BMDM with cells that had been stimulated with 10 ng/ml LPS overnight. LPS treatment induced a band of ∼95 kDa in WT BMDM (marked by an asterisk in Fig. 1A (top)) that was not present in iRhom2−/− BMDM. However, the anti-iRhom2 pAbs also reacted with several other proteins on blots of the BMDM lysates that were present in iRhom2-deficient samples, and thus nonspecific (Fig. 1A, top). The LPS treatment strongly induced the levels of pro- and mature ADAM17 in WT BMDM but only of pro-ADAM17 in iRhom2−/− BMDM (Fig. 1A (middle); tubulin served as loading control (bottom)).
Figure 1.
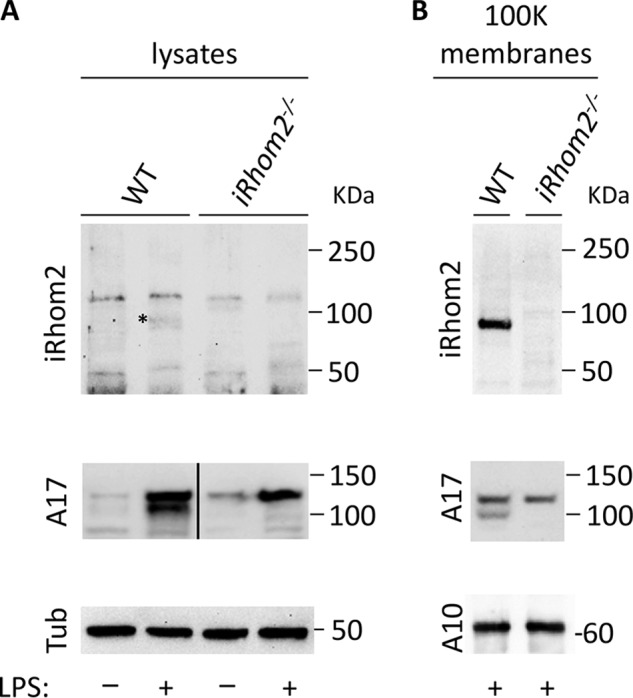
Western blot analysis of mouse iRhom2 in cell lysates and purified membranes isolated from mouse BMDM. A, primary BMDM isolated from WT or iRhom2−/− mice were either not treated or incubated with 10 ng/ml LPS for 14–18 h and then lysed and subjected to Western blot analysis. B, Western blotting of high-speed membrane preparations from LPS-treated WT or iRhom2−/− BMDM showed a strong enrichment of iRhom2 and much weaker nonspecific bands compared with the whole-cell lysate. The samples were either probed with rabbit polyclonal antibodies against the cytoplasmic domain of mouse iRhom2 (top panels), or antibodies against the cytoplasmic domain of ADAM17 (middle panels), or tubulin or ADAM10, as indicated (bottom panels). The heavy line in A (middle) indicates splicing of nonadjacent lanes. Each panel is representative of three separate experiments.
To improve detection of the seven-membrane–spanning iRhom2 and remove nonspecifically recognized soluble proteins, we purified cellular membranes by high-speed centrifugation to enrich for membrane proteins (see “Experimental procedures” for details). Western blot analysis of the purified material using the same anti-iRhom2(1–376) rabbit pAbs revealed a strong band of ∼95 kDa in the LPS-treated WT BMDM sample that was not present in the identically prepared iRhom2−/− control (Fig. 1B, top). Moreover, both pro- and mature ADAM17 were present in the whole lysate and membrane preparations from WT BMDM, whereas only pro-ADAM17 could be detected in the iRhom2−/− sample, but not mature ADAM17, as reported previously (24, 25) (Fig. 1, A and B, middle panels; the membrane-anchored ADAM10 (A10) served as a loading control in the bottom panel of Fig. 1B).
Cell-surface biotinylation of endogenous iRhom2 in LPS-stimulated BMDM
To establish whether the endogenous iRhom2 is present on the cell surface of LPS-treated BMDM, a non-membrane-permeable biotinylation reagent (Sulfo-NHS-LC-Biotin) was used to label cell-surface proteins on these cells (see “Experimental procedures” for details). When a Western blot of the purified biotinylated material was probed with anti-iRhom2 pAbs, a band of 95 kDa was detected in WT BMDM, but not in iRhom2−/− BMDM (Fig. 2, top). Endoglycosidase H (EndoH) typically cannot process N-linked carbohydrates from glycoproteins that have migrated through the medial Golgi apparatus. However, EndoH treatment of purified cell-surface iRhom2 led to slightly faster migration of cell surface–biotinylated iRhom2 (Fig. 2, top). When we instead treated these same samples with protein-N-glycanase F (PNGase F), which removes all N-linked carbohydrate residues, iRhom2 migrated faster than the EndoH-treated sample (Fig. 2, top). The partial susceptibility of N-linked carbohydrates in cell surface–biotinylated iRhom2 to treatment with EndoH was reminiscent of the effect of EndoH and PNGase F in Western blots of total iRhom2 in BMDM lysates in a prior report (25). The finding that the surface-labeled iRhom2 is partially susceptible to EndoH treatment demonstrates that iRhom2 progresses though the medial Golgi apparatus on the way to the cell surface and that at least one N-linked carbohydrate residue in iRhom2 does not acquire EndoH resistance (25). Similarly, blots of the cell surface–biotinylated ADAM17 in WT BMDM showed resistance to EndoH (Fig. 2, left), but sensitivity to PNGase F, consistent with previous studies on mature ADAM17 (25, 37). These results also corroborated that no mature ADAM17 could be biotinylated on the surface of iRhom2−/− BMDM (24, 25). Finally, cell surface–biotinylated ADAM9 was included as a control for the iRhom2−/− BMDM. Like iRhom2, cell surface–labeled ADAM9 is also partially sensitive to EndoH treatment, as described previously (38). To rule out nonspecific binding of iRhom2, ADAM17, or ADAM9 to the streptavidin-Sepharose beads used to precipitate cell surface–biotinylated proteins, we incubated extracts of equivalent cultures of WT cells that were either untreated or cell surface–biotinylated with streptavidin beads and performed a Western blot analysis on the bound proteins (Fig. 2B). These experiments confirmed that only the biotinylated forms of iRhom2, ADAM17, and ADAM9 bound to streptavidin beads under the conditions used here, whereas these proteins in the lysate of an equivalent number of untreated cells did not. The finding that endogenous iRhom2 can be biotinylated using a non-membrane-permeable biotinylation reagent demonstrates that endogenous iRhom2 is present on the cell surface of primary BMDM, similar to RAW264.7 cells (31).
Figure 2.

Labeling of endogenous iRhom2 on the cell surface of primary LPS-stimulated mouse BMDM. A, LPS-stimulated WT or iRhom2−/− BMDM were incubated with the non-membrane-permeable biotinylation reagent EZ-Link Sulfo-NHS-LC-Biotin. Western blots of biotinylated material isolated with streptavidin-Sepharose 4B beads were either not treated (left lane) or incubated with EndoH (middle lane) or PNGase F (right lane) and then probed with antibodies against the cytoplasmic domain of iRhom2, ADAM17 (A17), or ADAM9 (A9), as indicated. B, a control for the possible nonspecific binding of iRhom2, ADAM17, or ADAM9 to streptavidin-Sepharose 4B beads. Two confluent plates each of WT or iRhom2−/− BMDM were prepared, and one was subjected to cell-surface biotinylation as in A, whereas the other was left untreated. Following lysis in cell lysis buffer, both lysates were incubated in streptavidin beads under the same conditions as the samples in A. The Western blots are representative of three separate experiments with essentially identical results.
ADAM17 is required for the stabilization of iRhom2
iRhom2-deficient BMDM lack mature ADAM17, whereas the levels of pro-ADAM17 do not appear to be significantly affected (24, 25). This raises questions about whether the loss of ADAM17 would reciprocally affect the levels of iRhom2. Because Adam17−/− mice die at birth, we isolated macrophages from the livers of newborn Adam17−/− mice and their WT control littermates to assess the fate of iRhom2 in myeloid cells in the absence of ADAM17. iRhom2 was only weakly detectable in membrane preparations of unstimulated newborn liver-derived macrophages (NLDM) from WT mice (Fig. 3A, top, left lane). However, stimulation with 10 ng/ml LPS increased the production of both iRhom2 and ADAM17 in WT NLDM (Fig. 3A, top and middle panels, right lane), just as in BMDM from adult mice (Fig. 1). Interestingly, iRhom2 protein could not be detected by the polyclonal iRhom2 antibody in a Western blot analysis of membrane preparations of LPS-stimulated Adam17−/− NLDM (Fig. 3B, iRhom2 (top), ADAM17 (middle), and ADAM10 control (bottom)).
Figure 3.
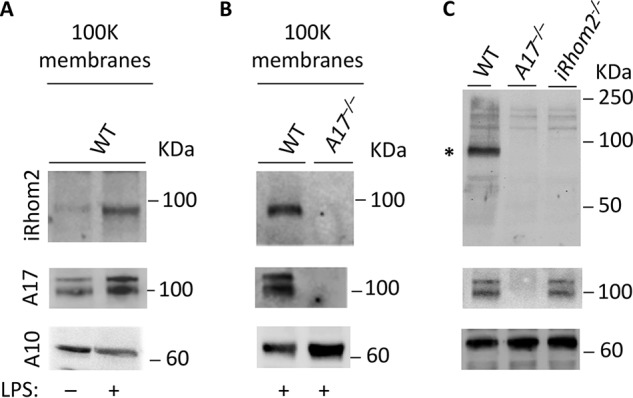
ADAM17 is required for the stabilization of iRhom2 in newborn mouse liver-derived macrophages and in newborn mice. A, Western blotting of a membrane preparation of NLDM demonstrates that LPS treatment enhances the production of iRhom2 and ADAM17. B, iRhom2 and ADAM17 can be detected in Western blots of LPS-stimulated WT, but not in Adam17−/− NLDM membrane preparations. C, Western blot analysis of membranes prepared from newborn WT mice show iRhom2 and pro- and mature ADAM17, whereas no iRhom2 or ADAM17 can be detected in identically prepared membranes from newborn Adam17−/− mice. Western blot analysis of membranes from iRhom2−/− mice shows no iRhom2, but pro- and mature ADAM17 are present. In each panel, ADAM10 serves as a loading control. Each panel is representative of three separate experiments.
To further explore the role of ADAM17 in stabilizing iRhom2 in mouse embryos, isolated membrane preparations from extracts of newborn mice (see “Experimental procedures” for details) were probed for iRhom2 or ADAM17. No iRhom2 could be detected in embryos lacking ADAM17 or iRhom2, although both ADAM17 and iRhom2 were present in WT control extracts (Fig. 3C). Moreover, pro- and mature ADAM17 were present in extracts of newborn iRhom2−/− mice, where maturation of ADAM17 is supported by iRhom1 (27).
Analysis of mRNA expression
The stabilization of iRhom2 by ADAM17 could depend on the requirement for a continuous interaction of the two heteromeric binding partners, or alternatively, ADAM17 may have a role in controlling the transcription of iRhom2. We therefore performed a qPCR analysis of the expression of iRhom2 in NLDM from WT, iRhom2−/−, or Adam17−/− mice. The results demonstrated that the levels of iRhom2 mRNA were similar in NLDM from WT and Adam17−/− mice, whereas no iRhom2 mRNA was detected in NLDM from iRhom2−/− mice (Fig. 4A). These findings argue against a role of ADAM17 in regulating the gene expression of iRhom2. We also addressed whether the catalytic activity of ADAM17 could have a role in regulating the levels of iRhom2, such as by affecting a putative signaling pathway with a role in regulating iRhom2 protein levels. However, when we incubated WT BMDM with the general metalloproteinase inhibitor marimastat, there was no significant effect on the levels of iRhom2 protein (Fig. 4B).
Figure 4.
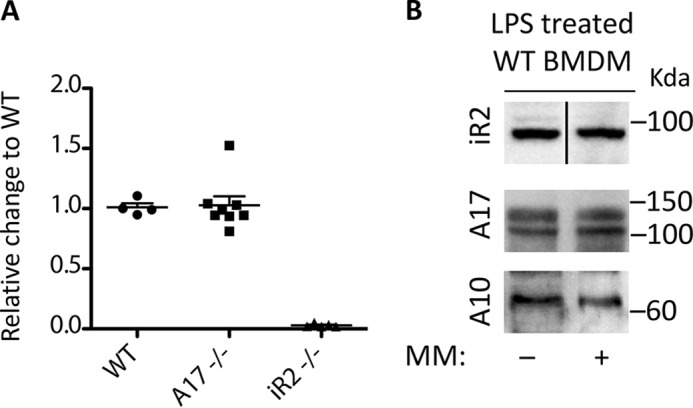
Expression analysis of iRhom2 by RT-qPCR and effect of treatment of WT cells with the metalloprotease inhibitor marimastat. A, an RT-qPCR analysis shows comparable expression of iRhom2 mRNA in WT and Adam17−/− BMDM (A17−/−), but not in iRhom2−/− BMDM (iR2−/−), used here as a control. B, LPS-stimulated BMDM from WT mice were treated with or without 5 μm marimastat for 18 h and then lysed, and the samples were subjected to Western blot analysis for iRhom2 or ADAM17, with ADAM10 as a loading control. The heavy line in the top panel (iR2 samples) indicates that these two lanes were spliced together. Each experiment was repeated three times with essentially similar outcomes, and one representative sample is shown.
Effect of protein degradation inhibitors on iRhom2 stability
We next considered possible degradation pathways for iRhom2 in the absence of ADAM17. We therefore incubated LPS-stimulated BMDM from mice, in which floxed alleles of ADAM17 were conditionally inactivated in myeloid cells through expression of LysM-Cre (A17LysM-Cre mice) with an inhibitor of proteasomal degradation (MG132, 10 μm) (30, 39), an inhibitor of ER-associated degradation (eeyarestatin, 10 μm) (40), and inhibitors of lysosomal acidification and autophagosome-lysosome fusion (chloroquine (100 μm) and bafilomycin (100 nm)) (41, 42). However, at these concentrations, which have been reported to be effective in cell-based assays (see “Experimental procedures” for details), neither of these degradation inhibitors had a significant effect on enhancing the stability of iRhom2, as detected by Western blotting (Fig. 5A, ADAM10 shown as loading control). Similar results were obtained when these inhibitors were tested on A17−/− mEFs (Fig. 5B). In a control experiment for the activity of the degradation inhibitors in WT mEFs, we found that 10 μm eeyarestatin or 10 μm MG132 effectively inhibited ubiquitin degradation (Fig. 5C) (43). Moreover, the addition of 100 nm bafilomycin or 100 μm chloroquine led to a strong increase in LC3-II, an accepted marker of autophagy inhibition (44) (Fig. 5D), corroborating the activity of these compounds.
Figure 5.
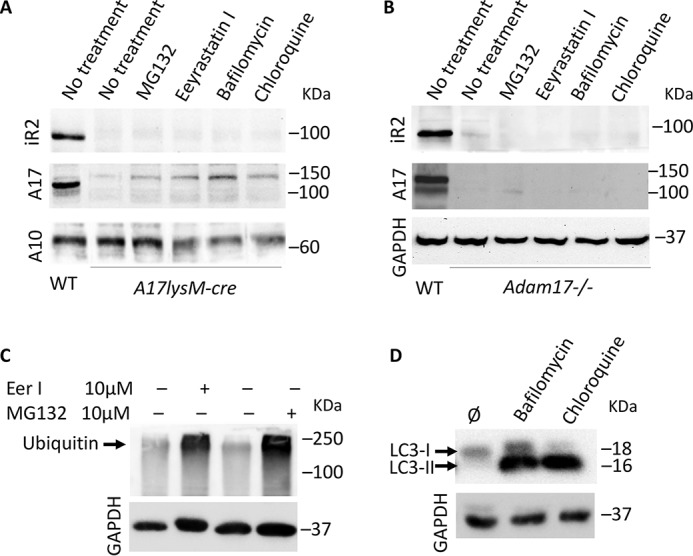
Effect of inhibitors of endocytosis or protein degradation on iRhom2 levels in ADAM17-deficient cells. BMDM from Adam17-LysM-cre mice (A) or Adam17−/− mEFs (B) were either left untreated or treated with 10 μm MG132, 100 μm chloroquine, 100 nm bafilomycin, or 10 μm eeyarestatin I, as indicated, for 18 h and then subjected to Western blot analysis for iRhom2, ADAM17, or ADAM10 and compared with untreated WT BMDM (A) or mEFs (B). As controls for the efficacy of the inhibitors, WT mEFs were incubated under identical conditions as the ADAM17-deficient BMDM (A) or mEFs (B). Whole-cell lysates of the WT mEFs treated with eeyarestatin I or MG132 were subjected to Western blot analysis with antibodies against ubiquitin (C), and Western blots of extracts from WT mEFs treated with bafilomycin or chloroquine were probed for LC3-II, a marker for inhibition of autophagy (D). The data are representative of three replicates with essentially similar results.
STING is not required for the stability of iRhom2
The multimembrane-spanning protein STING has been reported as an interacting partner of iRhom2 (35). To determine whether STING is required to stabilize iRhom2, we isolated BMDM from Sting−/− mice, or from WT controls. As shown in Fig. 6A, the lack of STING had no detectable effect on the protein levels of iRhom2 or ADAM17. Moreover, we found that inactivation of iRhom2 in BMDM also did not have a strong effect on the levels of STING that were detectable by Western blot analysis (Fig. 6B). Finally, the mRNA levels for iRhom2 were comparable in BMDM from WT and Sting−/− mice, but undetectable in BMDM from iRhom2−/− mice (Fig. 6C).
Figure 6.

Expression of STING, iRhom2, and ADAM17 in BMDM lacking STING or iRhom2. Western blot analysis for iRhom2, ADAM17, Sting, or ADAM10 was performed on unstimulated or LPS-stimulated BMDM from Sting−/− mice or wild type (WT) controls (A) or from iRhom2−/− mice and WT controls (B). The heavy line in B indicates splicing of nonadjacent lanes. C, an RT-qPCR analysis was used to confirm that the mRNA for iRhom2 was expressed at similar levels in Sting−/− BMDM compared with WT controls. Each experiment was repeated three times, and representative samples are shown. Error bars, S.E.M.
Analysis of the levels of iRhom2 and the related iRhom1 in mEFs lacking ADAM17
When we attempted to detect the endogenous iRhom2 in Western blots of mouse embryonic fibroblasts, we found that the rabbit anti-iRhom2(1–376) polyclonal antibodies against the cytoplasmic domain of iRhom2 were not reproducibly effective for this purpose (data not shown). Therefore, new rat mAbs against mouse iRhom1 or iRhom2 were generated (see “Experimental procedures” for details). As shown in Fig. 7A, the rat mAbs specifically recognized iRhom1 or -2 in WT mEFs, with mEFs lacking iRhom1 (iR1KO) or iRhom2 (iR2KO) or both iRhom1 and -2 (iR1/2DKO) serving as controls for the specificity of these mAbs. We noted that the levels of iRhom2 were not significantly affected by the absence of iRhom1 and that the anti-iRhom2 rat mAb recognized a small but detectable amount of iRhom2 in A17−/− mEFs. The rat mAb against iRhom1 demonstrated that the levels of iRhom1 were not significantly changed in iRhom2−/− mEFs, but they appeared slightly increased in the A17−/− mEFs. Cell-surface biotinylation of WT mEFs showed that both iRhom1 and iRhom2 could be detected on the cell surface of these cells (Fig. 7B). The cell surface biotinylation of only the mature form of ADAM17 and of ADAM9, used as loading control, but not their pro-forms served as an internal control that the biotinylation reagent was specific for cell-surface proteins and did not label intracellular proteins, in this case pro-ADAM17 or pro-ADAM9.
Figure 7.
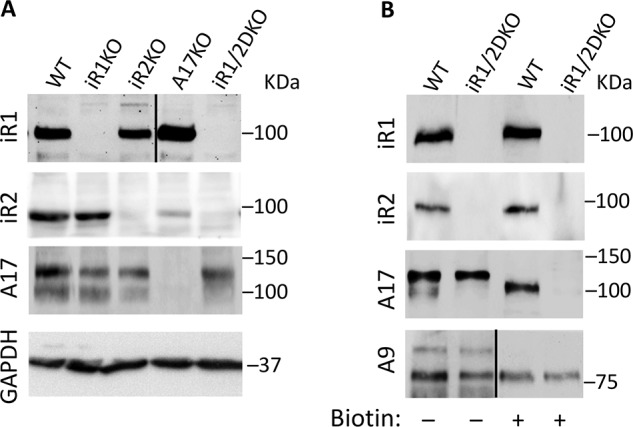
Rat monoclonal antibodies against iRhom1 or -2 allow detection of the endogenous proteins in mEFs. A, Western blot analysis of extracts of WT mEFs or mEFs lacking iRhom1 (iR1KO) or iRhom2 (iR2KO) or Adam17 (A17KO) or both iRhom1 and -2 (iR1/2DKO) with rat polyclonal antibodies against iRhom1 or iRhom2, or rabbit polyclonal antibodies against ADAM17, with GAPDH serving as a loading control. B, Western blots of extracts from nonbiotinylated WT or iR1/2DKO mEFs (left two lanes) or streptavidin-Sepharose 4B–precipitated samples from cell surface–biotinylated WT or iR1/2DKO mEFs were probed with antibodies against iRhom1, iRhom2, ADAM17, or ADAM9, which served as a loading control. The heavy lines in A (top) and B (bottom) indicate that these lanes were spliced together. These results are representative examples of three separate experiments.
Discussion
Previous studies have shown that pro-ADAM17 is synthesized in the absence of iRhom2 in BMDM, but not transported out of the ER to the trans-Golgi network, where its pro-domain is removed (24, 45). This finding raised questions about the fate of iRhom2 in the absence of ADAM17 or in the absence of a recently identified iRhom2-binding partner, STING. Moreover, because mEFs lacking both iRhom1 and iRhom2 only have pro-ADAM17, but no detectable mature ADAM17, we were interested in exploring whether endogenous iRhom2 and the related iRhom1 can be detected on the cell surface of WT mEFs, as would be predicted if they can function as regulators of endogenous mature ADAM17. Finally, we were interested in whether the lack of ADAM17 reciprocally affects the stability of iRhom1 and -2.
Our observation that little, if any, iRhom2 is detectable by Western blotting in myeloid cells or in embryos lacking ADAM17 under conditions where it can readily be detected in WT controls provides the first evidence that ADAM17 is required for the stabilization of endogenous iRhom2. Unlike pro-ADAM17, which is present at comparable levels in WT and iRhom2−/− BMDM and can be up-regulated by treatment of BMDM with LPS, we found no detectable iRhom2 in membrane preparations of LPS-treated Adam17−/− NLDM. Treatment with inhibitors of proteasomal degradation, ER-associated degradation, or lysosomal acidification and degradation did not restore the ability to detect iRhom2 in Adam17−/− NLDM or in Adam17−/− mEFs, arguing against a major role of these pathways in controlling the stability of iRhom2 in the absence of ADAM17. In addition, we found that the lack of ADAM17 did not affect mRNA levels of iRhom2 in LPS-treated NLDM. Finally, the catalytic activity of ADAM17 is most likely not required for the stabilization of iRhom2, because the general metalloprotease inhibitor marimastat had no detectable effects on the levels of iRhom2. Further studies will be necessary to better understand the mechanism of how the stability of iRhom2 is regulated in the absence of ADAM17. Taken together, these results suggest that the presence of the ADAM17 is required for the stabilization of iRhom2. Conversely, because only the mature form of ADAM17 is affected by the lack of iRhom2 in BMDM (24, 25) or NLDM, whereas the pro-form is not, these results suggest that pro-ADAM17 is produced at similar levels in the presence or absence of iRhom2 and does not require iRhom2 for stabilization.
The observation that pro-ADAM17 can exist in the absence of iRhom2 but requires iRhom2 to be converted into its mature, processed, and EndoH-resistant form supports a model in which pro-ADAM17 assembles with newly synthesized iRhom2 in the ER, allowing both to exit the ER and enter the secretory pathway together. The pro-domain of ADAM17 is then removed by pro-protein convertases in the trans-Golgi network (37, 46). This is a prerequisite for the activity of ADAM17, which can be rapidly enhanced by many different stimuli once ADAM17 has been processed by pro-protein convertases (19, 46). The inability to detect iRhom2 in the absence of ADAM17 in NLDM and in embryos and the very low levels of iRhom2 in A17−/− mEFs suggest that it is unstable without its binding partner. Moreover, because pro-ADAM17 is stable in the absence of iRhom2, this suggests that iRhom2 can associate with pre-existing molecules of ADAM17 to form an iRhom2/ADAM17 complex, presumably after pro-ADAM17 has been translocated across the ER membrane.
iRhom2 also interacts with another multimembrane-spanning protein, STING, which is involved in regulating innate immunity to DNA viruses (35). However, we found that STING is not essential for the stabilization of the iRhom2/ADAM17 complex. In addition, iRhom2 also interacts with several cytoplasmic molecules, including members of the 14-3-3 family of signaling adapters and FRMD8/iTAP (30–33). Moreover, because iRhom2 determines the substrate selectivity of ADAM17, the iRhom2/ADAM17 complex most likely also interacts with its substrates. This notion is further supported by the observation that the phenotype caused by the “curly-bare” (cub) mutation in the cytoplasmic domain of iRhom2 (curly hair and bare skin) is only seen in the presence of the iRhom2/ADAM17 substrate amphiregulin and not in mice lacking amphiregulin (47, 48). Presumably, the interaction between WT iRhom2/ADAM17 and its substrates is transient, so as to allow rapid substrate turnover upon activation of ADAM17.
Similar to iRhom2, the related iRhom1 can be detected on the surface of mouse embryonic fibroblasts. However, unlike iRhom2, the levels of iRhom1 are not significantly affected in the absence of ADAM17, suggesting that it interacts with ADAM17 differently from iRhom2 and can be stable on its own. The migration of the biotinylated iRhom1 or iRhom2 on SDS-PAGE was comparable with that of their nonbiotinylated counterparts that could be detected by Western in whole lysates, suggesting that the iRhom1 and -2 proteins do not undergo substantial proteolytic processing en route through the secretory pathway to the cell surface. It will be interesting to characterize the interaction between iRhom1 and ADAM17 in more detail in the future.
In summary, our results provide the first evidence that endogenous iRhom2 is stabilized by the presence of ADAM17, whereas iRhom1 is not. Nevertheless, both iRhom proteins can be detected on the cell surface of WT cells, where they presumably interact with ADAM17. Interestingly, a second interacting partner of iRhom2, STING, is not required for the stability of iRhom2. These findings support a model in which ADAM17 is a principal partner or client of iRhom2 and in which both must be present to support the maturation and function of the iRhom2/ADAM17 complex on the cell surface.
Experimental procedures
Reagents and antibodies
All reagents were purchased from Sigma–Aldrich unless specified otherwise. The rabbit antibodies against the cytoplasmic domain of iRhom2 were generated by immunizing female New Zealand White rabbits with a GSH S-transferase fusion protein with the cytoplasmic domain of mouse iRhom2 (amino acid residues 1–376, ProSci Inc., Poway, CA). The rat anti-iRhom1 and anti-iRhom2 monoclonal antibodies were generated using standard procedures (49). The iRhom1 antibody (RHF1A 20A8; IgG2a) was generated by immunization of ovalbumin-coupled peptide MSEARRDSTSSLQRKKPPW. For the anti-iRhom2 antibody (RHF2B 11H7; IgG2a) ovalbumin-coupled peptide GDWEGKRQNWHRRSL was used. Antibodies against the cytoplasmic domain of mouse ADAM9 and ADAM17 have been described previously (37, 50). Antibodies against A10 were from Abcam (Cambridge, MA) (catalog no. 1244695); antibodies against STING and antibodies against mouse tubulin were from Cell Signaling (Danvers, MA); and antibodies against GAPDH were from ABclonal (Woburn, MA). Anti-LC3 was from Novus Biological (Littleton, CO) (catalog no. Nb100-2220), and anti-ubiquitin clone P4G7 was from Biolegend (San Diego, CA) (catalog no. 838703). The deglycosylation enzymes EndoH and PNGase F were from New England Biolabs (Ipswich, MA). EZ-Link Sulfo-NHS-LC-Biotin was from Thermo Fisher Scientific (Waltham, MA) (catalog no. 21335). Streptavidin-Sepharose 4B beads were from Thermo Fisher Scientific (catalog no. 434341). Sigma–Aldrich was the source for chloroquine (catalog no. C6628), MG132 (catalog no. 474790), bafilomycin (catalog no. B1793), and eeyarestatin (catalog no. E1286). The inhibitors of protein degradation or endocytosis were used at the following concentrations and for the times indicated: MG132 at 10 μm for 18 h (33), chloroquine at 100 μm for 18 h (33), bafilomycin at 100 nm for 18 h (32), and eeyarestatin at 10 μm for 18 h (43). Marimastat was a gift from Dr. Ouathek Ouerfelli (Memorial Sloan Kettering Cancer Center, New York) (51).
Mouse lines
iRhom2−/− mice and Adam17−/− mice have been described previously (5, 24). Sting−/− mice were kindly provided by Dr. Liang Deng (Memorial Sloan Kettering Cancer Center) (52, 53). All animal experiments were approved by the Institutional Animal Use and Care Committee of the Hospital for Special Surgery and Weill Cornell Medicine.
Isolation and generation of mouse primary macrophages
To generate primary BMDM, we harvested and cultured the bone marrow cells of 4-week-old mice (equal distribution of male and female mice) as described previously (17). Briefly, femurs and tibiae were flushed with Hanks' balanced salt solution, and washed cells were plated on Petri dishes in RPMI medium supplemented with 20% fetal calf serum and murine macrophage colony-stimulating factor (Peprotech, Rocky Hill, NJ) at 10 ng/ml. After 7 days, macrophages were collected with a plastic tissue culture scraper, plated on fresh plates at 1 × 106 cells/10 cm2, and stimulated with 10 ng/ml LPS for 14–18 h. For the preparation of NLDM from mice at postnatal day 1 (P1), livers were removed after euthanasia and then dispersed through a 70-μm cell strainer (Denville Scientific, Holliston, MA). Red blood cells were removed with red blood cell lysis buffer (Sigma–Aldrich) according to the manufacturer's protocol, and cells were then cultured as described above. After 5 days in culture, 2 × 106 differentiated NLDM were stimulated with 10 ng/ml LPS for 14 h and processed for Western blot analysis.
Culture of mouse embryonic fibroblasts
Generation and culture of immortalized mouse embryonic fibroblasts used in this study has been described previously (5, 27, 29, 54).
Membrane preparations and lectin purification
For the preparation of membrane protein extracts from BMDM or NLDM, cells were washed twice with PBS and then scraped directly in membrane buffer (250 mm sucrose, 20 mm Hepes, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm EGTA, 1 mm EDTA) containing protease inhibitors plus a 5 mm concentration of the zinc-binding chelator 1,10-phenanthroline (24, 37). For membrane preparations from newborn mice (P1), the animals were euthanized, and the remaining tissues were minced with razor blades and then mechanically dispersed and subjected to two 30-s treatments with a Polytron homogenizer (Kinematica, Switzerland) in membrane buffer. The cell suspension or the homogenized tissues were then treated with 30 homogenization cycles in a Dounce homogenizer (Potter–Elvehjem, Sigma–Aldrich). The cell or tissue suspension was subjected to a low-speed spin at 720 × g for 10 min at 4 °C. The supernatant was subsequently transferred to a 13-ml ultracentrifuge tube (Ultra-Clear tubes, Beckman Coulter, Brea, CA) adjusted to 12.5 ml with membrane buffer and centrifuged for 1 h at 100,000 × g in a Beckmann Optima X Ultracentrifuge in an SW40 rotor. The resulting membrane pellets were resuspended by boiling for 5 min in 1× SDS-sample-loading buffer, separated on SDS-polyacrylamide gels, transferred to nitrocellulose membranes (see below), and then probed with antibodies against ADAM10, ADAM17, or iRhom2.
Controls for inhibitors of protein degradation
The patterns of ubiquitinated proteins and of a protein affected by autophagy (LC3 II) were evaluated using Western blot analysis. Cells from a 6-well culture dish (Nunc, Thermo Fisher Scientific) were incubated with inhibitors for 18 h and then washed in PBS and suspended in 1× SDS-containing sample loading buffer. The resulting whole-cell extracts were sonicated with a Bioruptor Pico sonicator (Diagenode, Denville, NJ) for three rounds of 10-s bursts at maximal strength and then boiled for 5 min and then separated on 10% Bis-Tris NuPage gels (Thermo Fisher Scientific, catalog no. NP0301) in MES buffer according to the manufacturer's instructions. Western blot analyses were performed as described, with anti-LC3 and anti-ubiquitin primary antibodies used at 1:1000.
Western blot analysis
To generate Western blots for iRhom2 and ADAM17 from BMDM samples from iRhom2−/− mice and WT controls (Fig. 1A) extracts from 1 × 106 cells were separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Pall Corp., Port Washington, NY). The Western blot analyses in Figs. 1B and 3 (A–C) were performed using extracts from 100,000 × g membrane preparations (see above). Western blots of lysates of mouse embryonic fibroblasts isolated from WT, iRhom1−/−, iRhom2−/−, and iRhom1/2−/− double knockout or A17−/− knockout E14.5 embryos were performed as described previously (27). After blocking in 5% nonfat milk at room temperature for 1 h, the nitrocellulose membranes were incubated in primary antibody overnight at 4 °C (1:2000 dilution for rabbit anti-iRhom2-cyto) or for 1 h at room temperature (rat monoclonal anti-iRhom1 and anti-iRhom2 antibodies at 1:10 culture supernatant dilution). STING antibodies (Cell Signaling, Danvers, MA) were each incubated overnight at 4 °C at 1:1000 dilution. The nitrocellulose membranes were then washed three times in PBS, 0.05% Tween 20 and then incubated in either HRP-labeled goat anti-rabbit secondary antibody, HRP-labeled goat anti-mouse secondary antibody (1:5000; Promega, Madison, WI), or HRP-labeled goat anti-rat secondary antibody (1:5000; Sigma). Bound antibodies were detected using the ECL system (Thermo Fisher Scientific) and a Chemdoc image analyzer (Bio-Rad), and the images were assembled using Microsoft Powerpoint software. Loading controls were generated either by Western blotting parallel samples or by stripping membranes for 15 min at 55 °C in stripping buffer (2% SDS, 50 mm 2-mercaptoethanol in 62 mm Tris, pH 6.7). These membranes were blocked as described and then incubated with anti-ADAM9, anti-ADAM10, anti-GAPDH, or anti-tubulin antibodies at 1:2000, and the bound antibodies were detected as described above.
Cell-surface biotinylation
Mouse embryonic fibroblasts or differentiated BMDM that had been left untreated or had been treated with 10 ng/ml LPS were cell surface–biotinylated by incubation with a 1 mg/ml solution of the non-membrane-permeable biotinylation reagent EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific) in sterile PBS for 45 min at 4 °C. The reaction was quenched with 0.1 m glycine in PBS, and then the cells were lysed in cell lysis buffer (PBS, 1% Triton X-100 containing protease inhibitors plus a 5 mm concentration of the zinc-binding chelator 1,10-phenanthroline (24, 37) and 5 μm marimastat). Extracts containing biotinylated proteins were incubated with 10 μl of washed streptavidin-Sepharose 4B beads for 30 min at 4 °C. The beads were then washed four times with cell lysis buffer, and bound proteins were eluted in boiling SDS-sample buffer and separated on NuPAGE Novex 3–8% Tris acetate protein gels (Thermo Fisher Scientific) and then subjected to Western blot analysis with antibodies against iRhom1 or -2, ADAM17, or ADAM9, as indicated. All other samples were separated on 10% SDS-polyacrylamide gels.
RT-qPCR analysis
Total RNA from cultured WT, Adam17−/−, or iRhom2−/− NLDM or from WT, iRhom2−/−, or Sting−/− BMDM was isolated with RNeasy (Qiagen, Germantown, MA) and subsequently reverse-transcribed (Oligo-dT/SuperScript RT III, Qiagen, Germantown, MA). Oligonucleotides for iR2 and GAPDH were purchased from Qiagen. Sequences for β-actin oligonucleotides were as follows: β-actin forward, 5′-AGGTGTGCACTTTTATTGGTCTCAA-3′; β-actin reverse, 5′-TGTATGAAGGCTTTGGTCTCCCT-3′. RT-qPCR was performed using SYBR Green on an ABI PRISM 7900HT cycler (both from Applied Biosystems, Thermo Fisher Scientific). GAPDH was used as an endogenous control to normalize each sample. Three independent experiments were performed in triplicate.
Author contributions
G. W., R. F., S. S., E. F.-P., S. F. L., and C. P. B. conceptualization; G. W., J. T., D. L., T. M., S. S., E. F.-P., J. E. S., S. F. L., and C. P. B. data curation; G. W., E. F.-P., D. R. M., J. E. S., S. F. L., and C. P. B. formal analysis; G. W., S. F. L., and C. P. B. supervision; G. W. validation; G. W., J. T., D. L., T. M., S. F. L., and C. P. B. investigation; G. W., J. T., D. L., R. F., S. S., S. F. L., and C. P. B. methodology; G. W., S. F. L., and C. P. B. writing-original draft; G. W., S. F. L., and C. P. B. project administration; G. W., J. T., R. F., T. M., S. S., E. F.-P., D. R. M., J. E. S., S. F. L., and C. P. B. writing-review and editing; D. R. M., T. W. M., S. F. L., and C. P. B. resources; J. E. S., S. F. L., and C. P. B. funding acquisition.
Acknowledgments
We thank Elin Mogollon, Sarah Loh and Katrin Moschke for technical assistance and Gregory Farber for help with the RT-qPCR analysis.
This work was supported by NIGMS, National Institutes of Health, Grants R01 GM64750 and R35 GM134907 (to C. P. B.). This work was also supported by the Deutsche Forschungsgemeinschaft (German Research Foundation), Grant FOR2290 (to S. F. L.) within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy, project ID 390857198) (to R. F., and S. F. L.). D. R. M., T. W. M., T. M., and C. P. B. hold a patent on a method of identifying agents for combination with inhibitors of iRhoms. C. P. B. and the Hospital for Special Surgery have identified iRhom2 inhibitors and have co-founded the start-up company SciRhom in Munich to commercialize these inhibitors. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ADAM17
- a disintegrin and metalloprotease 17
- Rhbdf1/2
- Rhomboid 5 homolog 1/2
- iRhom1/2
- inactive Rhomboid-like protein 1/2
- TNFα
- tumor necrosis factor α
- ER
- endoplasmic reticulum
- IL-6
- interleukin 6
- IL-6R
- interleukin-6 receptor
- TMD
- transmembrane domain
- LPS
- lipopolysaccharide
- BMDM
- bone marrow–derived macrophage(s)
- EndoH
- endoglycosidase H
- PNGase F
- N-glycosidase F
- EGFR
- epidermal growth factor receptor
- STING
- stimulator of interferon genes
- NLDM
- newborn liver-derived macrophage(s)
- pAb
- polyclonal antibody
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- P
- postnatal day
- E
- embryonic day
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- qPCR
- quantitative PCR
- HRP
- horseradish peroxidase.
References
- 1. Blobel C. P. (2005) ADAMs: key players in EGFR-signaling, development and disease. Nat. Rev. Mol. Cell Biol. 6, 32–43 10.1038/nrm1548 [DOI] [PubMed] [Google Scholar]
- 2. Weber S., and Saftig P. (2012) Ectodomain shedding and ADAMs in development. Development 139, 3693–3709 10.1242/dev.076398 [DOI] [PubMed] [Google Scholar]
- 3. Scheller J., Chalaris A., Garbers C., and Rose-John S. (2011) ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 32, 380–387 10.1016/j.it.2011.05.005 [DOI] [PubMed] [Google Scholar]
- 4. Peschon J. J., Slack J. L., Reddy P., Stocking K. L., Sunnarborg S. W., Lee D. C., Russell W. E., Castner B. J., Johnson R. S., Fitzner J. N., Boyce R. W., Nelson N., Kozlosky C. J., Wolfson M. F., Rauch C. T., et al. (1998) An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284 10.1126/science.282.5392.1281 [DOI] [PubMed] [Google Scholar]
- 5. Horiuchi K., Kimura T., Miyamoto T., Takaishi H., Okada Y., Toyama Y., and Blobel C. P. (2007) Cutting edge: TNF-α-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 179, 2686–2689 10.4049/jimmunol.179.5.2686 [DOI] [PubMed] [Google Scholar]
- 6. Black R. A., Rauch C. T., Kozlosky C. J., Peschon J. J., Slack J. L., Wolfson M. F., Castner B. J., Stocking K. L., Reddy P., Srinivasan S., Nelson N., Boiani N., Schooley K. A., Gerhart M., Davis R., et al. (1997) A metalloprotease disintegrin that releases tumour-necrosis factor-α from cells. Nature 385, 729–733 10.1038/385729a0 [DOI] [PubMed] [Google Scholar]
- 7. Moss M. L., Jin S.-L. C., Milla M. E., Burkhart W., Carter H. L., Chen W.-J., Clay W. C., Didsbury J. R., Hassler D., Hoffman C. R., Kost T. A., Lambert M. H., Leesnitzer M. A., McCauley P., McGeehan G., et al. (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 385, 733–736 10.1038/385733a0 [DOI] [PubMed] [Google Scholar]
- 8. Jackson L. F., Qiu T. H., Sunnarborg S. W., Chang A., Zhang C., Patterson C., and Lee D. C. (2003) Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J. 22, 2704–2716 10.1093/emboj/cdg264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sternlicht M. D., Sunnarborg S. W., Kouros-Mehr H., Yu Y., Lee D. C., and Werb Z. (2005) Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development 132, 3923–3933 10.1242/dev.01966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sahin U., Weskamp G., Kelly K., Zhou H. M., Higashiyama S., Peschon J., Hartmann D., Saftig P., and Blobel C. P. (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR-ligands. J. Cell Biol. 164, 769–779 10.1083/jcb.200307137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franzke C. W., Cobzaru C., Triantafyllopoulou A., Löffek S., Horiuchi K., Threadgill D. W., Kurz T., van Rooijen N., Bruckner-Tuderman L., and Blobel C. P. (2012) Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med. 209, 1105–1119 10.1084/jem.20112258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chalaris A., Adam N., Sina C., Rosenstiel P., Lehmann-Koch J., Schirmacher P., Hartmann D., Cichy J., Gavrilova O., Schreiber S., Jostock T., Matthews V., Häsler R., Becker C., Neurath M. F., Reiss K., Saftig P., Scheller J., and Rose-John S. (2010) Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 207, 1617–1624 10.1084/jem.20092366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blaydon D. C., Biancheri P., Di W. L., Plagnol V., Cabral R. M., Brooke M. A., van Heel D. A., Ruschendorf F., Toynbee M., Walne A., O'Toole E. A., Martin J. E., Lindley K., Vulliamy T., Abrams D. J., MacDonald T. T., Harper J. I., and Kelsell D. P. (2011) Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 365, 1502–1508 10.1056/NEJMoa1100721 [DOI] [PubMed] [Google Scholar]
- 14. Tholen S., Wolf C., Mayer B., Knopf J. D., Löffek S., Qian Y., Kizhakkedathu J. N., Biniossek M. L., Franzke C. W., and Schilling O. (2016) Skin barrier defects caused by keratinocyte-specific deletion of ADAM17 or EGFR are based on highly similar proteome and degradome alterations. J. Proteome Res. 15, 1402–1417 10.1021/acs.jproteome.5b00691 [DOI] [PubMed] [Google Scholar]
- 15. Rossello A., Nuti E., Ferrini S., and Fabbi M. (2016) Targeting ADAM17 sheddase activity in cancer. Curr. Drug Targets 17, 1908–1927 10.2174/1389450117666160727143618 [DOI] [PubMed] [Google Scholar]
- 16. Ardito C. M., Grüner B. M., Takeuchi K. K., Lubeseder-Martellato C., Teichmann N., Mazur P. K., Delgiorno K. E., Carpenter E. S., Halbrook C. J., Hall J. C., Pal D., Briel T., Herner A., Trajkovic-Arsic M., Sipos B., et al. (2012) EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 22, 304–317 10.1016/j.ccr.2012.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Issuree P. D., Maretzky T., McIlwain D. R., Monette S., Qing X., Lang P. A., Swendeman S. L., Park-Min K. H., Binder N., Kalliolias G. D., Yarilina A., Horiuchi K., Ivashkiv L. B., Mak T. W., Salmon J. E., and Blobel C. P. (2013) iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J. Clin. Invest. 123, 928–932 10.1172/JCI66168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scheller J., Garbers C., and Rose-John S. (2014) Interleukin-6: from basic biology to selective blockade of pro-inflammatory activities. Semin. Immunol. 26, 2–12 10.1016/j.smim.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 19. Le Gall S. M., Maretzky T., Issuree P. D. A., Niu X.-D., Reiss K., Saftig P., Khokha R., Lundell D., and Blobel C. P. (2010) ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 123, 3913–3922 10.1242/jcs.069997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maretzky T., Evers A., Zhou W., Swendeman S. L., Wong P. M., Rafii S., Reiss K., and Blobel C. P. (2011) Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat. Commun. 2, 229 10.1038/ncomms1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gschwind A., Hart S., Fischer O. M., and Ullrich A. (2003) TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 22, 2411–2421 10.1093/emboj/cdg231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Myers T. J., Brennaman L. H., Stevenson M., Higashiyama S., Russell W. E., Lee D. C., and Sunnarborg S. W. (2009) Mitochondrial reactive oxygen species mediate GPCR-induced TACE/ADAM17-dependent transforming growth factor-α shedding. Mol. Biol. Cell 20, 5236–5249 10.1091/mbc.e08-12-1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohtsu H., Dempsey P. J., Frank G. D., Brailoiu E., Higuchi S., Suzuki H., Nakashima H., Eguchi K., and Eguchi S. (2006) ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 26, e133–e137 10.1161/01.ATV.0000236203.90331.d0 [DOI] [PubMed] [Google Scholar]
- 24. McIlwain D. R., Lang P. A., Maretzky T., Hamada K., Ohishi K., Maney S. K., Berger T., Murthy A., Duncan G., Xu H. C., Lang K. S., Häussinger D., Wakeham A., Itie-Youten A., Khokha R., et al. (2012) iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232 10.1126/science.1214448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adrain C., Zettl M., Christova Y., Taylor N., and Freeman M. (2012) Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228 10.1126/science.1214400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Siggs O. M., Xiao N., Wang Y., Shi H., Tomisato W., Li X., Xia Y., and Beutler B. (2012) iRhom2 is required for the secretion of mouse TNFα. Blood 119, 5769–5771 10.1182/blood-2012-03-417949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li X., Maretzky T., Weskamp G., Monette S., Qing X., Issuree P. D., Crawford H. C., McIlwain D. R., Mak T. W., Salmon J. E., and Blobel C. P. (2015) iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 6080–6085 10.1073/pnas.1505649112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li X., Maretzky T., Perez-Aguilar J. M., Monette S., Weskamp G., Le Gall S., Beutler B., Weinstein H., and Blobel C. P. (2017) Structural modeling defines transmembrane residues in ADAM17 that are crucial for Rhbdf2/ADAM17-dependent proteolysis. J. Cell Sci. 130, 868–878 10.1242/jcs.196436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maretzky T., McIlwain D. R., Issuree P. D., Li X., Malapeira J., Amin S., Lang P. A., Mak T. W., and Blobel C. P. (2013) iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. U.S.A. 110, 11433–11438 10.1073/pnas.1302553110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grieve A. G., Xu H., Künzel U., Bambrough P., Sieber B., and Freeman M. (2017) Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE-dependent inflammatory and growth factor signalling. Elife 6, e23968 10.7554/eLife.23968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cavadas M., Oikonomidi I., Gaspar C. J., Burbridge E., Badenes M., Félix I., Bolado A., Hu T., Bileck A., Gerner C., Domingos P. M., von Kriegsheim A., and Adrain C. (2017) Phosphorylation of iRhom2 controls stimulated proteolytic shedding by the metalloprotease ADAM17/TACE. Cell Rep. 21, 745–757 10.1016/j.celrep.2017.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oikonomidi I., Burbridge E., Cavadas M., Sullivan G., Collis B., Naegele H., Clancy D., Brezinova J., Hu T., Bileck A., Gerner C., Bolado A., von Kriegsheim A., Martin S. J., Steinberg F., et al. (2018) iTAP, a novel iRhom interactor, controls TNF secretion by policing the stability of iRhom/TACE. Elife 7, e35032 10.7554/eLife.35032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Künzel U., Grieve A. G., Meng Y., Sieber B., Cowley S. A., and Freeman M. (2018) FRMD8 promotes inflammatory and growth factor signalling by stabilising the iRhom/ADAM17 sheddase complex. Elife 7, e35012 10.7554/eLife.35012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maney S. K., McIlwain D. R., Polz R., Pandyra A. A., Sundaram B., Wolff D., Ohishi K., Maretzky T., Brooke M. A., Evers A., Vasudevan A. A., Aghaeepour N., Scheller J., Münk C., Häussinger D., et al. (2015) Deletions in the cytoplasmic domain of iRhom1 and iRhom2 promote shedding of the TNF receptor by the protease ADAM17. Sci. Signal. 8, ra109 10.1126/scisignal.aac5356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luo W. W., Li S., Li C., Lian H., Yang Q., Zhong B., and Shu H. B. (2016) iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 17, 1057–1066 10.1038/ni.3510 [DOI] [PubMed] [Google Scholar]
- 36. Adrain C., Strisovsky K., Zettl M., Hu L., Lemberg M. K., and Freeman M. (2011) Mammalian EGF receptor activation by the rhomboid protease RHBDL2. EMBO Rep. 12, 421–427 10.1038/embor.2011.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schlöndorff J., Becherer J. D., and Blobel C. P. (2000) Intracellular maturation and localization of the tumour necrosis factor α convertase (TACE). Biochem. J. 347, 131–138 10.1042/bj3470131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roghani M., Becherer J. D., Moss M. L., Atherton R. E., Erdjument-Bromage H., Arribas J., Blackburn R. K., Weskamp G., Tempst P., and Blobel C. P. (1999) Metalloprotease-disintegrin MDC9: intracellular maturation and catalytic activity. J. Biol. Chem. 274, 3531–3540 10.1074/jbc.274.6.3531 [DOI] [PubMed] [Google Scholar]
- 39. Lee D. H., and Goldberg A. L. (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403 10.1016/S0962-8924(98)01346-4 [DOI] [PubMed] [Google Scholar]
- 40. Fiebiger E., Hirsch C., Vyas J. M., Gordon E., Ploegh H. L., and Tortorella D. (2004) Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol. Biol. Cell 15, 1635–1646 10.1091/mbc.e03-07-0506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mauvezin C., and Neufeld T. P. (2015) Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11, 1437–1438 10.1080/15548627.2015.1066957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pasquier B. (2016) Autophagy inhibitors. Cell Mol. Life Sci. 73, 985–1001 10.1007/s00018-015-2104-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Q., Li L., and Ye Y. (2008) Inhibition of p97-dependent protein degradation by eeyarestatin I. J. Biol. Chem. 283, 7445–7454 10.1074/jbc.M708347200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Myeku N., and Figueiredo-Pereira M. E. (2011) Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J. Biol. Chem. 286, 22426–22440 10.1074/jbc.M110.149252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adrain C., and Freeman M. (2012) New lives for old: evolution of pseudoenzyme function illustrated by iRhoms. Nat. Rev. Mol. Cell Biol. 13, 489–498 10.1038/nrm3392 [DOI] [PubMed] [Google Scholar]
- 46. Wong E., Maretzky T., Peleg Y., Blobel C. P., and Sagi I. (2015) The functional maturation of a disintegrin and metalloproteinase (ADAM) 9, 10, and 17 requires processing at a newly identified proprotein convertase (PC) cleavage site. J. Biol. Chem. 290, 12135–12146 10.1074/jbc.M114.624072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hosur V., Johnson K. R., Burzenski L. M., Stearns T. M., Maser R. S., and Shultz L. D. (2014) Rhbdf2 mutations increase its protein stability and drive EGFR hyperactivation through enhanced secretion of amphiregulin. Proc. Natl. Acad. Sci. U.S.A. 111, E2200–E2209 10.1073/pnas.1323908111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Siggs O. M., Grieve A., Xu H., Bambrough P., Christova Y., and Freeman M. (2014) Genetic interaction implicates iRhom2 in the regulation of EGF receptor signalling in mice. Biol. Open 3, 1151–1157 10.1242/bio.201410116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Köhler G., and Milstein C. (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 10.1038/256495a0 [DOI] [PubMed] [Google Scholar]
- 50. Weskamp G., Krätzschmar J., Reid M. S., and Blobel C. P. (1996) MDC9, a widely expressed cellular disintegrin containing cytoplasmic SH3 ligand domains. J. Cell Biol. 132, 717–726 10.1083/jcb.132.4.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maretzky T., Yang G., Ouerfelli O., Overall C. M., Worpenberg S., Hassiepen U., Eder J., and Blobel C. P. (2009) Characterization of the catalytic activity of the membrane-anchored metalloproteinase ADAM15 in cell-based assays. Biochem. J. 420, 105–113 10.1042/BJ20082127 [DOI] [PubMed] [Google Scholar]
- 52. Dai P., Wang W., Yang N., Serna-Tamayo C., Ricca J. M., Zamarin D., Shuman S., Merghoub T., Wolchok J. D., and Deng L. (2017) Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci. Immunol. 2, eaal1713 10.1126/sciimmunol.aal1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sauer J. D., Sotelo-Troha K., von Moltke J., Monroe K. M., Rae C. S., Brubaker S. W., Hyodo M., Hayakawa Y., Woodward J. J., Portnoy D. A., and Vance R. E. (2011) The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79, 688–694 10.1128/IAI.00999-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sahin U., Weskamp G., Zheng Y., Chesneau V., Horiuchi K., and Blobel C. P. (2006) A sensitive method to monitor ectodomain shedding of ligands of the epidermal growth factor receptor. in Epidermal Growth Factor: Methods and Protocols (Patel T. B., and Bertics P. J., eds) pp. 99–113, Humana Press Inc., Totowa, NJ: [DOI] [PubMed] [Google Scholar]