

Abstract
Tricellular tight junctions (tTJs) create paracellular barriers at tricellular contacts (TCs), where the vertices of three polygonal epithelial cells meet. tTJs are marked by the enrichment of two types of membrane proteins, tricellulin and angulin family proteins. However, how TC geometry is recognized for tTJ formation remains unknown. In the present study, we examined the molecular mechanism for the assembly of angulin-1 at the TCs. We found that clusters of cysteine residues in the juxtamembrane region within the cytoplasmic domain of angulin-1 are highly palmitoylated. Mutagenesis analyses of the cysteine residues in this region revealed that palmitoylation is essential for localization of angulin-1 at TCs. Consistently, suppression of Asp-His-His-Cys motif–containing palmitoyltransferases expressed in EpH4 cells significantly impaired the TC localization of angulin-1. Cholesterol depletion from the plasma membrane of cultured epithelial cells hampered the localization of angulin-1 at TCs, suggesting the existence of a lipid membrane microdomain at TCs that attracts highly palmitoylated angulin-1. Furthermore, the extracellular domain of angulin-1 was also required for its TC localization, irrespective of the intracellular palmitoylation. Taken together, our findings suggest that both angulin-1's extracellular domain and palmitoylation of its cytoplasmic region are required for its assembly at TCs.
Keywords: cell junction, tight junction, epithelial cell, lipid raft, cell-cell interaction, cholesterol, post-translational modification (PTM), angulin, tricellular tight junction, palmitoylation, membrane domain
Introduction
Tight junctions (TJs)5 play a crucial role in epithelial barrier function by regulating diffusion of solutes through the paracellular pathway in vertebrate epithelial cells (1, 2). TJs contain at least three types of membrane proteins: occludin, claudin family proteins, and JAM family proteins (1, 2). The functional units of TJs are fibril-like plasma membrane contacts formed by assembly of claudin family membrane proteins from both adjacent cells (3). The structures can be visualized by freeze-fracture replica EM (4) and are designated TJ strands. A network of TJ strands circumscribes each cell at the most apical part of the lateral membrane to form a belt that acts as a continuous paracellular diffusion barrier (4). However, the paracellular space at tricellular contacts (TCs), where the vertices of three epithelial cells meet, cannot theoretically be sealed by continuous TJs. At a TC, the most apical TJ strands of bicellular TJs from each of three directions meet at the center, turn, and extend in the basal direction attached to one another (4). Consequently, a very narrow tube of extracellular space surrounded by three vertical TJ strands is formed at the center of a TC. This structure is considered to act as a paracellular diffusion barrier at TCs, and the vertical TJs are designated tricellular TJs (tTJs) (4, 5).
Two types of membrane proteins were identified as the molecular constituents of tTJs: tricellulin and angulin family proteins, including angulin-1/LSR, angulin-2/ILDR1, and angulin-3/ILDR2, herein designated angulins (5–7). Analyses of angulins in cultured epithelial cells revealed that they recruited tricellulin to TCs and that both tricellulin and angulins were required for full barrier function of an epithelial cellular sheet (5–7). Furthermore, mutations in the human tricellulin and angulin-2 genes caused nonsyndromic familial deafness (DFNB49 and DFNB42, respectively) (8, 9). Consistently, mutant mice lacking these genes showed profound deafness (10–14). Angulin-2–deficient mice also exhibited polyuria accompanied by impaired urine concentration (15), whereas angulin-1–deficient mice showed embryonic lethality at embryonic days 13–15 (16). These observations suggest crucial roles of tTJs in vivo.
Despite the physiological importance of tTJs, the underlying mechanism for their correct formation at TCs remains unclear. It is reasonable to speculate that the process for tTJ formation can be divided into two steps: recognition of TC regions and assembly of vertical TJ strands at these regions. Previous reports suggested that tricellulin is involved in the latter step. In tricellulin knock-in mice mimicking a human tricellulin gene mutation in familial deafness, tTJ organization was impaired in the inner ear epithelial cells (10). Consistently, the assembly of TJs visualized by immunofluorescence staining of occludin was abnormal around TCs in tricellulin knockdown epithelial cells (5). Tricellulin bound to a Cdc42 guanine nucleotide exchange factor, Tuba, and activated Cdc42 to induce F-actin organization during cell-cell junction formation in cultured epithelial cells (17). This cytoskeletal regulation by tricellulin may play a role in the organization of tTJs. Meanwhile, the mechanism by which TCs are recognized as the proper sites for tTJ formation is unknown. Considering that angulins recruit tricellulin to TCs (6), clarification of the mechanism for how angulins are localized at TCs (i.e. what types of cues direct the assembly of angulins at TCs) would help to address this issue.
Post-translational modifications of proteins, including phosphorylation and lipid modification, are potential mechanisms that regulate protein localization. Among several types of lipid modifications, palmitoylation involves the addition of a C16 acyl chain to cytoplasmic cysteine residues in proteins through thioester bonds (18–21). Palmitoylation is catalyzed by a family of Asp-His-His-Cys motif–containing palmitoyltransferases (DHHCs) (19, 21). Palmitoylation occurs in cytoplasmic proteins as well as integral membrane proteins and influences their behaviors in terms of trafficking, membrane association, and localization at certain membrane domains (18–21). Among the TJ-associated proteins, claudin-3, claudin-5, claudin-7, claudin-14, and JAM-C were reported to undergo palmitoylation (22–26). Palmitoylation of claudin-14 was required for its effective localization at TJs in epithelial cells without affecting the stability and assembly of TJ strands (22), whereas palmitoylation of claudin-7 accelerated its distribution in glycolipid-enriched membrane domains (25). However, it remains unknown whether tTJ-associated proteins undergo lipid modifications.
In the present study, we investigated the mechanism for TC localization of angulin-1. We show that angulin-1 undergoes high levels of palmitoylation and that full palmitoylation is necessary for its localization at TCs. We further demonstrate that the extracellular domain is required for angulin-1 assembly at TCs. Finally, we discuss why the combination of these two elements is required for proper localization of angulin-1 to TCs.
Results
The cytoplasmic domain of angulin-1 is required for its localization at TCs
To elucidate the mechanism for TC localization of angulin-1, we examined the responsible domains in angulin-1. A conventional method for this type of analysis is the generation of various deletion mutants and assessment of their subcellular localizations after exogenous expression in cultured epithelial cells. However, endogenous angulin-1 in cultured epithelial cells may influence the behavior of exogenous angulin-1 mutants in such analyses, possibly through homotypic or homomeric assembly. To avoid this problem, we initially generated angulin-1–deficient cells from EpH4 mouse mammary epithelial cells by CRISPR/Cas9-mediated genome editing and used one of the established angulin-1–deficient EpH4 clones, designated Ang1KO, throughout the study (Fig. S1A). The Ang1KO cell genome contained frameshift mutations at two or four nucleotides downstream of the first ATG in each allele (Fig. S1A), and angulin-1 expression was not detected in Ang1KO cells by either immunoblotting or immunofluorescence staining (Fig. 1, A and B).
Figure 1.

Role of the C-terminal cytoplasmic domain of angulin-1 in the localization of angulin-1 at TCs. A, generation of angulin-1–deficient EpH4 cells. Shown is immunoblotting (IB) of parent EpH4 cells (EpH4) and Ang1KO cells with anti-angulin-1 and anti-occludin antibodies. The membrane was also immunoblotted with an anti-α-tubulin antibody as a loading control. B, double immunofluorescence staining of EpH4 cells and Ang1KO cells with antibodies against angulin-1 and occludin. C, schematic drawings of the full-length and C-terminal cytoplasmic domain–deleted constructs of angulin-1. The full-length mouse angulin-1 contains 575 amino acids, including the first methionine. In this study, we used two C-terminal deletion constructs of angulin-1 containing 258 amino acids and 211 amino acids. The constructs were tagged with GFP at their C terminus and designated Ang1G, Ang1–258G, and Ang1–211G, respectively. SP, signal peptide; Ig, immunoglobulin-like domain; TM, transmembrane domain. D, immunoblotting of Ang1KO cells (−) and cells stably expressing (+) Ang1G, Ang1–211G, and Ang1–258G with an anti-GFP antibody. The protein band indicated by the asterisk is probably caused by degradation of Ang1–258G. E, immunofluorescence staining of Ang1KO cells stably expressing Ang1G, Ang1–258G, or Ang1–211G with an anti-occludin antibody. The localizations of Ang1G, Ang1–258G, and Ang1–211G were detected by the fluorescent signals of GFP. Ang1G and Ang1–258G are detected at TCs (arrows), whereas Ang1–211G is not (arrowheads). An enhanced image of the GFP signal for Ang1–211G is also shown. Scale bar, 20 μm. F, quantitation of the TC enrichment of Ang1G, Ang1–258G, and Ang1–211G at TCs. The graph represents mean ± S.D. (error bars) (n = 4–8 each). ***, p < 0.0005, compared by t test. a.u., arbitary units.
Subsequently, we investigated the role of the cytoplasmic domain of mouse angulin-1 in its localization. We previously reported that a splicing isoform of mouse angulin-1 with 575 amino acids, including the N-terminal signal peptide (GenBankTM AK146807), was localized at tTJs (6). We also showed that an angulin-1 mutant lacking amino acids 259–575 fused with GFP (Ang1–258G) was able to assemble at TCs (6). Because the bioinformatics tool TMHMM (27) predicted that the membrane-spanning region would end at amino acid 210, we generated a plasmid construct for expression of a GFP-tagged angulin-1 mutant lacking amino acids 212–575, designated Ang1–211G, that also lacked most of the cytoplasmic domain (Fig. 1C). Full-length mouse angulin-1 fused with GFP (Ang1G), Ang1–258G, or Ang1–211G was then stably expressed in Ang1KO cells. Immunoblotting of lysates from these cells with an anti-GFP antibody confirmed reduced molecular weights of the mutants in accordance with their deletions (Fig. 1D). When their localizations were analyzed by fluorescence microscopy, Ang1G and Ang1–258G assembled at TCs (Fig. 1, E and F), consistent with our previous observations (6), although the TC localization index in Ang1–258G was lower than that in Ang1G. In contrast, most of the Ang1–211G appeared to remain in intracellular compartments and was not observed at TCs (Fig. 1, E and F). These results suggest that amino acids 212–258 in the cytoplasmic region of angulin-1 play an important role in its transport to or retention in the plasma membrane and localization at TCs.
Angulin-1 undergoes palmitoylation
Based on the prediction that the transmembrane domain of angulin-1 would end at amino acid 210, amino acids 212–258 of angulin-1, which are absent from Ang1–211, are likely to be located just beneath the plasma membrane. We found that this region and part of the connected transmembrane domain contained a high concentration of cysteine residues: 13 cysteines within amino acids 208–232 (Fig. 2A). This feature was also conserved in angulin-2 and angulin-3 (Fig. 2A). Because cysteine residues located beneath the plasma membrane frequently undergo post-translational palmitoylation (18–21), we examined whether angulin-1 was palmitoylated using the acyl-PEGyl exchange gel shift (APEGS) method (28). In the APEGS method, the palmitoyl groups in a given protein are substituted by PEG with a specific high-molecular weight through a series of organic chemical reactions, and the extent of the original palmitoylation is subsequently evaluated by the molecular weight shift on SDS-PAGE followed by immunoblotting. Using a 2-kDa PEG in the APEGS method, we detected a broad band of angulin-1 at more than 100 kDa in immunoblotting, whereas the original angulin-1 band without PEG modification had a molecular mass of around 60 kDa (Fig. 2B). These results indicate that angulin-1 is highly palmitoylated.
Figure 2.

Palmitoylation of angulin-1. A, amino acid sequence of the last half of the transmembrane domain and the following juxtamembrane region of mouse angulin-1 (GenBankTM AK146807). The predicted transmembrane domain is shown by the dashed line. The 13 cysteine residues (red) within amino acids 208–232 of angulin-1 are conserved within amino acids 187–211 of angulin-2 (GenBankTM K136284) and amino acids 188–212 of angulin-3 (GenBankTM FJ024498). B, palmitoylation of endogenous angulin-1 in EpH4 cells detected by the APEGS method. After the reduction and alkylation of free cysteine thiols of proteins in cell lysates, palmitoyl-thioester linkages of the protein palmitoylation sites in cell lysates are cleaved by hydroxylamine treatment, and newly formed thiols are labeled with mPEG-2k. As a negative control, NEM of 125 Da was added instead of mPEG-2k. The rightmost two lanes show the results for the original lysates without any treatments. The remarkable mobility shift of angulin-1 in the hydroxylamine(+)/mPEG-2k lane compared with the negative control lanes, hydroxylamine(−)/mPEG-2k and hydroxylamine(+)/NEM, in EpH4 cells demonstrates that angulin-1 is highly palmitoylated. The mobility shift observed in the hydroxylamine(+)/NEM lane compared with the hydroxylamine(−)/mPEG-2k lane (asterisk) is probably caused by the addition of NEM (125 Da) to multiple palmitoyl cysteine residues. Because replacement of a palmitoyl group (239 Da) with NEM reduces the molecular weight of angulin-1, the slower mobility of the hydroxylamine(+)/NEM sample may be caused by structural change of angulin-1 protein by the NEM blinding. The bottom panel shows immunoblotting of the corresponding samples with an anti-Gαq antibody as a positive control for protein palmitoylation. IB, immunoblotting. C, screening for DHHCs responsible for palmitoylation of angulin-1. Each DHHC family member with an HA tag was cotransfected with Ang1G into HEK293T cells. After metabolic labeling with [3H]palmitate, proteins in the cell lysates were separated by SDS-PAGE and subjected to autoradiography and immunoblotting with anti-GFP and anti-HA antibodies. The results are representative of two independent experiments. D, expression of DHHC transcripts in EpH4 cells. The expression of DHHCs capable of Ang1G palmitoylation in C was analyzed by RT-PCR using total RNA from EpH4 cells as a template. RT(+) and RT(−) indicates reverse transcriptase–positive and –negative reactions, respectively.
Protein palmitoylation is catalyzed by protein palmitoyltransferases belonging to the DHHC family, which contains 24 subtypes in mice and humans (19, 21). To investigate the DHHC subtypes that catalyze the palmitoylation of angulin-1, we overexpressed Ang1G and each of the HA-tagged mouse DHHC family proteins in HEK293T cells with tritium-labeled palmitate and detected palmitoylation of Ang1G by autoradiography. As shown in Fig. 2C, DHHC2, -3, -4, -6, -7, -10, and -15 clearly palmitoylated angulin-1. Among these DHHCs, expression of DHHC2, -3, -4, -6, and -7 was detected in EpH4 cells by RT-PCR (Fig. 2D).
We further examined which cysteine residues in angulin-1 were palmitoylated. The region of angulin-1 comprising amino acids 210–232 contains 12 cysteine residues. We divided these residues into four convenient clusters (1–4) from the plasma membrane side, with each cluster containing three cysteine residues that were continuously arranged or closely located in the vicinity of one another (Fig. 3A). We then constructed expression vectors for these GFP-tagged angulin-1 mutants in which all three cysteine residues in each cluster or in multiple clusters were replaced with serine residues (Fig. 3A). The cysteine-to-serine mutation has been utilized to analyze protein palmitoylation in previous studies, including those on tight junction–associated membrane proteins (22, 26). The angulin-1 mutants with cysteine-to-serine substitution in cluster 1, cluster 2, cluster 3, and cluster 4 were designated CS1, CS2, CS3, and CS4, respectively. Similarly, the angulin-1 mutants with cysteine-to-serine substitution in clusters 1 and 2, clusters 1–3, and clusters 1–4 were designated CS1–2, CS1–3, and CS1–4, respectively. We introduced the expression vectors for the mutants into Ang1KO cells, established stable clones expressing each of the mutants, and analyzed their palmitoylation status by the APEGS method (Fig. 3B). Compared with WT angulin-1, the palmitoylation levels of CS1, CS1–2, and CS1–3 showed stepwise reductions according to the extent of cysteine-to-serine substitution. The palmitoylation level in CS1–3 was the same as that in CS1–4, suggesting that the cysteine residues in cluster 4 in CS1–3 were not palmitoylated. Meanwhile, the palmitoylation levels in CS2, CS3, and CS4 were similar to that in CS1, suggesting that all four cysteine clusters underwent palmitoylation. CS1–4 lacking all 12 cysteine residues within amino acids 210–232 still showed a mobility shift, suggesting that cysteine 208 in the membrane-spanning region was also palmitoylated.
Figure 3.
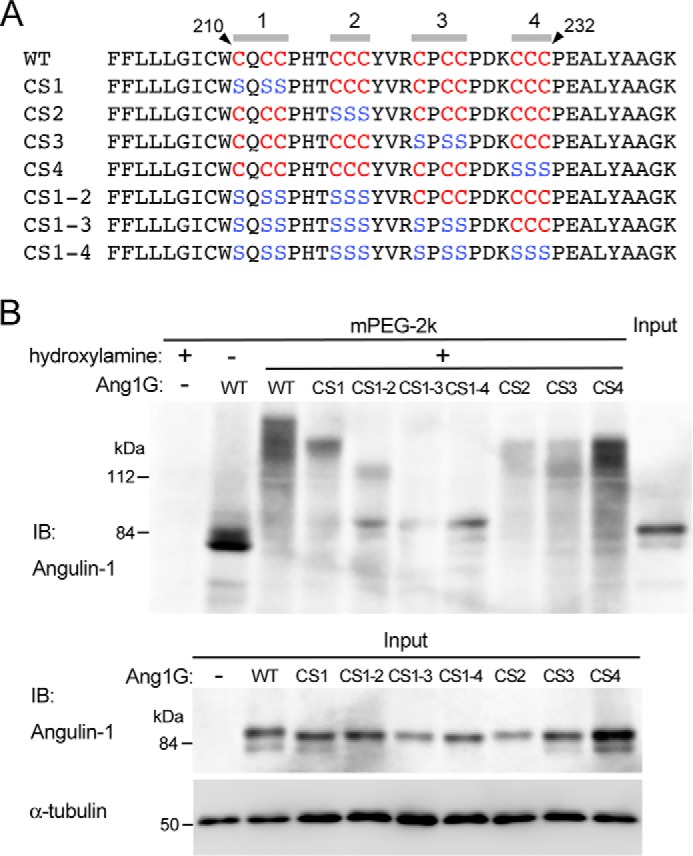
Determination of palmitoylated cysteine residues in angulin-1. A, amino acid sequences of the juxtamembrane regions in mouse angulin-1 (WT) and its mutants with serine substitution of the cytoplasmic cysteine residues (CS1, CS2, CS3, CS4, CS1–2, CS1–3, and CS1–4). The cysteine and serine residues within amino acids 210–232 are shown in red and blue letters, respectively. B, the top panel shows palmitoylation of GFP-tagged WT angulin-1 and its cysteine-to-serine–substituted mutants detected by the APEGS method. Lysates of Ang1KO cells stably expressing Ang1G (WT) or mutants were processed for the APEGS method. Mobility shifts of the bands to higher molecular weights indicate protein palmitoylation. The bottom panel shows immunoblotting of lysates from Ang1KO cells (−) and cells expressing Ang1G (WT) or cysteine-to-serine–substituted mutants before the procedure for the APEGS method with an anti-angulin-1 antibody and anti-α-tubulin antibody. IB, immunoblotting.
Full palmitoylation is required for TC localization of angulin-1
To examine whether cytoplasmic palmitoylation influenced the assembly of angulin-1 at TCs, the localizations of the CS mutants of angulin-1 were analyzed by fluorescence microscopy based on the signals of their GFP tags. As shown in Fig. 4 (A–C), none of the CS mutants were localized at TCs, but instead they were distributed throughout the lateral plasma membrane. These results suggest that palmitoylation of all four cysteine clusters is required for localization of angulin-1 at TCs.
Figure 4.

Role of palmitoylated cysteine residues in the localization of angulin-1 at TCs. A, immunofluorescence staining of Ang1KO cells stably expressing Ang1G (WT) or cysteine-to-serine–substituted mutants with an anti-occludin antibody. Ang1G or the mutants in the same field were also visualized by fluorescence of GFP. Scale bar, 20 μm. B, Z-stack sections of immunofluorescence staining of Ang1KO cells stably expressing Ang1G (WT) or cysteine-to-serine–substituted mutants. The GFP signal (green) from each of the cysteine-to-serine–substituted mutants is connected to the occludin signal (red) and extends in the basal direction, indicating that these mutants are localized along the lateral plasma membrane. Scale bar, 5 μm. C, quantitation of the TC enrichment of Ang1G (WT) or cysteine-to-serine–substituted mutants. The graph represents mean ± S.D. (error bars) (n = 2–3 each). **, p < 0.005, compared by t test. a.u., arbitary units.
To confirm that DHHCs are responsible for TC localization of angulin-1, we performed loss-of-function studies of DHHCs in EpH4 cells. First, we established a DHHC6-deficient EpH4 cell clone (DHHC6 KO cells) by CRISPR/Cas9-mediated genome editing (Fig. S1, B and C). In these cells, angulin-1 was clearly localized at TCs in immunofluorescence staining (Fig. S1D). When the expression of either DHHC2, -3, -4, or -7 was suppressed by siRNA in DHHC6 KO cells, localization of angulin-1 never changed (Fig. S2, A and B). However, simultaneous treatment of DHHC6 KO cells with all of these siRNAs significantly reduced the expression of these DHHCs and impaired the TC localization of angulin-1 (Fig. 5, A–C). These results suggest that DHHCs are responsible for TC localization of angulin-1.
Figure 5.
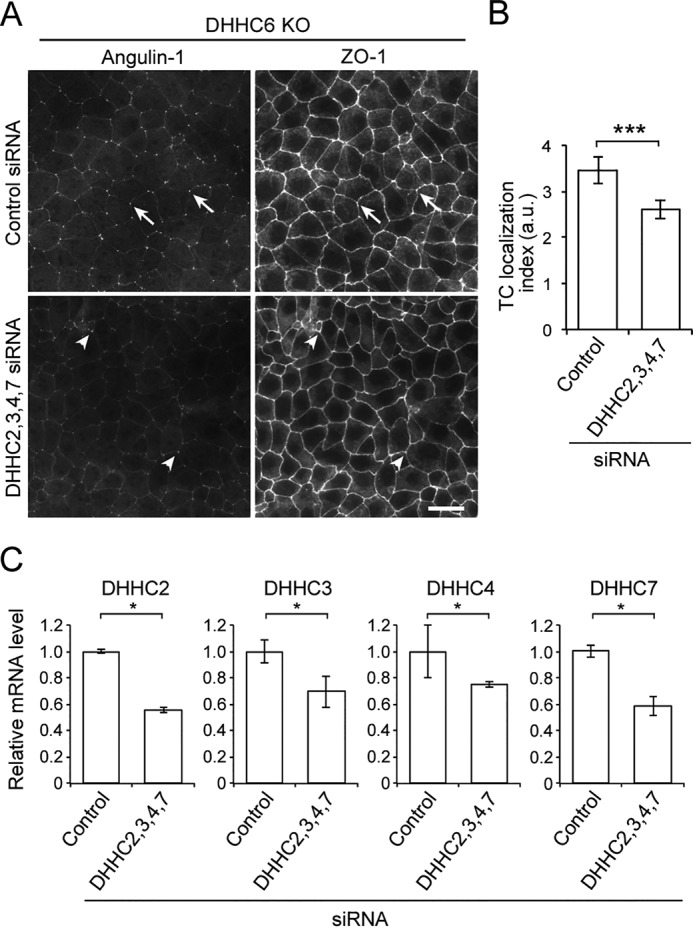
Role of DHHCs in the localization angulin-1 at TCs. A, immunofluorescence staining of DHHC6 KO cells treated with control siRNA or the mixture of DHHC2, -3, -4, -7 siRNAs with anti-angulin-1 and anti-ZO-1 antibodies. Compared with clear localization of angulin-1 at TCs in control siRNA-treated cells (arrows), the angulin-1 signal at TCs in DHHC2, -3, -4, -7 siRNA–treated cells was faint overall, although part of the TCs showed significant localization of angulin-1 (arrowheads). Scale bar, 20 μm. B, quantitation of the TC enrichment of angulin-1 in control and DHHC2, -3, -4, and -7 siRNA–treated DHHC6 KO cells. The graph represents mean ± S.D. (error bars) (n = 6 each). a.u., arbitary units. C, relative mRNA expression level of DHHC2, -3, -4, or -7 in DHHC6 KO cells treated with control siRNA or siRNAs for DHHC2, -3, -4, and -7 was measured by qPCR. Ribosomal protein L5 expression was used for normalization. The graph represents mean ± S.D. (n = 3 each). *, p < 0.05; ***, p < 0.0005, compared by t test.
It was reported that palmitoylation often regulates the incorporation of proteins into membrane domains, such as lipid rafts, wherein sphingolipids and cholesterol are highly enriched (18, 29). Our observations suggested that TCs may be specialized lipid microdomains in the plasma membrane that attract the highly palmitoylated angulin-1. To examine this notion, we disrupted the lipid domain of EpH4 cells by treatment with methyl-β-cyclodextrin (MβCD), which extracts cholesterol from the plasma membrane (30), and analyzed the behavior of angulin-1. When EpH4 cells were treated with 15 mm MβCD at 37 °C for 60 min, the localization of angulin-1 at TCs was impaired, whereas the localizations of occludin and ZO-1 at TJs were hardly affected (Fig. 6, A and B). Claudin-3 staining at cell-cell contacts appeared to be weakened with increased staining in the lateral membrane domain but was still clearly detected (Fig. 6A). Tricellulin was retained at TCs following the loss of angulin-1 by MβCD treatment, although angulins were previously shown to recruit tricellulin to TCs (6) (Fig. 6, A and B). It could be possible that angulin is required for initial recruitment of tricellulin but not for its maintenance at TCs (see “Discussion”). Under these conditions, no remarkable differences in the protein levels of angulin-1, occludin, claudin-3, ZO-1, and tricellulin were detected by immunoblotting (Fig. 6C). To investigate whether the distribution of cholesterol in the plasma membrane is biased at TCs, EpH4 cells were labeled with filipin, a fluorescent dye of cholesterol binding. However, we could not observe remarkable enrichment of filipin at TCs (Fig. S2C). It should be noted that the lack of enrichment does not rule out the presence of cholesterol at TCs. These results suggest that TCs contain a specialized membrane domain mediated by cholesterol and that localization of angulin-1 is sensitive to disruption of this domain.
Figure 6.

Effect of cholesterol depletion on the localization of angulin-1. A, immunofluorescence staining of EpH4 cells (control) and cholesterol-depleted cells after MβCD treatment using antibodies against tTJ-associated proteins (angulin-1, tricellulin) and TJ-associated proteins (occludin, claudin-3, and ZO-1). Scale bar, 20 μm. B, quantitation of the TC enrichment of angulin-1 or tricellulin upon MβCD treatment. The graph represents mean ± S.D. (error bars) (n = 3–4 each). **, p < 0.005, compared by t test; n.s., not significant; a.u., arbitary units. C, immunoblotting of lysates from EpH4 cells (control) and cholesterol-depleted cells after MβCD treatment using antibodies against tTJ-associated proteins (angulin-1 and tricellulin) and TJ-associated proteins (occludin, claudin-3, and ZO-1). IB, immunoblotting.
The extracellular domain of angulin-1 is required for its localization at TCs
We further investigated whether the extracellular domain is involved in TC localization of angulin-1 in addition to the cytoplasmic palmitoylation. We previously described several splicing isoforms of mouse angulin-1 based on databases (7). Among them, we examined isoform 2 with 575 amino acids (Fig. 7A; GenBankTM AK146807) as a prototype for angulin-1/LSR and confirmed its localization at TCs after expression in EpH4 cells and angulin-1-knockdown EpH4 cells in our previous studies (6, 7) and in Ang1KO cells in the present study (Fig. 1E). In this study, we focused on isoform 1 of mouse angulin-1 (Fig. 7A; GenBankTM BC004672). Compared with isoform 2, isoform 1 has an additional 19-amino acid insertion in the extracellular domain just upstream of the plasma membrane, but its cytoplasmic domain is identical. To examine whether this difference in the extracellular domain affected the localization of angulin-1, we transfected expression vectors for FLAG-tagged isoforms 1 and 2 of angulin-1 into Ang1KO cells and established stable clones (Fig. 7B). Immunofluorescence analyses confirmed that FLAG-tagged isoform 2 was concentrated at TCs (Fig. 7C). In contrast, FLAG-tagged isoform 1 was localized throughout the lateral membrane, but not at TCs (Fig. 7, C and D). A palmitoylation assay by the APEGS method revealed that FLAG-tagged isoforms 1 and 2 were both highly palmitoylated to similar levels (Fig. 7E). These observations suggest that the palmitoylated cytoplasmic domain is not sufficient on its own and that the extracellular domain is also required for TC localization of angulin-1.
Figure 7.

Role of the extracellular domain of angulin-1 in its localization at TCs. A, exon organization in the two isoforms of mouse angulin-1. The squares with numbers indicate the exons in the angulin-1 isoforms. Compared with isoform 1, isoform 2, which is known to localize at TCs, lacks exon 4 in the extracellular region close to the transmembrane domain. Gray lines, protein domains; SP, signal peptide; Ig-like, immunoglobulin-like domain; TM, transmembrane domain. B, immunoblotting of lysates from Ang1KO cells (control) and cells stably expressing FLAG-tagged angulin-1 isoform 2 or isoform 1 with an angulin-1 antibody (top), anti-FLAG antibody (middle), and anti-α-tubulin antibody (bottom). C, double immunofluorescence staining of Ang1KO cells stably expressing FLAG-tagged angulin-1 isoform 2 or isoform 1 with an anti-FLAG antibody and anti-occludin antibody. Scale bar, 20 μm. D, quantitation of the TC enrichment of Angulin-1 isoform 2 or isoform 1 in Ang1KO cells. The graph represents mean ± S.D. (error bars) (n = 3 each). *, p < 0.05, compared by t test. a.u., arbitary units. E, palmitoylation assay of FLAG-tagged angulin-1 isoform 2 and isoform 1 by the APEGS method performed using the same protocol as in Fig. 2B. F, immunofluorescence staining of cocultured EpH4 cells expressing GFP and Ang1KO cells with anti-angulin-1 and anti-ZO-1 antibodies. Among the TCs formed by two EpH4 cells and one Ang1KO cell, the arrow indicates an angulin-1–positive TC, whereas the arrowheads indicate angulin-1–negative TCs. Scale bar, 10 μm. G, quantification of the ratios of angulin-1–positive TCs in TCs formed by three EpH4 cells, by two EpH4 cells and one Ang1KO cell, by one EpH4 cell and two Ang1KO cells, or by three Ang1KO cells in three experiments. The numbers of analyzed TCs for those formed by three EpH4 cells, by two EpH4 cells and one Ang1KO cell, by one EpH4 cell and two Ang1KO cells, and by three Ang1KO cells in experiments 1, 2, and 3 were 200/120/97/214, 85/113/84/214, and 173/171/124/88, respectively. Throughout the experiments, angulin-1 was negative in TCs formed by one EpH4 cell and two Ang1KO cells or formed by three Ang1KO cells.
To obtain further insights into the role of the extracellular domain of angulin-1 in its TC localization, we cocultured EpH4 cells and Ang1KO cells and analyzed the localization of endogenously expressed angulin-1 in EpH4 cells. GFP was introduced into EpH4 cells in advance to distinguish them from Ang1KO cells by fluorescence microscopy. In these coculture experiments, four types of TCs were generated: TCs with three EpH4 cells, with two EpH4 cells and one Ang1KO cell, with one EpH4 cell and two Ang1KO cells, and with three Ang1KO cells. The immunofluorescence staining revealed concentrated localization of angulin-1 in >90% of TCs formed by three EpH4 cells. Clear dotlike localization of angulin-1 was also observed in about 20% of TCs composed of two EpH4 cells and one Ang1KO cell (Fig. 7, F and G). However, no concentration of angulin-1 was detected in TCs composed of one EpH4 cell and two Ang1KO cells (Fig. 7, F and G). These results suggest that extracellular interactions of angulin-1 between two cells play a role in its accumulation at TCs.
Discussion
One of the intriguing questions about tTJs is how they are correctly formed at TCs, which are considerably limited areas of cell-cell contacts in epithelial cells. Clarification of the underlying mechanism for the TC localization of angulins would help to provide an answer for this issue, because angulins are positioned upstream in terms of TC localization among the known tTJ-associated proteins. Herein, we showed that angulin-1 is highly palmitoylated and that full palmitoylation is required for its TC localization. We further demonstrated that the extracellular domain of angulin-1 is also involved in its localization at TCs. Our observations indicate that the TC localization of angulin-1 is determined by the combination of multiple factors and imply that TCs are specialized membrane domains.
In the present study, we divided the cytoplasmic 12 cysteine residues within amino acids 210–232 of angulin-1 into four clusters of three cysteines. We then expressed angulin mutants in which all three cysteines in each cluster or in multiple clusters were substituted with serine residues and analyzed their palmitoylation levels. The CS1, CS2, CS3, and CS4 mutants, in which all three cysteine residues in the corresponding clusters were substituted with serine residues, showed reduced palmitoylation compared with WT angulin-1 but still exhibited relatively high palmitoylation at similar levels to one another, suggesting that all four cysteine clusters underwent palmitoylation. Judging from the broad band for WT angulin-1 observed with the APEGS method, many of the 12 cysteine residues appear to be stochastically palmitoylated, although we did not identify which residues were actually palmitoylated. Meanwhile, the palmitoylation levels in the CS1–3 and CS1–4 mutants were similar, indicating that the cysteine residues in cluster 4 were not palmitoylated in the CS1–3 mutant. Because the cysteine residues in cluster 4 are located more than 20 amino acids downstream from the end of the membrane-spanning domain, they may only be able to access the responsible DHHCs when cluster 1, 2, or 3 becomes attached to the membrane by palmitoylation, leading to recruitment of cluster 4 to the membrane. Consistent with this notion, it was recently reported that protein palmitoylation was not determined by sequence motifs around susceptible cysteine residues but occurred stochastically in cytoplasmic cysteines close to the membrane or in the membrane of the inner leaflet at depths of up to 8 Å (23). EpH4 cells express DHHC2, -3, -4, -6, and -7, which were able to palmitoylate angulin-1 in HEK293 cells in overexpression studies. siRNA-mediated knockdown of any of DHHC2, -3, -4, or -7 in DHHC6 KO cells did not impair the localization of angulin-1 at TCs, but simultaneous knockdown of all of DHHC2, -3, -4, and -7 in DHHC6 KO cells did, suggesting that multiple DHHCs regulate TC localization of angulin-1. The modest effect on the localization of angulin-1 at TCs in our experiments may reflect that only partial DHHC mRNA depletion was achieved in siRNA-mediated knockdown. This is in agreement with the previous studies that have shown that multiple DHHCs are often redundantly involved in the palmitoylation of a certain protein (18, 19). In addition, DHHCs show distinct subcellular localization depending on their subtype (18, 19). Which DHHC directly palmitoylates angulin-1, and at which cellular compartment this occurs, remain to be solved in future studies.
Blaskovic et al. (18) proposed four mechanistic consequences of membrane protein palmitoylation: alterations in conformation of transmembrane domains, association with specific membrane domains, controlled interactions with other proteins, and controlled interplay with other post-translational modifications. In our study, the localization of angulin-1 at TCs was impaired in EpH4 cells after treatment with MβCD, which extracts cholesterol from the plasma membrane (30). This observation suggests that TCs are cholesterol-mediated membrane domains that attract the highly palmitoylated angulin-1. The high curvature of the membrane convex to the extracellular side in TCs may be involved in the formation of specific membrane domains with a unique lipid composition. Among the TJ-associated proteins, claudins, including claudin-3 and claudin-14, were reported to undergo palmitoylation in their cytoplasmic turns and C-terminal cytoplasmic domains. The cysteine residues in these regions are conserved in other claudin subtypes (22). A claudin-14 mutant with alanine substitution of four cysteine residues in these regions was less well-localized to TJs and was found in lysosomes in cultured epithelial cells (22). Furthermore, Shigetomi et al. (31) reported that removal of cholesterol from the plasma membrane of EpH4 cells by treatment with 50 mm MβCD for 30 min impaired the assembly of claudin-3 and occludin in cell-cell contacts, suggesting that TJs are cholesterol-mediated membrane domains. In the present study, the TC localization of angulin-1 was hampered by treatment with 15 mm MβCD for 60 min, whereas the claudin-3 and occludin localization in the TJ area was less affected, suggesting that the membrane domains at TCs differ from those at TJs. After the MβCD treatment, tricellulin remained localized at TCs despite the loss of angulin-1. These results appear to contradict our previous finding that angulins recruited tricellulin to TCs (6, 7). However, it should be noted that the previous studies utilized stable knockdown of angulin-1, whereas the present study involves acute depletion of cholesterol from cells with established tTJs. Thus, it is reasonable to assume that whereas angulins are important for the recruitment of tricellulin to TCs, tricellulin localization at tTJs may be stabilized upon localization to the TCs due to its affinity with TJ strands at tTJs, resulting in their differential sensitivity to acute cholesterol depletion. Consistently, tricellulin has affinity for claudin-based TJ strands within the plasma membrane (32), and claudin-3 was still clearly localized at bicellular contacts under the condition of MβCD treatment in this study.
Our study demonstrated that full palmitoylation of the cytoplasmic domain of angulin-1 was not sufficient for its localization at TCs and that the extracellular domain of angulin-1 was also required. Angulin family proteins contain single immunoglobulin-like domains in their extracellular part (7). When overexpressed in mouse L fibroblasts, angulin-1 often assembled into cell-cell contacts weakly on both sides of the adjacent cells, suggesting that the extracellular domains of two angulin-1 molecules can weakly interact with one another in a homophilic manner (6). This notion was supported by our coculture experiments with EpH4 cells and Ang1KO cells: angulin-1 was able to assemble into TCs formed by two EpH4 cells and one Ang1KO cell, but not into those formed by one EpH4 cell and two Ang1KO cells. Thus, the question arises as to why angulin-1 requires both cytoplasmic palmitoylation and the extracellular domain for its TC localization. We speculate that the fully palmitoylated cytoplasmic domain of angulin-1 has weak affinity for cholesterol-mediated membrane domains at TCs based on their high curvature convex to the outside, but palmitoylation is not sufficient for recruitment of angulin-1 to TCs by itself. We also assume that the extracellular domains of angulin-1 molecules have weak homophilic interaction, but the affinity is too weak for angulin-1 proteins to assemble into conventional cell-cell contacts by themselves. Only when these two properties—the curvature-based formation of cholesterol-mediated membrane domains recognized by the palmitoylated cytoplasmic region of angulin-1 and the cell-cell contact recognized by the homophilic interaction of angulin-1 extracellular region—are combined may angulin-1 be able to undergo assembly. TCs are regions that fulfill these two geometric features: plasma membrane domains with high curvature convex to the outside and the ends of cell-cell contacts corresponding to the sides of polygonal epithelial cells. Thus, the combinatorial action of the curvature-dependent assembly of plasma membrane domains and the extracellular domain–mediated intercellular interaction may ensure the tricellular localization of angulin-1, a prerequisite for tTJ formation. The two determinants for TC localization of angulin-1 could be required for targeting of angulin-1 to TCs or its retention at these sites. One possible idea is that palmitoylation is required for the access of angulin-1 in TCs, whereas the trans-interaction between two extracellular domains stabilizes angulin-1 there.
It was reported that single knockdown of JNK1 or JNK2 as well as double knockdown of both JNK1 and JNK2 affect the behavior of angulin-1 in EpH4 cells: angulin-1 was localized in bicellular contacts and/or the cytoplasm as dots in addition to TCs (33). Moreover, an angulin-1 mutant with alanine substitution for serine 288, which is a JNK1 phosphorylation site, was clearly localized along apical cell-cell junctions (33). These observations suggest that phosphorylation also influences the localization of angulin-1. However, angulin-1 should have an intrinsic mechanism for TC localization other than phosphorylation of serine 288 because Ang1–258G, which does not contain serine 288, was localized at TCs, albeit at lower efficiency.
Similar to vertebrate tTJs, invertebrate epithelial cells have specialized cell-cell junctions at TCs, namely tricellular junctions (TCJs) (34, 35). Drosophila TCJs are associated with septate junctions, the functional counterpart of vertebrate TJs. Two types of membrane proteins, Anakonda and Gliotactin, have been identified as the molecular constituents of TCJs in Drosophila, and Anakonda recruits Gliotactin to TCs (36, 37). Unlike angulin-1, the expression of Anakonda in all three cells forming a TC is required for its TC localization, whereas the cytoplasmic domain is not essential (37). Based on these observations, it has been proposed that the interaction between the extracellular domains of the Anakonda molecules from all three cells arranged at a certain angle is required for Anakonda assembly at TCs (37). These findings imply that vertebrates and invertebrates have developed different mechanisms for recognition of TCs by membrane proteins during the evolution of occluding junctions.
Experimental procedures
Cells and antibodies
A mouse mammary epithelial cell line, EpH4, was kindly provided by Dr. Ernst Reichmann (University-Children's Hospital, Zurich, Switzerland). EpH4 cells and their derivatives were cultured in Dulbecco's modified Eagle's medium (DMEM) (Nissui Pharmaceutical Co. Ltd.) supplemented with 10% fetal bovine serum (Biowest). HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum. The rat anti-occludin mAb (MOC37), rat anti-angulin-1 mAb (MS), rat anti-tricellulin mAb, and mouse anti-ZO-1 mAb (T8754) were raised and characterized as described previously (5, 7, 38, 39). A rabbit anti-GFP polyclonal antibody (598, MBL), a mouse anti-α-tubulin mAb (14-4502-82, eBioscience), a mouse anti-FLAG mAb (014-22383, Wako), and a rabbit anti-tricellulin polyclonal antibody (488400, Thermo Fisher Scientific) were commercially obtained. To generate a rabbit anti-angulin-1 polyclonal antibody, partial cDNA of dog angulin-1 corresponding to its C-terminal 230 amino acids was amplified by PCR with primer set #1 in Table 1 using a cDNA library of MDCK II cells as a template. A silence mutation was introduced into the amplified DNA fragment with the primer pair 5′-CCACACCCCCGGGACCCCCACTACGATGAC-3′ and 5′-GTCATCGTAGTGGGGGTCCCGGGGGTGTGG-3′ to remove the internal BamHI site, and the obtained DNA was digested with BamHI and EcoRI and subcloned into pGEX-2T vector (GE Healthcare). The GST-dog angulin-1 fusion protein was generated in Escherichia coli BL21 and purified with GSH-Sepharose (GE Healthcare). Against this fusion protein as antigens, rabbit polyclonal antibodies were raised by Kiwa Laboratory Animals (Wakayama, Japan). For affinity purification of the antibody, the cDNA encoding the C-terminal 230 amino acids of dog angulin-1 was subcloned into the pMAL-c2x vector (New England Biolabs) to produce the MBP-dog angulin-1 fusion protein in E. coli. The fusion protein was conjugated to CNBr-activated Sepharose 4B (GE Healthcare) according to the manufacturer's instructions and was used for affinity purification of the antibody. To generate anti-mouse DHHC6 polyclonal antibodies, a GST fusion protein with amino acids 233–414 of mouse DHHC6 was produced in E. coli and purified with GSH-Sepharose, followed by immunization to rabbits by Keary Co. Ltd.
Table 1.
PCR primers used in this study
| Primer no. | Target DNA | Forward | Reverse |
|---|---|---|---|
| #1 | Angulin-1 (dog) | atggatccGGCGCCCCCAATGGCCGTGTAG | aatgaatTCAGACGATTAAACTCTCCCGAC |
| #2 | Angulin-1 | TGGTTTTCGGGCGGGGCCCTTGGAAGATTGGCTCTTTGTGGTCGTG | GGTAGGAGCTCAGGGGCTTCTGAGGTCCTGCCAAGGACAATGAGCTCTGCG |
| #3 | Angulin-1 | GCTCCCACCCTTGTGTACCTGGGCG | GGAGATCTTTAATCTGCACTCCCGC |
| #4 | DHHC6 | GGGGTCGACGTTTTCCCATCTTGTTTTCTTTAG | GGGGAATTCGGACAGAGGCTGATTAGGTAAAAACC |
| #5 | DHHC2 | ATGGCGCCCTCGGGCTCGGGCG | GTCTCGTTCTCCATAGTTAATGC |
| #6 | DHHC3 | CCGAGACATTGAGCGGAAACC | ACCACATACTGGTACGGGTCTG |
| #7 | DHHC4 | ATGGACTTCCTGGTTCTCTTCTTG | TTTCTCCTTTTTCTTGTAACTGG |
| #8 | DHHC6 | ATGGGCATATTCTGCTCAGTAATCAAG | CTATCTGTTCTTCTTCTCCCCCTCTG |
| #9 | DHHC7 | CCGGGACATCGAGCACCATCCTC | CCTTTCCTGGTCCTCATCTGCAG |
| #10 | DHHC15 | ATGAAAGAGATGAACATCTGTGG | GAGGTGAAGCCACAAAACACACAC |
| #11 | Ribosomal protein L5 | GAAGCACATCATGGGTCAGA | TACGCATCTTCATCTTCCTCCATT |
| #12 | DHHC2 | GAGCAGACATCCGCACAATTC | AAGCGTGAGAGACGAGGAAG |
| #13 | DHHC3 | CCAAACCTGCTTCTGCTCTG | GGCCATCCATAGCCTTTAAGAAC |
| #14 | DHHC4 | ACCGGTTCGACCATCACTG | AGCAGTCACGGTGGCTATG |
| #15 | DHHC7 | GCTCCTGTGCTACTGAGGTG | ACGCAGAGACAATCTGAGGC |
Construction of expression vectors for angulin-1
The expression vectors for the full-length mouse angulin-1 and Ang1–258G were described previously (6). The expression vector for Ang1–211G was generated by PCR amplification of the amino acids 1–211 of mouse angulin-1, which was subcloned into pCAGGS (40) with a C-terminal GFP tag. The expression vector for isoform 2 of angulin-1 with FLAG tag was described previously (7). To generate the expression vector for isoform 1 of mouse angulin-1 (GenBankTM BC004672), the isoform 1-specific exon was inserted into isoform 2 of mouse angulin-1 (7) by inverse PCR as follows. The whole of the expression vector for FLAG-tagged isoform 2 of mouse angulin-1 (7) was amplified using primer set #2 in Table 1. Both ends of the amplified vector were ligated with T4 polynucleotide kinase (2021A, Takara) and Ligation high version 2 (LGK-201, TOYOBO).
The expression vectors for cysteine-serine–substituted mutants of mouse angulin-1 were generated by site-directed mutagenesis using inverse PCR with corresponding primer sets and the expression vectors for the full-length mouse angulin-1.
Genome editing, transfection, and establishment of stable cell clones
Ang1KO cells were established from parental EpH4 cells using the CRISPR/Cas9 system. pSpCas9(BB)-2A-Puro(PX459) version 2.0 (62988, Addgene) was used to construct CRISPR/Cas9 vectors. The sense and antisense DNA strands of the targeting site were annealed with each other in KOD FX Neo buffer (TOYOBO). The following DNAs were used for sense and antisense DNA strands of angulin-1: 5′-CACCGACGGCCGCGATGGCGCCGG-3′, 5′-AAACCCGGCGCCATCGCGGCCGTC-3′. The DNA duplexes were ligated to pSpCas9(BB)-2A-Puro(PX459) V2.0 digested by BpiI (FD1014; Thermo Fisher Scientific). Parental EpH4 cells were transfected with the CRISPR/Cas9 vectors using Lipofectamine LTX with Plus Reagent (Thermo Fisher Scientific) and cloned by limiting dilution. The cloned cells were picked up and immunostained with anti-angulin-1 antibody to confirm the depletion of angulin-1. In the angulin-1–deficient cells, mutations in the targeting sites were confirmed by Sanger sequencing of genomic DNA amplified by PCR with primer set #3 in Table 1.
DHHC6 KO cells were established from EpH4 cells by the same protocol as Ang1KO cells except for cell cloning. Two oligonucleotides, 5′-CACCGTTAAGGAGACTGTGTCATTG-3′ (sense strand) and 5′-AAACCAATGACACAGTCTCCTTAAC-3′ (antisense strand), were annealed to generate a DNA fragment containing the targeting site in the mouse Zdhhc6 gene. EpH4 cells were transfected with the CRISPR/Cas9 vectors for Zdhhc6 with pGKpuro, and cell clones that were resistant in the culture medium containing 3 μg/ml puromycin (Sigma–Aldrich) were picked up. The clones lacking expression of DHHC6 were further selected by immunoblotting using anti-DHHC6 antibody. Mutations in the targeting site were confirmed by Sanger sequencing of genomic DNA amplified by PCR with primer set #4 in Table 1.
Transfection of the expression plasmid constructs for the full-length angulin-1 and its mutants with epitope tags into Ang1KO cells was performed by lipofection using Lipofectamine LTX with Plus Reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. Transfected cells were cultured in the medium containing 400 μg/ml G418. The G418-resistant cell colonies were picked up as stable clones and propagated.
Immunofluorescence microscopy
The whole procedure was performed at room temperature. Cells cultured on coverslips at confluent condition were fixed with 1% formaldehyde in PBS containing 1 mm CaCl2 for 15 min, permeabilized with 0.2% (w/v) Triton X-100 in PBS for 15 min, and washed with PBS three times. After blocking with 1% BSA in PBS for 30 min, the cells were incubated with a primary antibody for 30 min, followed by PBS wash three times and incubation with a secondary antibody for 30 min. After washing with PBS three times, the samples were embedded in FluorSave reagent (Calbiochem) and observed with fluorescent microscopes IX70 and IX71 (Olympus) equipped with a complementary metal-oxide semiconductor–cooled camera and a charge-coupled camera (ORCA-ER, Hamamatsu Photonics K.K.), respectively. To obtain Z-stacked sections, we used a confocal microscope (TCS-SPE, Leica Microsystems) equipped with an HCX PL APO ×63/1.40 numerical aperture objective. Images were processed using Fiji/ImageJ software 2.0.0-rc-65/1.51w (National Institutes of Health).
Immunoblotting
Cells were lysed with Laemmli SDS sample buffer supplemented with 100 mm DTT and boiled at 100 °C for 5 min. The proteins were separated by SDS-PAGE using a 10, 12.5, or 15% polyacrylamide gel. In immunoblotting for except the APEGS analysis, one-fortieth of cell lysates prepared from confluent cells in a 35-mm dish were loaded per lane. The proteins in the gels were transferred to a polyvinylidene difluoride membrane with 0.45-μm pore size (Millipore). The membrane was blocked with 5% skim milk in TBS supplemented with 0.1% Tween 20 (TBS-T) and incubated with primary antibodies diluted with 5% skim milk in TBS-T or immunoreaction enhancer solution of Can Get Signal® (NKB-101, TOYOBO) for 60 min at 37 °C. The bound primary antibodies were detected using horseradish peroxidase–linked secondary antibodies and enhanced chemiluminescence (ECL prime, GE Healthcare). Images were obtained using LAS3000 mini (Fujifilm) and processed with Fiji/ImageJ software 2.0.0-rc-65/1.51w (National Institutes of Health).
APEGS assay
The method was basically followed as described previously (28). The indicated EpH4 cells were homogenized with Buffer A (4% SDS, 5 mm EDTA, 50 μg/ml phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml pepstatin A in PBS). After sonication and centrifugation at 100,000 × g for 15 min, supernatant proteins (200 μg) were reduced with 25 mm tris-(2-carboxyethyl)phosphine (Thermo Fisher Scientific) for 1 h at 55 °C and alkylated with 50 mm N-ethylmaleimide (NEM; Wako) for 3 h at room temperature. After chloroform/methanol precipitation (CM ppt), resuspended proteins were incubated in Buffer H (1% SDS, 0.2% Triton X-100, 5 mm EDTA, 1 m hydroxylamine (NH2OH; Sigma-Aldrich), pH 7.0) for 1 h at 37 °C to cleave thioester bonds. As a negative control, 1 m Tris was used. After CM ppt, resuspended proteins (75 μg) were PEGylated with 20 mm maleimide-conjugated PEG of 2 kDa (mPEG-2k; NOF) for 1 h. As a negative control, 20 mm NEM was used. After CM ppt, protein precipitates were resuspended with SDS-sample buffer (0.8–1.0 mg/ml) and boiled at 100 °C for 3 min. Protein concentration was measured by BCA protein assay after individual steps.
Metabolic labeling with [3H]palmitate
Individual DHHC clones were transfected together with Ang1G into HEK293T cells. Transfected HEK293T cells were preincubated for 30 min in serum-free DMEM with fatty acid–free BSA (5 mg/ml; Sigma–Aldrich). Cells were metabolically labeled with 0.4 mCi/ml [3H]palmitic acid (PerkinElmer Life Sciences) for 4 h in the preincubation buffer. Then the whole-cell lysate was separated by SDS-PAGE for fluorography and immunoblotting as described previously (41).
RT-PCR
Total RNA was prepared from EpH4 cells with TRIzol® reagent (Thermo Fisher Scientific) according to the manufacturer's instruction. Using the total RNA, cDNA was synthesized with SuperScript® III reverse transcriptase (Thermo Fisher Scientific) according to the manufacturer's instructions. For negative control, the identical aliquots of total RNA were incubated in the incubation buffer without the reverse transcriptase. 0.5 μl of the reaction product was used as a template for PCR. Conventional PCR was performed using a DNA polymerase KOD-Plus version 2 (TOYOBO) according to the manufacturer's instructions in 35 cycles of denaturation at 98 °C for 10 s, annealing at 58 °C for 20 s, and extension at 68 °C for 90 s with primer sets #5, #6, #7, #8, #9, and #10 in Table 1 to amplify the cDNAs of mouse DHHC2, DHHC3, DHHC4, DHHC6, DHHC7, and DHHC15, respectively. After PCR, the samples were separated by gel electrophoresis with 1% agarose gel. The expected sizes of the amplified DNA fragments were as follows: DHHC2, 1097 bp; DHHC3, 871 bp; DHHC4, 1029 bp; DHHC6, 1242 bp; DHHC7, 882 bp; DHHC10, 1018 bp; DHHC15, 1011 bp. For DHHC2, -3, -4, -6, and -7, each amplified DNA fragment was confirmed to be the correct PCR product for the corresponding DHHC by direct sequencing.
Real-time qPCR
Total RNA was isolated from cells using TRIzol® reagent (Thermo Fisher Scientific) following the manufacturer's instructions. The residual DNA was removed by RQ1 DNase (Promega). First-strand cDNA was synthesized from 2 μg of total RNA with oligo(dT) primers using SuperScriptIII reverse transcriptase (Thermo Fisher Scientific). cDNA samples were diluted 3-fold for qPCR. Real-time qPCR was conducted using a Light Cycler 96® system (Roche Applied Sciences). For each reaction, a 20-μl mixture containing 2.5 μl of diluted cDNA, 0.25 μm each of forward and reverse primers, and 10 μl of Luna® Universal qPCR mix (New England Biolabs) was prepared. The amplification program was as follows: 95 °C for 60 s, 55 cycles of denaturation at 95 °C for 15 s, followed by annealing and amplification at 60 °C for 30 s. Reactions for individual samples were carried out in triplicate. Ribosomal protein L5 expression was used for normalization. -Fold changes were calculated using the ΔΔCt method. The primer sets #11, #12, #13, #14, and #15 in Table 1 were used for the real-time qPCRs to amplify the cDNAs of ribosomal protein L5, DHHC2, DHHC3, DHHC4, and DHHC7, respectively.
Co-culture experiments
For co-culture experiments, EpH4 cells expressing GFP and Ang1 KO cells (1.5 × 105 cells each) were mixed in a 35-mm dish with coverslips and incubated for 3 days. The cells were fixed and immunostained with anti-GFP antibody, anti-angulin-1 antibody, and anti-ZO-1 antibody. Angulin-1–positive dots were counted at TCs composed of three EpH4 cells, of two EpH4 cells and one Ang1 KO cell, of one EpH4 cell and two Ang1 KO cells, or of three Ang1 KO cells. When mean fluorescence intensity of angulin-1 at a TC composed of two EpH4 cells and one Ang1 KO cell or of one EpH4 cell and two Ang1 KO cells was more than two-thirds or one-third of the mean fluorescence intensity of angulin-1 at TCs composed of three EpH4 cells, respectively, tricellular localization of angulin-1 was considered to be positive.
Image analysis
TC enrichment was quantified. The overall workflow was as follows: skeletonization of TJs, definition of vertices using the skeletonized image to generate a mask, and measurement of the ratio of the intensity of vertices versus total intensity. First, the vertices were defined using the images of counterstaining by occludin or ZO-1. In brief, noise reduction was performed by ImageJ by sequential application of the subtraction of background, median filter, and band-pass filter, and the junctional signals were extracted by thresholding. After removing outliers, the image was binarized and skeletonized. Next, to extract the vertices, an averaging filter was applied to the skeletonized image, followed by thresholding. The vertices were expanded by dilation to generate a mask for tricellular TJs. Misannotated vertices were manually removed. Finally, tricellular TJ signals were extracted from the angulin/tricellulin channel using the mask by the image calculator. After background subtraction, the mean intensity and area fraction were measured from the original image and the tricellular TJ image, and the signal density was determined by dividing the mean intensity with area fraction. The tricellular enrichment index was defined as the ratio between tricellular TJ signal density and total signal density. Image analysis was performed by Fiji/ImageJ 1.52f software, followed by analysis in Excel (Microsoft). Statistical significance was evaluated by Student's t test (two-tailed).
Author contributions
Y. O., Y. F., Y. I., T. H., M. Fukata, and M. Furuse conceptualization; Y. O., T. S., Y. F., Y. I., T. O., T. H., M. Fukata, and M. Furuse investigation; Y. O., T. S., Y. F., Y. I., and M. Fukata methodology; Y. O., T. S., Y. F., Y. I., T. O., T. H., M. Fukata, and M. Furuse writing-original draft; T. S., T. H., and M. Furuse data curation; T. S. formal analysis; Y. I., T. O., T. H., and M. Fukata resources; Y. I., T. H., M. Fukata, and M. Furuse supervision; Y. I., T. H., M. Fukata, and M. Furuse validation; T. O., T. H., and M. Furuse writing-review and editing; T. H. and M. Furuse funding acquisition; T. H. and M. Furuse visualization; T. H. and M. Furuse project administration.
Supplementary Material
Acknowledgments
We thank Hidekazu Hiroaki for providing a key idea for the experimental design; Mie Kubo and Mika Watanabe for excellent technical assistance; and Shigenobu Yonemura, Akira Nagafuchi, and all of the members of the Furuse laboratory for helpful discussions. We also thank the staff at the Center for Radioisotope Facilities at National Institutes for Basic Biology. Finally, we thank Alison Sherwin from the Edanz Group for editing a draft of the manuscript.
This work was supported in part by MEXT Program Grant LS084 (to M. Furuse), Grants-in-Aid for Scientific Research (B) 26291043 and 18H02440 (to M. Furuse) from the Japan Society for the Promotion of Science (JSPS), and a Life Science Aid from the Takeda Science Foundation (to M. Furuse). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2.
- TJ
- tight junction; tricellular contact
- TC
- tricellular contact
- tTJ
- tricellular tight junction
- DHHC
- Asp-His-His-Cys motif–containing palmitoyltransferase
- APEGS
- acyl-PEGyl exchange gel shift
- MβCD
- methyl-β-cyclodextrin
- TCJ
- tricellular junction
- DMEM
- Dulbecco's modified Eagle's medium
- MDCK
- Madin–Darby canine kidney
- NEM
- N-ethylmaleimide
- CM ppt
- chloroform/methanol precipitation
- mPEG-2k
- maleimide-conjugated PEG of 2 kDa
- qPCR
- quantitative PCR
- HA
- influenza hemagglutinin.
References
- 1. Tsukita S., Furuse M., and Itoh M. (2001) Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2, 285–293 10.1038/35067088 [DOI] [PubMed] [Google Scholar]
- 2. Zihni C., Mills C., Matter K., and Balda M. S. (2016) Tight junctions: from simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 17, 564–580 10.1038/nrm.2016.80 [DOI] [PubMed] [Google Scholar]
- 3. Furuse M., Sasaki H., and Tsukita S. (1999) Manner of interaction of heterogeneous claudin species within and between tight junction strands. J. Cell Biol. 147, 891–903 10.1083/jcb.147.4.891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Staehelin L. A. (1973) Further observations on the fine structure of freeze-cleaved tight junctions. J. Cell Sci. 13, 763–786 [DOI] [PubMed] [Google Scholar]
- 5. Ikenouchi J., Furuse M., Furuse K., Sasaki H., Tsukita S., and Tsukita S. (2005) Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J. Cell Biol. 171, 939–945 10.1083/jcb.200510043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Masuda S., Oda Y., Sasaki H., Ikenouchi J., Higashi T., Akashi M., Nishi E., and Furuse M. (2011) LSR defines cell corners for tricellular tight junction formation in epithelial cells. J. Cell Sci. 124, 548–555 10.1242/jcs.072058 [DOI] [PubMed] [Google Scholar]
- 7. Higashi T., Tokuda S., Kitajiri S., Masuda S., Nakamura H., Oda Y., and Furuse M. (2013) Analysis of the “angulin” proteins LSR, ILDR1 and ILDR2—tricellulin recruitment, epithelial barrier function and implication in deafness pathogenesis. J. Cell Sci. 126, 966–977 10.1242/jcs.116442 [DOI] [PubMed] [Google Scholar]
- 8. Riazuddin S., Ahmed Z. M., Fanning A. S., Lagziel A., Kitajiri S., Ramzan K., Khan S. N., Chattaraj P., Friedman P. L., Anderson J. M., Belyantseva I. A., Forge A., Riazuddin S., and Friedman T. B. (2006) Tricellulin Is a tight-junction protein necessary for hearing. Am. J. Hum. Genet. 79, 1040–1051 10.1086/510022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borck G., Ur Rehman A., Lee K., Pogoda H. M., Kakar N., von Ameln S., Grillet N., Hildebrand M. S., Ahmed Z. M., Nürnberg G., Ansar M., Basit S., Javed Q., Morell R. J., Nasreen N., et al. (2011) Loss-of-function mutations of ILDR1 cause autosomal-recessive hearing impairment DFNB42. Am. J. Hum. Genet. 88, 127–137 10.1016/j.ajhg.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nayak G., Lee S. I., Yousaf R., Edelmann S. E., Trincot C., Van Itallie C. M., Sinha G. P., Rafeeq M., Jones S. M., Belyantseva I. A., Anderson J. M., Forge A., Frolenkov G. I., and Riazuddin S. (2013) Tricellulin deficiency affects tight junction architecture and cochlear hair cells. J. Clin. Invest. 123, 4036–4049 10.1172/JCI69031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kamitani T., Sakaguchi H., Tamura A., Miyashita T., Yamazaki Y., Tokumasu R., Inamoto R., Matsubara A., Mori N., Hisa Y., and Tsukita S. (2015) Deletion of tricellulin causes progressive hearing loss associated with degeneration of cochlear hair cells. Sci. Rep. 5, 18402 10.1038/srep18402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morozko E. L., Nishio A., Ingham N. J., Chandra R., Fitzgerald T., Martelletti E., Borck G., Wilson E., Riordan G. P., Wangemann P., Forge A., Steel K. P., Liddle R. A., Friedman T. B., and Belyantseva I. A. (2015) ILDR1 null mice, a model of human deafness DFNB42, show structural aberrations of tricellular tight junctions and degeneration of auditory hair cells. Hum. Mol. Genet. 24, 609–624 10.1093/hmg/ddu474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Higashi T., Katsuno T., Kitajiri S., and Furuse M. (2015) Deficiency of angulin-2/ILDR1, a tricellular tight junction-associated membrane protein, causes deafness with cochlear hair cell degeneration in mice. PLoS ONE 10, e0120674 10.1371/journal.pone.0120674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sang Q., Li W., Xu Y., Qu R., Xu Z., Feng R., Jin L., He L., Li H., and Wang L. (2015) ILDR1 deficiency causes degeneration of cochlear outer hair cells and disrupts the structure of the organ of Corti: a mouse model for human DFNB42. Biol. Open 4, 411–418 10.1242/bio.201410876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gong Y., Himmerkus N., Sunq A., Milatz S., Merkel C., Bleich M., and Hou J. (2017) ILDR1 is important for paracellular water transport and urine concentration mechanism. Proc. Natl. Acad. Sci. U.S.A. 114, 5271–5276 10.1073/pnas.1701006114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mesli S., Javorschi S., Bérard A. M., Landry M., Priddle H., Kivlichan D., Smith A. J. H., Yen F. T., Bihain B. E., and Darmon M. (2004) Distribution of the lipolysis stimulated receptor in adult and embryonic murine tissues and lethality of LSR−/− embryos at 12.5 to 14.5 days of gestation. Eur. J. Biochem. 271, 3103–3114 10.1111/j.1432-1033.2004.04223.x [DOI] [PubMed] [Google Scholar]
- 17. Oda Y., Otani T., Ikenouchi J., and Furuse M. (2014) Tricellulin regulates junctional tension of epithelial cells at tricellular contacts through Cdc42. J. Cell Sci. 127, 4201–4212 10.1242/jcs.150607 [DOI] [PubMed] [Google Scholar]
- 18. Blaskovic S., Blanc M., and van der Goot F. G. (2013) What does S-palmitoylation do to membrane proteins? FEBS J. 280, 2766–2774 10.1111/febs.12263 [DOI] [PubMed] [Google Scholar]
- 19. Fukata Y., and Fukata M. (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 11, 161–175 10.1038/nrn2788 [DOI] [PubMed] [Google Scholar]
- 20. Linder M. E., and Deschenes R. J. (2007) Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 8, 74–84 10.1038/nrm2084 [DOI] [PubMed] [Google Scholar]
- 21. Chamberlain L. H., and Shipston M. J. (2015) The physiology of protein S- acylation. Physiol. Rev. 95, 341–376 10.1152/physrev.00032.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Itallie C. M., Gambling T. M., Carson J. L., Anderson J. M. (2005) Palmitoylation of claudins is required for efficient tight-junction localization. J. Cell Sci. 118, 1427–1436 10.1242/jcs.01735 [DOI] [PubMed] [Google Scholar]
- 23. Rodenburg R. N. P., Snijder J., van de Waterbeemd M., Schouten A., Granneman J., Heck A. J. R., and Gros P. (2017) Stochastic palmitoylation of accessible cysteines in membrane proteins revealed by native mass spectrometry. Nat. Commun. 8, 1280 10.1038/s41467-017-01461-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rajagopal N., Irudayanathan F. J., and Nangia S. (2019) Palmitoylation of claudin-5 proteins influences their lipid domain affinity and tight junction assembly at the blood-brain barrier interface. J. Phys. Chem. B 123, 983–993 10.1021/acs.jpca.8b10224, 10.1021/acs.jpcb.8b09535 [DOI] [PubMed] [Google Scholar]
- 25. Heiler S., Mu W., Zöller M., and Thuma F. (2015) The importance of claudin-7 palmitoylation on membrane subdomain localization and metastasis-promoting activities. Cell Commun. Signal. 13, 29 10.1186/s12964-015-0105-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aramsangtienchai P., Spiegelman N. A., Cao J., and Lin H. (2017) S-Palmitoylation of junctional adhesion molecule C regulates its tight junction localization and cell migration. J. Biol. Chem. 292, 5325–5334 10.1074/jbc.M116.730523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krogh A., Larsson B., von Heijne G., and Sonnhammer E. L. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 28. Yokoi N., Fukata Y., Sekiya A., Murakami T., Kobayashi K., and Fukata M. (2016) Identification of PSD-95 depalmitoylating enzymes. J. Neurosci. 36, 6431–6444 10.1523/JNEUROSCI.0419-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levental I., Lingwood D., Grzybek M., Coskun U., and Simons K. (2010) Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. U.S.A. 107, 22050–22054 10.1073/pnas.1016184107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zidovetzki R., and Levitan I. (2007) Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim. Biophys. Acta 1768, 1311–1324 10.1016/j.bbamem.2007.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shigetomi K., Ono Y., Inai T., and Ikenouchi J. (2018) Adherens junctions influence tight junction formation via changes in membrane lipid composition. J. Cell Biol. 217, 2373–2381 10.1083/jcb.201711042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ikenouchi J., Sasaki H., Tsukita S., Furuse M., and Tsukita S. (2008) Loss of occludin affects tricellular localization of tricellulin. Mol. Biol. Cell 19, 4687–4693 10.1091/mbc.e08-05-0530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakatsu D., Kano F., Taguchi Y., Sugawara T., Nishizono T., Nishikawa K., Oda Y., Furuse M., and Murata M. (2014) JNK1/2-dependent phosphorylation of angulin-1/LSR is required for the exclusive localization of angulin-1/LSR and tricellulin at tricellular contacts in EpH4 epithelial sheet. Genes Cells 19, 565–581 10.1111/gtc.12158 [DOI] [PubMed] [Google Scholar]
- 34. Noirot-Timothée C., Graf F., and Noirot C. (1982) The specialization of septate junctions in regions of tricellular junctions. II. Pleated septate junctions. J. Ultrastruct. Res. 78, 152–165 10.1016/S0022-5320(82)80020-8 [DOI] [PubMed] [Google Scholar]
- 35. Graf F., Noirot-Timothée C., and Noirot C. (1982) The specialization of septate junctions in regions of tricellular junctions. I. Smooth septate junctions (=Continuous junctions). J. Ultrastruct. Res. 78, 136–151 10.1016/S0022-5320(82)80019-1 [DOI] [PubMed] [Google Scholar]
- 36. Schulte J., Tepass U., and Auld V. J. (2003) Gliotactin, a novel marker of tricellular junctions, is necessary for septate junction development in Drosophila. J. Cell Biol. 161, 991–1000 10.1083/jcb.200303192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Byri S., Misra T., Syed Z. A., Bätz T., Shah J., Boril L., Glashauser J., Aegerter-Wilmsen T., Matzat T., Moussian B., Uv A., and Luschnig S. (2015) The triple-repeat protein Anakonda controls epithelial tricellular junction formation in Drosophila. Dev. Cell 33, 535–548 10.1016/j.devcel.2015.03.023 [DOI] [PubMed] [Google Scholar]
- 38. Saitou M., Ando-Akatsuka Y., Itoh M., Furuse M., Inazawa J., Fujimoto K., and Tsukita S. (1997) Mammalian occludin in epithelial cells: its expression and subcellular distribution. Eur. J. Cell Biol. 73, 222–231 [PubMed] [Google Scholar]
- 39. Itoh M., Yonemura S., Nagafuchi A., Tsukita S., and Tsukita S. (1991) A 220-kD undercoat-constitutive protein: its specific localization at cadherin-based cell-cell adhesion sites. J. Cell Biol. 115, 1449–1462 10.1083/jcb.115.5.1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Niwa H., Yamamura K., and Miyazaki J. (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 10.1016/0378-1119(91)90434-D [DOI] [PubMed] [Google Scholar]
- 41. Fukata M., Fukata Y., Adesnik H., Nicoll R. A., and Bredt D. S. (2004) Identification of PSD-95 palmitoylating enzymes. Neuron 44, 987–996 10.1016/j.neuron.2004.12.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.