

Abstract
Excitatory amino acid transporters (EAATs) represent a protein family that is an emerging drug target with great therapeutic potential for managing central nervous system disorders characterized by dysregulation of glutamatergic neurotransmission. As such, it is of significant interest to discover selective modulators of EAAT2 function. Here, we applied computational methods to identify specific EAAT2 inhibitors. Utilizing a homology model of human EAAT2, we identified a binding pocket at the interface of the transport and trimerization domain. We next conducted a high-throughput virtual screen against this site and identified a selective class of EAAT2 inhibitors that were tested in glutamate uptake and whole-cell electrophysiology assays. These compounds represent potentially useful pharmacological tools suitable for further exploration of the therapeutic potential of EAAT2 and may provide molecular insights into mechanisms of allosteric modulation for glutamate transporters.
Keywords: drug discovery, homology modeling, allosteric regulation, glutamate, neurological disease, neurotransmitter, excitatory amino acid transporters, glutamate transporter, high throughput virtual screening, SLC1A
Introduction
Sodium-dependent glutamate transporters (SLC1A1–3, 6, or 7 or excitatory amino acid transporters EAAT1–5)3 play essential roles in the maintenance and regulation of glutamatergic neurotransmission (1–3) and have emerged as important targets for drug discovery (4). EAAT1–5 transporters rapidly bind and buffer glutamate, removing it from the synaptic cleft. This removal contributes to the termination of synaptic activity and to the clearance of potentially cytotoxic extracellular glutamate. EAAT1–5 transports the bound glutamate into cells where it can be recycled for use in subsequent synaptic events (3). Glutamate transporters can also influence membrane potential and cellular excitability, either as a result of the electrogenic cotransport of 3Na+ and 1H+ and coupled counter-transport of 1K+ for each glutamate molecule transported or via the generation of an uncoupled chloride conductance (5). Importantly, major neuropsychiatric and neurological disorders such as schizophrenia, bipolar disorder, depression, and memory impairment are characterized by dysregulation of glutamatergic neurotransmission (6–8). Slowing glutamate uptake with selective EAAT inhibitors could improve transmission at glutamatergic synapses and provide therapeutic benefit under some conditions (1, 3, 9–13). Understanding the precise role of EAAT2 in health and disease requires identification of new selective modulators of EAAT2 function. However, to date the availability of pharmacological tools for EAAT2 is limited (14, 15). The aim of the present study, therefore, was to identify novel selective tools for the study of EAAT2.
Computational methods are routinely utilized in drug discovery to identify diverse sets of bioactive compounds for pharmaceutically relevant targets (16–19). They can be largely classified as either structure-based or ligand-based techniques and have played a major role in the discovery and development of new drug molecules for over three decades. In this study, we applied structure-based tools to the discovery of novel modulators of the human excitatory amino acid transporter 2 (hEAAT2).
Much of what is known of the structure of glutamate transporters comes from crystal structures of the prokaryotic homolog from Pyrococcus horikoshii (GltPh). GltPh has been crystallized in an apo form, in complex with a substrate (l-aspartate), and in complex with the competitive inhibitor TBOA (20–22). These structures show that GltPh exists as a homotrimer, with each monomer of the trimeric structure consisting of two domains: a trimerization domain formed by transmembrane helices 1, 2, 4, and 5 and a transport domain formed by transmembrane helices 3, 6, 7, and 8 and two re-entrant loops (helical hairpins 1 and 2). The structures capture two distinct conformations, inward-facing and outward-facing, where individual transport domains undergo relocations ∼15 Å normal to the membrane and provides substrate and ions alternating access to the extracellular (outward) and intracellular (inward) regions (23). Because interdomain interactions determine the transport rate of glutamate uptake (24), domain unlocking by disruption of interdomain interactions should modulate the movement of the transport domain and, hence, the glutamate transport rate.
Here, an hEAAT2 homology model built from GltPh was generated to identify novel allosteric site(s) and assist in the identification of selective hEAAT2 modulators. A virtual screen was completed of a component of the Janssen inventory, and our study led to the discovery of a novel and selective hEAAT2 inhibitor. To our knowledge, this is the first selective, allosteric hEAAT2 inhibitor described in the literature.
Results and discussion
Homology model generation
The homology modeling tool Prime (25, 26) was used to generate two homology models built from GltPh crystal structures: one model in an inward-facing conformation (PDB code 4P19 (21) as template) and one in an outward-facing conformation (PDB code 1XFH (22) as template). Because of the low sequence identity between GltPh and hEAAT2 (∼30% sequence identity), the predicted sequence alignment from Prime required manual intervention. Data from a multiple sequence alignment generated by Yernool et al. (22) between GltPh, hEAAT3, and additional homologs was useful to guide the manual alignment as hEAAT2 and hEAAT3 have ∼55% sequence identity. Additionally, there are many functionally important amino acids that are highly conserved across the SLC family and were used to guide the sequence alignment (the sequence alignment is provided in the Fig. S1).
The resulting models appeared to be robust; tertiary and secondary structures were maintained with small differences in flexible loop areas. Furthermore, the RMSD between the inward-facing model and 4P19 was found to be 0.38 Å, and the RMSD between the outward-facing model and 1XFH was 0.39 Å. Both models are provided in the Fig. S7. There is a large insertion in eukaryotic transporters between helices 4b and 4c (∼50-residue insertion in hEAAT2) that was not modeled because it is very difficult to accurately predict the structure of these residues de novo. To assess the structural quality of the model, protein reports were generated within Maestro (27) to examine steric clashes, bond length deviations, etc. Based on this assessment, the models appear to be of good structural quality and suitable for structure-based applications (details provided in the supporting data).
During the study, hEAAT1 structures were solved in the outward conformation in complex with a substrate (l-aspartate), the competitive inhibitor (2S, 3S)-3-[3-[4-(trifluoromethyl)benzoylamino]benzyloxy]aspartate, and a novel allosteric inhibitor, UCPH101 (28). The structures were found to be very similar to GltPh. Additionally, there is good agreement between the hEAAT2 model and hEAAT1 crystal structure in the outward-facing conformation (PDB codes 5LLM (28), 5LLU (28), 5LM4 (28), and 5MJU (28)), provided in the Figs. S2 and S3; the RMSD between the model and 5LLM crystal structure is 0.52 Å. The insertion residues are not fully resolved in the hEAAT1 structures; hence, it is still unclear whether they take on any secondary structure and how they impact the overall fold.
Pocket identification and evaluation
The two hEAAT2 homology models (inward-facing and outward-facing conformations) were utilized to predict allosteric sites that could potentially modulate the transport rate of glutamate. A binding pocket evaluation using SiteMap (29, 30) identified a site at the interface of the trimerization and transport domains (site A, inward-facing conformation), located in a region previously identified as important for Parawixin 1 binding (15), shown in Fig. 1. The volume of the site is 956.8 Å3 and the propensity for ligand binding (PLB) score (31) was 3.93, the highest predicted index for a cavity on the model. The PLB is based off amino acid composition of the predicted site, and a validation study was found for 79% of their test data set; the site with the highest PLB index was indeed the true binding site (31). Additional sites were observed but 1) the PLB index was very low (<1), and 2) the PLB index did not appear to be in a region that would have a functional effect on the transporter. FTMap was also utilized to confirm that site A contained hot spots for ligand binding; 9 of the 16 probes used by FTMap mapped to this region.
Figure 1.

Predicted binding sites on hEAAT2 homology models. Yellow, transport domain. Blue, trimerization domain. Top panel, site A, inward-facing conformation (shown in spherical representation). Bottom panel, site B, outward-facing conformation (shown in spherical representation) located on the back side of hEAAT2 as compared with known glutamate- and UCPh101-binding sites. Green, TBOA. Cyan, UCPH101.
A second pocket evaluation was conducted for the outward-facing homology model and identified two high-ranking sites. The top ranked site was the orthosteric binding site and has a volume of 932.4 Å3; the PLB index was 2.99. Identification of this site validated the use of SiteMap.
The second ranked site (site B, outward-facing conformation) was located on the opposite side of the transporter compared with the glutamate-binding pocket and at the interface of the trimerization and transport domains (Fig. 1). The volume of this site is 1278.0 Å3, and the PLB index was 1.53. FTMap also identified this site with all 16 probes being represented. An overlay of the two hEAAT2 conformations showed that the site is not present in the inward-facing conformation because it is occupied by the transport domain (provided in the Fig. S4). We hypothesized that binding to this region could potentially result in negative allosteric modulation because compound binding would sterically block the conformational change of the transport domain that is necessary for glutamate uptake.
HTVS led to NAM discovery
Approximately 350,000 unique compounds, a component of the Janssen corporate library, were virtually screened using the two identified binding sites of the EAAT2 homology models (site A, inward-facing conformation; site B, outward-facing conformation). High-throughput virtual screening (HTVS) has been shown to be a promising and resource-efficient tool for hit identification (32–39) with a number of recent studies illustrating its successful application to drug discovery (40–45). Because there was no available literature data for benchmarking, constraints were not utilized in either the site A or site B HTVS run. Various filters were applied to prioritize hits such as removing compounds with a molecular weight of <150 and reactive functionalities, quality control filters, and a GlideScore cutoff of −6.5 for site A and −7 for site B (because site A is smaller than site B, fewer compounds were scored). Additionally, post-docking hydrogen bond constraint filters were applied to ensure that at least one hydrogen bond was made to a residue in the predicted binding site. An additional filter based on availability was applied resulting in a high priority hit list of 2360. All 2360 compounds were tested for EAAT2 modulatory activity using a 384-well fluorescent membrane potential assay. Based on these results, 26 compounds were selected for follow-up in orthogonal assays of EAAT2 activity (glutamate uptake and whole-cell electrophysiology).
The compounds were tested at 25 μm, and only one compound, compound 1, significantly decreased EAAT2-mediated glutamate uptake as demonstrated by Fig. 2A (one-way analysis of variance; Dunnett test *, p < 0.0332). To confirm those data, we also assessed the potency and selectivity of compound 1 at hEAAT2 compared with the closely related hEAAT1. Compound 1 decreased hEAAT2-mediated glutamate uptake with an IC50 of 6.6 ± 0.6 μm (Fig. 2B) and was selective for hEAAT2 over hEAAT1 (EAAT1 IC50 value was >50 μm; Table 1) The inhibitory activity of compound 1 was further confirmed in whole-cell electrophysiology (Fig. 2D), considered a measure of direct binding; the inset shows inhibition of glutamate-induced current in a cell expressing hEAAT2. Inhibition was reversible upon washout of the compound.
Figure 2.

A, uptake of 10 μm [14C]glutamic acid in HEK293TRex-TO/hEAAT2 cells in the presence of either control (DMSO) or 25 μm compounds. B, compound 1 dose-dependently inhibits the uptake of 10 μm [14C]glutamic acid in hEAAT2-expressing cells (6.6 ± 0.6 μm). C, compound 1v decreases the maximal transport capacity of the EAAT2 transporter without affecting glutamate Kd, suggesting an allosteric noncompetitive mode of action. D, compound 1 dose-dependently inhibits EAAT2-meduated currents in hEAAT2-expressing cells. The inhibition is reversible, as demonstrated in the inset.
Table 1.
Structure and potency of compounds at inhibiting either EAAT2- or EAAT1-mediated glutamate uptake in stable cell lines stably expressing either EAAT2 or EAAT1
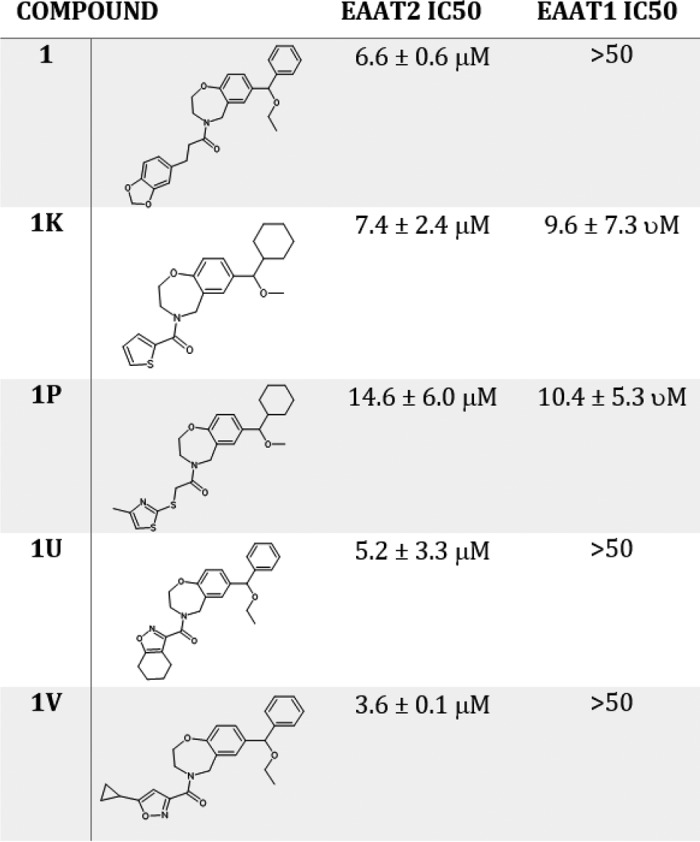
The confirmed inhibitor, compound 1, was identified by allosteric site B on the outward-facing conformation of hEAAT2 and is provided in Table 1. A similarity search of compound 1 against the Janssen inventory identified 28 analogs available for experimental testing. Four of the 28 compounds: 1k, 1p, 1u, and 1v, exhibited significant hEAAT2 inhibitory activity, shown in Table 1. The molecular formula strings, an NMR analysis and a compound purity assessment for the compounds are provided in the Figs. S6, S8, and S9a–e, respectfully. Additionally, the four analogs were tested against hEAAT1, and two were selective for hEAAT2. We performed mechanistic studies with those compounds. As shown in Fig. 2C, compound 1v decreased the maximal transport capacity of the EAAT2 transporter without affecting glutamate Kd, suggesting an allosteric noncompetitive mode of action, further highlighted by the Eadie–Hofstee and Lineweaver–Burk analyses (provided in the Fig. S10).
Although the number of compounds tested is limited, some initial structure–activity relationship has emerged. Compounds with a single carbon linker between the amide and left-hand substituent were not active. However, substituents that are directly linked or with a two-atom linker (similar to compound 1) are active. The linker number does not appear to affect selectivity over hEAAT1. Cyclohexyl and benzyl groups are both tolerated as right-hand substituents of the benzoxazepine core. Interestingly, compounds with cyclohexyl are not selective for hEAAT2, whereas those with benzyl are selective. Both cyclohexyl compounds also have an ethyl rather than methyl ether, which could contribute to the loss of selectivity for hEAAT2 over hEAAT1. The ClogP for this series ranges from 3.55 to 4.81 (and AlogP from 3.54 to 4.28); as such, it is highly probable that the compounds are able to penetrate the membrane.
Predicted binding mode of compound 1
Compound 1 was identified through a virtual screen of site B from the outward facing conformation. Fig. 3 shows the predicted binding modes that compound 1 could likely adopt. The poses are reasonable and consistent across varying parameters of the Glide docking program. However, although Trp-355 and Phe-348 are predicted to be important residues for binding in both poses, we have been unable to confirm the importance of these residues using site-directed mutagenesis (because single-point mutations, F348A and W355L did not reduce compound 1 activity; data not shown). Moreover, Fig. 3 shows the major differences between EAAT2 and EAAT1 in compound 1's predicted EAAT2-binding site. Although Gly-329 in EAAT2 is a leucine in EAAT1 that would clash with one of the predicted binding modes of compound 1, mutating EAAT2 Gly-329 into leucine did not have any significant effect on compound 1 potency at inhibiting EAAT2-mediated glutamate uptake (not shown).
Figure 3.

Top panel, predicted binding modes of compound 1 (orange and yellow) in site B of the hEAAT2 outward facing conformation model (green). Bottom panel, relevant residue differences between hEAAT2 (green) and hEAAT1 (purple) in site B, shown in stick representation.
Because site B is a large, flexible pocket, it is possible that it might rearrange in an induced-fit manner to accommodate ligand binding, and a variation of these binding poses would likely be the actual binding mode. For example, there are many Phe residues in the pocket that could rearrange to π–π stack with the aromatic groups of compound 1. It is well-known that apo structures are notoriously challenging in structure-based drug design. Furthermore, because compound 1 is a μm level inhibitor, it is also possible that the contributions made by each residue are not significant enough to show an effect as a single-point mutation, and double mutations may be necessary to result in a loss of activity. Given the flexibility and conformational heterogeneity of hEAAT2, it is very difficult to make definitive statements concerning the actual binding pose and the basis of the EAAT subtype selectivity for this series (relevant residue differences between hEAAT1 and hEAAT2 are shown in Fig. 3). More experimental work, including crystallography, multiresidue mutations, or experiments with more potent compounds, is required to better understand the nature of the compound interactions.
Although site B is the most likely binding site for compound 1, we did entertain the possibility that it could bind instead to other sites on hEAAT2. To test this, the series was docked into site A from the inward facing conformation, and it was found that the compounds were too large for this site. Additionally, the binding site of the hEAAT1 NAM (28), UCPH101, was explored. It was determined that this was unlikely the binding site because the GlideScore was quite low; there was no consistency in the poses or specific interactions being made. Both static and induced fit docking were utilized for docking into site 1 and the UCPH101 site.
Conclusion
At the onset of a drug discovery project, virtual screens can be effectively employed to identify novel chemical leads. In cases where high-resolution structural data, such as crystals structures, is not available, homology models can be applied with success.
Here, we utilized binding site identification tools and identified a novel, allosteric pocket on hEAAT2. A virtual screen was conducted against this pocket and resulted in the discovery of an hEAAT2 inhibitor selective over hEAAT1. To our knowledge this is the first example of a selective allosteric hEAAT2 inhibitor described in the literature. Analogs of the chemical series were identified through hit expansion techniques and provided initial structure–activity relationship. The compound may be useful as a novel molecular pharmacology tool and provide insight into how EAATs can be allosterically modulated. Furthermore, SLC transporters are an emerging drug target family with great therapeutic potential (2); as such, an optimized hEAAT2 inhibitor may help characterize and validate the role of EAAT2 in the development of numerous neurological and neuropsychiatric disorders.
Materials and methods
Homology modeling
The homology modeling tool Prime (25, 26), from the Schrödinger suite of programs (27), was used to generate a model of the inward-facing conformation and outward-facing conformation of hEAAT2. A BLAST (46) search was conducted in the PDB (47) using the hEAAT2 sequence to determine which homologs were available in the public domain with structural information. The Glu transporter homolog from P. horikoshii (∼30% sequence identity) crystal structure PDB code 4P19 (21) was utilized as the template for the inward-facing conformation, and PDB code 1XFH (22) for the outward-facing conformation, using default parameters. The crystal structures were first prepared using the Protein Preparation Wizard within Maestro (27) including adding hydrogens, filling in missing side chains, optimizing hydrogen bonds, and a restrained minimization of all protein atoms. Upon completion of the model-building calculations, the final models were optimized, and energy was minimized with a truncated-Newton energy minimization using OPLS 2000 all-atom force field (48).
Data from a multiple sequence alignment generated by Yernool et al. (22) between GltPh, hEAAT3, and additional homologs was used to guide the manual alignment because hEAAT2 and hEAAT3 have ∼55% sequence identity. The sequence alignment is provided in the Fig. S1. A homology model of hEAAT2 was also generated using the hEAAT1 crystal structure PDB code 5LLM (28) (outward-facing conformation) again using the homology modeling tool Prime (25, 26) with default parameters and following the protocol discussed above. The amino acid sequences between EAAT1 and EAAT2 are 65% identical; hence, the alignment was straightforward.
Pocket identification and evaluation
SiteMap (29, 30), a grid-based method for quick calculation and comparison of pocket volumes, was used to assess the hEAAT2 inward- and outward-facing conformation homology models and to identify pockets amenable for small molecule binding. Default parameters were employed to analyze the pocket volume and chemical environment for both conformations. Additionally, FTMap (49) was utilized to detect hot spots, areas that have a high propensity for ligand binding, on the protein surface of both models again using default parameters.
Ligand database preparation
A virtual database was previously created from a portion of the Janssen corporate library using default LigPrep (27) parameters to generate tautomers, ionization states (using Epik (50, 51)), and stereoisomers. The final virtual data set contained 350,000 unique compounds. Because the library was prefiltered, no compounds were flagged as PAINS (52). At the time of the study, there were no known hEAAT2 compounds for benchmarking.
High-throughput virtual screen
The virtual data set of 350,000 unique compounds from the Janssen corporate library was docked into the two identified binding sites of the EAAT2 homology models (site A, inward-facing conformation; site B, outward-facing conformation) using Glide SP (53, 54) from the Schrödinger suite of programs (55). Default parameters were utilized for grid generation (site A grid center: 3.43, −4.62, 18.89; and site B: −9.63, 51.55, −49.39). Analysis of the HTVS results was performed using Pipeline Pilot (56). All scored compounds were filtered using the following criteria: GlideScore of −6.5 for site A and −7 for site B, availability of the molecule for testing, in-house quality-control criteria, and filters for molecular weight of <150 and reactive functionalities. Additionally, post-docking hydrogen-bond constraint filters were applied to ensure that at least one hydrogen bond was made to a residue in the predicted binding site. Compounds in common between the screens were pooled, and duplicates were removed and clustered using proprietary clustering methodology (57), resulting in 2,360 hits.
Cell culture and stable cell line generation
The cDNA for hEAAT2 and hEAAT1 were subcloned into pcDNA4-TO (Invitrogen) using EcoRV and XhoI restriction sites. The HEK293-TRex cells were maintained at 37 °C in a humidified 5% CO2 incubator in culture medium (Dulbecco's modified Eagle's medium supplemented with penicillin (100 units/ml), streptomycin (100 mg/ml), and 10% tetracycline-free fetal bovine serum). For the stable expression of both human SLC1A2 (EAAT2) and SLC1A3 (EAAT1), the cells were transfected using FuGENE according to the protocol of the manufacturer (Promega, Madison, WI) and maintained for 2 weeks in selection medium containing 200 μg/ml Zeocin and 5 μg/ml Blasticidin. Antibiotic-resistant colonies were isolated and screened individually in the [14C]citrate uptake assay after overnight incubation with 1 μg/ml tetracycline. Noninduced cells were used as a negative control. Characterization of EAAT1 and EAAT2 stable cell lines provided in the Fig. S5.
Glutamate uptake assay
HEK293TRex-TO/hEAAT2 cells were plated and induced overnight with 1 μg/ml tetracycline at a density of 100,000 cells/well in PDL-pretreated 96-well plates (PerkinElmer). On the day of the experiment, the cells were washed twice with assay buffer (Hanks' balanced salt solution supplemented with 20 mm HEPES, 2 mm CaCl2, 1 mm MgSO4, and 5 mm glucose, pH 7.4) before adding 100 μl of assay buffer. Inhibitors were added and incubated for 5 min at room temperature before addition of [14C]glutamic acid on ice for 5 min. Glutaraldehyde (0.5% final) was then added to block uptake. The experiments were performed in quadruplets three or four times for each compound. The data are presented as averages ± S.E., and statistical analysis, when presented, was performed as indicated using GraphPad Prism software version 6.02. Saturation experiments were performed to confirm the known Kd of glutamate at both EAAT2 and EAAT1 (supporting data S1 and not shown). WAY213613 (Tocris) was also used to confirm that the uptake measured was EAAT2-mediated.
Whole cell electrophysiology
HEK TREX cells stably expressing hEAAT2 were plated at ∼10,000 cells/well on coverslips in 24-well plates and induced with 1 μg/ml tetracycline overnight. On the day of the experiment, glass coverslips were placed in a bath on the stage of an inverted microscope and perfused (∼1 ml/min) with extracellular solution (149 mm NaCl, 4 mm KCl, 10 mm HEPES, 5 mm glucose, 2 mm CaCl2, 1 mm MgCl2, 311 mOsm, pH 7.4). Pipettes were filled with an intracellular solution of the following composition: 40 mm KCl, 100 mm KF, 10 mm EGTA, 10 mm HEPES, 2 mm MgCl2, pH 7.3–7.4, with KOH, ∼290 mOsm (sucrose) and had a resistance of 2–4 mΩ. All recordings were made at room temperature (22–24 °C) using a Multiclamp 700A amplifier and pClamp 9 software (Axon Instruments). Current records were acquired at 2KHz and filtered at 1 KHz. The drugs were applied using an SF-77B Fast-Step Perfusion device (Warner Instruments). Transporter currents were elicited by 10 μm glutamate at holding potential of −60 mV. Compounds were applied in the continued presence of glutamate.
Author contributions
K. L. D.-G., M.-L. R., A. D. W., and T. M. conceptualization; K. L. D.-G., M.-L. R., A. D. W., and H. M. M. data curation; K. L. D.-G. and M.-L. R. formal analysis; K. L. D.-G., M.-L. R., A. D. W., and H. M. M. investigation; K. L. D.-G. writing-original draft; K. L. D.-G., M.-L. R., A. D. W., and T. M. writing-review and editing; T. M. project administration.
Supplementary Material
Acknowledgments
We acknowledge Jack Kauffman and other members of Janssen Discovery Sciences for help with compound testing. Additionally, we thank Mark Seierstad for help in creating the Glide version of the Janssen corporate database that was used for the virtual screen.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supporting data and Figs. S1–S10.
- SLC
- solute carrier
- EAAT
- excitatory amino acid transporter
- hEAAT
- human EAAT
- GltPh
- glutamate transporter P. horikoshii
- HTVS
- high-throughput virtual screening
- PLB
- propensity for ligand binding
- TBOA
- dl-threo-β-benzyloxyaspartic acid
- PDB
- Protein Data Bank
- RMSD
- root-mean-square deviation.
References
- 1. Nakagawa T., and Kaneko S. (2013) SLC1 glutamate transporters and diseases: psychiatric diseases and pathological pain. Curr. Mol. Pharmacol. 6, 66–73 10.2174/18744672113069990033 [DOI] [PubMed] [Google Scholar]
- 2. Rives M. L., Javitch J. A., and Wickenden A. D. (2017) Potentiating SLC transporter activity: emerging drug discovery opportunities. Biochem. Pharmacol. 135, 1–11 10.1016/j.bcp.2017.02.010 [DOI] [PubMed] [Google Scholar]
- 3. Takahashi K., Foster J. B., and Lin C. L. (2015) Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell. Mol. Life Sci. 72, 3489–3506 10.1007/s00018-015-1937-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Colas C., Ung P. M., and Schlessinger A. (2016) SLC transporters: structure, function, and drug discovery. Medchemcomm. 7, 1069–1081 10.1039/C6MD00005C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fairman W. A., Vandenberg R. J., Arriza J. L., Kavanaugh M. P., and Amara S. G. (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375, 599–603 10.1038/375599a0 [DOI] [PubMed] [Google Scholar]
- 6. Abdallah C. G., Averill C. L., Salas R., Averill L. A., Baldwin P. R., Krystal J. H., Mathew S. J., and Mathalon D. H. (2017) Prefrontal connectivity and glutamate transmission: relevance to depression pathophysiology and ketamine treatment. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2, 566–574 10.1016/j.bpsc.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goff D. C., and Coyle J. T. (2001) The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am. J. Psychiatry 158, 1367–1377 10.1176/appi.ajp.158.9.1367 [DOI] [PubMed] [Google Scholar]
- 8. Tang Y. P., Shimizu E., Dube G. R., Rampon C., Kerchner G. A., Zhuo M., Liu G., and Tsien J. Z. (1999) Genetic enhancement of learning and memory in mice. Nature 401, 63–69 10.1038/43432 [DOI] [PubMed] [Google Scholar]
- 9. Behrens P. F., Franz P., Woodman B., Lindenberg K. S., and Landwehrmeyer G. B. (2002) Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125, 1908–1922 10.1093/brain/awf180 [DOI] [PubMed] [Google Scholar]
- 10. Fontana A. C. (2015) Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 134, 982–1007 10.1111/jnc.13200 [DOI] [PubMed] [Google Scholar]
- 11. Rajkowska G., and Stockmeier C. A. (2013) Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr. Drug Targets 14, 1225–1236 10.2174/13894501113149990156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reissner K. J., and Kalivas P. W. (2010) Using glutamate homeostasis as a target for treating addictive disorders. Behav. Pharmacol. 21, 514–522 10.1097/FBP.0b013e32833d41b2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rothstein J. D., Van Kammen M., Levey A. I., Martin L. J., and Kuncl R. W. (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84 10.1002/ana.410380114 [DOI] [PubMed] [Google Scholar]
- 14. Falcucci R. M., Wertz R., Green J. L., Meucci O., Salvino J., and Fontana A. C. K. (2019) Novel positive allosteric modulators of glutamate transport have neuroprotective properties in an in vitro excitotoxic model. ACS Chem. Neurosci. 10, 3437–3453 10.1021/acschemneuro.9b00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fontana A. C., de Oliveira Beleboni R., Wojewodzic M. W., Ferreira Dos Santos W., Coutinho-Netto J., Grutle N. J., Watts S. D., Danbolt N. C., and Amara S. G. (2007) Enhancing glutamate transport: mechanism of action of Parawixin1, a neuroprotective compound from Parawixia bistriata spider venom. Mol. Pharmacol. 72, 1228–1237 10.1124/mol.107.037127 [DOI] [PubMed] [Google Scholar]
- 16. Baig M. H., Ahmad K., Roy S., Ashraf J. M., Adil M., Siddiqui M. H., Khan S., Kamal M. A., Provazník I., and Choi I. (2016) Computer aided drug design: success and limitations. Curr. Pharm. Des. 22, 572–581 10.2174/1381612822666151125000550 [DOI] [PubMed] [Google Scholar]
- 17. Duarte Y., Márquez-Miranda V., Miossec M. J., and González-Nilo F. (2019) Integration of target discovery, drug discovery and drug delivery: a review on computational strategies. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 11, e1554 [DOI] [PubMed] [Google Scholar]
- 18. Macalino S. J., Gosu V., Hong S., and Choi S. (2015) Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 38, 1686–1701 10.1007/s12272-015-0640-5 [DOI] [PubMed] [Google Scholar]
- 19. Sliwoski G., Kothiwale S., Meiler J., and Lowe E. W. Jr. (2014) Computational methods in drug discovery. Pharmacol. Rev. 66, 334–395 10.1124/pr.112.007336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boudker O., Ryan R. M., Yernool D., Shimamoto K., and Gouaux E. (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445, 387–393 10.1038/nature05455 [DOI] [PubMed] [Google Scholar]
- 21. Verdon G., Oh S., Serio R. N., and Boudker O. (2014) Coupled ion binding and structural transitions along the transport cycle of glutamate transporters. eLife 3, e02283 10.7554/eLife.02283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yernool D., Boudker O., Jin Y., and Gouaux E. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818 10.1038/nature03018 [DOI] [PubMed] [Google Scholar]
- 23. Reyes N., Ginter C., and Boudker O. (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462, 880–885 10.1038/nature08616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reyes N., Oh S., and Boudker O. (2013) Binding thermodynamics of a glutamate transporter homolog. Nat. Struct. Mol. Biol. 20, 634–640 10.1038/nsmb.2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacobson M. P., Friesner R. A., Xiang Z., and Honig B. (2002) On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320, 597–608 10.1016/S0022-2836(02)00470-9 [DOI] [PubMed] [Google Scholar]
- 26. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J., Honig B., Shaw D. E., and Friesner R. A. (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367 10.1002/prot.10613 [DOI] [PubMed] [Google Scholar]
- 27. Maestro (2018) release 10.6, Schrödinger, LLC, New York [Google Scholar]
- 28. Canul-Tec J. C., Assal R., Cirri E., Legrand P., Brier S., Chamot-Rooke J., and Reyes N. (2017) Structure and allosteric inhibition of excitatory amino acid transporter 1. Nature 544, 446–451 10.1038/nature22064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Halgren T. (2007) New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 69, 146–148 10.1111/j.1747-0285.2007.00483.x [DOI] [PubMed] [Google Scholar]
- 30. Halgren T. A. (2009) Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 49, 377–389 10.1021/ci800324m [DOI] [PubMed] [Google Scholar]
- 31. Soga S., Shirai H., Kobori M., and Hirayama N. (2007) Use of amino acid composition to predict ligand-binding sites. J. Chem. Inf. Model. 47, 400–406 10.1021/ci6002202 [DOI] [PubMed] [Google Scholar]
- 32. Cerqueira N. M., Sousa S. F., Fernandes P. A., and Ramos M. J. (2009) Virtual screening of compound libraries. Methods Mol. Biol. 572, 57–70 [DOI] [PubMed] [Google Scholar]
- 33. Danishuddin M., and Khan A. U. (2015) Structure based virtual screening to discover putative drug candidates: necessary considerations and successful case studies. Methods 71, 135–145 10.1016/j.ymeth.2014.10.019 [DOI] [PubMed] [Google Scholar]
- 34. Fradera X., and Babaoglu K. (2017) Overview of methods and strategies for conducting virtual small molecule screening. Curr. Protoc. Chem. Biol. 9, 196–212 10.1002/cpch.27 [DOI] [PubMed] [Google Scholar]
- 35. Irwin J. J., and Shoichet B. K. (2016) Docking screens for novel ligands conferring new biology. J. Med. Chem. 59, 4103–4120 10.1021/acs.jmedchem.5b02008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lavecchia A., and Di Giovanni C. (2013) Virtual screening strategies in drug discovery: a critical review. Curr. Med. Chem. 20, 2839–2860 10.2174/09298673113209990001 [DOI] [PubMed] [Google Scholar]
- 37. Lionta E., Spyrou G., Vassilatis D. K., and Cournia Z. (2014) Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr. Top. Med. Chem. 14, 1923–1938 10.2174/1568026614666140929124445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lounnas V., Ritschel T., Kelder J., McGuire R., Bywater R. P., and Foloppe N. (2013) Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 5, e201302011 10.5936/csbj.201302011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schneider G. (2010) Virtual screening: an endless staircase? Nat. Rev. Drug Discov. 9, 273–276 10.1038/nrd3139 [DOI] [PubMed] [Google Scholar]
- 40. Chen S., Feng Z., Wang Y., Ma S., Hu Z., Yang P., Chai Y., and Xie X. (2017) Discovery of novel ligands for TNF-α and TNF receptor-1 through structure-based virtual screening and biological assay. J. Chem. Inf. Model. 57, 1101–1111 10.1021/acs.jcim.6b00672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Damm-Ganamet K. L., Bembenek S. D., Venable J. W., Castro G. G., Mangelschots L., Peeters D. C., McAllister H. M., Edwards J. P., Disepio D., and Mirzadegan T. (2016) A prospective virtual screening study: enriching hit rates and designing focus libraries to find inhibitors of PI3Kδ and PI3Kγ. J. Med. Chem. 59, 4302–4313 10.1021/acs.jmedchem.5b01974 [DOI] [PubMed] [Google Scholar]
- 42. Kumarasiri M., Teo T., Yu M., Philip S., Basnet S. K., Albrecht H., Sykes M. J., Wang P., and Wang S. (2017) In search of novel CDK8 inhibitors by virtual screening. J. Chem. Inf. Model. 57, 413–416 10.1021/acs.jcim.6b00711 [DOI] [PubMed] [Google Scholar]
- 43. Li N., Ainsworth R. I., Ding B., Hou T., and Wang W. (2015) Using hierarchical virtual screening to combat drug resistance of the HIV-1 protease. J. Chem. Inf. Model. 55, 1400–1412 10.1021/acs.jcim.5b00056 [DOI] [PubMed] [Google Scholar]
- 44. Rettenmaier T. J., Fan H., Karpiak J., Doak A., Sali A., Shoichet B. K., and Wells J. A. (2015) Small-molecule allosteric modulators of the protein kinase PDK1 from structure-based docking. J. Med. Chem. 58, 8285–8291 10.1021/acs.jmedchem.5b01216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Veljkovic V., Loiseau P. M., Figadere B., Glisic S., Veljkovic N., Perovic V. R., Cavanaugh D. P., and Branch D. R. (2015) Virtual screen for repurposing approved and experimental drugs for candidate inhibitors of EBOLA virus infection. F1000Research 4, 34 10.12688/f1000research.6110.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 47. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jorgensen W. L., and Tirado-Rives J. (1988) The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666 10.1021/ja00214a001 [DOI] [PubMed] [Google Scholar]
- 49. Kozakov D., Grove L. E., Hall D. R., Bohnuud T., Mottarella S. E., Luo L., Xia B., Beglov D., and Vajda S. (2015) The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 10, 733–755 10.1038/nprot.2015.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Greenwood J. R., Calkins D., Sullivan A. P., and Shelley J. C. (2010) Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 24, 591–604 10.1007/s10822-010-9349-1 [DOI] [PubMed] [Google Scholar]
- 51. Shelley J. C., Cholleti A., Frye L. L., Greenwood J. R., Timlin M. R., and Uchimaya M. (2007) Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 21, 681–691 10.1007/s10822-007-9133-z [DOI] [PubMed] [Google Scholar]
- 52. Baell J. B., and Holloway G. A. (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740 10.1021/jm901137j [DOI] [PubMed] [Google Scholar]
- 53. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., and Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring: 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 10.1021/jm0306430 [DOI] [PubMed] [Google Scholar]
- 54. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., and Banks J. L. (2004) Glide: a new approach for rapid, accurate docking and scoring: 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 10.1021/jm030644s [DOI] [PubMed] [Google Scholar]
- 55. Glide (2018) release 10.6, Schrödinger, LLC, New York [Google Scholar]
- 56. BIOVIA Pipeline Pilot, Accelrys, San Diego, CA [Google Scholar]
- 57. Hack M. D., Rassokhin D. N., Buyck C., Seierstad M., Skalkin A., ten Holte P., Jones T. K., Mirzadegan T., and Agrafiotis D. K. (2011) Library enhancement through the wisdom of crowds. J. Chem. Inf. Model. 51, 3275–3286 10.1021/ci200446y [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.