

Abstract
Expansions of simple tandem repeats are responsible for almost 50 human diseases, the majority of which are severe, degenerative, and not currently treatable or preventable. In this review, we first describe the molecular mechanisms of repeat-induced toxicity, which is the connecting link between repeat expansions and pathology. We then survey alternative DNA structures that are formed by expandable repeats and review the evidence that formation of these structures is at the core of repeat instability. Next, we describe the consequences of the presence of long structure-forming repeats at the molecular level: somatic and intergenerational instability, fragility, and repeat-induced mutagenesis. We discuss the reasons for gender bias in intergenerational repeat instability and the tissue specificity of somatic repeat instability. We also review the known pathways in which DNA replication, transcription, DNA repair, and chromatin state interact and thereby promote repeat instability. We then discuss possible reasons for the persistence of disease-causing DNA repeats in the genome. We describe evidence suggesting that these repeats are a payoff for the advantages of having abundant simple-sequence repeats for eukaryotic genome function and evolvability. Finally, we discuss two unresolved fundamental questions: (i) why does repeat behavior differ between model systems and human pedigrees, and (ii) can we use current knowledge on repeat instability mechanisms to cure repeat expansion diseases?
Keywords: trinucleotide repeat disease, DNA replication, DNA repair, DNA recombination, genomic instability, gene expression, DNA structure, G-quadruplex, R-loop, S-DNA, triplex H-DNA, Huntington disease, amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease), hairpin
Introduction
In 1905, British ophthalmologist Edward Nettleship made a peculiar observation: children that suffered from certain degenerative genetic diseases exhibited pathological symptoms earlier than their parents. Nettleship termed this phenomenon “genetic anticipation” (1). This concept was further substantiated by Bruno Fleischer in his genetic studies of myotonic dystrophy (DM)2 with cataracts (2).
For most of the 20th century, the idea of genetic anticipation was highly controversial due to its adoption, misinterpretation, and abuse by eugenicists. Most notably, Nazi officials used the concept to justify the sterilization of people who exhibited mild symptoms of mental disorders, believing that these symptoms were harbingers of severe psychopathology in later generations (3).
The post-war political backlash against eugenics spelled the near demise for genetic anticipation as a serious scientific concept. The scientific community firmly rejected the existence of non-Mendelian inheritance of this type. Nonetheless, a growing body of evidence began to show that patterns of inheritance in certain types of human diseases, such as DM or Huntington's disease (HD) are hard to explain using conventional genetics (3). Furthermore, in 1985, Stephany Sherman convincingly demonstrated that penetrance and expressivity of fragile X syndrome alleles increase as the disease alleles pass through generations. This phenomenon became known as the Sherman paradox (4).
In 1991, two disease-causing genes were cloned: the fragile X gene FMR1 in the laboratories of Stephen Warren, Robert Richards, and Jean-Louis Mandel (5–9) and the X-linked spinal and bulbar muscular atrophy (SBMA) gene AR in the laboratory of Kenneth Fischbeck (10). These feats revealed the molecular basis of the genetic anticipation. It turned out that fragile X syndrome results from an expansion of the CGG repeats in the 5′-untranslated region (UTR) of the FMR1 gene (5–9). The number of CGG repeats expands during parental transmission, leading to increased disease severity. These observations resolved Sherman's paradox and provided a molecular explanation for genetic anticipation. Similarly to fragile X, SBMA was found to be caused by an expansion of CAG repeats in the coding region of the AR gene (10). These discoveries were rapidly followed by the recognition of repeat expansion as the cause of DM1 (11) and HD (12). In the blink of an eye, the infamous concept of genetic anticipation was reestablished as a valid scientific phenomenon (3).
Today, we know 13 different types of tandem repeats whose expansions cause various human diseases (Table 1). The majority of these repeat expansion diseases (REDs) are neither curable nor preventable at present. The most common cause of REDs is an expansion of CAG repeats (or complementary CTG repeats), which are responsible for 16 conditions, including HD and multiple spinocerebellar ataxias (SCAs). CGG repeat expansions cause six different conditions, including fragile X syndrome. Next, two disorders are caused by GAA repeat expansion. In addition to the expansions of these trinucleotide repeats, expansions of one tetranucleotide (CCTG), five pentanucleotide (ATTCT, TGGAA, TTTTA, TTTCA, and AAGGG), three hexanucleotide (GGCCTG, CCCTCT, and GGGGCC), and one dodecanucleotide (CCCCGCCCCGCG) repeat cause 13 other diseases. A separate class of REDs, so-called polyalanine (poly(A)) diseases, are caused by an in-frame expansion of imperfect GCN repeats. This expansion results in abnormally long stretches of alanine in the corresponding proteins. Altogether, nearly 50 REDs are currently known. Beyond these monogenic diseases, some specific repeat expansions might contribute to the etiology of various complex polygenic psychiatric and brain disorders, such as autism spectrum disorder (13), bipolar spectrum disorders, schizophrenia, and others (reviewed in Ref. 14). The number of known REDs will likely grow, as more than 100 human genes contain DNA repeats that are known to expand in some REDs. These repeats, therefore, can a priori expand, causing a disease (database of genes related to REDs RRID:SCR_018086).
Table 1.
Currently known REDs
| Repeat unit | Gene and position of the repeat in the gene | Disease | Year of discovery | Highlight paper | Inheritance pattern | Healthy range, no. of repeats | Symptomatic range, no. of repeats |
|---|---|---|---|---|---|---|---|
| ATTCT | ATXN10, intron | Spinocerebellar ataxia 10 (SCA10) | 2000 | 535 | Autosomal dominant | 10–22 | 500–4500 |
| DAB1; intron | Spinocerebellar ataxia 37 (SCA37) | 2017 | 22 | Autosomal dominant | <30 | 46–71 | |
| CAG | ATXN1, coding sequence | Spinocerebellar ataxia1 (SCA1) | 1993 | 536 | Autosomal dominant | 6–39 | 41–83 |
| ATXN2, coding sequence | Spinocerebellar ataxia2 (SCA2) | 1996 | 21, 537 | Autosomal dominant | <31 | 33–200 | |
| ATXN3, coding sequence | Spinocerebellar ataxia3 (SCA3) | 1994 | 18 | Autosomal dominant | <44 | 52–86 | |
| CACNA1A, coding sequence | Spinocerebellar ataxia6 (SCA6) | 1997 | 538 | Autosomal dominant | <18 | 20–33 | |
| TPB, coding sequence | Spinocerebellar ataxia 17 (SCA17) | 2001 | 539 | Autosomal dominant | 25–44 | 47–63 | |
| ATN1, coding sequence | Dentatorubral-pallidoluysian atrophy, Naito-Oyanagi disease (DRPLA) | 1994 | 29, 30 | Autosomal dominant | 6–35 | 49–88 | |
| HTT, coding sequence | Huntington disease (HD) | 1993 | 12 | Autosomal dominant | 9–29 | 36–121 | |
| ATXN8, coding sequence | Spinocerebellar ataxia8 (SCA8) | 1999 | 114 | Autosomal dominant | 15–50 | 71–1300 | |
| PPP2R2B, 5′ UTR | Spinocerebellar ataxia 12 (SCA12) | 1999 | 540 | Autosomal dominant | 7–32 | 51–78 | |
| ATXN7, coding sequence | Spinocerebellar ataxia7 (SCA7) | 1997 | 17 | Autosomal dominant | 7–17 | 38–130 | |
| AR, coding sequence | Spinal and bulbar muscular atrophy (SBMA) | 1991 | 10 | X-linked recessive | <34 | >38 | |
| GLS, 5′-UTR | Glutaminase deficiency (GD) | 2019 | 470 | Autosomal recessive | 8–16 | 400–1500 | |
| CCCCGCCCCGCG | CSTB, 5′-UTR | Progressive myoclonus epilepsy of the Unverricht--Lundborg type (EPM1) | 1997 | 541 | Autosomal recessive | 2–3 | 38–77 |
| CCCTCT | TAF1, intron | X-linked dystonia parkinsonism (XDP) | 2019 | 31 | X-linked recessive | N/A | 30–60 |
| CCTG | CNBP, intron | Myotonic mystrophy2 (DM2) | 2001 | 54 | Autosomal dominant | <30 | 75–11,000 |
| CTG | DM1, 3′-UTR | Myotonic mystrophy1 (DM1) | 1992 | 11 | Autosomal dominant | 5–37 | 50–5,000 |
| JPH3, 3′-UTR | Huntington disease-like 2 | 2001 | 542 | Autosomal dominant | 6–28 | >41 | |
| ATXN8, intron | Spinocerebellar ataxia8 (SCA8) | 1999 | 114 | Autosomal dominant | 15–34 | 90–250 | |
| TCF4, intron | Fuchs endothelial corneal dystrophy | 2012 | 543 | Autosomal dominant | <40 | >50 | |
| GAA | DMD, intron | Duchenne muscular dystrophy (DMD) | 2016 | 64 | X-linked recessive | 11–33 | 59–82 |
| FXN, intron | Friedreich ataxia 1 (FRDA) | 1996 | 62 | Autosomal recessive | 5–30 | >70 | |
| CGG | XYLT1, 5′-UTR | Baratela--Scott syndrome (BSS) | 2019 | 544 | Autosomal recessive | 9–20 | 100–800 |
| FMR2, 5′-UTR | Mental retardation, X-linked, associated with fragile site FRAXE | 1993 | 545 | X-linked recessive | 4–39 | >200 | |
| DIP2B, 5′-UTR | Mental retardation, associated with fragile site FRA12A | 2007 | 546 | Autosomal dominant | 12–26 | >150 | |
| FMR1, 5′-UTR | Fragile X mental retardation syndrome | 1991 | 5–9 | X-linked dominant | 6–52 | 231–2000 | |
| CBL, 5′-UTR | Jacobsen syndrome | 1998 | 547 | Not inherited | 11 | >100 | |
| NOTCH2NLC | Neuronal intranuclear inclusion disease (NIID) | 2019 | 380 | Autosomal dominant | 13–30 | 60–959 | |
| GCN (polyAla) | RUNX2, coding sequence | Cleidocranial dysplasia | 1997 | 42 | Autosomal dominant | 17 | 27 |
| SOX3, coding sequence | Mental retardation, X-linked | 2002 | 43 | X-linked recessive | 11 | 15–26 | |
| PRDM12, coding sequence | Congenital insensitivity to pain (CIP) | 2015 | 44 | Autosomal recessive | 12 | 18–19 | |
| PABPN1, coding sequence | Oculopharyngeal muscular dystrophy | 1998 | 45 | Autosomal dominant | 6 | 7–17 | |
| HOXD13, coding sequence | Synpolydactyly 1 | 1996 | 46, 47 | Autosomal dominant | 15 | 22–29 | |
| HOXA13, coding sequence | Hand-foot-genital (HFG) syndrome | 2000 | 48 | Autosomal dominant | 18 | 24–26 | |
| ARX, coding sequence | Epileptic encephalopathy, early infantile, 1 | 2007 | 49, 50 | X-linked recessive | 10–16 | 17–23 | |
| PHOX2B, coding sequence | Central hypoventilation syndrome, congenital | 2003 | 51 | Autosomal dominant | 20 | 24–33 | |
| ZIC2, coding sequence | Holoprosencephaly 5 | 2001 | 52 | Autosomal dominant | 15 | 25 | |
| FOXL2, coding sequence | Blepharophimosis, ptosis, and epicanthus inversus syndrome | 2001 | 53 | Autosomal dominant | 14 | 22–24 | |
| GGCCTG | NOP56, intron | Spinocerebellar ataxia 36 (SCA36) | 2011 | 93 | Autosomal dominant | 3–14 | 650–2500 |
| GGGGCC | C9orf72, intron | Amyotrophic lateral sclerosis and frontotemporal degeneration (ALS/FTD) | 2011 | 37, 100 | Autosomal dominant | 2–19 | 250–1600 |
| TGGAA | BEAN1, intron | Spinocerebellar ataxia 31 (SCA31) | 2009 | 548 | Autosomal dominant | 26 | 500–760 |
| TTTCA/TTTTA | SAMD12, intron | Familial adult myoclonic epilepsy1 (FAME1) | 2018 | 67, 68 | Autosomal dominant | 7–20 | 440–3680 |
| TNRC6A, intron | Familial adult myoclonic epilepsy6 (FAME6) | 2018 | 67, 68 | Autosomal dominant | 18 | >22 | |
| RAPGEF2, intron | Familial adult myoclonic epilepsy7 (FAME7) | 2018 | 67, 68 | Autosomal dominant | 18 | >22 | |
| MARCH6, intron | Familial adult myoclonic epilepsy3 (FAME3) | 2019 | 32 | Autosomal dominant | 9–20 | 791–1035 | |
| STARD7, intron | Familial adult myoclonic epilepsy2 (FAME2) | 2019 | 69 | Autosomal dominant | 5–35 | 40–1000 | |
| AAGGG | RFC1, intron | Cerebellar ataxia, neuropathy and vestibular areflexia syndrome | 2019 | 291 | Autosomal recessive | 11 | 400–2000 |
The majority of REDs share two common features. First, the number of inherited repeats typically positively correlates with disease severity and negatively correlates with age of onset. Diseases with strong correlations between the number of repeats and age of onset are SCA7 (16, 17), SCA3 (18), SCA2 (19–21), SCA37 (22), HD (23–26), DM1 (27, 28), dentatorubral-pallidoluysian atrophy (DRPLA) (29, 30), X-linked dystonia parkinsonism (XDP) (31), familial adult myoclonic epilepsy 3 (FAME3) (32), and Friedreich's ataxia (FRDA) (33, 34). For some disorders, the correlation between the number of repeats and symptom manifestation is less clear, although there typically is a marginally significant trend (35–39). Second, expansion of one disease-causing repeat does not promote expansions of other repeats in the patient's genome. In other words, each RED patient typically has only a single repeat type expanded (40, 41).
Other characteristics differ among REDs. First, even though repeat expansion is necessary for some RED manifestation, there are REDs in which mutations other than repeat expansions can cause the same disease. Second, the RED mode of inheritance can be either autosomal recessive, dominant, or X-linked. Third, REDs can demonstrate a clear pattern of genetic anticipation (11, 23) or a complete lack thereof (34). Fourth, expandable repeats can be located in the coding part of a gene, in an intron, or in the 5′- or 3′-UTR. Fifth, the size of repeat expansion sufficient to cause pathological symptoms ranges from several repeat units for poly(A) diseases (42–53) to thousands for DM1 (11) and DM2 (54). Sixth, the origin of expanded alleles varies across repeats. For example, single common ancestors had a pathogenic amplification of ATTCT repeats in SCA10 (55–57), CCTG repeats in DM2 (58), and TTTTA/TTTCA repeats in FAME3 (32). Contrastingly, de novo repeat expansions were reported for the CAG repeat in HD (59), the CGG repeat in fragile X syndrome (60), and the GCN repeat in hand-foot-genital (HFG) syndrome (61). Last, the mechanisms of repeat toxicity vary among different repeats and include both a loss and gain of function.
The goal of this review is to depict the molecular mechanisms implicated in REDs. We first describe models of repeat-induced pathogenicity. We then discuss how repeat length instability and the propensity to induce various types of mutations compromises genome integrity. The focal point of the review is the description of the molecular mechanisms responsible for repeat instability and repeat-induced mutagenesis (RIM). Whereas these mechanisms were primarily established in model experimental systems, we discuss their relevance to human REDs whenever it is possible. Then we reflect on the existence of expandable repeats from the standpoints of genome function and evolvability. As for future directions, we discuss possible reasons for the differences in expandable repeats' behavior in model systems versus human pedigrees as well as how we could use our current knowledge about characteristics and instability of expandable repeats to develop therapies for REDs.
From expanded DNA repeats to disease
Repeat expansions induce changes in cell metabolism that become detrimental to the function of specific tissues. What are these changes? First, an expanded repeat located within a gene may lead to a transcription defect and consequently a loss of function of the carrier gene. Such a loss-of-function mechanism predicts an autosomal recessive or X-linked mode of inheritance. It also predicts that in a subset of patients, an inactivation of the same gene could happen due to a missense, indel, or frameshift mutation, instead of repeat expansion. Both of these predictions are true for autosomal recessive FRDA (62), progressive myoclonus epilepsy of the Unverricht–Lundborg type (EPM1) (63), Congenital insensitivity to pain (44), X-linked Duchenne muscular dystrophy (64), and fragile X syndrome (65, 66).
The majority of REDs exhibit an autosomal dominant mode of inheritance and are caused exclusively by repeat expansions. Moreover, an expansion of a particular repeat may have the same detrimental consequences regardless of the gene in which this expansion happens. For example, expansion of (CAG)n repeats in 13 different genes causes SCAs. Similarly, recently discovered expansions of (TTTTA)n(TTTCA)m repeats in the introns of five different genes are all linked to FAME (32, 67–69). Therefore, researchers have proposed toxic gain-of-function mechanisms, which could be either at the RNA or protein levels.
Loss of function of a gene with an expanded repeat tract
Long repeat tracts can impede transcription of their corresponding gene (70) and therefore lead to decreased production of an essential protein (31, 62, 71). This process could occur in several ways. Formation of RNA-DNA hybrids (R-loops) potentially combined with formation of an unusual secondary structure within the expanded repeats may physically stall transcription (72). For example, in FRDA, RNA polymerase fails to successfully transcribe through expanded GAA repeats located in the intron of the FXN gene (62, 71), likely because of the formation of R-loop–containing structures (73, 74), such as an H-loop (75). This transcription hindrance leads to the appearance of repressive chromatin marks at and around an expanded repeat, ultimately leading to local heterochromatinization and gene repression (reviewed in Ref. 76) (Fig. 1). Reduced FXN expression causes mitochondrial dysfunction, which eventually leads to cell death (77, 78). Similarly, R-loop formation within expanded CGG (72, 79) repeats in the 5′-UTR of the FMR1 gene leads to promoter DNA methylation at the repeat and the adjacent promoter (80), followed by histone and DNA methylation, resulting in massive heterochromatinization that can spread up to 1.8 Mb (reviewed in Ref. 81) (Fig. 1). It is, in fact, this constitutive heterochromatinization that slows down DNA replication through this region, leading to the characteristic fragile X phenotype (82).
Figure 1.

Loss-of-function mechanisms caused by expanded repeats exemplified by GAA repeats in FXN and CGG repeats in FMR1 genes. Repeats are shown in blue and red, and the flanking DNA sequences are shown in black. Top, when repeats are short, transcription is unimpeded. Bottom left, expansion of GAA repeats leads to formation of an R-loop at the repeat, which triggers histone deacetylation and the presence of repressive histone marks (such as histone H3 Lys-9 methylation), heterochromatization, and gene silencing (reviewed in Ref. 76). Bottom right, expansion of CGG repeats leads to formation of an R-loop at the repeat, which triggers methylation of the repeat as well as of the CpG islands upstream of the FMR1 promoter, followed by histone methylation. This results in heterochromatization and, ultimately, gene silencing (reviewed in Ref. 81).
Expansion of the dodecamer CCCCGCCCCGCG repeat in the 5′-UTR of the CSTB gene seems to decrease CSTB gene expression by a mechanism different from that discussed above. This repeat is located between two cis-regulatory elements that activate CSTB transcription. Expansion of the repeat increases the distance between the two regulatory elements such that it prevents efficient transcription initiation of the CSTB gene (63).
Toxic gain of function at the level of RNA
Growing evidence suggests that some expanded repeats exhibit cellular toxicity on their own, rather than in a context of their carrier gene. One example is the expansion of CCTG repeats in an intron of the CNBP gene (in DM2). This expansion does not directly affect gene expression: it changes neither the methylation pattern of the gene promoter (83) nor the splicing pattern of CNBP mRNA nor the CNBP protein level (84). Instead, expanded CCTG repeats are transcribed into toxic repetitive RNA. The toxicity of expanded RNA repeats is likely associated with their ability to fold into unusual RNA secondary structures. The list of known repetitive RNAs with confirmed toxicity includes CUG, CCUG, CGG, CAG, AUUCU, UGGAA, AUUUC, GGCCUG, and GGGGCC repeats (22, 85–94).
What are the molecular mechanisms of this toxicity? Expanded repetitive RNAs can engage in multivalent base pairing, which leads to their gelation and aggregation into visible nuclear foci (95). As a consequence, these RNAs remain in the nucleus and might sequester RNA-binding proteins. In the best-studied cases of DM1 and DM2 diseases, expanded RNA repeats sequester Muscleblind (Mbnl) proteins, whose normal function is to regulate splicing of muscle-specific genes (96) (Fig. 2). This mechanism is evidenced by the fact that disruption of the MBNL1 gene leads to the same symptoms as the expression of expanded repeats alone (97). For other repeats, the precise toxicity mechanism is less understood. Expression of r(GGGGCC)exp causes length-dependent cognitive impairment in mice (98), but the mechanistic connection between RNA expression and symptoms remains unclear. This repetitive RNA folds into G-quadruplexes (99), forms nuclear foci (98, 100), sequesters important RNA-binding proteins (101–104), and impairs mRNA transport (105) likely by damaging the nuclear pore complex (106). However, it is not yet clear which of these features of the r(GGGGCC)exp repeats is primarily responsible for toxicity.
Figure 2.
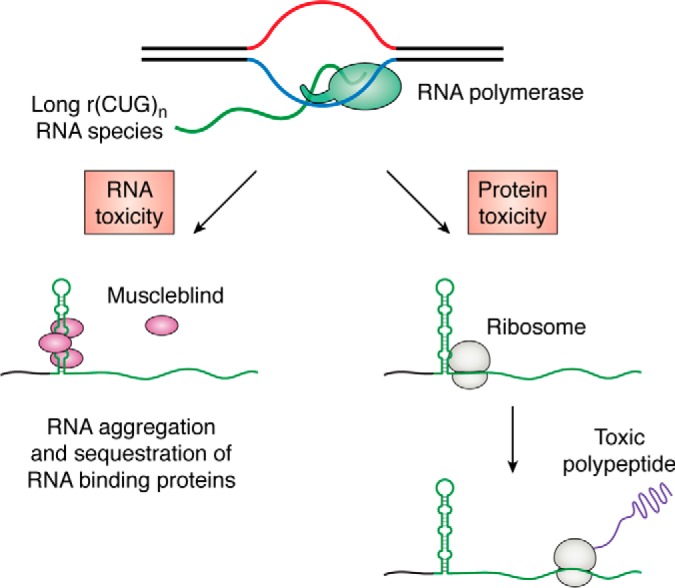
Toxic gain-of-function mechanisms in REDs exemplified by an expansion of a CTG repeat. Left, transcription of expanded CTG repeats produces long r(CUG)n RNA species that fold into RNA secondary structures, aggregate, and sequester the Muscleblind protein. This is the main source of toxicity in DM1 disease (reviewed in Ref. 15). Right, RAN translation of r(CUG)n. Expanded RNA repeats recruit ribosomes. This recruitment is likely mediated by the formation of an RNA secondary structure. Translation of r(CUG)n results in accumulation of toxic repetitive polypeptides in all three reading frames (reviewed in Refs. 123–125). Note that antisense transcription of the same repeat might also undergo RAN translation (not shown).
Toxic gain of function at the level of protein
A group of REDs are caused by expansions of GCN or CAG repeats located in coding parts of various genes. Translation of these in-frame repeats results in abnormally long poly(A) or polyglutamine poly(Q) tracts in their corresponding proteins. These long tracts could abrogate function of a protein and/or exhibit toxic gain of function.
Poly(A) tracts are common protein motifs that were suggested to promote protein interactions (107) or serve as nuclear export signals (108). At the molecular level, poly(A) peptides undergo a conformational change as their length increases. Whereas short poly(A) tracts preferably form monomeric α-helices, longer tracts fold into polymeric β-sheets (reviewed in Ref. 109) and into coiled coils (110). These conformational changes can lead to a toxic gain of function by introducing novel types of protein interactions, aggregation, mislocalization, and sequestration of other essential proteins. Commonly, proteins with poly(A) expansions co-aggregate with their WT versions, leading to effective haploinsufficiency, which explains their dominant inheritance patterns (reviewed in Ref. 109).
Similar to expanded poly(A) tracts, long poly(Q) tracts misfold into β-sheet structures that aggregate into toxic inclusion bodies in neurons. These structures might act as so-called “polar zippers,” which exhibit nonspecific affinity to various regulatory proteins in a cell (111). Accumulation of poly(Q) runs is believed to cause neurodegeneration (reviewed in Ref. 112). First discovered in 1997 for CAG repeats in HD (113), poly(Q) accumulation was used to explain symptoms of SCAs and other diseases caused by expansions of translated CAG repeats. However, there was one mysterious exception: SCA8. Unlike other SCAs, SCA8 is caused by an expansion of transcribed CTG rather than CAG repeats. Nonetheless, its symptoms resemble other SCAs (114). This conundrum was partially resolved after the discovery of bidirectional transcription of the SCA8 locus and subsequent translation of antisense transcripts (115). Yet one question remained unanswered: how can CAG repeats from antisense transcripts be translated without a start codon?
The discovery of an unusual phenomenon called repeat-associated non-ATG (RAN) translation shed some light on this mystery (116). RAN translation requires neither the canonical nor an alternative start codon for initiation and results in production of repetitive proteins in all possible reading frames. Taking into consideration bidirectional transcription, one expanded repeat may produce up to six repetitive polypeptides. To illustrate, RAN translation of the GGGGCC repeat sense transcript, r(GGGGCC)exp, results in poly(GA), poly(GP), and poly(AR) peptides, all of which were detected in ALS/FTD patient–derived cells (117). The antisense transcription of the same repeat leads to accumulation of polyGP, polyAP, and polyPR peptides (118, 119). These polypeptides aggregate into high-molecular weight insoluble clusters (120) and seem to be neurotoxic (98, 121), although it is not yet clear to what extent different polypeptides contribute to overall toxicity (122).
Length-dependent RAN translation has been reported for CAG, CGG, GGGGCC, GGCCUG, and TGGAA repeats (reviewed in Refs. 123–125). Nonetheless, we still lack a good understanding of RAN's mechanistic details. One hypothesis is that formation of unusual secondary RNA structures, such as hairpins (114) or G-quadruplexes (G4s) (126), might somehow recruit ribosomes and trigger this peculiar mode of translation (Fig. 2).
Taken together, mechanisms by which expanded repeats lead to cellular toxicity include loss of function of a protein and a toxic gain of function on the RNA or protein levels. These mechanisms might take place in combination with each other. Unfortunately, for many REDs, there is still no consensus on what mechanism plays a major role in disease progression. For example, accumulation of toxic poly(Q) proteins was historically used to explain HD's phenotype. However, evidence accumulated in the last decade suggests that gain of function at the RNA level might also contribute to disease manifestation. This is supported by experiments in model systems where CAG repeats interrupted by the CAA codon express decreased cellular toxicity when compared with pure CAG repeat tracts (reviewed in Ref. 127). This is unexpected because both CAA and CAG codons code for glutamine. As such, the length of transcribed poly(Q) tracts does not depend on the presence of a CAA interruption. Additionally, it was recently documented that HD age of onset is better predicted by the length of uninterrupted CAG repeat tracts rather than by the sheer number of consecutive glutamines in the Htt protein (128–130). However, there exists an alternative explanation of the same phenomenon: the disease age of onset might depend on somatic expansions of CAG repeats during the patient's lifetime, a process that requires repeat integrity (see below).
Overall, due to technical obstacles, it is highly challenging to decipher the precise role of various toxicity mechanisms in a specific RED (125). Therefore, we have a good understanding of possible mechanisms of expanded repeats' toxicity, even though the precise mechanisms are established for only few REDs.
Dynamic DNA structures as the key to repeat instability
Tandem repeats are abundant in the human genome (131), particularly in centromeres and telomeres, as well as in regulatory regions (132). High variability of tandem repeats between different individuals (133) makes analysis of tandem repeat polymorphisms useful in forensics and paternity testing (134). Nonetheless, most of the known tandem repeats do not expand as dramatically as disease-causing repeats, which may gain up to thousands of repeat units in affected individuals (11, 54).
What are the main features that make certain repeats prone to large-scale expansions resulting in REDs? Numerous studies that we review below attempted to resolve this fundamental question. Initially, the DNA slippage model was proposed to explain instability of tandem repeats. It postulated that repeats may be lost or gained during local misalignment of DNA during replication or repair (135). Indeed, formation of slipped DNA structures followed by repeat unit gains was detected in in vitro replication experiments (136). These structures were also observed in DM1 patient samples (137).
The DNA slippage model is beautiful in its simplicity and sufficient to explain the small-scale instability of any tandem repeat. However, it does not easily address several questions. First, why do some repeats gain thousands of repetitive units and others do not? Second, why does the instability of expandable repeats increase exponentially but not linearly with the size of the repeat? And last, why do repeat interruptions dramatically stabilize expanded repeat tracts?
Relationship between repeat size and instability
Length-dependent instability is one of the most universal properties of expandable repeats. What is the nature of this relationship? The DNA slippage model predicts a linear correlation between a repeat's length and its propensity to expand or contract: the number of repetitive units should reflect the number of opportunities for local DNA strand misalignments. However, the expandable repeats' behavior is inconsistent with this prediction.
First, for the majority of expandable repeats, there exists a repeat number threshold length after which the repeat starts to exhibit dramatic intergenerational and somatic instability. Curiously, this threshold length is comparable with the average Okazaki fragment size in eukaryotes (138). Moreover, disease-causing repeats show a nonlinear relationship between the repeat length and instability in experimental systems. Depending on an experimental system, doubling the repeat tract length results in a 6.5-fold increase in instability for GGGGCC repeats (139), up to a 100-fold increase for CGG repeats (140, 141), and up to a 1000-fold increase for GAA repeats (142). Together, data from both patient samples and experimental systems demonstrate that there is a qualitative difference between the instability of long and short repeats. This difference is evident from the differential propensity of repeats to expand or contract, the scale of expansions and contractions, and, likely, mechanisms of expansions and contractions (reviewed in Ref. 143).
Role of repeat interruptions in repeat stability
The DNA slippage model predicts that a small number of repeat interruptions should not dramatically change the overall instability of a repeat. However, there appears to be a causal connection between repeat purity and length. Analysis of DNA samples derived from RED patients revealed that the expanded repeat tracts commonly lose repeat interruptions found in their ancestral alleles (also called long normal alleles). For example, (CGG)20–52 repeat alleles located in the FMR1 promoter are typically randomly interrupted by AGG triplets, whereas expanded alleles consist of almost pure CGG repeats (144). The presence of AGG interruptions stabilizes both normal and premutational CGG repeat alleles during intergenerational transmission (145–150) and in experimental systems (151). Overall, it seems like the total length of a pure uninterrupted CGG tract rather than the repeat length overall predicts the level of CGG repeat instability. In other words, a loss of AGG interruption likely predisposes CGG repeat tracts to large-scale expansions (149, 152). Generally, the stabilizing role of interruptions is common for expandable repeats and has been reported for GAA (153, 154), CAG (38, 128, 129, 155–162), CCTG (54, 58, 163–165), and other repeats in somatic tissues, intergenerational transmission, and experimental systems.
Taken together, it appears that DNA slippage cannot be solely responsible for the repeat instability observed in REDs. On the contrary, more complex mechanisms, likely involving formation of alternative DNA structures, should be involved. It is generally believed that formation of such structures allows certain repetitive sequences to escape faithful DNA replication or repair and to undergo large-scale expansion. These structures include DNA triplexes (also known as H-DNA), G4s, and imperfect hairpins. Some AT-rich sequences can also undergo major DNA unwinding; hence, they are called DNA-unwinding elements (DUEs) (Fig. 3).
Figure 3.

Types of dynamic DNA structures and repeat sequences that form them. *, complement of CCTG; **, complement of CCCTCT.
H-DNA
As discovered in 1987, homopurine/homopyrimidine mirror repeats can adopt an intramolecular triple-helical DNA structure called H-DNA. In H-DNA, one of the DNA strands corresponding to one-half of the mirror repeat folds back, forming a triple helix with the remaining duplex part of the mirror repeat. The resulting triple helix constitutes a stack of base triads, which are stabilized by Hoogsteen or reversed Hoogsteen base pairing within the triplets and by π-π interactions between the stacks (Fig. 3). Two types of triplexes exist (166). First, a pyrimidine triplex, or H-y, consists of TA·T and protonated CG·C+ triads and requires a mild acidic environment to form (167). Second, a purine triplex, or H-r, consists of CG·G and TA·A triads and is stable at a physiological pH in the presence of bivalent cations (168).
H-DNA formation is favored in negatively supercoiled DNA, as it is topologically equivalent to DNA unwinding (169). Triplex-forming motifs are grossly overrepresented in eukaryotic genomes (170), which might point to their evolutionarily conserved role in genome function. On the other hand, formation of triplexes can pose a threat to a genome: triplexes impede DNA replication (171, 172) and transcription (73, 173, 174), promote formation of double-strand breaks (DSBs) (175), and trigger nucleotide excision repair (NER) (176). Three perfect homopurine-homopyrimidine repeats are known in REDs: GAA, AAGGG, and CCCTCT. GAA repeats are the best studied of the three and were reported to form both H-y and H-r triplexes in vitro and in vivo (177–182). Importantly, homopurine/homopyrimidine repeats that lack mirror symmetry (and therefore are unlikely to form stable triplexes) are dramatically more stable than (GAA)n repeats (183). This indicates that formation of a triplex might be central to repeat instability.
G4-DNA
One year after the discovery of H-DNA, a G4 structure was described for a GC-rich sequence from the immunoglobulin switch region (184). G4 is a four-stranded DNA structure, which consists of several G-quartets stacked upon each other and held together by π-π interactions. Each quartet is formed by four guanines connected by Hoogsteen hydrogen bonds (Fig. 3). Monovalent cations, specifically sodium and potassium, stabilize the G-quartets and their stacking interaction. The consensus motif is typically represented as G3N1–7G3N1–7G3N1–7G3, although G4-DNA with loops longer than 7 bp, with two G-tetrads, or with a bulged nonguanine base inside the three guanine tracts also is possible (reviewed in Ref. 185). G4 can exist in a large variety of isoforms differing in the number of DNA strands (one, two, or four) and relative strand polarity. In the context of this review, an intramolecular G4 structure is of prime relevance and is presented in Fig. 3.
G4 motifs are highly abundant in eukaryotic and some prokaryotic genomes (186). G4s were detected in vivo using G4-specific antibodies and small molecules. They are found in telomeres and multiple other regions in the genome (reviewed in Ref. 186) and are enriched in promoters and 5′-UTR sequences of genes. It is believed that G4s are likely to form in nucleosome-depleted, highly transcribed regions (187, 188). G4s interfere with regulation of transcription, genome integrity, and telomere maintenance (reviewed in Refs. 186 and 189–191). Importantly, they can constitute potent barriers for replication (reviewed in Ref. 192) and, in certain circumstances, promote DSB formation (193–195). Because these structures are highly stable at physiological conditions, a number of helicases, including Pif1, Rtel1, FANCJ, Bloom (BLM), and Werner (WRN), have evolved to unfold them (reviewed in Ref. 191). Additionally, various proteins, for example topoisomerase I (196–198) or nucleolin (199), can bind to and/or interact with G4 motifs (reviewed in Ref. 200).
Based on our knowledge of G-quadruplex structures and in vitro data, expandable repeats that should be able to readily adopt G4-DNA are GGGGCC (201–203), CCCCGCCCCGCG, AAGGG, and AGAGGG—the complement of CCCTCT. Other repeats, such as CGG (204), form a peculiar version of G4 composed of G-quartets separated by hydrogen-bonded cytosines. CAGG (the complement of CCTG) and GGCCTG repeats also have a potential to form a weak G4, in theory (Fig. 3).
Imperfect hairpins
An inverted DNA repeat may form a hairpin: a DNA secondary structure in which an upstream region anneals to a complementary downstream region via normal Watson–Crick base pairing. When hairpins form on either side of a double helix directly opposite to each other, the overall structure is called DNA cruciform. If two opposite hairpins are shifted relative to each other, the resulting structure is called slipped DNA or S-DNA (Fig. 4A). No expandable repeats can form a perfect hairpin or a cruciform. That said, many expandable repeats, such as CAG (205–207), CGG (204, 207), CCTG (208), GGGGCC (203), CCCCGCCCCGCG (209), and TGGAA (210), form hairpins containing mismatches—imperfect hairpins (Fig. 4A)—as was first demonstrated in a groundbreaking paper from the McMurray laboratory (207). Based on its sequence, another repeat, GGCCTG, also theoretically adopts an imperfect hairpin. Similar to perfect hairpins, complementary imperfect hairpins can also form an S-DNA (Fig. 4A).
Figure 4.

A, DNA cruciform versus S-DNA structures. B, difference in the kinetics of perfect versus imperfect hairpin formation (see “Imperfect hairpins” in the “Dynamic DNA structures as the key to repeat instability” section).
Both DNA cruciform and S-DNA are topologically equivalent to completely unwound DNA and, thus, favored by negative DNA supercoiling. There is a principal difference, however, in their formation kinetics. In the case of a DNA cruciform, the first step is slow: it involves the unwinding of a ∼10-bp DNA segment around the pseudosymmetry axis of an inverted repeat to form a central bubble. Self-annealing of separated DNA strands in this bubble results in cruciform nucleation. The nucleus then quickly extrudes into the full-size cruciform via branch migration. Upon DNA linearization, DNA branch migration would instantly convert the cruciform back to a regular DNA duplex (211) (Fig. 4B).
The kinetics of S-DNA formation is different, owing to the fact that the presence of mismatches completely blocks spontaneous branch migration, thus precluding the extrusion step (212). Consequently, S-DNA formation must be preceded by the unwinding of a long DNA segment followed by the out-of-register self-annealing of individual DNA strands. In vitro, this could be achieved upon denaturing and renaturing of repetitive DNA segments. In vivo, this could happen during DNA replication, transcription, or repair, all of which involve unwinding of duplex DNA. After an S-DNA is formed, the same kinetic principles apply in reverse: the S-DNA cannot easily convert into duplex DNA via branch migration. In other words, such S-DNA is kinetically trapped and, thus, relatively stable even though this conformation is thermodynamically unfavorable (Fig. 4B).
DUEs
Highly AT-rich repeats ATTCT, TTTTA, and TTTCA are similar in their sequence composition to DUEs, which are involved in DNA unwinding at replication origins. Indeed, the DNA-unwinding capacity of the ATTCT repeat was confirmed experimentally. In supercoiled plasmids, this repeat is highly unwound or even completely unpaired, thus being accessible to small molecules, oligonucleotides, and DNA polymerase (213). In fact, long ATTCT can function as aberrant replication origins in human cells (214), which might contribute to their instability.
Taken together, all expandable DNA repeats can form dynamic DNA structures during biological transactions that involve either extensive DNA unwinding or accumulation of negative supercoiling (169, 215). Surprisingly, mechanisms of repeat instability are not the same for different repeats. These mechanisms depend on the repeat sequence, length, purity, and location within a genome, as well as cell type, developmental stage, and replicative and transcriptional status. Rephrasing Leo Tolstoy: stable DNA sequences are all alike; every unstable DNA sequence is unstable in its own way. In the next section, we review how the presence of expandable repeats affects genome integrity or, in other words, types of repeat-mediated genome instability.
Types of repeat-mediated genome instability
Length instability during intergenerational transmission
Expandable repeats change their length dramatically while being transmitted between generations. Some of them preferentially expand, which leads to the genetic anticipation phenomenon described above. Contrary to initial beliefs, however, most REDs do not exhibit genetic anticipation, and the repeat length changes in both directions between generations.
Remarkably, parental gender is the most significant determinant of the direction of intergenerational instability. For example, there is a strong bias for expansions of GAA (FRDA), CGG (fragile X), or CCCTCT (XDP) repeats during maternal transmission, whereas paternal transmission typically results in a contraction of the same repeats (31, 34, 140, 216–219). The opposite pattern is observed for the majority of diseases caused by CAG repeat expansions: repeats are stable or slightly biased toward contractions during maternal transmissions, whereas paternal transmissions are biased toward expansions (23, 24, 30, 159, 220–225). Note, however, that CAG repeats in SCA8 show the opposite tendency (114, 226), whereas CAG repeats in SCA2 (20) and SCA7 (16, 17) lack an apparent parental gender bias. Another interesting example is the ATTCT repeat: this repeat expands or contracts dramatically during paternal transmission but stays virtually unchanged during maternal transmission (227, 228).
The second important factor is parental age. Intergenerational instability of GAA (FRDA) (216), CTG (DM1) (229), and CAG (SCA17 (225) and SCA2 (224)) repeats directly correlates with parental age. DRPLA, SCA1, and DM1 mice also show age-dependent intergenerational CAG repeat instability (230–232). Having said that, age dependence is not a universal rule: for example, intergenerational instability of CAG repeats in HD depends neither on the mother's nor the father's age both in human (221) and mice (233) pedigrees.
Why does intergenerational repeat instability depend on parental gender and age? The increase of instability with age indicates that repeat expansions and contractions might take place before fertilization. This may simply be because a longer lifetime allows more chances for a repeat to contract or expand. Why repeat instability also depends on parental gender is a more difficult question for which we do not yet have a full answer. Several hypotheses that incorporate the differences between male and female germ cells were proposed to explain this phenomenon. First, the differential bias for expansions and contractions could be due to the differences in duration between spermatogenesis and oogenesis (230, 232, 234, 235). Second, the counterselection for repeat expansions could differ between sperm and oocytes (16, 218). For example, large-scale expansions of CGG repeats in the FMR1 gene seem to be counterselected in sperm (218, 236), possibly because FMR1 loss of function reduces sperm mobility, at least in Drosophila (237). Third, the difference in the expression level of certain DNA repair proteins such as Msh3 during spermatogenesis and oogenesis might explain different levels of repeat instability (238).
We hypothesize that two other mechanisms might account for the difference in repeat instability during spermatogenesis or oogenesis. First, a differential pattern of origin firing and replication timing might affect repeats' propensity to expand or contract (as will be discussed below). Second, the fate of chromatin, which is dramatically different between the two gamete types, could differentially affect repeat instability. Mature oocyte chromatin is very loose, to allow rapid production of RNAs and proteins required for successful fertilization and development into an embryo (reviewed in Ref. 239). In contrast, mature sperm chromatin is compact, owing to the temporary replacement of core histones with sperm-specific histone variants, followed by transition proteins and, finally, protamines (reviewed in Refs. 239 and 240).
Length instability in somatic tissues
The majority of REDs are not congenital; instead, they have a fairly late age of onset. It was initially thought that this is because repeat-associated toxicity is low and results in a slow, gradual degradation of patients' tissues that becomes symptomatic later in life (241, 242). Although certainly attractive, this hypothesis fails to explain two groups of data. First, it does not explain why the age of onset of dominant HD is well-predicted by the length of the longer repeat expansion allele in homozygous patients (243, 244) or why the age of onset of recessive FRDA is determined solely by the shorter GAA expansion allele (245–247). Second, it fails to explain why the age of onset of HD is better predicted by the length of noninterrupted CAG repeats in DNA rather than by the length of the polyQ tract in the protein (128–130).
The currently accepted view is that the age of onset and disease progression are governed by a combination of two different mechanisms (243). This hypothesis states that the number of repeats in inherited alleles is typically insufficient to exhibit significant cellular toxicity. However, repeats expand in some somatic tissues (16, 17, 27, 31, 32, 35, 36, 54, 80, 160, 248–251), which causes cell toxicity when a repeat length exceeds a disease-specific threshold. Subsequent disease progression is determined by the mechanism of toxicity, which is specific for each disease (252). This model provides an elegant explanation for the dependence of age of onset on the number of inherited repeats in both heterozygous and homozygous patients.
The level of somatic instability is tissue-specific (253, 254), likely because of differential expression of DNA replication and repair genes across various cells types (255). Somatic instability also depends on the type and length of the repeat (256) and tends to increase with age (54, 257, 258). Somatic instability can be so dramatic that it may preclude precise estimation of the initial repeat tract length (259): for example, the number of CTG repeats (DM1) may differ by 40–400 repeat units within one tissue and up to 5770 repeats between different tissues in a single person (253). In sum, it is the combination of the number of inherited repeats and the level of somatic instability that determine the age of onset for a particular disease and a particular patient.
Fragility and repeat-induced mutagenesis
DNA fragility is the propensity of a certain DNA sequence to promote chromosomal breakage (i.e. produce DSBs). Expanded CGG repeats located in the FRAXA, FRAXE, and FRA11B loci are renowned for their fragility (141). In fact, they were initially described as rare, folate-sensitive fragile sites (260–262). This is because they exhibit fragility under low-dNTP conditions, likely because DNA replication through expanded CGG repeats cannot compete with DNA replication of other genomic regions when the dNTP pool is limited (260). A less known fact is that many other repeats are also fragile. In model experimental systems GAA, CAG, and ATTCT repeats exhibit length-dependent fragility (263–268), whereas triplex-forming DNA sequences promote DSB formation in human cells (175). Additionally, breakpoints of genomic rearrangements in cancer are enriched for structure-forming tandem DNA repeats, such as (AT)n, (GAA)n, and (GAAA)n (269).
What could be the consequences of a DSB formation? Generally, error-prone repair of a DSB provides many opportunities for mutagenesis. Notably, mutations may arise kilobases away from the DSB site (reviewed in Ref. 270). As such, fragile, repetitive tracts could induce mutagenesis at a distance, a phenomenon that we have termed RIM (271). Long CGG repeats are highly prone to RIM. Based on patient data, they facilitate large chromosomal deletions (272, 273), chromosomal arm loss (262, 274), and even a loss of a whole X chromosome. The latter is evidenced by an unusually large proportion of fragile X females with mosaic Turner syndrome (273, 275). Furthermore, using an experimental system to detect CGG-mediated repeat instability in human cells, we recently confirmed accumulation of point mutations up to 3 kb away from the repeat (276). Other structure-forming repeats also seem to exhibit RIM. Data from FRDA patients also show that GAA repeats elevate the number of point mutations in the genomic areas surrounding the repeat locus (277). In yeast, repair of DSBs formed within expanded GAA repeats results in the accumulation of point mutations located more than 1 kb away from the repeat (138, 264, 268), large deletions (264), and gross chromosomal rearrangements (278). Finally, a recent study has found that somatic expansions of TTTCA repeats in FAME3 patients are concurrent with small and large genomic rearrangements (32).
As of today, most research on REDs focuses exclusively on changes in repeat lengths. However, two different repeats, GAA and TTTCA, whose fragility has never been directly observed in humans, likely promote RIM in RED patients (32, 277). Therefore, we believe that repeat fragility and RIM are important and overlooked factors in RED progression and pathology.
In sum, expandable DNA repeats demonstrate profound length instability during both intergenerational transmissions and somatic cell divisions. They also cause chromosomal fragility, induce mutagenesis at a distance, and trigger formation of complex genome rearrangements. In the next section, we discuss the role of DNA replication, transcription, repair, and chromatin environment in these phenomena.
Role of DNA replication in repeat instability
Duplicating long structure-forming repetitive sequences presents a significant challenge for the replication machinery. This phenomenon is well-established and has been observed in virtually all experimental systems. In this section, we review how replication contributes to repeat instability.
Evidence that replication promotes repeat instability
In experiments with mammalian cells, long GAA (279) or CAG (280) repeats cloned into plasmids expand and contract more frequently if the plasmids are able to replicate upon transfection. This indicated that replication per se promotes repeat instability.
DNA replication is inherently asymmetrical. Whereas the nascent leading strand is synthesized more or less continuously in the direction of replication, the nascent lagging strand is replicated in the opposite direction and in short patches known as Okazaki fragments. Consequently, a floating zone in the lagging strand template called the Okazaki initiation zone stays single-stranded, facilitating the formation of DNA secondary structures. There are also differences in DNA polymerases that synthesize the two DNA strands. The leading strand DNA polymerase ϵ (Pol ϵ) is a highly processive and precise enzyme. The lagging strand DNA polymerase δ (Pol δ) is less processive, and its fidelity is an order of magnitude lower than that of Pol ϵ, which is compensated by a higher activity of mismatch repair (MMR) on the lagging strand. Unlike Pol ϵ, Pol δ has strand displacement activity, which is needed for Okazaki fragment processing (reviewed in Ref. 281). To additionally complicate the picture, it was recently demonstrated that leading strand DNA synthesis is, in fact, initiated by Pol δ followed by a switch to Pol ϵ (282).
Not surprisingly therefore, the asymmetry of the replication fork leads to orientation-dependent repeat instability. Indeed, CAG/CTG repeats tend to contract if the CTG tract is in the lagging strand template and tend to expand if the CAG tract is in the lagging strand template in bacterial (35, 283), yeast (161, 162, 284, 285), and mammalian cells (206, 280, 286). Typically, this orientation dependence is explained by the fact that an imperfect hairpin formed by a (CTG)n tract is more stable than the one formed by a (CAG)n tract. Thus, when a (CTG)n tract is located in the lagging strand template, DNA polymerase might somehow jump through a hairpin formed by this repeat, resulting in a contraction. Alternatively, when the (CTG)n tract is in the nascent DNA strand during lagging strand replication, it can form slipped hairpins that, if unrepaired, would lead to expansions (reviewed in Ref. 287). Similarly to CAG repeats, the CGG and GGGGCC repeats form imperfect hairpins, and the hairpins formed by the G-rich strands are more stable (287). In addition, the G-rich strand of these two repeats may fold into a G4 (201–204). Consistent with the formation of a hairpin or a G4 during lagging strand synthesis, several studies have found that when located in the lagging strand template, CGG (141, 151, 288, 289) and CCGGGG (139) tracts are prone to repeat contraction. GAA repeats do not fold into a hairpin or a G4 and instead form an H-DNA. They also undergo orientation-dependent instability. In a majority of studies, GAA repeats are more unstable when (GAA)n tracts are located in the lagging strand template (73, 142, 153, 290), although we recently found that there is almost no difference in the repeat contraction rate among the two orientations (183).
Altogether, the dependence of repeat instability on the orientation relative to the origin of replication led us to propose the “ori-switch” model. It postulates that inactivation of a replication origin on one side of a repeat and the subsequent switch to the origin located on its opposite side changes the pattern of repeat instability, predisposing them to either expansions or contractions (292).
Repeat instability also strongly depends on the distance between the repeat and the nearest origin of replication (206, 208, 214, 284, 286, 293). The mechanisms of this dependence are not well-understood. Our “ori-shift” model (292) posits that this may be caused by the differences in repeat position within the Okazaki initiation zone. Given recent observations (282), this may also be caused by the repeat position relative to the location where Pol δ switches to Pol ϵ during leading strand synthesis.
Overall, it seems highly likely that a repeat's position within the replicon should play a big role in repeat-induced pathogenesis in humans. First, the “ori-switch” and “ori-shift” models explain why the same repeat tract exhibits dramatically different patterns of instability in two different locations in the genome (250, 279). Second, it could explain time- and tissue-specific differences in repeat instability. For example, the fragile X CGG repeat is believed to expand and contract during gametogenesis and early embryogenesis but stays relatively stable later in a person's life (60, 272, 294–297). Tissue-specific differences in origin-firing patterns might explain why CGG instability is limited to such a narrow developmental window. It is possible that during embryonal development, the CGG repeat is in an unstable position relative to the active origin of replication. During later cell division, a different origin might be utilized, thus fixating repeat length (298, 299). Additionally, expanded CGG repeats may themselves influence activation of the near origins and, as such, put themselves in an unstable position (82). This hypothesis explains the existence of a length threshold required for CGG repeat instability.
Role of Okazaki fragment processing in repeat instability
Another piece of evidence for the role of replication in repeat instability comes from the observation that impaired Okazaki fragment processing destabilizes various types of repeats, at least in yeast. During lagging strand replication, Pol δ partially displaces the 5′-end of a preceding Okazaki fragment while replicating the succeeding fragment. Specific 5′ flap endonucleases then process the flap, which is followed by the ligation of two fragments. In yeast, two endonucleases do the bulk of the job: Rad27, which only cuts short flaps (300–302), and Dna2, which cuts longer flaps (301–306). Mutations in either of these nucleases lead to dramatic increases in the expansions of various repeats, including CAG, GAA, CGG, G, GT, CAGT, CAACG, CAATCGGT, yeast telomeric repeats, and several minisatellites (142, 289, 307–318). In other words, the effect of rad27 or dna2 mutations is virtually independent of the repeat sequence. The classic interpretation of these data involves the incorporation of a long repetitive flap into the nascent strand during DNA replication, resulting in an elongated repeat tract (319) (Fig. 5A). However, the same mutations also increase repeat contraction rates (183, 289, 307, 309, 310, 312–314, 316–318), a phenomenon that cannot be explained by the flap incorporation model. A more general model suggests that in the absence of fully functional Rad27 or Dna2, the genome-wide accumulation of long ssDNA species (314, 320, 321), such as flaps or gaps, titrates the ssDNA-binding protein replication protein A (RPA) from the repetitive regions (322). Due to the lack of RPA, transiently formed ssDNA within the repetitive tracts is likely to equilibrate into DNA secondary structures, which trigger repeat instability (Fig. 5B) (183).
Figure 5.

Two models depicting the potential role of Rad27 flap endonuclease in the instability of structure-forming repeats. A, flap ligation model. In the absence of Rad27, long flaps that formed during Okazaki maturation might become incorporated into the nascent DNA during lagging strand synthesis, resulting in repeat expansion. B, RPA depletion model. In the absence of Rad27, long flaps that accumulate genome-wide during Okazaki fragment maturation titrate RPA out from the repetitive DNA. This promotes formation of DNA secondary structures (the hairpin is shown) within the repeat and can result in repeat contraction or expansion.
Are these models applicable to humans? The yeast Rad27 and human Fen1 proteins are highly conserved, such that Fen1 overexpression rescues the rad27Δ phenotype in yeast (323). However, despite these similarities, attempts to find mutations in the FEN1 gene that modify the phenotype of CAG repeat expansion in HD patients were fruitless (324–327). Surely, failure to identify such modifiers could potentially be explained by the high toxicity or lethality of FEN1 mutations. However, experiments with siRNA knockdown of Fen1 in a cell line system (328–330) and with Fen1-deficient mice generally do not detect a role for Fen1 in CAG repeat stability (331), except for one study where the nuclease activity of Fen1 was found to counteract CAG repeat instability, although the effect was not as dramatic as in yeast (332). Additionally, knockdown of Fen1 in mammalian cells does not promote GAA repeat instability.3 Therefore, there could exist fundamental differences between yeast and humans, either in the Okazaki fragment processing pathways or in the regulation of RPA homeostasis.
Direct evidence that expandable repeats are hard to replicate
Contemporary techniques, such as quantification of nascent strand abundance, two-dimensional gel electrophoresis, EM, and single-molecule visualization of replicating DNA allow researchers to directly measure replication fork progression. These techniques unconditionally proved that expanded tracts of CAG, GAA, CGG, and GGGGCC repeats physically stall the replication fork in a length-dependent manner in every experimental system studied, including bacterial, yeast, and mammalian cells. Notably, the effect is most pronounced for the CGG and GGGGCC repeats (73, 139, 290, 333–341). How do expanded repeats stall the replication fork? First, replication through a long repetitive sequence might exhaust the local dNTP pool, which slows down the replisome (342). Second, and even more important, DNA sequences prone to form alternative DNA structures block replication both in vivo and in vitro, particularly when their purine-rich strands are in the lagging template (171, 343). Third, these alternative DNA structures may recruit various proteins, which could create “protein bumps” that impede replisome progression (335).
What happens after the replication fork has stalled depends on the repeat sequence, replication mode, and other cellular factors. Consequently, the amount of fork stall may (290) or may not reflect the amount of repeat instability (139, 335, 340). In the best-case scenario, the replication fork can temporarily slow down but quickly recover and continue DNA replication as if nothing had happened. However, a longer pause may lead to the formation of single-stranded gaps, which might result in the accumulation of mutations around the repeat tract (Fig. 6). A prolonged stall can, in turn, facilitate DNA slippage or template switching, both of which can lead to repeat length instability. During template switching, a stalled replicative polymerase, either Pol δ or Pol ϵ, temporarily switches its template and replicates from the nascent strand to bypass a lesion. If a polymerase stalls at a repetitive template, it might invade the opposite strand out of register and thus end up synthesizing the wrong number of repeats (reviewed in Ref. 344) (Fig. 6). Data from our laboratory revealed that this process could be responsible for GAA repeat expansion in yeast (142).
Figure 6.

Possible mutagenic consequences of fork stalling within structure-forming repeats (see “Direct evidence that expandable repeats are hard to replicate” for details).
A dramatic consequence resulting from the replication fork colliding with a potent barrier is fork reversal (345). Fork reversal is a process in which two nascent strands anneal to each other, whereas their template strands reanneal back (Fig. 6), resulting in the formation of the so-called “chicken foot structure” containing a four-way junction. The biological role of fork reversal is to allow a bypass of a DNA lesion in a template strand using an unperturbed nascent strand as a temporal template (reviewed in Ref. 345). Formation of dynamic DNA structures could serve as such a lesion, as reversed forks were detected in vivo for CAG (346) and GAA repeats (338). Furthermore, formation of a reversed fork could promote repeat instability. Indeed, transition of a normal replication fork into a chicken foot structure involves several strand-reannealing steps and as such provides multiple opportunities for potential misalignment of tandem repeats. Furthermore, chicken foot structures are substrates for structure-specific nucleases, which can transform reversed forks into one-ended DSBs. These DSBs must be repaired through break-induced replication (BIR), a conservative mode of DNA replication, which leaves its mutagenic trace as much as several kilobases away from the initial break site (reviewed in Ref. 347) (Fig. 6).
If the replication fork cannot recover from a stall, it leaves unreplicated ssDNA regions that can be broken into a two-ended DSB (Fig. 6), which would need to be repaired via homologous replication (HR). However, mitotic cell division may start before a DSB is formed. This is common for late replicating fragile sites, like the CGG repeat in the FMR1 promoter (348). In this scenario, ssDNA regions transform into anaphase bridges, which may lead a permanent loss of a part of or even a whole chromosome (261, 341).
How to maintain hard-to-replicate expandable repeats?
Cells have evolved a number of mechanisms to minimize fork stalling and ensure, as much as possible, smooth progression of the replication fork through repetitive DNA. Replication fork–stabilizing factors, Tof1 and Mrc1, prevent CAG repeat instability and fragility of CAG, ATTCT, and GAA repeats in yeast (142, 263, 308, 349). In addition, specific helicases assist replisomes by unwinding unwanted DNA secondary structures.
Yeast helicases Srs2 and Sgs1 unwind hairpins formed by CTG and CGG repeats in vitro, with Srs2 being more efficient (350). In vivo, knocking out SRS2, but not SGS1, promotes instability and fragility of CAG and CGG repeats (351–353), although some reports found that Sgs1 also maintains CAG (354) and CGG repeat stability (289). Despite this disagreement between studies, the amounts of CAG repeat instability in srs2Δ or sgs1Δ mutants reflect the amount of fork stalling. Furthermore, human helicase Rtel1, which unwinds hairpins formed by CAG repeats and complements the yeast srs2Δ mutant, prevents CAG repeat expansions in human cells (355). These observations support the model in which Srs2/Rtel1 and, to a lesser extent, Sgs1 helicases unwind DNA hairpins during DNA replication to prevent fork stalling. Interestingly, the secondary structure–unwinding activity of Srs2 and Sgs1 seems to be specific to hairpin unwinding. Indeed, the instability and fragility of non-hairpin-forming repeats, such as telomeric or GAA repeats, does not depend on Srs2 (142, 183, 351) and is mildly affected by Sgs1, if at all (142). It is not yet known whether there are other helicases that unwind DNA hairpins. Recently, for example, human DNA helicase B was reported to localize to replication stalls at CGG repeats (356), although direct evidence that this helicase unwinds hairpins is so far absent.
Can other DNA secondary structures such as G4 or triplex also be unwound by specialized helicases? Because G4 is so stable under physiological conditions, there is a team of helicases that unwind this structure. This team includes FANCJ, Rtel1, BLM, and Pif1, to name just a few (reviewed in Refs. 191, 192, and 357). Triplex-unwinding helicases, on the other hand, are still largely unknown. Reports have demonstrated in vitro triplex-unwinding activity for several proteins, namely human Dhx9 (358), Ddx11 (359), and BLM and WRN helicases (360) as well as the yeast Stm1 protein (361). However, in vivo evidence for the involvement of these helicases in triplex unwinding is scarce. In particular, data from our laboratory did not show evidence for Stm1 and Chl1 (Saccharomyces cerevisiae Ddx11) triplex-unwinding activity in yeast in vivo (183).
Role of transcription in repeat instability
Remarkably, expandable repeats can exhibit instability in the absence of replication (257, 362), and, in some circumstances, transcription through expandable repeats is sufficient to destabilize them. As a matter of fact, transcription through expanded repeats promotes their fragility (257, 363), instability (154, 330, 364–367), and RIM (368). On the face of it, this phenomenon seems unexpected, considering that there is no DNA synthesis involved in transcription. Nonetheless, transcription can affect repeat instability via at least three different mechanisms. First, transcription can change the chromatin landscape and, as such, alter repeat stability indirectly (see below). Second, the Smith laboratory recently showed that DNA replication often initiates at transcription start sites in human cells (369). This means that transcription determines origin-firing patterns, which could directly control repeat instability (see above). Third, formation of transcription-dependent R-loops directly influences repeat instability.
An R-loop is a DNA-RNA hybrid that forms during transcription when a nascent RNA strand invades the DNA duplex and reanneals to its template DNA via normal Watson–Crick base pairing. In a eukaryotic cell, R-loops act as a double-edged sword. On the one hand, they play an important role in the regulation of gene expression and transcription termination. On the other hand, they can stall replication and transcription, promote replication-transcription collisions, and, as such, lead to DSB formation and genome instability (reviewed in Refs. 370 and 371).
In vitro, transcription through CAG, CGG, and GAA repeats induces single and double R-loops. The amount and size of these R-loops correlate with an increase in negative supercoiling upstream of an elongating RNA polymerase (365, 372). R-loops are also detected at expanded GAA, CGG, GGGGCC, and CAG (72, 373) repeats in patient samples as well as in several experimental systems (79, 365). Most importantly, R-loop formation within long repeats likely promotes their instability. This is evidenced by the fact that RNase H, which degrades R-loops, counteracts repeat instability (75, 365, 374).
How can formation of R-loops lead to repeat destabilization? When an R-loop forms on a template strand, it leaves the complementary part of the DNA duplex single-stranded, thus promoting the formation of dynamic DNA structures. The result is a composite DNA structure consisting of an R-loop and an alternative DNA structure (Fig. 7) that facilitates repeat instability. For example, formation of a DNA hairpin opposite to an R-loop strand (S-loop) was suggested to explain the R-loop–induced instability of CAG and CGG repeats (79, 365, 374). In a G4-forming repeat, a structure called a G-loop is possible. It can come in two flavors: either G4-DNA is formed in the nontemplate strand, or a hybrid G4-DNA-RNA structure is formed between the nontemplate DNA and a nascent RNA strand. Such structures might form in human cells with expanded CGG and GGGGCC repeats (373, 375). Likewise, a triplex-forming repeat might form an H-loop; in this structure, an R-loop is formed between an RNA transcript and a single-stranded portion of H-DNA (74, 75) (Fig. 7). Collision between a replication fork and an H-loop formed by a GAA repeat might lead to the formation of a DSB, followed by repeat expansion in the course of BIR (75).
Figure 7.
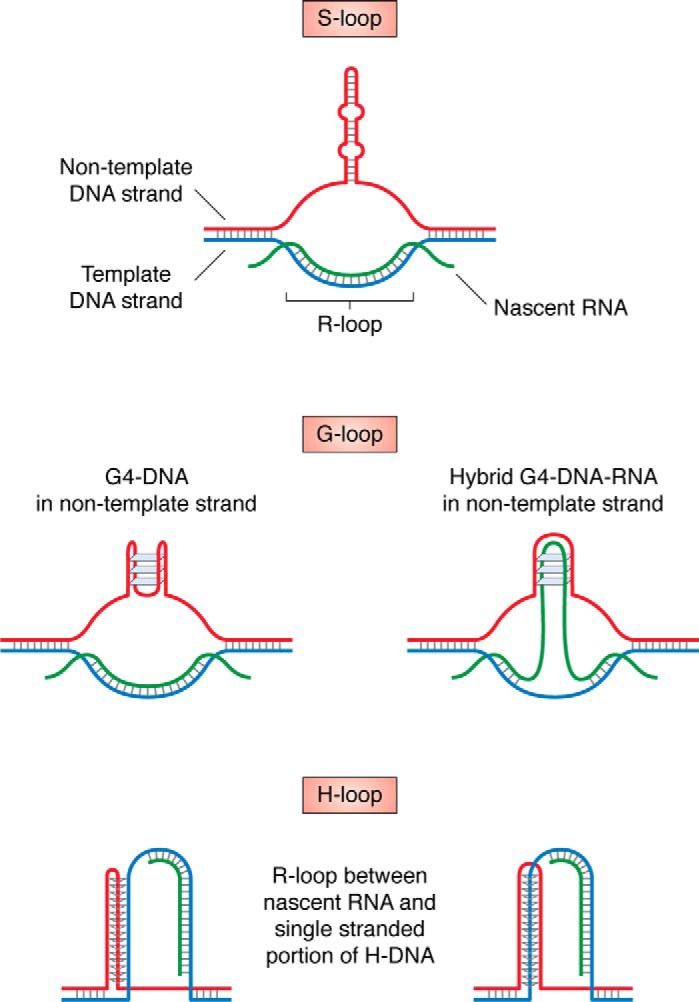
Various R-loop-based structures formed by expandable DNA repeats (see “Role of transcription in repeat instability” for details).
Another level of complexity is added by the fact that most of the mammalian genome is bidirectionally transcribed (376, 377), and repetitive regions are no exception (115, 378–380). Bidirectional transcription was detected for CAG (381–384), CGG (380, 385), GGGGCC (379), and other repeats (reviewed in Ref. 378) and can synergistically destabilize them (386), likely due to formation of double R-loops (387).
Role of DNA repair in repeat instability
Replication and/or transcription through expanded repeats fosters the formation of DNA secondary structures, which might lead to the formation of a DNA nick or a DSB. This DNA damage then serves as a substrate for various pathways of DNA repair. Consequently, DNA repair plays an important role in repeat instability.
HR
As discussed above, structure-forming repeats promote DSBs either during replication or transcription through the repeat. Thus, it is not surprising that these repeats serve as hotspots for homologous recombination in Escherichia coli (388–393) and yeast (394). These recombination events are often accompanied by a repeat length change (388, 390, 392, 395–398), likely because of an out-of-register strand invasion during the classical HR. However, in yeast, the majority of unstable repeats are not hotspots for recombination during meiosis (289, 394, 399), and their barely detectable meiotic instability depends on the Spo11 nuclease (267, 400). Data from human (401, 402) and mouse (403, 404) pedigrees also agree that repeat instability generally does not arise from unequal crossing over. A notable exception to this rule is poly(A) diseases, which are caused by expansions of GCN repeats (46, 47, 50, 52, 53). In this case, expanded alleles likely originate via unequal crossing over between parental repeats (44, 51, 52, 109, 398) (Fig. 8A). This explains the lack of detectable somatic instability of GCN repeats as well as their small scale of expansion.
Figure 8.

HR mechanisms implemented in repeat instability. A, as a result of an unequal crossing over during meiosis, one homologous chromosome inherits a contracted repeat tract, whereas another inherits an expanded repeat tract. This mechanism is typical for poly(A) diseases. B, a DSB initiated at a repetitive sequence is followed by end resection and reannealing of the repetitive ends. This can ultimately result in repeat contraction. C, a one-ended DSB can be repaired via BIR. Out-of-register invasion into a repetitive template can give rise to both expansions and contractions, while point mutations accumulated in the course of BIR are responsible for the RIM phenomenon.
Besides canonical HR, two other pathways, single-strand annealing (SSA) and BIR, were implicated in repeat instability. The SSA pathway involves the resection of DSB ends followed by the annealing of flanking direct repeats, which normally results in a deletion. SSA does not require strand invasion performed by Rad51, but it does require strand annealing by Rad52 or Rad59. As such, one would expect this pathway to give rise to repeat contractions, rather than expansions (Fig. 8B). Indeed, an artificial induction of a DSB within a repetitive tract drives repeat contractions via SSA (396, 405, 406).
The BIR pathway is a highly error-prone, conservative mode of DNA replication that specifically repairs one-ended DSBs. BIR is identified by its mutational signature and dependence on Rad52 recombinase and a processivity subunit of DNA polymerase δ, Pol32 (PolD3 and PolD4 in mammals) (reviewed in Ref. 347). We have recently shown that in yeast, large-scale expansions of CAG repeats (407) and transcription-induced expansions of GAA repeats (75) happen through BIR. Moreover, BIR drives instability of carrier-size CGG repeats in a mammalian cell culture system (276). In the latter case, small-scale expansions and contractions of CGG repeats are accompanied by point mutations and complex rearrangements in the reporter gene. In all of these scenarios, BIR is likely triggered by a secondary structure formation at the repeat locus, and mutations accumulate during error-prone synthesis by Pol δ (Fig. 8C). Aside from BIR, RIM spanning to kilobases away from the repeat can also arise via a different mechanism. Replication through a repeat can leave behind a long single-stranded gap to be filled in by an error-prone translesion polymerase (264, 268, 389).
Importantly, structure-forming repeats not only promote recombination, but can also interfere with it. Any recombination event has to start with resection and typically involves ssDNA end invasion. If this end readily folds into a secondary structure, it could impede the recombination process (408). Unusual DNA structures may inhibit later stages of HR as well. For example, triplexes inhibit branch migration of Holliday junctions in vitro (409).
End-joining (EJ) pathways
In addition to various HR pathways, cells have evolved several EJ pathways to repair DSBs. As follows from the name, EJ pathways involve the direct fusion of two broken DNA ends. The classic pathway, named nonhomologous EJ (NHEJ), involves threading the DNA into Ku70/80 protein rings followed by the direct ligation of the two DNA ends with DNA ligase IV. This pathway leads to a virtually error-free mode of repair. However, when ends cannot be directly ligated, they need to be processed. This can result in small (up to 4-bp) deletions. In a situation when NHEJ cannot be executed, cells employ more error-prone, noncanonical variations of EJ pathways, such as alternative NHEJ, microhomology-mediated EJ (MMEJ), or synthesis-mediated MMEJ (reviewed in Ref. 270). We do not yet know all of the details and differences between these mechanisms, but what they have in common is that they (i) are highly error-prone and typically result in indels up to 20 bp in size, (ii) do not require classic NHEJ mediators such as Ku70/80 proteins or DNA ligase IV, and (iii) typically act when classic NHEJ is compromised (reviewed in Refs. 270 and 410).
The classic NHEJ seems to be protective against repeat instability, as evidenced by experiments with CAG repeats in yeast (395, 411) and CGG repeats in mice (412). Probably, NHEJ directly ligates a subset of DNA ends originated from a DSB within a repeat, without an alteration in repeat length. Otherwise, these ends would have been repaired via repeat-destabilizing HR or noncanonical EJ pathways.
How can noncanonical EJ pathways destabilize repetitive sequences? It was found that Pol β, which might participate in MMEJ (413), promotes CAG repeat expansions in vitro (414). Knocking down key alternative NHEJ modulators, namely XRCC1, LIG3, and PARP1, suppresses CAG repeat contraction induced by environmental stresses, such as cold, heat, and hypoxia, in human cell cultures (329).
At first glance, it might seem that HR plays a much bigger role in repeat instability than EJ pathways. However, this may simply reflect the fact that most mechanistic studies regarding repeat instability were primarily conducted in replicating yeast or mammalian cells. In replicating cells, DSBs typically arise during DNA replication and are predominantly repaired via HR pathways. Independent of replication, specifically in G0 and G1 cell cycle stages, DSBs originate in the course of transcription and result from oxidative damage to DNA. These breaks are typically repaired via EJ pathways (reviewed in Refs. 415 and 416). To fully elucidate the relative contributions of HR and EJ in repeat instability, one needs to carefully compare repeat instability between isogenic replicating and nonreplicating cells.
MMR
MMR recognizes and fixes mismatches as well as small loop-outs that distort the double-helix structure. In eukaryotes, mismatch repair starts by the binding of MutS complexes that recognize a lesion. Mismatches and 1–3-bp loop-outs are recognized by the MutSα (Msh2 and Msh6), and larger loop-outs are recognized by MutSβ (Msh2 and Msh3). Upon mismatch recognition, one of several MutL complexes excises the lesion to allow Pol δ to fill the resulting gap (reviewed in Ref. 417). Mutations in MMR give rise to Lynch syndrome, an autosomal dominant disease associated with a high risk of colon cancer. On the molecular level, Lynch syndrome is characterized by high microsatellite instability: the patients have a higher rate of length instability among various short tandem repeats (reviewed in Ref. 418).
Because MMR protects from microsatellite instability in colon cancer, does it also suppress repeat expansion in REDs? Surprisingly, both somatic and intergenerational expansion of CAG and CGG repeats are virtually absent in mice lacking Mlh1, Mlh3, Msh2, or Msh3 (257, 419–427). In other words, MMR promotes rather than suppresses repeat expansions. The profound effect of MMR on repeat instability could explain the strain-specific differences in the amount of repeat instability: differences in the expression level of Mlh1 and/or Msh3 in different mice strains correlate with the amount of repeat somatic instability in those mice (419, 428, 429). Interestingly, the effect of knocking out Msh6 or Exo1 has either a much smaller (420, 423) or an opposite effect (422, 430) on repeat instability in mice. In agreement with mouse data, knocking out components of mismatch repair in other systems decreases CAG repeat instability rates (335, 389, 431, 432), although some studies have failed to detect an effect (161, 280, 433). Instability of other repeats, such as GAA, also seems to be promoted by components of MMR, but to a lesser extent, and not specifically by MutSβ (434–439).
Does MMR play as big of a role in mediating repeat expansions in RED patients as it does in mice and other experimental systems? Given the lack of experimental data, analysis of big data sets can address this question. Numerous genome-wide association studies have found that SNPs in the MLH1 and MSH3 genes act as genetic modifiers of HD's age of onset (128, 130, 325–327, 440). Likewise, an SNP in PMS2 modifies age of onset in several other poly(Q) diseases (441). Furthermore, an SNP in MSH3 affects the CTG repeat somatic instability level in DM1 patients (28, 440).
What molecular mechanism underlies MMR's propensity to promote repeat expansion? Based on in vitro data, it seems like MutSβ is very efficient at repairing short slip-outs of 1–3 CAG/CTG repeats, but it cannot handle long or clustered slip-outs (442). Other studies have found that the MutSβ (420, 424) and, possibly, MutSα (430) complexes bind to a CAG or CGG hairpin DNA efficiently but unproductively, meaning that cleavage does not follow binding (420, 424). Overall, it seems that secondary structures formed by expanded repeats, such as hairpins, can escape repair by MMR. Consequently, they might become incorporated into the DNA, resulting in a repeat expansion.
There are still plenty of unanswered questions in regard to the interaction between MMR and structure-forming repeats. What are the direct consequences of MutSβ's unproductive binding to a hairpin? What is the precise contribution of MutSα and MutL complexes in this process? What are the mechanistic details of MMR's interaction with non-hairpin-forming repeats, such as GAA? Future studies focusing on the role of MMR in repeat instability will shed light on these mysteries.
Base excision repair (BER)
BER is a conserved DNA repair pathway that corrects modified or damaged DNA bases. In the course of BER, a specialized DNA glycosylase removes the damaged DNA base to create an abasic site, after which AP endonuclease 1 makes a nick. Then DNA Pol β substitutes the abasic site for the correct nucleotide. In a case in which the abasic site cannot be easily removed, long patch BER comes into play. In this case, other DNA polymerases, including Pol δ, perform strand displacement synthesis, followed by flap removal by Fen1 and DNA ligation (reviewed in Ref. 443).
Substantial evidence suggests that BER promotes somatic CAG repeat instability. First, components of BER machinery, namely Neil1 (444), Ogg1 (445), and Pol β (446, 447), promote CAG repeat instability in mouse tissues or cell extracts. In yeast, BER promotes R-loop–mediated contractions of CAG repeats (374). Finally, Pol β is enriched at CAG repeats in a tissue-specific manner, with tissues prone to higher CAG repeat somatic instability showing a larger accumulation of the protein (448).
What is the molecular mechanism of BER-promoted repeat instability? At least in vitro, Pol β is inherently slippery when synthesizing over CAG repeats and often adds (449) or skips over (450, 451) a portion of the repeat. Importantly, purified MutSβ complex additionally promotes Polβ-mediated repeat instability (449). Furthermore, in vivo, MutSβ and Pol β colocalize as a response to DNA damage (451) and together accumulate at CAG repeats during DNA replication (449). Overall, it appears that BER and MMR cooperate together to promote CAG repeat instability (449, 451). It is possible that during BER, CAG repeats may fold into a hairpin or a loop-out structure, which, if incorporated into the nascent strand, would lead to a repeat expansion. MutSβ likely stabilizes this structure, thus enhancing the chances for expansion (449, 452) (Fig. 9A). Notably, the oxidative damage that accumulates in neurons with age creates more opportunities for repeat-destabilizing BER repair, establishing a so-called “toxic cycle” of oxidative damage and repeat elongation. This model explains age-dependent CAG repeat instability in post-mitotic tissues (445).
Figure 9.

Two hypothetical hybrid DNA repair pathways that could promote repeat instability. A, an oxidized nucleotide within a repetitive tract is being removed by the BER pathway to create a nick. In the course of strand displacement synthesis, the displaced flap might form a DNA secondary structure (e.g. a hairpin). The structure is stabilized by MutSβ and gets incorporated into the DNA, resulting in a repeat expansion. B, when a repeat is located in an actively transcribed gene, RNA polymerase might promote formation of a DNA secondary structure, such as S-DNA. These structures, when additionally stabilized by MutSβ, stall the next RNA polymerase, triggering NER. The NER repair might lead to either excision or incorporation of a secondary structure, leading to repeat contraction (this scenario is shown) or expansion (not shown), respectively.
NER
NER is another conserved DNA repair pathway. It removes DNA lesions that distort the DNA double helix: bulky DNA adducts, abasic sites, nicks, gaps, and DNA secondary structures. In the course of NER, the recognition of a lesion is followed by local DNA unwinding. Then the lesion-containing oligonucleotide is excised from the DNA, the remaining gap is filled, and the DNA ends are ligated back together. Two major NER pathways exist. First, global genomic NER removes DNA lesions genome-wide. Second, transcription-coupled NER (TC-NER) specializes in removing lesions that occur during transcription and impede RNA polymerase progression (453).
Remarkably, TC-NER appears to be responsible for transcription-induced repeat instability in dividing yeast (454, 455), human cell cultures (330, 456), and mouse neurons (457). The latter might reflect a role for this pathway in somatic expansions of CAG repeats (Fig. 9B). It remains unclear, however, whether NER also plays a role in intergenerational repeat instability. On the one hand, NER indeed promotes germline CAG repeat instability in flies (366). Additionally, the Cockayne syndrome group B (CSB) protein, which is required for TC-NER, promotes CGG repeat expansion during maternal, but not paternal, transmission in mice (458). On the other hand, intergenerational CAG repeat expansion is inhibited by CSB (459). We do not yet know how to explain this contradiction.
In sum, an emerging consensus in the field is that several hybrid pathways that combine components of MMR, BER, and NER could be responsible for repeat instability (Fig. 9). The length and type of a repetitive sequence, the replicational and transcriptional state of a cell, and the relative concentration of various repair proteins likely determine the choice of pathway and, ultimately, the repair outcome: repeat expansion, contraction, or no change (459).
Other DNA repair pathways
Genome-wide association studies have also identified the FAN1 gene, which encodes a flap endonuclease with a poorly understood biological role, as an age-of-onset modifier in several polyQ diseases (128, 130, 325–327, 441, 460). Moreover, the study of prediagnostic carriers of expanded CAG alleles in HD revealed that FAN1 SNPs can reliably predict age of onset (461). These SNPs likely affect the FAN1 transcription level. This is supported by the fact that an increased Fan1 expression level negatively correlates with HD age of onset and prevents CAG repeat expansion in human cell cultures (462). Thus, high expression of the FAN1 gene probably prevents CAG repeat somatic instability, thus delaying age of onset. This effect might not be exclusive to CAG repeats, as Fan1 also inhibits somatic expansions of CGG repeats in mice (463).
We do not yet know how Fan1 protects from repeat expansions mechanistically, as its normal biological role is not clear. Fan1 is a conserved flap endonuclease (464) that participates in interstrand cross-link repair either independently or as a part of the Falconi anemia repair pathway (reviewed in Ref. 465). Importantly, Fan1 might interact with MMR (466–468) and play a role in the processing of stalled replication forks (reviewed in Ref. 465). One proposed hypothesis is that Fan1 either prevents MMR proteins from binding to CAG repeat loop-outs or sequesters MMR proteins, thus counteracting MMR's promotion of CAG repeat expansion (462). Another hypothesis is that Fan1 may substitute for Exo1 in the course of an MMR-like process to promote an error-free repair of a repeat loop-out (463). It would be intriguing to investigate these possibilities in further detail.
Role of the chromatin environment in repeat instability
Expanded tracts of structure-forming repeats may dramatically alter the surrounding chromatin. They can lead to the formation of repressive histone marks, such as histone H3 Lys-9 and histone H4 Lys-20 trimethylation, decreased histone acetylation, and changes in DNA methylation. These epigenetic changes often contribute to disease phenotypes, as they can alter gene expression patterns (469, 470). The chromatin environment, in turn, can promote or inhibit repeat instability. For example, decreased nucleosome occupancy likely promotes large-scale GAA repeat expansion in yeast (367). Consistently, bioinformatic analysis revealed that, in human cells, expandable CAG repeats are located in open chromatin regions, unlike stable CAG repeats (471).
How can an open chromatin region facilitate repeat instability? First, the loss of nucleosomes undoubtedly facilitates formation of dynamic DNA structures, due to the release of negative DNA supercoils previously stored at nucleosomes (472). Second, large nucleosome-free regions might provide more opportunities for template switching during DNA replication (367). Third, open chromatin regions are more accessible to various DNA repair factors, particularly MMR (reviewed in Refs. 417 and 473). As such, open chromatin might facilitate error-prone DNA repair within repeats (417).
Furthermore, patterns of nucleosome assembly might be an important factor controlling repeat expansion (455, 474). The fact that ATTCT (475) and CAG (474, 476, 477) repeats are potent nucleosome-positioning elements in vitro likely contributes to their instability. Types of nucleosomes also can affect repeat instability, as evidenced by the recent finding that the presence of a specific histone variant contributes to stable maintenance of the CAG repeat (478). Importantly, other DNA-binding proteins also might alter repeat stability. For example, binding of the CCCTC-binding factor to the flanking regions of several CAG repeat diseases might stabilize this repeat (479).
A recent finding suggested a more general model for the role of chromatin structure on repeat expansions. Based on Hi-C data, it was suggested that the position of a repeat relative to the 3D genome architecture influences its propensity to expand. This was based on the observation that most disease-causing repeats are located at 3D chromatin boundaries. Moreover, CGG repeat expansion in the FMR1 promoter shifts the gene's position relative to the chromatin boundaries, which results in its complete silencing and ultimately causes the disease (480). This discovery might explain why some human tandem repeats are relatively stable and others are prone to dramatic expansions.
Why do disease-causing DNA repeats persist in the genome?
The majority of REDs are severe, debilitating disorders, like fragile X, FRDA, ALS, Huntington's disease, etc. Why are expandable DNA repeats not eliminated from the human genome if they are so detrimental to human health? Several hypotheses address this conundrum.
The most intuitive explanation is based on the fact that the majority of REDs have a late onset and typically exhibit symptoms when a person already has passed reproductive age. Thus, long repeat alleles are not subject to purifying selection. In support of this hypothesis, repeats in coding areas never expand as much as repeats in noncoding areas, likely because long repeats within a coding region are toxic and quickly counterselected. However, this hypothesis does not consider the existence of REDs with childhood or even congenital onset.
An alternative explanation could be that the existence of REDs is the price that eukaryotes pay for the use of tandem repeats in genome function and evolution. Numerous tandem repeats appear to be inherent to the eukaryotic genome: in humans, tandem repeats constitute nearly 3% of the genome, a similar proportion as protein-coding sequences (131). All tandem repeats are unstable in length, and the rate of their length instability exceeds the rate of point mutations by several orders of magnitude (132, 481).
What could be the advantages of having so many tandem repeats in a genome?
One well-known advantage is that tandem repeats serve as principal genomic structural elements, such as telomeres (482), centromeres (483), and regions of constitutive heterochromatin (484). Importantly, the same mechanisms that lead to repeat expansions in human disease are involved in the maintenance of those structural elements (reviewed in Ref. 344).
Another popular hypothesis is that the abundance of tandem repeats increases genome evolvability (132). Several pieces of evidence support this idea. First, tandem repeat loci (but not the lengths of the repeats) are conserved in related species (471, 485). At least some of these repeats seem to be actively maintained (486), which indicates that the existence of tandem repeats in these loci could be advantageous from an evolutionary standpoint. Sometimes, tandem repeat sites even evolve independently in homologous genes (132).
Second, about 15% of all human genes contain tandem repeats in their promoters or coding regions (reviewed in Ref. 132). Importantly, length variation of tandem repeats located in regulatory regions correlates with variance in the expression of these genes (487, 488). On top of that, tandem repeats located in gene promoters were shown to drive evolution of gene expression in yeast (489). Moreover, genes that contain tandem repeats in their coding or promoter regions are typically regulatory genes or transcription factors (reviewed in Ref. 132). Furthermore, a recent study found that DNA secondary structures, which commonly form within tandem repeats, are abundant in vivo and may control gene regulation (490). Taken together, these observations point to a potential role of tandem repeats in regulatory evolution.
Third, tandem repeats located in coding regions, typically in-frame trinucleotide repeats, could drive protein evolution. There are well-documented examples of tandem repeat tracts within protein-coding sequences whose length variation permits quick adaptation to a particular environmental change (132, 491, 492). One such example is the tandem repeats located in coding regions of circadian clock genes. The length of these repeats directly determines the circadian rhythms of various species, including fungi, flies, and, possibly, birds. In other words, these repeats act as a tool used when a species has to quickly change its regimen in response to habitat change (reviewed in Ref. 132). Another example is repetitive protein motifs located in genes that define vertebrate body morphology. Length variation of these repetitive sequences likely allows for rapid evolution of a body shape without dramatically altering overall body topology (493). For example, the ratio of poly(A)/polyQ repeat tract lengths in the canine RUNX-2 gene defines head morphology in dogs. Along the same line, deletion of a portion of a polyQP tandem repeat in the canine ALX4 gene causes dogs to grow an extra claw (493). In humans, on the other hand, an expansion of poly(A) repeats in the coding part of the HOXD13 and HOXA13 genes causes REDs, characterized by abnormal limb morphology, synpolydactyly 1 (46, 47), and HFG syndrome, respectively (48).
An additional proof of the tandem repeats' role in genome evolvability is their contribution to cancer progression. For example, it was recently found that tandem repeat length variation allows cells to quickly evolve during the early stages of malignant transformation (494). Furthermore, as discussed above, translocation junctions in cancer genomes often coincide with structure-forming tandem repeats (269).
Finally, we cannot resist mentioning a radical hypothesis suggested by a titan in the field of molecular evolution stipulating that expansions of unstable triplet repeats were at the heart of early life evolution (495).
Unanswered questions
To what extent do model experimental systems reflect repeat instability seen in humans?
The elephant in the room of the RED field is the substantial difference in how expandable repeats behave in humans as compared to model experimental systems. In humans, intergenerational transmissions of premutational or full mutation repeat alleles lead to a dramatic change in length with nearly 100% probability. As discussed above, the direction of instability (contraction or expansion) and its scale depend on multiple factors, including parental gender and age. There are likely different mechanisms that are responsible for various types of instability: contractions or expansions, small-scale or large-scale. Each of these types of instability seem to predominantly happen in a given germ cell type and within a given developmental window (496). That being said, expansions are prevalent over contractions in multiple circumstances (see above for examples).
Unlike human pedigrees, repeats studied in commonly used simple experimental systems, such as yeast or mammalian cell cultures, exhibit instability with a probability on the order of 10−3 to 10−6 (263) and predominantly contract rather than expand (142, 183, 497), with rare exceptions (154, 498). Furthermore, the expansions that are observed in these systems are smaller in scale compared with those observed in human pedigrees, even under selection for expansions (142, 161, 263). Mouse models, at first glance, better recapitulate the patterns of repeat instability in humans. Yet even mice rarely, if at all, exhibit the addition of hundreds of repeats characteristic of human pedigrees (230, 233, 499, 500). This is particularly surprising given the fact that mice generally tolerate longer tandem repeats better than humans, as illustrated by extremely long mouse telomeres (501). There is no coherent explanation for this difference between humans and model experimental systems. Understanding the reasons for this difference is an important goal for future studies.
Despite this difference, we are convinced that investigating mechanisms of repeat instability in model systems provides critical insights. One example is the role of MMR in somatic repeat instability, which became evident from experiments in yeast and mice and was then confirmed by genome-wide association studies in RED patients (see above). Other examples are the “ori-shift” and “ori-switch” models (292), discussed above. The inspiration for these models came from the observation that repeat instability in yeast, bacteria, and mammalian episomes uniformly depends on the repeat orientation and distance relative to the nearest origin of replication (see above). A decade later, an SNP in the closest origin of replication to the CGG repeat in fragile X locus was found to correlate with this repeat's replication program and propensity to expand in human pedigrees (502).
Overall, simple model systems provide the advantage of conducting detailed molecular genetic analysis of repeat instability mechanisms, which is impossible to do in humans and difficult in mice. The remaining challenge, however, is to relate a mechanism unraveled in a model system with a mode of repeat instability observed in a particular cell type and/or developmental window in humans. Overcoming this challenge would shed light on the origin of REDs and, in the long run, help with developing therapies.
Can REDs be cured?
The most important and urgent question facing the field of REDs is whether we can start to use the extensive knowledge about expandable repeats to fight these debilitating and currently incurable diseases. In attempts to develop therapies for REDs, researchers try to obviate repeats' toxicity on different levels. Typically, potential therapies aim at the downstream effects arising from repeat expansion to reduce their toxic consequences. The goal is to either reverse gene silencing (70, 81, 469, 503–505) or alleviate adverse effects of toxic RNA (98, 101, 203, 506–514) and/or toxic proteins (98, 101, 203, 506, 515). These methods, while worthy and promising, warrant several caveats. First, undergoing a therapy that targets downstream repeat–mediated toxicity would likely be a lifelong commitment for a patient. Second, the precise toxicity mechanisms are unestablished for the majority of REDs. Thus, it is unclear which toxic factor is the best therapeutic target. Third, targeting downstream effects of repeat expansions could alleviate disease symptoms post-onset but is unlikely to postpone disease onset.
Another direction could be inhibiting cellular pathways that promote somatic repeat expansion (252). For example, inhibiting MMR or BER could reduce somatic expansions of CAG repeats (426, 439). An obvious drawback of this approach is that inhibition of DNA repair pathways could dramatically increase cancer risk, as indicated by Lynch syndrome and other cancers linked to mutations in MMR (reviewed in Ref. 418).
Yet another promising therapeutic approach could be to prevent the formation of dynamic DNA structures that are at the heart of repeat expansions. One way to achieve this is to introduce endogenous oligonucleotides. The binding of an oligonucleotide to a single-stranded repetitive strand would compete with the formation of DNA secondary structure and therefore shift the equilibrium away from secondary structure formation. Indeed, the addition of (CTG)n or (CAG)n oligonucleotides complementary to the lagging strand template rescues replication fork stalling within the repeats in HeLa cells (336). The downside of this approach is that the half-life of DNA oligonucleotides is short in the human body. To address this problem, chemically modified oligonucleotides that do not easily degrade could be used. Alternatively, one could use peptide nucleic acids or polyamides that recognize expandable DNA repeats and preclude them from forming an alternative DNA conformation. As a proof of principle, GAA-specific polyamides were recently shown to rescue fork stalling at GAA repeats in patient-derived samples (339). Additionally, another recent report showed that a small molecule binding to an S-DNA structure promotes CAG repeat contractions in vivo (549).
Finally, directly promoting repeat contractions in somatic cells might present a powerful therapeutic approach. Several teams have achieved progress in this direction using transcription activator-like effector nucleases, zinc finger nucleases, or CRISPR-Cas9 to induce a DSB within or adjacent to a repeat. This approach promoted repeat contractions in bacterial (406), yeast (433, 516), primate (517), and human (206, 397, 518–524) cells (also reviewed in Ref. 525). However, at this stage of research, the safety of such technologies remains unclear. As with any other genome-editing techniques, the absence of off-target effects should be thoroughly verified. An additional concern is that besides repeat contractions, DSBs might also drive repeat expansions, which could counteract their therapeutic effects. Instead of inducing a DSB within a repetitive DNA, it might be safer to create a nick using Cas9 nickases. Indeed, the Dion laboratory has recently found that creating a nick within an expanded CAG repeat facilitates its contraction but not expansion (519).
Several methods can be utilized to drive repeat contractions in somatic tissues of RED patients. One possibility could be viral delivery of a Cas9 or related nuclease that make a targeted DSB or a nick at a repeat. Recently, for example, delivery of Cas9 targeted to the mutated HTT gene via adeno-associated virus reduced HD symptoms in mice (526). Alternatively, repeats may be shortened in situ in patient-derived iPSCs, followed by their differentiation. This approach allows one to scrupulously select for cells in which the repeat contracted successfully and to ensure the absence of off-target effects. The resultant cells can be transplanted back into the patient. A challenge to all of these approaches is that it probably would be very hard to “correct” the repeat's length in all affected cells. Thus, additional studies are needed to evaluate whether contracting a repeat in a subset of affected tissues could prove sufficient to reverse disease progression.
As of today, there is still no effective way to manipulate repeat length in RED patients. However, contemporary diagnostic methods allow one to precisely estimate the length of all known expandable repeats in a given person. As such, future parents might choose to know their carrier status for the most common REDs, such as HD, fragile X, or DM1. This type of genetic testing is especially encouraged for people with a family history of REDs. For example, screening programs that aim to identify carriers of DM1 have existed in Canada since 1988 (527). Aspiring parents who appear to be carriers of an RED can then choose to perform in vitro fertilization (IVF) with preimplantation genetic diagnosis (PGD) of the repeat length to ensure that their offspring do not inherit a disease-size repeat allele (528–532). Note, however, that the procedure of IVF/PGD comes with serious ethical and legal challenges (533). For example, a mother-to-be might want to perform prenatal testing of a fetus, because her male partner has a family history of HD. However, that might reveal his carrier status, even if he would prefer not to know it (534).
Concluding remarks
Almost 30 years have passed from the day when the first RED was discovered (6). Since then, scientists have unraveled the basic principles that underlie mechanisms of repeat instability. First, the repetitive tract forms a DNA secondary structure—typically transiently—during DNA unwinding in the course of replication, transcription, or repair. Then, after a biological transaction that often involves some complex interplay between various modes of DNA repair, the DNA secondary structure gets excised or incorporated into the DNA. As a result, the repetitive tract either contracts or expands, respectively. This paradigm applies to the majority of repeat instabilities, although the detailed mechanisms depend on the repeat sequence, length, and location within a genome as well as the organism or cell type examined.
As of now, the majority of REDs are incurable and nonpreventable. Because structure-forming DNA repeats are central to the development of these diseases, direct targeting of those repeats could be a promising therapeutic strategy. In the last few years, researchers have undertaken the first attempts aimed at developing such therapies (206, 339, 397, 518–522, 524, 525, 549). Hopefully, one day, these approaches will be used to combat REDs.
Acknowledgments
We thank Jorge Cebrian for sharing unpublished results and Julia Hisey and Ethan Hoffman for proofreading the manuscript.
This work was supported by National Institutes of Health Grant R35GM130322 and by a contribution from the White family (to S. M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
J. Cebrian, unpublished results.
- DM
- myotonic dystrophy
- HD
- Huntington's disease
- SBMA
- spinal and bulbar muscular atrophy
- RED
- repeat expansion disease
- SCA
- spinocerebellar ataxia
- DRPLA
- dentatorubral-pallidoluysian atrophy
- XDP
- X-linked dystonia parkinsonism
- FAME3
- familial adult myoclonic epilepsy 3
- FRDA
- Friedreich's ataxia
- HFG
- hand-foot-genital
- polyQ
- polyglutamine
- RAN
- repeat-associated non-ATG
- G4
- G-quadruplex
- DUE
- DNA-unwinding element
- DSB
- double-strand break
- RIM
- repeat-induced mutagenesis
- Pol
- polymerase
- ssDNA
- single-stranded DNA
- RPA
- replication protein A
- BIR
- break-induced replication
- SSA
- single-strand annealing
- EJ
- end-joining
- NHEJ
- nonhomologous EJ
- MMEJ
- microhomology-mediated EJ
- MMR
- mismatch repair
- BER
- base excision repair
- TC-NER
- transcription-coupled NER
- CSB
- Cockayne syndrome group B
- IVF
- in vitro fertilization
- PGD
- preimplantation genetic diagnosis.
References
- 1. Nettleship E. (1905) On heredity in the various forms of cataract. Rep. R. Lond. Ophthal. Hosp. 16, 179–246 [Google Scholar]
- 2. Fleischer B. (1918) Über myotonische Dystrophie mit Katarakt. Graefes. Arch. Klein. Ophthalmol. 96, 91–133 10.1007/BF02018704 [DOI] [Google Scholar]
- 3. Friedman J. E. (2011) Anticipation in hereditary disease: the history of a biomedical concept. Hum. Genet. 130, 705–714 10.1007/s00439-011-1022-9 [DOI] [PubMed] [Google Scholar]
- 4. Sherman S. L., Jacobs P. A., Morton N. E., Froster-Iskenius U., Howard-Peebles P. N., Nielsen K. B., Partington M. W., Sutherland G. R., Turner G., and Watson M. (1985) Further segregation analysis of the fragile X syndrome with special reference to transmitting males. Hum. Genet. 69, 289–299 10.1007/BF00291644 [DOI] [PubMed] [Google Scholar]
- 5. Yu S., Pritchard M., Kremer E., Lynch M., Nancarrow J., Baker E., Holman K., Mulley J. C., Warren S. T., Schlessinger D., Warren S. T., and Schlessinger D. (1991) Fragile X genotype characterized by an unstable region of DNA. Science 252, 1179–1181 10.1126/science.252.5009.1179 [DOI] [PubMed] [Google Scholar]
- 6. Fu Y. H., Kuhl D. P., Pizzuti A., Pieretti M., Sutcliffe J. S., Richards S., Verkerk A. J., Holden J. J., Fenwick R. G. Jr., and Warren S. T. (1991) Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67, 1047–1058 10.1016/0092-8674(91)90283-5 [DOI] [PubMed] [Google Scholar]
- 7. Oberlé I., Rousseau F., Heitz D., Kretz C., Devys D., Hanauer A., Boué J., Bertheas M. F., and Mandel J. L. (1991) Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102 10.1126/science.252.5009.1097 [DOI] [PubMed] [Google Scholar]
- 8. Verkerk A. J., Pieretti M., Sutcliffe J. S., Fu Y. H., Kuhl D. P., Pizzuti A., Reiner O., Richards S., Victoria M. F., and Zhang F. P. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 10.1016/0092-8674(91)90397-H [DOI] [PubMed] [Google Scholar]
- 9. Kremer E. J., Pritchard M., Lynch M., Yu S., Holman K., Baker E., Warren S. T., Schlessinger D., Sutherland G. R., and Richards R. I. (1991) Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252, 1711–1714 10.1126/science.1675488 [DOI] [PubMed] [Google Scholar]
- 10. La Spada A. R., Wilson E. M., Lubahn D. B., Harding A. E., and Fischbeck K. H. (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79 10.1038/352077a0 [DOI] [PubMed] [Google Scholar]
- 11. Harley H. G., Rundle S. A., Reardon W., Myring J., Crow S., Brook J. D., Harper P. S., and Shaw D. J. (1992) Unstable DNA sequence in myotonic dystrophy. Lancet 339, 1125–1128 10.1016/0140-6736(92)90729-M [DOI] [PubMed] [Google Scholar]
- 12. The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 10.1016/0092-8674(93)90585-e [DOI] [PubMed] [Google Scholar]
- 13. Metsu S., Rainger J. K., Debacker K., Bernhard B., Rooms L., Grafodatskaya D., Weksberg R., Fombonne E., Taylor M. S., Scherer S. W., Kooy R. F., and FitzPatrick D. R. (2014) A CGG-repeat expansion mutation in ZNF713 causes FRA7A: association with autistic spectrum disorder in two families. Hum. Mutat. 35, 1295–1300 10.1002/humu.22683 [DOI] [PubMed] [Google Scholar]
- 14. Hannan A. J. (2010) Tandem repeat polymorphisms: modulators of disease susceptibility and candidates for “missing heritability”. Trends Genet. 26, 59–65 10.1016/j.tig.2009.11.008 [DOI] [PubMed] [Google Scholar]
- 15. Sznajder Ł. J., and Swanson M. S. (2019) Short tandem repeat expansions and RNA-mediated pathogenesis in myotonic dystrophy. Int. J. Mol. Sci. 20, E3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gouw L. G., Castañeda M. A., McKenna C. K., Digre K. B., Pulst S. M., Perlman S., Lee M. S., Gomez C., Fischbeck K., Gagnon D., Storey E., Bird T., Jeri F. R., and Ptácek L. J. (1998) Analysis of the dynamic mutation in the SCA7 gene shows marked parental effects on CAG repeat transmission. Hum. Mol. Genet. 7, 525–532 10.1093/hmg/7.3.525 [DOI] [PubMed] [Google Scholar]
- 17. David G., Abbas N., Stevanin G., Dürr A., Yvert G., Cancel G., Weber C., Imbert G., Saudou F., Antoniou E., Drabkin H., Gemmill R., Giunti P., Benomar A., Wood N., Ruberg M., Agid Y., Mandel J. L., and Brice A. (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70 10.1038/ng0997-65 [DOI] [PubMed] [Google Scholar]
- 18. Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., Kawakami H., Nakamura S., Nishimura M., and Akiguchi I. (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 8, 221–228 10.1038/ng1194-221 [DOI] [PubMed] [Google Scholar]
- 19. Giuffrida S., Lanza S., Restivo D. A., Saponara R., Valvo S. C., Le Pira F., Trovato Salinaro A., Spinella F., Nicoletti A., and Condorelli D. F. (1999) Clinical and molecular analysis of 11 Sicilian SCA2 families: influence of gender on age at onset. Eur. J. Neurol. 6, 301–307 10.1046/j.1468-1331.1999.630301.x [DOI] [PubMed] [Google Scholar]
- 20. Cancel G., Dürr A., Didierjean O., Imbert G., Burk K., Lezin A., Belal S., Benomar A., Abada-Bendib M., Vial C., Guimaraes J., Chneiweiss H., Stevanin G., Yvert G., Abbas N., et al. (1997) Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. Hum. Mol. Genet. 6, 709–715 10.1093/hmg/6.5.709 [DOI] [PubMed] [Google Scholar]
- 21. Imbert G., Saudou F., Yvert G., Devys D., Trottier Y., Garnier J. M., Weber C., Mandel J. L., Cancel G., Abbas N., Dürr A., Didierjean O., Stevanin G., Agid Y., and Brice A. (1996) Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 14, 285–291 10.1038/ng1196-285 [DOI] [PubMed] [Google Scholar]
- 22. Seixas A. I., Loureiro J. R., Costa C., Ordóñez-Ugalde A., Marcelino H., Oliveira C. L., Loureiro J. L., Dhingra A., Brandão E., Cruz V. T., Timóteo A., Quintáns B., Rouleau G. A., Rizzu P., Carracedo A., Bessa J., Heutink P., Sequeiros J., Sobrido M. J., Coutinho P., and Silveira I. (2017) A pentanucleotide ATTTC repeat insertion in the non-coding region of DAB1, mapping to SCA37, causes spinocerebellar ataxia. Am. J. Hum. Genet. 101, 87–103 10.1016/j.ajhg.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ranen N. G., Stine O. C., Abbott M. H., Sherr M., Codori A. M., Franz M. L., Chao N. I., Chung A. S., Pleasant N., and Callahan C. (1995) Anticipation and instability of IT-15 (CAG)n repeats in parent-offspring pairs with Huntington disease. Am. J. Hum. Genet. 57, 593–602 [PMC free article] [PubMed] [Google Scholar]
- 24. Trottier Y., Biancalana V., and Mandel J. L. (1994) Instability of CAG repeats in Huntington's disease: relation to parental transmission and age of onset. J. Med. Genet. 31, 377–382 10.1136/jmg.31.5.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vital M., Bidegain E., Raggio V., and Esperon P. (2016) Molecular characterization of genes modifying the age at onset in Huntington's disease in Uruguayan patients. Int. J. Neurosci. 126, 510–513 10.3109/00207454.2015.1036422 [DOI] [PubMed] [Google Scholar]
- 26. Andrew S. E., Goldberg Y. P., Kremer B., Telenius H., Theilmann J., Adam S., Starr E., Squitieri F., Lin B., and Kalchman M. A. (1993) The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat. Genet. 4, 398–403 10.1038/ng0893-398 [DOI] [PubMed] [Google Scholar]
- 27. Morales F., Couto J. M., Higham C. F., Hogg G., Cuenca P., Braida C., Wilson R. H., Adam B., del Valle G., Brian R., Sittenfeld M., Ashizawa T., Wilcox A., Wilcox D. E., and Monckton D. G. (2012) Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum. Mol. Genet. 21, 3558–3567 10.1093/hmg/dds185 [DOI] [PubMed] [Google Scholar]
- 28. Morales F., Vásquez M., Santamaría C., Cuenca P., Corrales E., and Monckton D. G. (2016) A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst.) 40, 57–66 10.1016/j.dnarep.2016.01.001 [DOI] [PubMed] [Google Scholar]
- 29. Koide R., Ikeuchi T., Onodera O., Tanaka H., Igarashi S., Endo K., Takahashi H., Kondo R., Ishikawa A., and Hayashi T. (1994) Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 6, 9–13 10.1038/ng0194-9 [DOI] [PubMed] [Google Scholar]
- 30. Nagafuchi S., Yanagisawa H., Sato K., Shirayama T., Ohsaki E., Bundo M., Takeda T., Tadokoro K., Kondo I., and Murayama N. (1994) Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat. Genet. 6, 14–18 10.1038/ng0194-14 [DOI] [PubMed] [Google Scholar]
- 31. Westenberger A., Reyes C. J., Saranza G., Dobricic V., Hanssen H., Domingo A., Laabs B. H., Schaake S., Pozojevic J., Rakovic A., Grütz K., Begemann K., Walter U., Dressler D., Bauer P., et al. (2019) A hexanucleotide repeat modifies expressivity of X-linked dystonia parkinsonism. Ann. Neurol. 85, 812–822 10.1002/ana.25488 [DOI] [PubMed] [Google Scholar]
- 32. Florian R. T., Kraft F., Leitão E., Kaya S., Klebe S., Magnin E., van Rootselaar A. F., Buratti J., Kühnel T., Schröder C., Giesselmann S., Tschernoster N., Altmueller J., Lamiral A., Keren B., et al. (2019) Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat. Commun. 10, 4919 10.1038/s41467-019-12763-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Al-Mahdawi S., Ging H., Bayot A., Cavalcanti F., La Cognata V., Cavallaro S., Giunti P., and Pook M. A. (2018) Large interruptions of GAA repeat expansion mutations in Friedreich ataxia are very rare. Front. Cell Neurosci. 12, 443 10.3389/fncel.2018.00443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Monrós E., Moltó M. D., Martínez F., Cañizares J., Blanca J., Vílchez J. J., Prieto F., de Frutos R., and Palau F. (1997) Phenotype correlation and intergenerational dynamics of the Friedreich ataxia GAA trinucleotide repeat. Am. J. Hum. Genet. 61, 101–110 10.1086/513887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fournier C., Barbier M., Camuzat A., Anquetil V., Lattante S., Clot F., Cazeneuve C., Rinaldi D., Couratier P., Deramecourt V., Sabatelli M., Belliard S., Vercelletto M., Forlani S., Jornea L., et al. (2019) Relations between C9orf72 expansion size in blood, age at onset, age at collection and transmission across generations in patients and presymptomatic carriers. Neurobiol. Aging 74, 234.e1–234.e8 10.1016/j.neurobiolaging.2018.09.010 [DOI] [PubMed] [Google Scholar]
- 36. Nordin A., Akimoto C., Wuolikainen A., Alstermark H., Jonsson P., Birve A., Marklund S. L., Graffmo K. S., Forsberg K., Brännstrom T., and Andersen P. M. (2015) Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet. 24, 3133–3142 10.1093/hmg/ddv064 [DOI] [PubMed] [Google Scholar]
- 37. Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A. M., Kaganovich A., et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wiethoff S., O'Connor E., Haridy N. A., Nethisinghe S., Wood N., Giunti P., Bettencourt C., and Houlden H. (2018) Sequencing analysis of the SCA6 CAG expansion excludes an influence of repeat interruptions on disease onset. J. Neurol. Neurosurg. Psychiatry 89, 1226–1227 10.1136/jnnp-2017-317253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hyppönen J., Äikiä M., Joensuu T., Julkunen P., Danner N., Koskenkorva P., Vanninen R., Lehesjoki A. E., Mervaala E., and Kälviäinen R. (2015) Refining the phenotype of Unverricht-Lundborg disease (EPM1): a population-wide Finnish study. Neurology 84, 1529–1536 10.1212/WNL.0000000000001466 [DOI] [PubMed] [Google Scholar]
- 40. Ida C. M., Butz M. L., Lundquist P. A., and Dawson D. B. (2018) C9orf72 repeat expansion frequency among patients with Huntington disease genetic testing. Neurodegener. Dis. 18, 239–253 10.1159/000492499 [DOI] [PubMed] [Google Scholar]
- 41. Aydin G., Dekomien G., Hoffjan S., Gerding W. M., Epplen J. T., and Arning L. (2018) Frequency of SCA8, SCA10, SCA12, SCA36, FXTAS and C9orf72 repeat expansions in SCA patients negative for the most common SCA subtypes. BMC Neurol. 18, 3 10.1186/s12883-017-1009-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mundlos S., Otto F., Mundlos C., Mulliken J. B., Aylsworth A. S., Albright S., Lindhout D., Cole W. G., Henn W., Knoll J. H., Owen M. J., Mertelsmann R., Zabel B. U., and Olsen B. R. (1997) Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 89, 773–779 10.1016/S0092-8674(00)80260-3 [DOI] [PubMed] [Google Scholar]
- 43. Laumonnier F., Ronce N., Hamel B. C., Thomas P., Lespinasse J., Raynaud M., Paringaux C., Van Bokhoven H., Kalscheuer V., Fryns J. P., Chelly J., Moraine C., and Briault S. (2002) Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am. J. Hum. Genet. 71, 1450–1455 10.1086/344661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen Y. C., Auer-Grumbach M., Matsukawa S., Zitzelsberger M., Themistocleous A. C., Strom T. M., Samara C., Moore A. W., Cho L. T., Young G. T., Weiss C., Schabhüttl M., Stucka R., Schmid A. B., Parman Y., et al. (2015) Transcriptional regulator PRDM12 is essential for human pain perception. Nat. Genet. 47, 803–808 10.1038/ng.3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brais B., Bouchard J. P., Xie Y. G., Rochefort D. L., Chrétien N., Tomé F. M., Lafrenière R. G., Rommens J. M., Uyama E., Nohira O., Blumen S., Korczyn A. D., Heutink P., Mathieu J., Duranceau A., et al. (1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 18, 164–167 10.1038/ng0298-164 [DOI] [PubMed] [Google Scholar]
- 46. Muragaki Y., Mundlos S., Upton J., and Olsen B. R. (1996) Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science 272, 548–551 10.1126/science.272.5261.548 [DOI] [PubMed] [Google Scholar]
- 47. Akarsu A. N., Stoilov I., Yilmaz E., Sayli B. S., and Sarfarazi M. (1996) Genomic structure of HOXD13 gene: a nine polyalanine duplication causes synpolydactyly in two unrelated families. Hum. Mol. Genet. 5, 945–952 10.1093/hmg/5.7.945 [DOI] [PubMed] [Google Scholar]
- 48. Goodman F. R., Bacchelli C., Brady A. F., Brueton L. A., Fryns J. P., Mortlock D. P., Innis J. W., Holmes L. B., Donnenfeld A. E., Feingold M., Beemer F. A., Hennekam R. C., and Scambler P. J. (2000) Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am. J. Hum. Genet. 67, 197–202 10.1086/302961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guerrini R., Moro F., Kato M., Barkovich A. J., Shiihara T., McShane M. A., Hurst J., Loi M., Tohyama J., Norci V., Hayasaka K., Kang U. J., Das S., and Dobyns W. B. (2007) Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology 69, 427–433 10.1212/01.wnl.0000266594.16202.c1 [DOI] [PubMed] [Google Scholar]
- 50. Kato M., Saitoh S., Kamei A., Shiraishi H., Ueda Y., Akasaka M., Tohyama J., Akasaka N., and Hayasaka K. (2007) A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet. 81, 361–366 10.1086/518903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weese-Mayer D. E., Berry-Kravis E. M., Zhou L., Maher B. S., Silvestri J. M., Curran M. E., and Marazita M. L. (2003) Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am. J. Med. Genet. A 123A, 267–278 10.1002/ajmg.a.20527 [DOI] [PubMed] [Google Scholar]
- 52. Brown L. Y., Odent S., David V., Blayau M., Dubourg C., Apacik C., Delgado M. A., Hall B. D., Reynolds J. F., Sommer A., Wieczorek D., Brown S. A., and Muenke M. (2001) Holoprosencephaly due to mutations in ZIC2: alanine tract expansion mutations may be caused by parental somatic recombination. Hum. Mol. Genet. 10, 791–796 10.1093/hmg/10.8.791 [DOI] [PubMed] [Google Scholar]
- 53. De Baere E., Dixon M. J., Small K. W., Jabs E. W., Leroy B. P., Devriendt K., Gillerot Y., Mortier G., Meire F., Van Maldergem L., Courtens W., Hjalgrim H., Huang S., Liebaers I., Van Regemorter N., et al. (2001) Spectrum of FOXL2 gene mutations in blepharophimosis-ptosis-epicanthus inversus (BPES) families demonstrates a genotype–phenotype correlation. Hum. Mol. Genet. 10, 1591–1600 10.1093/hmg/10.15.1591 [DOI] [PubMed] [Google Scholar]
- 54. Liquori C. L., Ricker K., Moseley M. L., Jacobsen J. F., Kress W., Naylor S. L., Day J. W., and Ranum L. P. (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867 10.1126/science.1062125 [DOI] [PubMed] [Google Scholar]
- 55. Gheno T. C., Furtado G. V., Saute J. A. M., Donis K. C., Fontanari A. M. V., Emmel V. E., Pedroso J. L., Barsottini O., Godeiro-Junior C., van der Linden H., Ternes Pereira E., Cintra V. P., Marques W. Jr., de Castilhos R. M., Alonso I., et al. (2017) Spinocerebellar ataxia type 10: common haplotype and disease progression rate in Peru and Brazil. Eur. J. Neurol 24, 892–e36 10.1111/ene.13281 [DOI] [PubMed] [Google Scholar]
- 56. Almeida T., Alonso I., Martins S., Ramos E. M., Azevedo L., Ohno K., Amorim A., Saraiva-Pereira M. L., Jardim L. B., Matsuura T., Sequeiros J., and Silveira I. (2009) Ancestral origin of the ATTCT repeat expansion in spinocerebellar ataxia type 10 (SCA10). PLoS ONE 4, e4553 10.1371/journal.pone.0004553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Landrian I., McFarland K. N., Liu J., Mulligan C. J., Rasmussen A., and Ashizawa T. (2017) Inheritance patterns of ATCCT repeat interruptions in spinocerebellar ataxia type 10 (SCA10) expansions. PLoS ONE 12, e0175958 10.1371/journal.pone.0175958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bachinski L. L., Udd B., Meola G., Sansone V., Bassez G., Eymard B., Thornton C. A., Moxley R. T., Harper P. S., Rogers M. T., Jurkat-Rott K., Lehmann-Horn F., Wieser T., Gamez J., Navarro C., et al. (2003) Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am. J. Hum. Genet. 73, 835–848 10.1086/378566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. De Rooij K. E., De Koning Gans P. A., Skraastad M. I., Belfroid R. D., Vegter-Van Der Vlis M., Roos R. A., Bakker E., Van Ommen G. J., Den Dunnen J. T., and Losekoot M. (1993) Dynamic mutation in Dutch Huntington's disease patients: increased paternal repeat instability extending to within the normal size range. J. Med. Genet. 30, 996–1002 10.1136/jmg.30.12.996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maia N., Loureiro J. R., Oliveira B., Marques I., Santos R., Jorge P., and Martins S. (2017) Contraction of fully expanded FMR1 alleles to the normal range: predisposing haplotype or rare events? J. Hum. Genet. 62, 269–275 10.1038/jhg.2016.122 [DOI] [PubMed] [Google Scholar]
- 61. Owens K. M., Quinonez S. C., Thomas P. E., Keegan C. E., Lefebvre N., Roulston D., Larsen C. A., Stadler H. S., and Innis J. W. (2013) Analysis of de novo HOXA13 polyalanine expansions supports replication slippage without repair in their generation. Am. J. Med. Genet. A 161A, 1019–1027 10.1002/ajmg.a.35843 [DOI] [PubMed] [Google Scholar]
- 62. Campuzano V., Montermini L., Moltò M. D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., Zara F., Cañizares J., Koutnikova H., Bidichandani S. I., Gellera C., et al. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427 10.1126/science.271.5254.1423 [DOI] [PubMed] [Google Scholar]
- 63. Lalioti M. D., Scott H. S., and Antonarakis S. E. (1999) Altered spacing of promoter elements due to the dodecamer repeat expansion contributes to reduced expression of the cystatin B gene in EPM1. Hum. Mol. Genet. 8, 1791–1798 10.1093/hmg/8.9.1791 [DOI] [PubMed] [Google Scholar]
- 64. Kekou K., Sofocleous C., Papadimas G., Petichakis D., Svingou M., Pons R. M., Vorgia P., Gika A., Kitsiou-Tzeli S., and Kanavakis E. (2016) A dynamic trinucleotide repeat (TNR) expansion in the DMD gene. Mol. Cell Probes 30, 254–260 10.1016/j.mcp.2016.07.001 [DOI] [PubMed] [Google Scholar]
- 65. Zink A. M., Wohlleber E., Engels H., Rødningen O. K., Ravn K., Heilmann S., Rehnitz J., Katzorke N., Kraus C., Blichfeldt S., Hoffmann P., Reutter H., Brockschmidt F. F., Kreiss-Nachtsheim M., Vogt P. H., et al. (2014) Microdeletions including FMR1 in three female patients with intellectual disability—further delineation of the phenotype and expression studies. Mol. Syndromol. 5, 65–75 10.1159/000357962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hammond L. S., Macias M. M., Tarleton J. C., and Shashidhar Pai G. (1997) Fragile X syndrome and deletions in FMR1: new case and review of the literature. Am. J. Med. Genet. 72, 430–434 10.1002/(SICI)1096-8628(19971112)72:4<430::AID-AJMG11>3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- 67. Ishiura H., Doi K., Mitsui J., Yoshimura J., Matsukawa M. K., Fujiyama A., Toyoshima Y., Kakita A., Takahashi H., Suzuki Y., Sugano S., Qu W., Ichikawa K., Yurino H., Higasa K., et al. (2018) Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 50, 581–590 10.1038/s41588-018-0067-2 [DOI] [PubMed] [Google Scholar]
- 68. Zeng S., Zhang M. Y., Wang X. J., Hu Z. M., Li J. C., Li N., Wang J. L., Liang F., Yang Q., Liu Q., Fang L., Hao J. W., Shi F. D., Ding X. B., Teng J. F., et al. (2019) Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J. Med. Genet. 56, 265–270 10.1136/jmedgenet-2018-105484 [DOI] [PubMed] [Google Scholar]
- 69. Corbett M. A., Kroes T., Veneziano L., Bennett M. F., Florian R., Schneider A. L., Coppola A., Licchetta L., Franceschetti S., Suppa A., Wenger A., Mei D., Pendziwiat M., Kaya S., Delledonne M., et al. (2019) Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun. 10, 4920 10.1038/s41467-019-12671-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Soragni E., Herman D., Dent S. Y., Gottesfeld J. M., Wells R. D., and Napierala M. (2008) Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia. Nucleic Acids Res. 36, 6056–6065 10.1093/nar/gkn604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li Y., Lu Y., Polak U., Lin K., Shen J., Farmer J., Seyer L., Bhalla A. D., Rozwadowska N., Lynch D. R., Butler J. S., and Napierala M. (2015) Expanded GAA repeats impede transcription elongation through the FXN gene and induce transcriptional silencing that is restricted to the FXN locus. Hum. Mol. Genet. 24, 6932–6943 10.1093/hmg/ddv397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Groh M., Lufino M. M., Wade-Martins R., and Gromak N. (2014) R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 10, e1004318 10.1371/journal.pgen.1004318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ohshima K., Montermini L., Wells R. D., and Pandolfo M. (1998) Inhibitory effects of expanded GAA.TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo. J. Biol. Chem. 273, 14588–14595 10.1074/jbc.273.23.14588 [DOI] [PubMed] [Google Scholar]
- 74. Grabczyk E., Mancuso M., and Sammarco M. C. (2007) A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 35, 5351–5359 10.1093/nar/gkm589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Neil A. J., Liang M. U., Khristich A. N., Shah K. A., and Mirkin S. M. (2018) RNA-DNA hybrids promote the expansion of Friedreich's ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res. 46, 3487–3497 10.1093/nar/gky099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kumari D., and Usdin K. (2012) Is Friedreich ataxia an epigenetic disorder? Clin. Epigenetics 4, 2 10.1186/1868-7083-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Koutnikova H., Campuzano V., Foury F., Dollé P., Cazzalini O., and Koenig M. (1997) Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 16, 345–351 10.1038/ng0897-345 [DOI] [PubMed] [Google Scholar]
- 78. Anzovino A., Lane D. J., Huang M. L., and Richardson D. R. (2014) Fixing frataxin: “ironing out” the metabolic defect in Friedreich's ataxia. Br. J. Pharmacol. 171, 2174–2190 10.1111/bph.12470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Loomis E. W., Sanz L. A., Chédin F., and Hagerman P. J. (2014) Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 10, e1004294 10.1371/journal.pgen.1004294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pretto D. I., Mendoza-Morales G., Lo J., Cao R., Hadd A., Latham G. J., Durbin-Johnson B., Hagerman R., and Tassone F. (2014) CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet. 51, 309–318 10.1136/jmedgenet-2013-102021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kumari D., Gazy I., and Usdin K. (2019) Pharmacological reactivation of the silenced FMR1 gene as a targeted therapeutic approach for fragile X syndrome. Brain Sci. 9, E39 10.3390/brainsci9020039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hansen R. S., Canfield T. K., Lamb M. M., Gartler S. M., and Laird C. D. (1993) Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell 73, 1403–1409 10.1016/0092-8674(93)90365-W [DOI] [PubMed] [Google Scholar]
- 83. Santoro M., Fontana L., Maiorca F., Centofanti F., Massa R., Silvestri G., Novelli G., and Botta A. (2018) Expanded [CCTG]n repetitions are not associated with abnormal methylation at the CNBP locus in myotonic dystrophy type 2 (DM2) patients. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 917–924 10.1016/j.bbadis.2017.12.037 [DOI] [PubMed] [Google Scholar]
- 84. Botta A., Caldarola S., Vallo L., Bonifazi E., Fruci D., Gullotta F., Massa R., Novelli G., and Loreni F. (2006) Effect of the [CCTG]n repeat expansion on ZNF9 expression in myotonic dystrophy type II (DM2). Biochim. Biophys. Acta 1762, 329–334 10.1016/j.bbadis.2005.11.004 [DOI] [PubMed] [Google Scholar]
- 85. Qawasmi L., Braun M., Guberman I., Cohen E., Naddaf L., Mellul A., Matilainen O., Roitenberg N., Share D., Stupp D., Chahine H., Cohen E., Garcia S. M. D. A., and Tabach Y. (2019) Expanded CUG repeats trigger disease phenotype and expression changes through the RNAi machinery in C. elegans. J. Mol. Biol. 431, 1711–1728 10.1016/j.jmb.2019.03.003 [DOI] [PubMed] [Google Scholar]
- 86. Yu Z., Teng X., and Bonini N. M. (2011) Triplet repeat-derived siRNAs enhance RNA-mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet. 7, e1001340 10.1371/journal.pgen.1001340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hu J., Rong Z., Gong X., Zhou Z., Sharma V. K., Xing C., Watts J. K., Corey D. R., and Mootha V. V. (2018) Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis-splicing in Fuchs' dystrophy. Hum. Mol. Genet. 27, 1015–1026 10.1093/hmg/ddy018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Meola G., and Cardani R. (2015) Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta 1852, 594–606 10.1016/j.bbadis.2014.05.019 [DOI] [PubMed] [Google Scholar]
- 89. Cerro-Herreros E., Chakraborty M., Pérez-Alonso M., Artero R., and Llamusí B. (2017) Expanded CCUG repeat RNA expression in Drosophila heart and muscle trigger myotonic dystrophy type 1-like phenotypes and activate autophagocytosis genes. Sci. Rep. 7, 2843 10.1038/s41598-017-02829-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pascual M., Vicente M., Monferrer L., and Artero R. (2006) The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation 74, 65–80 10.1111/j.1432-0436.2006.00060.x [DOI] [PubMed] [Google Scholar]
- 91. Wojciechowska M., and Krzyzosiak W. J. (2011) Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum. Mol. Genet. 20, 3811–3821 10.1093/hmg/ddr299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Niimi Y., Takahashi M., Sugawara E., Umeda S., Obayashi M., Sato N., Ishiguro T., Higashi M., Eishi Y., Mizusawa H., and Ishikawa K. (2013) Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology 33, 600–611 10.1111/neup.12032 [DOI] [PubMed] [Google Scholar]
- 93. Kobayashi H., Abe K., Matsuura T., Ikeda Y., Hitomi T., Akechi Y., Habu T., Liu W., Okuda H., and Koizumi A. (2011) Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 89, 121–130 10.1016/j.ajhg.2011.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yuva-Aydemir Y., Almeida S., and Gao F. B. (2018) Insights into C9ORF72-related ALS/FTD from Drosophila and iPSC models. Trends Neurosci. 41, 457–469 10.1016/j.tins.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jain A., and Vale R. D. (2017) RNA phase transitions in repeat expansion disorders. Nature 546, 243–247 10.1038/nature22386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Miller J. W., Urbinati C. R., Teng-Umnuay P., Stenberg M. G., Byrne B. J., Thornton C. A., and Swanson M. S. (2000) Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448 10.1093/emboj/19.17.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kanadia R. N., Johnstone K. A., Mankodi A., Lungu C., Thornton C. A., Esson D., Timmers A. M., Hauswirth W. W., and Swanson M. S. (2003) A muscleblind knockout model for myotonic dystrophy. Science 302, 1978–1980 10.1126/science.1088583 [DOI] [PubMed] [Google Scholar]
- 98. Jiang J., Zhu Q., Gendron T. F., Saberi S., McAlonis-Downes M., Seelman A., Stauffer J. E., Jafar-Nejad P., Drenner K., Schulte D., Chun S., Sun S., Ling S. C., Myers B., Engelhardt J., et al. (2016) Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550 10.1016/j.neuron.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Reddy K., Zamiri B., Stanley S. Y., Macgregor R. B. Jr., and Pearson C. E. (2013) The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem. 288, 9860–9866 10.1074/jbc.C113.452532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G. Y., Karydas A., et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Conlon E. G., Lu L., Sharma A., Yamazaki T., Tang T., Shneider N. A., and Manley J. L. (2016) The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 5, e17820 10.7554/eLife.17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shen J., Zhang Y., Zhao S., Mao H., Wang Z., Li H., and Xu Z. (2018) Purα repaired expanded hexanucleotide GGGGCC repeat noncoding RNA-caused neuronal toxicity in Neuro-2a cells. Neurotox. Res. 33, 693–701 10.1007/s12640-017-9803-0 [DOI] [PubMed] [Google Scholar]
- 103. Xu Z., Poidevin M., Li X., Li Y., Shu L., Nelson D. L., Li H., Hales C. M., Gearing M., Wingo T. S., and Jin P. (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 7778–7783 10.1073/pnas.1219643110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mori K., Lammich S., Mackenzie I. R., Forné I., Zilow S., Kretzschmar H., Edbauer D., Janssens J., Kleinberger G., Cruts M., Herms J., Neumann M., Van Broeckhoven C., Arzberger T., and Haass C. (2013) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 125, 413–423 10.1007/s00401-013-1088-7 [DOI] [PubMed] [Google Scholar]
- 105. Burguete A. S., Almeida S., Gao F. B., Kalb R., Akins M. R., and Bonini N. M. (2015) GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife 4, e08881 10.7554/eLife.08881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Freibaum B. D., Lu Y., Lopez-Gonzalez R., Kim N. C., Almeida S., Lee K. H., Badders N., Valentine M., Miller B. L., Wong P. C., Petrucelli L., Kim H. J., Gao F. B., and Taylor J. P. (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133 10.1038/nature14974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lavoie H., Debeane F., Trinh Q. D., Turcotte J. F., Corbeil-Girard L. P., Dicaire M. J., Saint-Denis A., Pagé M., Rouleau G. A., and Brais B. (2003) Polymorphism, shared functions and convergent evolution of genes with sequences coding for polyalanine domains. Hum. Mol. Genet. 12, 2967–2979 10.1093/hmg/ddg329 [DOI] [PubMed] [Google Scholar]
- 108. Li L., Ng N. K., Koon A. C., and Chan H. Y. (2017) Expanded polyalanine tracts function as nuclear export signals and promote protein mislocalization via eEF1A1 factor. J. Biol. Chem. 292, 5784–5800 10.1074/jbc.M116.763599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Messaed C., and Rouleau G. A. (2009) Molecular mechanisms underlying polyalanine diseases. Neurobiol. Dis. 34, 397–405 10.1016/j.nbd.2009.02.013 [DOI] [PubMed] [Google Scholar]
- 110. Pelassa I., Corà D., Cesano F., Monje F. J., Montarolo P. G., and Fiumara F. (2014) Association of polyalanine and polyglutamine coiled coils mediates expansion disease-related protein aggregation and dysfunction. Hum. Mol. Genet. 23, 3402–3420 10.1093/hmg/ddu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Perutz M. F., Johnson T., Suzuki M., and Finch J. T. (1994) Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. U.S.A. 91, 5355–5358 10.1073/pnas.91.12.5355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Takeuchi T., and Nagai Y. (2017) Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci. 7, E128 10.3390/brainsci7100128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., and Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 10.1126/science.277.5334.1990 [DOI] [PubMed] [Google Scholar]
- 114. Koob M. D., Moseley M. L., Schut L. J., Benzow K. A., Bird T. D., Day J. W., and Ranum L. P. (1999) An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat. Genet. 21, 379–384 10.1038/7710 [DOI] [PubMed] [Google Scholar]
- 115. Moseley M. L., Zu T., Ikeda Y., Gao W., Mosemiller A. K., Daughters R. S., Chen G., Weatherspoon M. R., Clark H. B., Ebner T. J., Day J. W., and Ranum L. P. (2006) Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 38, 758–769 10.1038/ng1827 [DOI] [PubMed] [Google Scholar]
- 116. Zu T., Gibbens B., Doty N. S., Gomes-Pereira M., Huguet A., Stone M. D., Margolis J., Peterson M., Markowski T. W., Ingram M. A., Nan Z., Forster C., Low W. C., Schoser B., Somia N. V., et al. (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U.S.A. 108, 260–265 10.1073/pnas.1013343108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mori K., Weng S. M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H. A., Cruts M., Van Broeckhoven C., Haass C., and Edbauer D. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 10.1126/science.1232927 [DOI] [PubMed] [Google Scholar]
- 118. Gendron T. F., Bieniek K. F., Zhang Y. J., Jansen-West K., Ash P. E., Caulfield T., Daughrity L., Dunmore J. H., Castanedes-Casey M., Chew J., Cosio D. M., van Blitterswijk M., Lee W. C., Rademakers R., Boylan K. B., et al. (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844 10.1007/s00401-013-1192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Mori K., Arzberger T., Grässer F. A., Gijselinck I., May S., Rentzsch K., Weng S. M., Schludi M. H., van der Zee J., Cruts M., Van Broeckhoven C., Kremmer E., Kretzschmar H. A., Haass C., and Edbauer D. (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893 10.1007/s00401-013-1189-3 [DOI] [PubMed] [Google Scholar]
- 120. Ash P. E., Bieniek K. F., Gendron T. F., Caulfield T., Lin W. L., Dejesus-Hernandez M., van Blitterswijk M. M., Jansen-West K., Paul J. W. 3rd, Rademakers R., Boylan K. B., Dickson D. W., and Petrucelli L. (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 10.1016/j.neuron.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Freibaum B. D., and Taylor J. P. (2017) The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front. Mol. Neurosci. 10, 35 10.3389/fnmol.2017.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lopez-Gonzalez R., Lu Y., Gendron T. F., Karydas A., Tran H., Yang D., Petrucelli L., Miller B. L., Almeida S., and Gao F. B. (2016) Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391 10.1016/j.neuron.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cleary J. D., Pattamatta A., and Ranum L. P. W. (2018) Repeat-associated non-ATG (RAN) translation. J. Biol. Chem. 293, 16127–16141 10.1074/jbc.R118.003237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cleary J. D., and Ranum L. P. (2017) New developments in RAN translation: insights from multiple diseases. Curr. Opin. Genet. Dev. 44, 125–134 10.1016/j.gde.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Swinnen B., Robberecht W., and Van Den Bosch L. (2020) RNA toxicity in non-coding repeat expansion disorders. EMBO J. 39, e101112 10.15252/embj.2018101112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Fay M. M., Lyons S. M., and Ivanov P. (2017) RNA G-quadruplexes in biology: principles and molecular mechanisms. J. Mol. Biol. 429, 2127–2147 10.1016/j.jmb.2017.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Martí E. (2016) RNA toxicity induced by expanded CAG repeats in Huntington's disease. Brain Pathol. 26, 779–786 10.1111/bpa.12427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium and Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium (2019) CAG repeat not polyglutamine length determines timing of Huntington's disease onset. Cell 178, 887–900.e14 10.1016/j.cell.2019.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wright G. E. B., Collins J. A., Kay C., McDonald C., Dolzhenko E., Xia Q., Bečanović K., Drögemöller B. I., Semaka A., Nguyen C. M., Trost B., Richards F., Bijlsma E. K., Squitieri F., Ross C. J. D., et al. (2019) Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am. J. Hum. Genet. 104, 1116–1126 10.1016/j.ajhg.2019.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ciosi M., Maxwell A., Cumming S. A., Hensman Moss D. J., Alshammari A. M., Flower M. D., Durr A., Leavitt B. R., Roos R. A. C., Track-HD team, Enroll-HD team, Holmans P., Jones L., Langbehn D. R., Kwak S., et al. (2019) A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine 48, 568–580 10.1016/j.ebiom.2019.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Treangen T. J., and Salzberg S. L. (2011) Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46 10.1038/nrg3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Gemayel R., Vinces M. D., Legendre M., and Verstrepen K. J. (2010) Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 44, 445–477 10.1146/annurev-genet-072610-155046 [DOI] [PubMed] [Google Scholar]
- 133. Tautz D. (1989) Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res. 17, 6463–6471 10.1093/nar/17.16.6463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Panneerchelvam S., and Norazmi M. N. (2003) Forensic DNA profiling and database. Malays. J. Med. Sci. 10, 20–26 [PMC free article] [PubMed] [Google Scholar]
- 135. Kunkel T. A. (1993) Nucleotide repeats: slippery DNA and diseases. Nature 365, 207–208 10.1038/365207a0 [DOI] [PubMed] [Google Scholar]
- 136. Lyons-Darden T., and Topal M. D. (1999) Effects of temperature, Mg2+ concentration, and mismatches on triplet-repeat expansion during DNA replication in vitro. Nucleic Acids Res. 27, 2235–2240 10.1093/nar/27.11.2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Axford M. M., Wang Y. H., Nakamori M., Zannis-Hadjopoulos M., Thornton C. A., and Pearson C. E. (2013) Detection of slipped-DNAs at the trinucleotide repeats of the myotonic dystrophy type I disease locus in patient tissues. PLoS Genet. 9, e1003866 10.1371/journal.pgen.1003866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Shah K. A., Shishkin A. A., Voineagu I., Pavlov Y. I., Shcherbakova P. V., and Mirkin S. M. (2012) Role of DNA polymerases in repeat-mediated genome instability. Cell Rep. 2, 1088–1095 10.1016/j.celrep.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Thys R. G., and Wang Y. H. (2015) DNA Replication dynamics of the GGGGCC repeat of the C9orf72 gene. J. Biol. Chem. 290, 28953–28962 10.1074/jbc.M115.660324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Rifé M., Badenas C., Quintó L., Puigoriol E., Tazón B., Rodriguez-Revenga L., Jiménez L., Sánchez A., and Milà M. (2004) Analysis of CGG variation through 642 meioses in Fragile X families. Mol. Hum. Reprod 10, 773–776 10.1093/molehr/gah102 [DOI] [PubMed] [Google Scholar]
- 141. Balakumaran B. S., Freudenreich C. H., and Zakian V. A. (2000) CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in Saccharomyces cerevisiae. Hum. Mol. Genet. 9, 93–100 10.1093/hmg/9.1.93 [DOI] [PubMed] [Google Scholar]
- 142. Shishkin A. A., Voineagu I., Matera R., Cherng N., Chernet B. T., Krasilnikova M. M., Narayanan V., Lobachev K. S., and Mirkin S. M. (2009) Large-scale expansions of Friedreich's ataxia GAA repeats in yeast. Mol. Cell 35, 82–92 10.1016/j.molcel.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kim J. C., and Mirkin S. M. (2013) The balancing act of DNA repeat expansions. Curr. Opin. Genet. Dev. 23, 280–288 10.1016/j.gde.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhong N., Yang W., Dobkin C., and Brown W. T. (1995) Fragile X gene instability: anchoring AGGs and linked microsatellites. Am. J. Hum. Genet. 57, 351–361 [PMC free article] [PubMed] [Google Scholar]
- 145. Kunst C. B., and Warren S. T. (1994) Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell 77, 853–861 10.1016/0092-8674(94)90134-1 [DOI] [PubMed] [Google Scholar]
- 146. Nolin S. L., Brown W. T., Glicksman A., Houck G. E. Jr., Gargano A. D., Sullivan A., Biancalana V., Bröndum-Nielsen K., Hjalgrim H., Holinski-Feder E., Kooy F., Longshore J., Macpherson J., Mandel J. L., Matthijs G., et al. (2003) Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 72, 454–464 10.1086/367713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Crawford D. C., Zhang F., Wilson B., Warren S. T., and Sherman S. L. (2000) Fragile X CGG repeat structures among African-Americans: identification of a novel factor responsible for repeat instability. Hum. Mol. Genet. 9, 1759–1769 10.1093/hmg/9.12.1759 [DOI] [PubMed] [Google Scholar]
- 148. Domniz N., Ries-Levavi L., Cohen Y., Marom-Haham L., Berkenstadt M., Pras E., Glicksman A., Tortora N., Latham G. J., Hadd A. G., Nolin S. L., and Elizur S. E. (2018) Absence of AGG interruptions is a risk factor for full mutation expansion among Israeli FMR1 premutation carriers. Front. Genet. 9, 606 10.3389/fgene.2018.00606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Eichler E. E., Holden J. J., Popovich B. W., Reiss A. L., Snow K., Thibodeau S. N., Richards C. S., Ward P. A., and Nelson D. L. (1994) Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 8, 88–94 10.1038/ng0994-88 [DOI] [PubMed] [Google Scholar]
- 150. Kunst C. B., Leeflang E. P., Iber J. C., Arnheim N., and Warren S. T. (1997) The effect of FMR1 CGG repeat interruptions on mutation frequency as measured by sperm typing. J. Med. Genet. 34, 627–631 10.1136/jmg.34.8.627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Shimizu M., Gellibolian R., Oostra B. A., and Wells R. D. (1996) Cloning, characterization and properties of plasmids containing CGG triplet repeats from the FMR-1 gene. J. Mol. Biol. 258, 614–626 10.1006/jmbi.1996.0273 [DOI] [PubMed] [Google Scholar]
- 152. Snow K., Tester D. J., Kruckeberg K. E., Schaid D. J., and Thibodeau S. N. (1994) Sequence analysis of the fragile X trinucleotide repeat: implications for the origin of the fragile X mutation. Hum. Mol. Genet. 3, 1543–1551 10.1093/hmg/3.9.1543 [DOI] [PubMed] [Google Scholar]
- 153. Pollard L. M., Sharma R., Gómez M., Shah S., Delatycki M. B., Pianese L., Monticelli A., Keats B. J., and Bidichandani S. I. (2004) Replication-mediated instability of the GAA triplet repeat mutation in Friedreich ataxia. Nucleic Acids Res. 32, 5962–5971 10.1093/nar/gkh933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ditch S., Sammarco M. C., Banerjee A., and Grabczyk E. (2009) Progressive GAA.TTC repeat expansion in human cell lines. PLoS Genet. 5, e1000704 10.1371/journal.pgen.1000704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Braida C., Stefanatos R. K., Adam B., Mahajan N., Smeets H. J., Niel F., Goizet C., Arveiler B., Koenig M., Lagier-Tourenne C., Mandel J. L., Faber C. G., de Die-Smulders C. E., Spaans F., and Monckton D. G. (2010) Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum. Mol. Genet. 19, 1399–1412 10.1093/hmg/ddq015 [DOI] [PubMed] [Google Scholar]
- 156. Cumming S. A., Hamilton M. J., Robb Y., Gregory H., McWilliam C., Cooper A., Adam B., McGhie J., Hamilton G., Herzyk P., Tschannen M. R., Worthey E., Petty R., Ballantyne B., Scottish Myotonic Dystrophy, Consortium, et al. (2018) De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur. J. Hum. Genet. 26, 1635–1647 10.1038/s41431-018-0156-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Pesov̌ić J., Perić S., Brkušanin M., Brajušković G., Rakočević-Stojanović V., and Savić-Pavićević D. (2018) Repeat interruptions modify age at onset in myotonic dystrophy type 1 by stabilizing DMPK expansions in somatic cells. Front. Genet. 9, 601 10.3389/fgene.2018.00601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Musova Z., Mazanec R., Krepelova A., Ehler E., Vales J., Jaklova R., Prochazka T., Koukal P., Marikova T., Kraus J., Havlovicova M., and Sedlacek Z. (2009) Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. A 149A, 1365–1374 10.1002/ajmg.a.32987 [DOI] [PubMed] [Google Scholar]
- 159. Chung M. Y., Ranum L. P., Duvick L. A., Servadio A., Zoghbi H. Y., and Orr H. T. (1993) Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat. Genet. 5, 254–258 10.1038/ng1193-254 [DOI] [PubMed] [Google Scholar]
- 160. Menon R. P., Nethisinghe S., Faggiano S., Vannocci T., Rezaei H., Pemble S., Sweeney M. G., Wood N. W., Davis M. B., Pastore A., and Giunti P. (2013) The role of interruptions in polyQ in the pathology of SCA1. PLoS Genet. 9, e1003648 10.1371/journal.pgen.1003648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Miret J. J., Pessoa-Brandão L., and Lahue R. S. (1998) Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 95, 12438–12443 10.1073/pnas.95.21.12438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Dixon M. J., and Lahue R. S. (2004) DNA elements important for CAG*CTG repeat thresholds in Saccharomyces cerevisiae. Nucleic Acids Res. 32, 1289–1297 10.1093/nar/gkh292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Radvanszky J., Surovy M., Polak E., and Kadasi L. (2013) Uninterrupted CCTG tracts in the myotonic dystrophy type 2 associated locus. Neuromuscul. Disord. 23, 591–598 10.1016/j.nmd.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 164. Bachinski L. L., Czernuszewicz T., Ramagli L. S., Suominen T., Shriver M. D., Udd B., Siciliano M. J., and Krahe R. (2009) Premutation allele pool in myotonic dystrophy type 2. Neurology 72, 490–497 10.1212/01.wnl.0000333665.01888.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Mahyera A. S., Schneider T., Halliger-Keller B., Schrooten K., Hörner E. M., Rost S., and Kress W. (2018) Distribution and structure of DM2 repeat tract alleles in the German population. Front. Neurol. 9, 463 10.3389/fneur.2018.00463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Frank-Kamenetskii M. D., and Mirkin S. M. (1995) Triplex DNA structures. Annu. Rev. Biochem. 64, 65–95 10.1146/annurev.bi.64.070195.000433 [DOI] [PubMed] [Google Scholar]
- 167. Mirkin S. M., Lyamichev V. I., Drushlyak K. N., Dobrynin V. N., Filippov S. A., and Frank-Kamenetskii M. D. (1987) DNA H form requires a homopurine-homopyrimidine mirror repeat. Nature 330, 495–497 10.1038/330495a0 [DOI] [PubMed] [Google Scholar]
- 168. Kohwi Y., and Kohwi-Shigematsu T. (1988) Magnesium ion-dependent triple-helix structure formed by homopurine-homopyrimidine sequences in supercoiled plasmid DNA. Proc. Natl. Acad. Sci. U.S.A. 85, 3781–3785 10.1073/pnas.85.11.3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Mirkin S. M. (2008) Discovery of alternative DNA structures: a heroic decade (1979–1989). Front. Biosci. 13, 1064–1071 10.2741/2744 [DOI] [PubMed] [Google Scholar]
- 170. Cox R., and Mirkin S. M. (1997) Characteristic enrichment of DNA repeats in different genomes. Proc. Natl. Acad. Sci. U.S.A. 94, 5237–5242 10.1073/pnas.94.10.5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Liu G., Myers S., Chen X., Bissler J. J., Sinden R. R., and Leffak M. (2012) Replication fork stalling and checkpoint activation by a PKD1 locus mirror repeat polypurine-polypyrimidine (Pu-Py) tract. J. Biol. Chem. 287, 33412–33423 10.1074/jbc.M112.402503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Patel H. P., Lu L., Blaszak R. T., and Bissler J. J. (2004) PKD1 intron 21: triplex DNA formation and effect on replication. Nucleic Acids Res. 32, 1460–1468 10.1093/nar/gkh312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Bidichandani S. I., Ashizawa T., and Patel P. I. (1998) The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet. 62, 111–121 10.1086/301680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Krasilnikova M. M., Kireeva M. L., Petrovic V., Knijnikova N., Kashlev M., and Mirkin S. M. (2007) Effects of Friedreich's ataxia (GAA)n*(TTC)n repeats on RNA synthesis and stability. Nucleic Acids Res. 35, 1075–1084 10.1093/nar/gkl1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Wang G., and Vasquez K. M. (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 13448–13453 10.1073/pnas.0405116101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Faruqi A. F., Datta H. J., Carroll D., Seidman M. M., and Glazer P. M. (2000) Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol. Cell. Biol. 20, 990–1000 10.1128/MCB.20.3.990-1000.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Sakamoto N., Chastain P. D., Parniewski P., Ohshima K., Pandolfo M., Griffith J. D., and Wells R. D. (1999) Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich's ataxia. Mol. Cell 3, 465–475 10.1016/S1097-2765(00)80474-8 [DOI] [PubMed] [Google Scholar]
- 178. Ohshima K., Sakamoto N., Labuda M., Poirier J., Moseley M. L., Montermini L., Ranum L. P., Wells R. D., and Pandolfo M. (1999) A nonpathogenic GAAGGA repeat in the Friedreich gene: implications for pathogenesis. Neurology 53, 1854–1857 10.1212/WNL.53.8.1854 [DOI] [PubMed] [Google Scholar]
- 179. Grabczyk E., and Usdin K. (2000) The GAA*TTC triplet repeat expanded in Friedreich's ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 28, 2815–2822 10.1093/nar/28.14.2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. LeProust E. M., Pearson C. E., Sinden R. R., and Gao X. (2000) Unexpected formation of parallel duplex in GAA and TTC trinucleotide repeats of Friedreich's ataxia. J. Mol. Biol. 302, 1063–1080 10.1006/jmbi.2000.4073 [DOI] [PubMed] [Google Scholar]
- 181. Potaman V. N., Oussatcheva E. A., Lyubchenko Y. L., Shlyakhtenko L. S., Bidichandani S. I., Ashizawa T., and Sinden R. R. (2004) Length-dependent structure formation in Friedreich ataxia (GAA)n*(TTC)n repeats at neutral pH. Nucleic Acids Res. 32, 1224–1231 10.1093/nar/gkh274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Mariappan S. V., Catasti P., Silks L. A. 3rd, Bradbury E. M., and Gupta G. (1999) The high-resolution structure of the triplex formed by the GAA/TTC triplet repeat associated with Friedreich's ataxia. J. Mol. Biol. 285, 2035–2052 10.1006/jmbi.1998.2435 [DOI] [PubMed] [Google Scholar]
- 183. Khristich A. N., Armenia J. F., Matera R. M., Kolchinski A. A., and Mirkin S. M. (2020) Large-scale contractions of Friedreich's ataxia GAA repeats in yeast occur during DNA replication due to their triplex-forming ability. Proc. Natl. Acad. Sci. U.S.A. 117, 1628–1637 10.1073/pnas.1913416117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Sen D., and Gilbert W. (1988) Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 334, 364–366 10.1038/334364a0 [DOI] [PubMed] [Google Scholar]
- 185. Kwok C. K., and Merrick C. J. (2017) G-quadruplexes: prediction, characterization, and biological application. Trends Biotechnol. 35, 997–1013 10.1016/j.tibtech.2017.06.012 [DOI] [PubMed] [Google Scholar]
- 186. Murat P., and Balasubramanian S. (2014) Existence and consequences of G-quadruplex structures in DNA. Curr. Opin. Genet. Dev. 25, 22–29 10.1016/j.gde.2013.10.012 [DOI] [PubMed] [Google Scholar]
- 187. Hänsel-Hertsch R., Beraldi D., Lensing S. V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M., Tannahill D., and Balasubramanian S. (2016) G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 48, 1267–1272 10.1038/ng.3662 [DOI] [PubMed] [Google Scholar]
- 188. Huppert J. L., and Balasubramanian S. (2007) G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 35, 406–413 10.1093/nar/gkl1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Armas P., and Calcaterra N. B. (2018) G-quadruplex in animal development: Contribution to gene expression and genomic heterogeneity. Mech. Dev. 154, 64–72 10.1016/j.mod.2018.05.004 [DOI] [PubMed] [Google Scholar]
- 190. Schofield J. P., Cowan J. L., and Coldwell M. J. (2015) G-quadruplexes mediate local translation in neurons. Biochem. Soc. Trans. 43, 338–342 10.1042/BST20150053 [DOI] [PubMed] [Google Scholar]
- 191. Sauer M., and Paeschke K. (2017) G-quadruplex unwinding helicases and their function in vivo. Biochem. Soc. Trans. 45, 1173–1182 10.1042/BST20170097 [DOI] [PubMed] [Google Scholar]
- 192. Lerner L. K., and Sale J. E. (2019) Replication of G quadruplex DNA. Genes (Amst.) 10, E95 10.3390/genes10020095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Rodriguez R., Miller K. M., Forment J. V., Bradshaw C. R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., and Jackson S. P. (2012) Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 8, 301–310 10.1038/nchembio.780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. De S., and Michor F. (2011) DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat. Struct. Mol. Biol. 18, 950–955 10.1038/nsmb.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Kruisselbrink E., Guryev V., Brouwer K., Pontier D. B., Cuppen E., and Tijsterman M. (2008) Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr. Biol. 18, 900–905 10.1016/j.cub.2008.05.013 [DOI] [PubMed] [Google Scholar]
- 196. Arimondo P. B., Riou J. F., Mergny J. L., Tazi J., Sun J. S., Garestier T., and Hélène C. (2000) Interaction of human DNA topoisomerase I with G-quartet structures. Nucleic Acids Res. 28, 4832–4838 10.1093/nar/28.24.4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Ogloblina A. M., Khristich A. N., Karpechenko N. Y., Semina S. E., Belitsky G. A., Dolinnaya N. G., and Yakubovskaya M. G. (2018) Multi-targeted effects of G4-aptamers and their antiproliferative activity against cancer cells. Biochimie (Paris) 145, 163–173 10.1016/j.biochi.2017.11.020 [DOI] [PubMed] [Google Scholar]
- 198. Ogloblina A. M., Bannikova V. A., Khristich A. N., Oretskaya T. S., Yakubovskaya M. G., and Dolinnaya N. G. (2015) Parallel G-guadruplexes formed by guanine-rich microsatellite repeats inhibit human topoisomerase I. Biochemistry (Mosc.) 80, 1026–1038 10.1134/S0006297915080088 [DOI] [PubMed] [Google Scholar]
- 199. Lago S., Tosoni E., Nadai M., Palumbo M., and Richter S. N. (2017) The cellular protein nucleolin preferentially binds long-looped G-quadruplex nucleic acids. Biochim. Biophys. Acta Gen. Subj. 1861, 1371–1381 10.1016/j.bbagen.2016.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Brázda V., Hároníková L., Liao J. C., and Fojta M. (2014) DNA and RNA quadruplex-binding proteins. Int. J. Mol. Sci. 15, 17493–17517 10.3390/ijms151017493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Brčić J., and Plavec J. (2017) ALS and FTD linked GGGGCC-repeat containing DNA oligonucleotide folds into two distinct G-quadruplexes. Biochim. Biophys. Acta Gen. Subj. 1861, 1237–1245 10.1016/j.bbagen.2016.11.018 [DOI] [PubMed] [Google Scholar]
- 202. Brcic J., and Plavec J. (2018) NMR structure of a G-quadruplex formed by four d(G4C2) repeats: insights into structural polymorphism. Nucleic Acids Res. 46, 11605–11617 10.1093/nar/gky886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Su Z., Zhang Y., Gendron T. F., Bauer P. O., Chew J., Yang W. Y., Fostvedt E., Jansen-West K., Belzil V. V., Desaro P., Johnston A., Overstreet K., Oh S. Y., Todd P. K., Berry J. D., et al. (2014) Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 83, 1043–1050 10.1016/j.neuron.2014.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Darlow J. M., and Leach D. R. (1998) Secondary structures in d(CGG) and d(CCG) repeat tracts. J. Mol. Biol. 275, 3–16 10.1006/jmbi.1997.1453 [DOI] [PubMed] [Google Scholar]
- 205. Tam M., Erin Montgomery S., Kekis M., Stollar B. D., Price G. B., and Pearson C. E. (2003) Slipped (CTG).(CAG) repeats of the myotonic dystrophy locus: surface probing with anti-DNA antibodies. J. Mol. Biol. 332, 585–600 10.1016/S0022-2836(03)00880-5 [DOI] [PubMed] [Google Scholar]
- 206. Liu G., Chen X., Bissler J. J., Sinden R. R., and Leffak M. (2010) Replication-dependent instability at (CTG) × (CAG) repeat hairpins in human cells. Nat. Chem. Biol. 6, 652–659 10.1038/nchembio.416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Gacy A. M., Goellner G., Juranić N., Macura S., and McMurray C. T. (1995) Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81, 533–540 10.1016/0092-8674(95)90074-8 [DOI] [PubMed] [Google Scholar]
- 208. Dere R., Napierala M., Ranum L. P., and Wells R. D. (2004) Hairpin structure-forming propensity of the (CCTG.CAGG) tetranucleotide repeats contributes to the genetic instability associated with myotonic dystrophy type 2. J. Biol. Chem. 279, 41715–41726 10.1074/jbc.M406415200 [DOI] [PubMed] [Google Scholar]
- 209. Pataskar S. S., Dash D., and Brahmachari S. K. (2001) Progressive myoclonus epilepsy [EPM1] repeat d(CCCCGCCCCGCG)n forms folded hairpin structures at physiological pH. J. Biomol. Struct. Dyn. 19, 293–305 10.1080/07391102.2001.10506740 [DOI] [PubMed] [Google Scholar]
- 210. Huang T. Y., Chang C. K., Kao Y. F., Chin C. H., Ni C. W., Hsu H. Y., Hu N. J., Hsieh L. C., Chou S. H., Lee I. R., and Hou M. H. (2017) Parity-dependent hairpin configurations of repetitive DNA sequence promote slippage associated with DNA expansion. Proc. Natl. Acad. Sci. U.S.A. 114, 9535–9540 10.1073/pnas.1708691114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Murchie A. I., and Lilley D. M. (1987) The mechanism of cruciform formation in supercoiled DNA: initial opening of central basepairs in salt-dependent extrusion. Nucleic Acids Res. 15, 9641–9654 10.1093/nar/15.23.9641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Panyutin I. G., and Hsieh P. (1993) Formation of a single base mismatch impedes spontaneous DNA branch migration. J. Mol. Biol. 230, 413–424 10.1006/jmbi.1993.1159 [DOI] [PubMed] [Google Scholar]
- 213. Potaman V. N., Bissler J. J., Hashem V. I., Oussatcheva E. A., Lu L., Shlyakhtenko L. S., Lyubchenko Y. L., Matsuura T., Ashizawa T., Leffak M., Benham C. J., and Sinden R. R. (2003) Unpaired structures in SCA10 (ATTCT)n. (AGAAT)n repeats. J. Mol. Biol. 326, 1095–1111 10.1016/S0022-2836(03)00037-8 [DOI] [PubMed] [Google Scholar]
- 214. Liu G., Bissler J. J., Sinden R. R., and Leffak M. (2007) Unstable spinocerebellar ataxia type 10 (ATTCT*(AGAAT) repeats are associated with aberrant replication at the ATX10 locus and replication origin-dependent expansion at an ectopic site in human cells. Mol. Cell. Biol. 27, 7828–7838 10.1128/MCB.01276-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Vologodskii A. V., Anshelevich V. V., Lukashin A. V., and Frank-Kamenetskii M. D. (1979) Statistical mechanics of supercoils and the torsional stiffness of the DNA double helix. Nature 280, 294–298 10.1038/280294a0 [DOI] [PubMed] [Google Scholar]
- 216. De Michele G., Cavalcanti F., Criscuolo C., Pianese L., Monticelli A., Filla A., and Cocozza S. (1998) Parental gender, age at birth and expansion length influence GAA repeat intergenerational instability in the X25 gene: pedigree studies and analysis of sperm from patients with Friedreich's ataxia. Hum. Mol. Genet. 7, 1901–1906 10.1093/hmg/7.12.1901 [DOI] [PubMed] [Google Scholar]
- 217. Nolin S. L., Glicksman A., Tortora N., Allen E., Macpherson J., Mila M., Vianna-Morgante A. M., Sherman S. L., Dobkin C., Latham G. J., and Hadd A. G. (2019) Expansions and contractions of the FMR1 CGG repeat in 5,508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. A 179, 1148–1156 10.1002/ajmg.a.61165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Ashley-Koch A. E., Robinson H., Glicksman A. E., Nolin S. L., Schwartz C. E., Brown W. T., Turner G., and Sherman S. L. (1998) Examination of factors associated with instability of the FMR1 CGG repeat. Am. J. Hum. Genet. 63, 776–785 10.1086/302018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Reyniers E., Vits L., De Boulle K., Van Roy B., Van Velzen D., de Graaff E., Verkerk A. J., Jorens H. Z., Darby J. K., and Oostra B. (1993) The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat. Genet. 4, 143–146 10.1038/ng0693-143 [DOI] [PubMed] [Google Scholar]
- 220. Aziz N. A., van Belzen M. J., Coops I. D., Belfroid R. D., and Roos R. A. (2011) Parent-of-origin differences of mutant HTT CAG repeat instability in Huntington's disease. Eur. J. Med. Genet. 54, e413–e418 10.1016/j.ejmg.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 221. Wheeler V. C., Persichetti F., McNeil S. M., Mysore J. S., Mysore S. S., MacDonald M. E., Myers R. H., Gusella J. F., Wexler N. S., and US-Venezuela Collaborative Research Group (2007) Factors associated with HD CAG repeat instability in Huntington disease. J. Med. Genet. 44, 695–701 10.1136/jmg.2007.050930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Nørremølle A., Sørensen S. A., Fenger K., and Hasholt L. (1995) Correlation between magnitude of CAG repeat length alterations and length of the paternal repeat in paternally inherited Huntington's disease. Clin. Genet. 47, 113–117 [DOI] [PubMed] [Google Scholar]
- 223. Matilla T., Volpini V., Genís D., Rosell J., Corral J., Dávalos A., Molins A., and Estivill X. (1993) Presymptomatic analysis of spinocerebellar ataxia type 1 (SCA1) via the expansion of the SCA1 CAG-repeat in a large pedigree displaying anticipation and parental male bias. Hum. Mol. Genet. 2, 2123–2128 10.1093/hmg/2.12.2123 [DOI] [PubMed] [Google Scholar]
- 224. Almaguer-Mederos L. E., Mesa J. M. L., González-Zaldívar Y., Almaguer-Gotay D., Cuello-Almarales D., Aguilera-Rodríguez R., Falcón N. S., Gispert S., Auburger G., and Velázquez-Pérez L. (2018) Factors associated with ATXN2 CAG/CAA repeat intergenerational instability in spinocerebellar ataxia type 2. Clin. Genet. 94, 346–350 10.1111/cge.13380 [DOI] [PubMed] [Google Scholar]
- 225. Rasmussen A., De Biase I., Fragoso-Benítez M., Macías-Flores M. A., Yescas P., Ochoa A., Ashizawa T., Alonso M. E., and Bidichandani S. I. (2007) Anticipation and intergenerational repeat instability in spinocerebellar ataxia type 17. Ann. Neurol. 61, 607–610 10.1002/ana.21139 [DOI] [PubMed] [Google Scholar]
- 226. Day J. W., Schut L. J., Moseley M. L., Durand A. C., and Ranum L. P. (2000) Spinocerebellar ataxia type 8: clinical features in a large family. Neurology 55, 649–657 10.1212/WNL.55.5.649 [DOI] [PubMed] [Google Scholar]
- 227. Matsuura T., Fang P., Lin X., Khajavi M., Tsuji K., Rasmussen A., Grewal R. P., Achari M., Alonso M. E., Pulst S. M., Zoghbi H. Y., Nelson D. L., Roa B. B., and Ashizawa T. (2004) Somatic and germline instability of the ATTCT repeat in spinocerebellar ataxia type 10. Am. J. Hum. Genet. 74, 1216–1224 10.1086/421526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Grewal R. P., Achari M., Matsuura T., Durazo A., Tayag E., Zu L., Pulst S. M., and Ashizawa T. (2002) Clinical features and ATTCT repeat expansion in spinocerebellar ataxia type 10. Arch. Neurol. 59, 1285–1290 10.1001/archneur.59.8.1285 [DOI] [PubMed] [Google Scholar]
- 229. Morales F., Vásquez M., Cuenca P., Campos D., Santamaría C., Del Valle G., Brian R., Sittenfeld M., and Monckton D. G. (2015) Parental age effects, but no evidence for an intrauterine effect in the transmission of myotonic dystrophy type 1. Eur. J. Hum. Genet. 23, 646–653 10.1038/ejhg.2014.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Sato T., Oyake M., Nakamura K., Nakao K., Fukusima Y., Onodera O., Igarashi S., Takano H., Kikugawa K., Ishida Y., Shimohata T., Koide R., Ikeuchi T., Tanaka H., Futamura N., et al. (1999) Transgenic mice harboring a full-length human mutant DRPLA gene exhibit age-dependent intergenerational and somatic instabilities of CAG repeats comparable with those in DRPLA patients. Hum. Mol. Genet. 8, 99–106 10.1093/hmg/8.1.99 [DOI] [PubMed] [Google Scholar]
- 231. Kaytor M. D., Burright E. N., Duvick L. A., Zoghbi H. Y., and Orr H. T. (1997) Increased trinucleotide repeat instability with advanced maternal age. Hum. Mol. Genet. 6, 2135–2139 10.1093/hmg/6.12.2135 [DOI] [PubMed] [Google Scholar]
- 232. Zhang Y., Monckton D. G., Siciliano M. J., Connor T. H., and Meistrich M. L. (2002) Age and insertion site dependence of repeat number instability of a human DM1 transgene in individual mouse sperm. Hum. Mol. Genet. 11, 791–798 10.1093/hmg/11.7.791 [DOI] [PubMed] [Google Scholar]
- 233. Neto J. L., Lee J. M., Afridi A., Gillis T., Guide J. R., Dempsey S., Lager B., Alonso I., Wheeler V. C., and Pinto R. M. (2017) Genetic contributors to intergenerational CAG repeat instability in Huntington's disease knock-in mice. Genetics 205, 503–516 10.1534/genetics.116.195578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Entezam A., and Usdin K. (2008) ATR protects the genome against CGG.CCG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 36, 1050–1056 10.1093/nar/gkm1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Zhao X. N., and Usdin K. (2018) Timing of expansion of Fragile X premutation alleles during intergenerational transmission in a mouse model of the Fragile X-related disorders. Front. Genet. 9, 314 10.3389/fgene.2018.00314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Sullivan A. K., Crawford D. C., Scott E. H., Leslie M. L., and Sherman S. L. (2002) Paternally transmitted FMR1 alleles are less stable than maternally transmitted alleles in the common and intermediate size range. Am. J. Hum. Genet. 70, 1532–1544 10.1086/340846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Coffee R. L. Jr., Tessier C. R., Woodruff E. A. 3rd, and Broadie K. (2010) Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis. Model. Mech. 3, 471–485 10.1242/dmm.004598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Martins S., Coutinho P., Silveira I., Giunti P., Jardim L. B., Calafell F., Sequeiros J., and Amorim A. (2008) Cis-acting factors promoting the CAG intergenerational instability in Machado-Joseph disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 439–446 10.1002/ajmg.b.30624 [DOI] [PubMed] [Google Scholar]
- 239. Zheng H., and Xie W. (2019) The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 20, 535–550 10.1038/s41580-019-0132-4 [DOI] [PubMed] [Google Scholar]
- 240. Hao S. L., Ni F. D., and Yang W. X. (2019) The dynamics and regulation of chromatin remodeling during spermiogenesis. Gene 706, 201–210 10.1016/j.gene.2019.05.027 [DOI] [PubMed] [Google Scholar]
- 241. Perutz M. F., and Windle A. H. (2001) Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature 412, 143–144 10.1038/35084141 [DOI] [PubMed] [Google Scholar]
- 242. Legleiter J., Mitchell E., Lotz G. P., Sapp E., Ng C., DiFiglia M., Thompson L. M., and Muchowski P. J. (2010) Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 285, 14777–14790 10.1074/jbc.M109.093708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Squitieri F., Gellera C., Cannella M., Mariotti C., Cislaghi G., Rubinsztein D. C., Almqvist E. W., Turner D., Bachoud-Lévi A. C., Simpson S. A., Delatycki M., Maglione V., Hayden M. R., and Donato S. D. (2003) Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain 126, 946–955 10.1093/brain/awg077 [DOI] [PubMed] [Google Scholar]
- 244. Cubo E., Martinez-Horta S. I., Santalo F. S., Descalls A. M., Calvo S., Gil-Polo C., Muñoz I., Llano K., Mariscal N., Diaz D., Gutierrez A., Aguado L., Ramos-Arroyo M. A., and European HD Network (2019) Clinical manifestations of homozygote allele carriers in Huntington disease. Neurology 92, e2101–e2108 [DOI] [PubMed] [Google Scholar]
- 245. Mateo I., Llorca J., Volpini V., Corral J., Berciano J., and Combarros O. (2003) GAA expansion size and age at onset of Friedreich's ataxia. Neurology 61, 274–275 10.1212/01.WNL.0000073537.08141.77 [DOI] [PubMed] [Google Scholar]
- 246. De Michele G., Filla A., Criscuolo C., Scarano V., Cavalcanti F., Pianese L., Monticelli A., and Cocozza S. (1998) Determinants of onset age in Friedreich's ataxia. J. Neurol. 245, 166–168 10.1007/s004150050198 [DOI] [PubMed] [Google Scholar]
- 247. Mateo I., Llorca J., Volpini V., Corral J., Berciano J., and Combarros O. (2004) Expanded GAA repeats and clinical variation in Friedreich's ataxia. Acta Neurol. Scand. 109, 75–78 10.1034/j.1600-0404.2003.00190.x [DOI] [PubMed] [Google Scholar]
- 248. Wong L. J., Ashizawa T., Monckton D. G., Caskey C. T., and Richards C. S. (1995) Somatic heterogeneity of the CTG repeat in myotonic dystrophy is age and size dependent. Am. J. Hum. Genet. 56, 114–122 [PMC free article] [PubMed] [Google Scholar]
- 249. Monckton D. G., Wong L. J., Ashizawa T., and Caskey C. T. (1995) Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum. Mol. Genet. 4, 1–8 10.1093/hmg/4.1.1 [DOI] [PubMed] [Google Scholar]
- 250. Monckton D. G., Coolbaugh M. I., Ashizawa K. T., Siciliano M. J., and Caskey C. T. (1997) Hypermutable myotonic dystrophy CTG repeats in transgenic mice. Nat. Genet. 15, 193–196 10.1038/ng0297-193 [DOI] [PubMed] [Google Scholar]
- 251. Clever F., Cho I. K., Yang J., and Chan A. W. S. (2019) Progressive polyglutamine repeat expansion in peripheral blood cells and sperm of transgenic Huntington's disease monkeys. J. Huntingtons Dis. 8, 443–448 10.3233/JHD-190359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Kaplan S., Itzkovitz S., and Shapiro E. (2007) A universal mechanism ties genotype to phenotype in trinucleotide diseases. PLoS Comput. Biol. 3, e235 10.1371/journal.pcbi.0030235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. López Castel A., Nakamori M., Tomé S., Chitayat D., Gourdon G., Thornton C. A., and Pearson C. E. (2011) Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet. 20, 1–15 10.1093/hmg/ddq427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Mornet E., Chateau C., Hirst M. C., Thepot F., Taillandier A., Cibois O., and Serre J. L. (1996) Analysis of germline variation at the FMR1 CGG repeat shows variation in the normal-premutated borderline range. Hum. Mol. Genet. 5, 821–825 10.1093/hmg/5.6.821 [DOI] [PubMed] [Google Scholar]
- 255. Mason A. G., Tomé S., Simard J. P., Libby R. T., Bammler T. K., Beyer R. P., Morton A. J., Pearson C. E., and La Spada A. R. (2014) Expression levels of DNA replication and repair genes predict regional somatic repeat instability in the brain but are not altered by polyglutamine disease protein expression or age. Hum. Mol. Genet. 23, 1606–1618 10.1093/hmg/ddt551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Cannella M., Maglione V., Martino T., Ragona G., Frati L., Li G. M., and Squitieri F. (2009) DNA instability in replicating Huntington's disease lymphoblasts. BMC Med. Genet. 10, 11 10.1186/1471-2350-10-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Kovtun I. V., and McMurray C. T. (2001) Trinucleotide expansion in haploid germ cells by gap repair. Nat. Genet. 27, 407–411 10.1038/86906 [DOI] [PubMed] [Google Scholar]
- 258. Lia A. S., Seznec H., Hofmann-Radvanyi H., Radvanyi F., Duros C., Saquet C., Blanche M., Junien C., and Gourdon G. (1998) Somatic instability of the CTG repeat in mice transgenic for the myotonic dystrophy region is age dependent but not correlated to the relative intertissue transcription levels and proliferative capacities. Hum. Mol. Genet. 7, 1285–1291 10.1093/hmg/7.8.1285 [DOI] [PubMed] [Google Scholar]
- 259. Day J. W., Ricker K., Jacobsen J. F., Rasmussen L. J., Dick K. A., Kress W., Schneider C., Koch M. C., Beilman G. J., Harrison A. R., Dalton J. C., and Ranum L. P. (2003) Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 60, 657–664 10.1212/01.WNL.0000054481.84978.F9 [DOI] [PubMed] [Google Scholar]
- 260. Lukusa T., and Fryns J. P. (2008) Human chromosome fragility. Biochim. Biophys. Acta 1779, 3–16 10.1016/j.bbagrm.2007.10.005 [DOI] [PubMed] [Google Scholar]
- 261. Bjerregaard V. A., Garribba L., McMurray C. T., Hickson I. D., and Liu Y. (2018) Folate deficiency drives mitotic missegregation of the human FRAXA locus. Proc. Natl. Acad. Sci. U.S.A. 115, 13003–13008 10.1073/pnas.1808377115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Jones C., Slijepcevic P., Marsh S., Baker E., Langdon W. Y., Richards R. I., and Tunnacliffe A. (1994) Physical linkage of the fragile site FRA11B and a Jacobsen syndrome chromosome deletion breakpoint in 11q23.3. Hum. Mol. Genet. 3, 2123–2130 10.1093/hmg/3.12.2123 [DOI] [PubMed] [Google Scholar]
- 263. Cherng N., Shishkin A. A., Schlager L. I., Tuck R. H., Sloan L., Matera R., Sarkar P. S., Ashizawa T., Freudenreich C. H., and Mirkin S. M. (2011) Expansions, contractions, and fragility of the spinocerebellar ataxia type 10 pentanucleotide repeat in yeast. Proc. Natl. Acad. Sci. U.S.A. 108, 2843–2848 10.1073/pnas.1009409108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Tang W., Dominska M., Gawel M., Greenwell P. W., and Petes T. D. (2013) Genomic deletions and point mutations induced in Saccharomyces cerevisiae by the trinucleotide repeats (GAA.TTC) associated with Friedreich's ataxia. DNA Repair (Amst.) 12, 10–17 10.1016/j.dnarep.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Kumari D., Hayward B., Nakamura A. J., Bonner W. M., and Usdin K. (2015) Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia. Mutat. Res. 781, 14–21 10.1016/j.mrfmmm.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Schweitzer J. K., Reinke S. S., and Livingston D. M. (2001) Meiotic alterations in CAG repeat tracts. Genetics 159, 1861–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Jankowski C., Nasar F., and Nag D. K. (2000) Meiotic instability of CAG repeat tracts occurs by double-strand break repair in yeast. Proc. Natl. Acad. Sci. U.S.A. 97, 2134–2139 10.1073/pnas.040460297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Saini N., Zhang Y., Nishida Y., Sheng Z., Choudhury S., Mieczkowski P., and Lobachev K. S. (2013) Fragile DNA motifs trigger mutagenesis at distant chromosomal loci in saccharomyces cerevisiae. PLoS Genet. 9, e1003551 10.1371/journal.pgen.1003551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Bacolla A., Tainer J. A., Vasquez K. M., and Cooper D. N. (2016) Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 44, 5673–5688 10.1093/nar/gkw261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Rodgers K., and McVey M. (2016) Error-prone repair of DNA double-strand breaks. J. Cell. Physiol. 231, 15–24 10.1002/jcp.25053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Shah K. A., and Mirkin S. M. (2015) The hidden side of unstable DNA repeats: mutagenesis at a distance. DNA Repair (Amst.) 32, 106–112 10.1016/j.dnarep.2015.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. García Arocena D., de Diego Y., Oostra B. A., Willemsen R., and Mirta Rodriguez M. (2000) A fragile X case with an amplification/deletion mosaic pattern. Hum. Genet. 106, 366–369 10.1007/s004390051052 [DOI] [PubMed] [Google Scholar]
- 273. Verdyck P., Berckmoes V., De Vos A., Verpoest W., Liebaers I., Bonduelle M., and De Rycke M. (2015) Chromosome fragility at FRAXA in human cleavage stage embryos at risk for fragile X syndrome. Am. J. Med. Genet. A 167A, 2306–2313 10.1002/ajmg.a.37149 [DOI] [PubMed] [Google Scholar]
- 274. Jones C., Penny L., Mattina T., Yu S., Baker E., Voullaire L., Langdon W. Y., Sutherland G. R., Richards R. I., and Tunnacliffe A. (1995) Association of a chromosome deletion syndrome with a fragile site within the proto-oncogene CBL2. Nature 376, 145–149 10.1038/376145a0 [DOI] [PubMed] [Google Scholar]
- 275. Dobkin C., Radu G., Ding X. H., Brown W. T., and Nolin S. L. (2009) Fragile X prenatal analyses show full mutation females at high risk for mosaic Turner syndrome: fragile X leads to chromosome loss. Am. J. Med. Genet. A 149A, 2152–2157 10.1002/ajmg.a.33011 [DOI] [PubMed] [Google Scholar]
- 276. Kononenko A. V., Ebersole T., Vasquez K. M., and Mirkin S. M. (2018) Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat. Struct. Mol. Biol. 25, 669–676 10.1038/s41594-018-0094-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Bidichandani S. I., Purandare S. M., Taylor E. E., Gumin G., Machkhas H., Harati Y., Gibbs R. A., Ashizawa T., and Patel P. I. (1999) Somatic sequence variation at the Friedreich ataxia locus includes complete contraction of the expanded GAA triplet repeat, significant length variation in serially passaged lymphoblasts and enhanced mutagenesis in the flanking sequence. Hum. Mol. Genet. 8, 2425–2436 10.1093/hmg/8.13.2425 [DOI] [PubMed] [Google Scholar]
- 278. McGinty R. J., Rubinstein R. G., Neil A. J., Dominska M., Kiktev D., Petes T. D., and Mirkin S. M. (2017) Nanopore sequencing of complex genomic rearrangements in yeast reveals mechanisms of repeat-mediated double-strand break repair. Genome Res. 27, 2072–2082 10.1101/gr.228148.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Rindler P. M., Clark R. M., Pollard L. M., De Biase I., and Bidichandani S. I. (2006) Replication in mammalian cells recapitulates the locus-specific differences in somatic instability of genomic GAA triplet-repeats. Nucleic Acids Res. 34, 6352–6361 10.1093/nar/gkl846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Pelletier R., Farrell B. T., Miret J. J., and Lahue R. S. (2005) Mechanistic features of CAG*CTG repeat contractions in cultured cells revealed by a novel genetic assay. Nucleic Acids Res. 33, 5667–5676 10.1093/nar/gki880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Burgers P. M. J., and Kunkel T. A. (2017) Eukaryotic DNA replication fork. Annu. Rev. Biochem. 86, 417–438 10.1146/annurev-biochem-061516-044709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Garbacz M. A., Lujan S. A., Burkholder A. B., Cox P. B., Wu Q., Zhou Z. X., Haber J. E., and Kunkel T. A. (2018) Evidence that DNA polymerase δ contributes to initiating leading strand DNA replication in Saccharomyces cerevisiae. Nat. Commun. 9, 858 10.1038/s41467-018-03270-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Kang S., Jaworski A., Ohshima K., and Wells R. D. (1995) Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat. Genet. 10, 213–218 10.1038/ng0695-213 [DOI] [PubMed] [Google Scholar]
- 284. Maurer D. J., O'Callaghan B. L., and Livingston D. M. (1996) Orientation dependence of trinucleotide CAG repeat instability in Saccharomyces cerevisiae. Mol. Cell. Biol. 16, 6617–6622 10.1128/MCB.16.12.6617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Freudenreich C. H., Stavenhagen J. B., and Zakian V. A. (1997) Stability of a CTG/CAG trinucleotide repeat in yeast is dependent on its orientation in the genome. Mol. Cell. Biol. 17, 2090–2098 10.1128/MCB.17.4.2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Cleary J. D., Nichol K., Wang Y. H., and Pearson C. E. (2002) Evidence of cis-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat. Genet. 31, 37–46 10.1038/ng870 [DOI] [PubMed] [Google Scholar]
- 287. Lenzmeier B. A., and Freudenreich C. H. (2003) Trinucleotide repeat instability: a hairpin curve at the crossroads of replication, recombination, and repair. Cytogenet. Genome Res. 100, 7–24 10.1159/000072836 [DOI] [PubMed] [Google Scholar]
- 288. Hirst M. C., and White P. J. (1998) Cloned human FMR1 trinucleotide repeats exhibit a length- and orientation-dependent instability suggestive of in vivo lagging strand secondary structure. Nucleic Acids Res. 26, 2353–2358 10.1093/nar/26.10.2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. White P. J., Borts R. H., and Hirst M. C. (1999) Stability of the human fragile X (CGG)n triplet repeat array in Saccharomyces cerevisiae deficient in aspects of DNA metabolism. Mol. Cell. Biol. 19, 5675–5684 10.1128/MCB.19.8.5675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Krasilnikova M. M., and Mirkin S. M. (2004) Replication stalling at Friedreich's ataxia (GAA)n repeats in vivo. Mol. Cell. Biol. 24, 2286–2295 10.1128/MCB.24.6.2286-2295.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Cortese A., Simone R., Sullivan R., Vandrovcova J., Tariq H., Yau W. Y., Humphrey J., Jaunmuktane Z., Sivakumar P., Polke J., Ilyas M., Tribollet E., Tomaselli P. J., Devigili G., Callegari I., et al. (2019) Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 51, 649–658 10.1038/s41588-019-0372-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Mirkin S. M., and Smirnova E. V. (2002) Positioned to expand. Nat. Genet. 31, 5–6 10.1038/ng0502-5 [DOI] [PubMed] [Google Scholar]
- 293. Nichol Edamura K., Leonard M. R., and Pearson C. E. (2005) Role of replication and CpG methylation in fragile X syndrome CGG deletions in primate cells. Am. J. Hum. Genet. 76, 302–311 10.1086/427928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Schmucker B., and Seidel J. (1999) Mosaicism for a full mutation and a normal size allele in two fragile X males. Am. J. Med. Genet. 84, 221–225 10.1002/(SICI)1096-8628(19990528)84:3<221::AID-AJMG11>3.0.CO;2-M [DOI] [PubMed] [Google Scholar]
- 295. Manor E., Jabareen A., Magal N., Kofman A., Hagerman R. J., and Tassone F. (2017) Prenatal diagnosis of Fragile X: can a full mutation allele in the FMR1 gene contract to a normal size? Front. Genet. 8, 158 10.3389/fgene.2017.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Wöhrle D., Hennig I., Vogel W., and Steinbach P. (1993) Mitotic stability of fragile X mutations in differentiated cells indicates early post-conceptional trinucleotide repeat expansion. Nat. Genet. 4, 140–142 10.1038/ng0693-140 [DOI] [PubMed] [Google Scholar]
- 297. Nolin S. L., Ding X. H., Houck G. E., Brown W. T., and Dobkin C. (2008) Fragile X full mutation alleles composed of few alleles: implications for CGG repeat expansion. Am. J. Med. Genet. A. 146A, 60–65 10.1002/ajmg.a.32087 [DOI] [PubMed] [Google Scholar]
- 298. Brylawski B. P., Chastain P. D. 2nd, Cohen S. M., Cordeiro-Stone M., and Kaufman D. G. (2007) Mapping of an origin of DNA replication in the promoter of fragile X gene FMR1. Exp. Mol. Pathol. 82, 190–196 10.1016/j.yexmp.2006.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Gray S. J., Gerhardt J., Doerfler W., Small L. E., and Fanning E. (2007) An origin of DNA replication in the promoter region of the human fragile X mental retardation (FMR1) gene. Mol. Cell. Biol. 27, 426–437 10.1128/MCB.01382-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Kao H. I., Henricksen L. A., Liu Y., and Bambara R. A. (2002) Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J. Biol. Chem. 277, 14379–14389 10.1074/jbc.M110662200 [DOI] [PubMed] [Google Scholar]
- 301. Bae S. H., Bae K. H., Kim J. A., and Seo Y. S. (2001) RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature 412, 456–461 10.1038/35086609 [DOI] [PubMed] [Google Scholar]
- 302. Gloor J. W., Balakrishnan L., Campbell J. L., and Bambara R. A. (2012) Biochemical analyses indicate that binding and cleavage specificities define the ordered processing of human Okazaki fragments by Dna2 and FEN1. Nucleic Acids Res. 40, 6774–6786 10.1093/nar/gks388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Levikova M., and Cejka P. (2015) The Saccharomyces cerevisiae Dna2 can function as a sole nuclease in the processing of Okazaki fragments in DNA replication. Nucleic Acids Res. 43, 7888–7897 10.1093/nar/gkv710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Balakrishnan L., and Bambara R. A. (2013) Okazaki fragment metabolism. Cold Spring Harb. Perspect. Biol. 5, a010173 10.1101/cshperspect.a010173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Stith C. M., Sterling J., Resnick M. A., Gordenin D. A., and Burgers P. M. (2008) Flexibility of eukaryotic Okazaki fragment maturation through regulated strand displacement synthesis. J. Biol. Chem. 283, 34129–34140 10.1074/jbc.M806668200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Ayyagari R., Gomes X. V., Gordenin D. A., and Burgers P. M. (2003) Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J. Biol. Chem. 278, 1618–1625 10.1074/jbc.M209801200 [DOI] [PubMed] [Google Scholar]
- 307. Xie Y., Counter C., and Alani E. (1999) Characterization of the repeat-tract instability and mutator phenotypes conferred by a Tn3 insertion in RFC1, the large subunit of the yeast clamp loader. Genetics 151, 499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Zhang Y., Shishkin A. A., Nishida Y., Marcinkowski-Desmond D., Saini N., Volkov K. V., Mirkin S. M., and Lobachev K. S. (2012) Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol. Cell 48, 254–265 10.1016/j.molcel.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Refsland E. W., and Livingston D. M. (2005) Interactions among DNA ligase I, the flap endonuclease and proliferating cell nuclear antigen in the expansion and contraction of CAG repeat tracts in yeast. Genetics 171, 923–934 10.1534/genetics.105.043448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Lopes J., Debrauwère H., Buard J., and Nicolas A. (2002) Instability of the human minisatellite CEB1 in rad27Δ and dna2–1 replication-deficient yeast cells. EMBO J. 21, 3201–3211 10.1093/emboj/cdf310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Schweitzer J. K., and Livingston D. M. (1998) Expansions of CAG repeat tracts are frequent in a yeast mutant defective in Okazaki fragment maturation. Hum. Mol. Genet. 7, 69–74 10.1093/hmg/7.1.69 [DOI] [PubMed] [Google Scholar]
- 312. Omer S., Lavi B., Mieczkowski P. A., Covo S., and Hazkani-Covo E. (2017) Whole genome sequence analysis of mutations accumulated in rad27Δ yeast strains with defects in the processing of Okazaki fragments indicates template-switching events. G3 (Bethesda) 7, 3775–3787 10.1534/g3.117.300262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Maleki S., Cederberg H., and Rannug U. (2002) The human minisatellites MS1, MS32, MS205 and CEB1 integrated into the yeast genome exhibit different degrees of mitotic instability but are all stabilised by RAD27. Curr. Genet. 41, 333–341 10.1007/s00294-002-0307-x [DOI] [PubMed] [Google Scholar]
- 314. Parenteau J., and Wellinger R. J. (1999) Accumulation of single-stranded DNA and destabilization of telomeric repeats in yeast mutant strains carrying a deletion of RAD27. Mol. Cell. Biol. 19, 4143–4152 10.1128/MCB.19.6.4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Tsutakawa S. E., Thompson M. J., Arvai A. S., Neil A. J., Shaw S. J., Algasaier S. I., Kim J. C., Finger L. D., Jardine E., Gotham V. J. B., Sarker A. H., Her M. Z., Rashid F., Hamdan S. M., Mirkin S. M., et al. (2017) Phosphate steering by Flap endonuclease 1 promotes 5′-flap specificity and incision to prevent genome instability. Nat. Commun. 8, 15855 10.1038/ncomms15855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Johnson R. E., Kovvali G. K., Prakash L., and Prakash S. (1998) Role of yeast Rth1 nuclease and its homologs in mutation avoidance, DNA repair, and DNA replication. Curr. Genet. 34, 21–29 10.1007/s002940050362 [DOI] [PubMed] [Google Scholar]
- 317. Freudenreich C. H., Kantrow S. M., and Zakian V. A. (1998) Expansion and length-dependent fragility of CTG repeats in yeast. Science 279, 853–856 10.1126/science.279.5352.853 [DOI] [PubMed] [Google Scholar]
- 318. Kokoska R. J., Stefanovic L., Tran H. T., Resnick M. A., Gordenin D. A., and Petes T. D. (1998) Destabilization of yeast micro- and minisatellite DNA sequences by mutations affecting a nuclease involved in Okazaki fragment processing (rad27) and DNA polymerase δ (pol3-t). Mol. Cell. Biol. 18, 2779–2788 10.1128/MCB.18.5.2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Gordenin D. A., Kunkel T. A., and Resnick M. A. (1997) Repeat expansion—all in a flap? Nat. Genet. 16, 116–118 10.1038/ng0697-116 [DOI] [PubMed] [Google Scholar]
- 320. Parenteau J., and Wellinger R. J. (2002) Differential processing of leading- and lagging-strand ends at Saccharomyces cerevisiae telomeres revealed by the absence of Rad27p nuclease. Genetics 162, 1583–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Liu B., Hu J., Wang J., and Kong D. (2017) Direct visualization of RNA-DNA primer removal from Okazaki fragments provides support for flap cleavage and exonucleolytic pathways in eukaryotic cells. J. Biol. Chem. 292, 4777–4788 10.1074/jbc.M116.758599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Kahli M., Osmundson J. S., Yeung R., and Smith D. J. (2019) Processing of eukaryotic Okazaki fragments by redundant nucleases can be uncoupled from ongoing DNA replication in vivo. Nucleic Acids Res. 47, 1814–1822 10.1093/nar/gky1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Greene A. L., Snipe J. R., Gordenin D. A., and Resnick M. A. (1999) Functional analysis of human FEN1 in Saccharomyces cerevisiae and its role in genome stability. Hum. Mol. Genet. 8, 2263–2273 10.1093/hmg/8.12.2263 [DOI] [PubMed] [Google Scholar]
- 324. Otto C. J., Almqvist E., Hayden M. R., and Andrew S. E. (2001) The “flap” endonuclease gene FEN1 is excluded as a candidate gene implicated in the CAG repeat expansion underlying Huntington disease. Clin. Genet. 59, 122–127 10.1034/j.1399-0004.2001.590210.x [DOI] [PubMed] [Google Scholar]
- 325. Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium (2015) Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 162, 516–526 10.1016/j.cell.2015.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Lee J. M., Chao M. J., Harold D., Abu Elneel K., Gillis T., Holmans P., Jones L., Orth M., Myers R. H., Kwak S., Wheeler V. C., MacDonald M. E., and Gusella J. F. (2017) A modifier of Huntington's disease onset at the MLH1 locus. Hum. Mol. Genet. 26, 3859–3867 10.1093/hmg/ddx286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Moss D. J. H., Pardiñas A. F., Langbehn D., Lo K., Leavitt B. R., Roos R., Durr A., Mead S., Track-HD investigators, REGISTRY investigators, Holmans P., Jones L., and Tabrizi S. J. (2017) Identification of genetic variants associated with Huntington's disease progression: a genome-wide association study. Lancet Neurol. 16, 701–711 10.1016/S1474-4422(17)30161-8 [DOI] [PubMed] [Google Scholar]
- 328. Moe S. E., Sorbo J. G., and Holen T. (2008) Huntingtin triplet-repeat locus is stable under long-term Fen1 knockdown in human cells. J. Neurosci. Methods 171, 233–238 10.1016/j.jneumeth.2008.03.012 [DOI] [PubMed] [Google Scholar]
- 329. Chatterjee N., Lin Y., Yotnda P., and Wilson J. H. (2016) Environmental stress induces trinucleotide repeat mutagenesis in human cells by alt-nonhomologous end joining repair. J. Mol. Biol. 428, 2978–2980 10.1016/j.jmb.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Lin Y., Dion V., and Wilson J. H. (2006) Transcription promotes contraction of CAG repeat tracts in human cells. Nat. Struct. Mol. Biol. 13, 179–180 10.1038/nsmb1042 [DOI] [PubMed] [Google Scholar]
- 331. van den Broek W. J., Nelen M. R., van der Heijden G. W., Wansink D. G., and Wieringa B. (2006) Fen1 does not control somatic hypermutability of the (CTG)n*(CAG)n repeat in a knock-in mouse model for DM1. FEBS Lett. 580, 5208–5214 10.1016/j.febslet.2006.08.059 [DOI] [PubMed] [Google Scholar]
- 332. Spiro C., and McMurray C. T. (2003) Nuclease-deficient FEN-1 blocks Rad51/BRCA1-mediated repair and causes trinucleotide repeat instability. Mol. Cell. Biol. 23, 6063–6074 10.1128/MCB.23.17.6063-6074.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Samadashwily G. M., Raca G., and Mirkin S. M. (1997) Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 17, 298–304 10.1038/ng1197-298 [DOI] [PubMed] [Google Scholar]
- 334. Pelletier R., Krasilnikova M. M., Samadashwily G. M., Lahue R., and Mirkin S. M. (2003) Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 23, 1349–1357 10.1128/MCB.23.4.1349-1357.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Viterbo D., Michoud G., Mosbach V., Dujon B., and Richard G. F. (2016) Replication stalling and heteroduplex formation within CAG/CTG trinucleotide repeats by mismatch repair. DNA Repair (Amst.) 42, 94–106 10.1016/j.dnarep.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 336. Liu G., Chen X., and Leffak M. (2013) Oligodeoxynucleotide binding to (CTG). (CAG) microsatellite repeats inhibits replication fork stalling, hairpin formation, and genome instability. Mol. Cell. Biol. 33, 571–581 10.1128/MCB.01265-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Chandok G. S., Patel M. P., Mirkin S. M., and Krasilnikova M. M. (2012) Effects of Friedreich's ataxia GAA repeats on DNA replication in mammalian cells. Nucleic Acids Res. 40, 3964–3974 10.1093/nar/gks021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Follonier C., Oehler J., Herrador R., and Lopes M. (2013) Friedreich's ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat. Struct. Mol. Biol. 20, 486–494 10.1038/nsmb.2520 [DOI] [PubMed] [Google Scholar]
- 339. Gerhardt J., Bhalla A. D., Butler J. S., Puckett J. W., Dervan P. B., Rosenwaks Z., and Napierala M. (2016) Stalled DNA replication forks at the endogenous GAA repeats drive repeat expansion in Friedreich's ataxia cells. Cell Rep. 16, 1218–1227 10.1016/j.celrep.2016.06.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Voineagu I., Surka C. F., Shishkin A. A., Krasilnikova M. M., and Mirkin S. M. (2009) Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 16, 226–228 10.1038/nsmb.1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Yudkin D., Hayward B. E., Aladjem M. I., Kumari D., and Usdin K. (2014) Chromosome fragility and the abnormal replication of the FMR1 locus in fragile X syndrome. Hum. Mol. Genet. 23, 2940–2952 10.1093/hmg/ddu006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Kuzminov A. (2013) Inhibition of DNA synthesis facilitates expansion of low-complexity repeats: is strand slippage stimulated by transient local depletion of specific dNTPs? Bioessays 35, 306–313 10.1002/bies.201200128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Lapidot A., Baran N., and Manor H. (1989) (dT-dC)n and (dG-dA)n tracts arrest single stranded DNA replication in vitro. Nucleic Acids Res. 17, 883–900 10.1093/nar/17.3.883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Neil A. J., Kim J. C., and Mirkin S. M. (2017) Precarious maintenance of simple DNA repeats in eukaryotes. Bioessays 39, 10.1002/bies.201700077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Neelsen K. J., and Lopes M. (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16, 207–220 10.1038/nrm3935 [DOI] [PubMed] [Google Scholar]
- 346. Fouché N., Ozgür S., Roy D., and Griffith J. D. (2006) Replication fork regression in repetitive DNAs. Nucleic Acids Res. 34, 6044–6050 10.1093/nar/gkl757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Kramara J., Osia B., and Malkova A. (2018) Break-induced replication: the where, the why, and the how. Trends Genet. 34, 518–531 10.1016/j.tig.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Yeshaya J., Shalgi R., Shohat M., and Avivi L. (1999) FISH-detected delay in replication timing of mutated FMR1 alleles on both active and inactive X-chromosomes. Hum. Genet. 105, 86–97 10.1007/s004399900081 [DOI] [PubMed] [Google Scholar]
- 349. Gellon L., Kaushal S., Cebrián J., Lahiri M., Mirkin S. M., and Freudenreich C. H. (2019) Mrc1 and Tof1 prevent fragility and instability at long CAG repeats by their fork stabilizing function. Nucleic Acids Res. 47, 794–805 10.1093/nar/gky1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Bhattacharyya S., and Lahue R. S. (2005) Srs2 helicase of Saccharomyces cerevisiae selectively unwinds triplet repeat DNA. J. Biol. Chem. 280, 33311–33317 10.1074/jbc.M503325200 [DOI] [PubMed] [Google Scholar]
- 351. Anand R. P., Shah K. A., Niu H., Sung P., Mirkin S. M., and Freudenreich C. H. (2012) Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Res. 40, 1091–1105 10.1093/nar/gkr836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Bhattacharyya S., and Lahue R. S. (2004) Saccharomyces cerevisiae Srs2 DNA helicase selectively blocks expansions of trinucleotide repeats. Mol. Cell. Biol. 24, 7324–7330 10.1128/MCB.24.17.7324-7330.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Daee D. L., Mertz T., and Lahue R. S. (2007) Postreplication repair inhibits CAG.CTG repeat expansions in Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 102–110 10.1128/MCB.01167-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Kerrest A., Anand R. P., Sundararajan R., Bermejo R., Liberi G., Dujon B., Freudenreich C. H., and Richard G. F. (2009) SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat. Struct. Mol. Biol. 16, 159–167 10.1038/nsmb.1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Frizzell A., Nguyen J. H., Petalcorin M. I., Turner K. D., Boulton S. J., Freudenreich C. H., and Lahue R. S. (2014) RTEL1 inhibits trinucleotide repeat expansions and fragility. Cell Rep. 6, 827–835 10.1016/j.celrep.2014.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Guler G. D., Rosenwaks Z., and Gerhardt J. (2018) Human DNA helicase B as a candidate for unwinding secondary CGG repeat structures at the Fragile X mental retardation gene. Front. Mol. Neurosci. 11, 138 10.3389/fnmol.2018.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Lansdorp P., and van Wietmarschen N. (2019) Helicases FANCJ, RTEL1 and BLM act on guanine quadruplex DNA in vivo. Genes (Basel) 10, E870 10.3390/genes10110870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Jain A., Bacolla A., Chakraborty P., Grosse F., and Vasquez K. M. (2010) Human DHX9 helicase unwinds triple-helical DNA structures. Biochemistry 49, 6992–6999 10.1021/bi100795m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Guo M., Hundseth K., Ding H., Vidhyasagar V., Inoue A., Nguyen C. H., Zain R., Lee J. S., and Wu Y. (2015) A distinct triplex DNA unwinding activity of ChlR1 helicase. J. Biol. Chem. 290, 5174–5189 10.1074/jbc.M114.634923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Brosh R. M. Jr, Majumdar A., Desai S., Hickson I. D., Bohr V. A., and Seidman M. M. (2001) Unwinding of a DNA triple helix by the Werner and Bloom syndrome helicases. J. Biol. Chem. 276, 3024–3030 10.1074/jbc.M006784200 [DOI] [PubMed] [Google Scholar]
- 361. Nelson L. D., Musso M., and Van Dyke M. W. (2000) The yeast STM1 gene encodes a purine motif triple helical DNA-binding protein. J. Biol. Chem. 275, 5573–5581 10.1074/jbc.275.8.5573 [DOI] [PubMed] [Google Scholar]
- 362. Gonitel R., Moffitt H., Sathasivam K., Woodman B., Detloff P. J., Faull R. L., and Bates G. P. (2008) DNA instability in postmitotic neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 3467–3472 10.1073/pnas.0800048105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. McGinty R. J., Puleo F., Aksenova A. Y., Hisey J. A., Shishkin A. A., Pearson E. L., Wang E. T., Housman D. E., Moore C., and Mirkin S. M. (2017) A defective mRNA cleavage and polyadenylation complex facilitates expansions of transcribed (GAA)n repeats associated with Friedreich's ataxia. Cell Rep. 20, 2490–2500 10.1016/j.celrep.2017.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Rindler P. M., and Bidichandani S. I. (2011) Role of transcript and interplay between transcription and replication in triplet-repeat instability in mammalian cells. Nucleic Acids Res. 39, 526–535 10.1093/nar/gkq788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Lin Y., Dent S. Y., Wilson J. H., Wells R. D., and Napierala M. (2010) R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl. Acad. Sci. U.S.A. 107, 692–697 10.1073/pnas.0909740107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Jung J., and Bonini N. (2007) CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science 315, 1857–1859 10.1126/science.1139517 [DOI] [PubMed] [Google Scholar]
- 367. Shah K. A., McGinty R. J., Egorova V. I., and Mirkin S. M. (2014) Coupling transcriptional state to large-scale repeat expansions in yeast. Cell Rep. 9, 1594–1602 10.1016/j.celrep.2014.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Kosmider B., and Wells R. D. (2007) Fragile X repeats are potent inducers of complex, multiple site rearrangements in flanking sequences in Escherichia coli. DNA Repair (Amst.) 6, 1850–1863 10.1016/j.dnarep.2007.07.014 [DOI] [PubMed] [Google Scholar]
- 369. Chen Y. H., Keegan S., Kahli M., Tonzi P., Fenyö D., Huang T. T., and Smith D. J. (2019) Transcription shapes DNA replication initiation and termination in human cells. Nat. Struct. Mol. Biol. 26, 67–77 10.1038/s41594-018-0171-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Crossley M. P., Bocek M., and Cimprich K. A. (2019) R-loops as cellular regulators and genomic threats. Mol. Cell 73, 398–411 10.1016/j.molcel.2019.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Hegazy Y. A., Fernando C. M., and Tran E. J. (2020) The balancing act of R-loop biology: the good, the bad, and the ugly. J. Biol. Chem. 295, 905–913 10.1074/jbc.REV119.011353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Reddy K., Tam M., Bowater R. P., Barber M., Tomlinson M., Nichol Edamura K., Wang Y. H., and Pearson C. E. (2011) Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 39, 1749–1762 10.1093/nar/gkq935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Abu Diab M., Mor-Shaked H., Cohen E., Cohen-Hadad Y., Ram O., Epsztejn-Litman S., and Eiges R. (2018) The G-rich repeats in FMR1 and C9orf72 loci are hotspots for local unpairing of DNA. Genetics 210, 1239–1252 10.1534/genetics.118.301672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Su X. A., and Freudenreich C. H. (2017) Cytosine deamination and base excision repair cause R-loop-induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 114, E8392–E8401 10.1073/pnas.1711283114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Haeusler A. R., Donnelly C. J., Periz G., Simko E. A., Shaw P. G., Kim M. S., Maragakis N. J., Troncoso J. C., Pandey A., Sattler R., Rothstein J. D., and Wang J. (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200 10.1038/nature13124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Katayama S., Tomaru Y., Kasukawa T., Waki K., Nakanishi M., Nakamura M., Nishida H., Yap C. C., Suzuki M., Kawai J., Suzuki H., Carninci P., Hayashizaki Y., Wells C., Frith M., et al. (2005) Antisense transcription in the mammalian transcriptome. Science 309, 1564–1566 10.1126/science.1112009 [DOI] [PubMed] [Google Scholar]
- 377. Trinklein N. D., Aldred S. F., Hartman S. J., Schroeder D. I., Otillar R. P., and Myers R. M. (2004) An abundance of bidirectional promoters in the human genome. Genome Res. 14, 62–66 10.1101/gr.1982804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Budworth H., and McMurray C. T. (2013) Bidirectional transcription of trinucleotide repeats: roles for excision repair. DNA Repair (Amst.) 12, 672–684 10.1016/j.dnarep.2013.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Mizielinska S., Lashley T., Norona F. E., Clayton E. L., Ridler C. E., Fratta P., and Isaacs A. M. (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126, 845–857 10.1007/s00401-013-1200-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Sone J., Mitsuhashi S., Fujita A., Mizuguchi T., Hamanaka K., Mori K., Koike H., Hashiguchi A., Takashima H., Sugiyama H., Kohno Y., Takiyama Y., Maeda K., Doi H., Koyano S., et al. (2019) Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 51, 1215–1221 10.1038/s41588-019-0459-y [DOI] [PubMed] [Google Scholar]
- 381. Cho D. H., Thienes C. P., Mahoney S. E., Analau E., Filippova G. N., and Tapscott S. J. (2005) Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 20, 483–489 10.1016/j.molcel.2005.09.002 [DOI] [PubMed] [Google Scholar]
- 382. Wilburn B., Rudnicki D. D., Zhao J., Weitz T. M., Cheng Y., Gu X., Greiner E., Park C. S., Wang N., Sopher B. L., La Spada A. R., Osmand A., Margolis R. L., Sun Y. E., and Yang X. W. (2011) An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron 70, 427–440 10.1016/j.neuron.2011.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Sopher B. L., Ladd P. D., Pineda V. V., Libby R. T., Sunkin S. M., Hurley J. B., Thienes C. P., Gaasterland T., Filippova G. N., and La Spada A. R. (2011) CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron 70, 1071–1084 10.1016/j.neuron.2011.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Seixas A. I., Holmes S. E., Takeshima H., Pavlovich A., Sachs N., Pruitt J. L., Silveira I., Ross C. A., Margolis R. L., and Rudnicki D. D. (2012) Loss of junctophilin-3 contributes to Huntington disease-like 2 pathogenesis. Ann. Neurol. 71, 245–257 10.1002/ana.22598 [DOI] [PubMed] [Google Scholar]
- 385. Ladd P. D., Smith L. E., Rabaia N. A., Moore J. M., Georges S. A., Hansen R. S., Hagerman R. J., Tassone F., Tapscott S. J., and Filippova G. N. (2007) An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 16, 3174–3187 10.1093/hmg/ddm293 [DOI] [PubMed] [Google Scholar]
- 386. Lin Y., Leng M., Wan M., and Wilson J. H. (2010) Convergent transcription through a long CAG tract destabilizes repeats and induces apoptosis. Mol. Cell. Biol. 30, 4435–4451 10.1128/MCB.00332-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Reddy K., Schmidt M. H., Geist J. M., Thakkar N. P., Panigrahi G. B., Wang Y. H., and Pearson C. E. (2014) Processing of double-R-loops in (CAG).(CTG) and C9orf72 (GGGGCC).(GGCCCC) repeats causes instability. Nucleic Acids Res. 42, 10473–10487 10.1093/nar/gku658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Dere R., and Wells R. D. (2006) DM2 CCTG*CAGG repeats are crossover hotspots that are more prone to expansions than the DM1 CTG*CAG repeats in Escherichia coli. J. Mol. Biol. 360, 21–36 10.1016/j.jmb.2006.05.012 [DOI] [PubMed] [Google Scholar]
- 389. Blackwood J. K., Okely E. A., Zahra R., Eykelenboom J. K., and Leach D. R. (2010) DNA tandem repeat instability in the Escherichia coli chromosome is stimulated by mismatch repair at an adjacent CAG.CTG trinucleotide repeat. Proc. Natl. Acad. Sci. U.S.A. 107, 22582–22586 10.1073/pnas.1012906108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Pluciennik A., Iyer R. R., Napierala M., Larson J. E., Filutowicz M., and Wells R. D. (2002) Long CTG.CAG repeats from myotonic dystrophy are preferred sites for intermolecular recombination. J. Biol. Chem. 277, 34074–34086 10.1074/jbc.M202127200 [DOI] [PubMed] [Google Scholar]
- 391. Edwards S. F., Hashem V. I., Klysik E. A., and Sinden R. R. (2009) Genetic instabilities of (CCTG).(CAGG) and (ATTCT).(AGAAT) disease-associated repeats reveal multiple pathways for repeat deletion. Mol. Carcinog. 48, 336–349 10.1002/mc.20534 [DOI] [PubMed] [Google Scholar]
- 392. Hashem V. I., Rosche W. A., and Sinden R. R. (2004) Genetic recombination destabilizes (CTG)n.(CAG)n repeats in E. coli. Mutat. Res. 554, 95–109 10.1016/j.mrfmmm.2004.03.012 [DOI] [PubMed] [Google Scholar]
- 393. Napierala M., Dere R., Vetcher A., and Wells R. D. (2004) Structure-dependent recombination hot spot activity of GAA.TTC sequences from intron 1 of the Friedreich's ataxia gene. J. Biol. Chem. 279, 6444–6454 10.1074/jbc.M309596200 [DOI] [PubMed] [Google Scholar]
- 394. Tang W., Dominska M., Greenwell P. W., Harvanek Z., Lobachev K. S., Kim H. M., Narayanan V., Mirkin S. M., and Petes T. D. (2011) Friedreich's ataxia (GAA)n*(TTC)n repeats strongly stimulate mitotic crossovers in Saccharomyces cerevisae. PLoS Genet. 7, e1001270 10.1371/journal.pgen.1001270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Sundararajan R., Gellon L., Zunder R. M., and Freudenreich C. H. (2010) Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics 184, 65–77 10.1534/genetics.109.111039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Hebert M. L., Spitz L. A., and Wells R. D. (2004) DNA double-strand breaks induce deletion of CTG.CAG repeats in an orientation-dependent manner in Escherichia coli. J. Mol. Biol. 336, 655–672 10.1016/j.jmb.2003.12.038 [DOI] [PubMed] [Google Scholar]
- 397. Mittelman D., Moye C., Morton J., Sykoudis K., Lin Y., Carroll D., and Wilson J. H. (2009) Zinc-finger directed double-strand breaks within CAG repeat tracts promote repeat instability in human cells. Proc. Natl. Acad. Sci. U.S.A. 106, 9607–9612 10.1073/pnas.0902420106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Warren S. T. (1997) Polyalanine expansion in synpolydactyly might result from unequal crossing-over of HOXD13. Science 275, 408–409 10.1126/science.275.5298.408 [DOI] [PubMed] [Google Scholar]
- 399. Richard G. F., Cyncynatus C., and Dujon B. (2003) Contractions and expansions of CAG/CTG trinucleotide repeats occur during ectopic gene conversion in yeast, by a MUS81-independent mechanism. J. Mol. Biol. 326, 769–782 10.1016/S0022-2836(02)01405-5 [DOI] [PubMed] [Google Scholar]
- 400. Jankowski C., and Nag D. K. (2002) Most meiotic CAG repeat tract-length alterations in yeast are SPO11 dependent. Mol. Genet. Genomics 267, 64–70 10.1007/s00438-001-0635-4 [DOI] [PubMed] [Google Scholar]
- 401. Ashley C. T. Jr., and Warren S. T. (1995) Trinucleotide repeat expansion and human disease. Annu. Rev. Genet. 29, 703–728 10.1146/annurev.ge.29.120195.003415 [DOI] [PubMed] [Google Scholar]
- 402. La Spada A. R., Paulson H. L., and Fischbeck K. H. (1994) Trinucleotide repeat expansion in neurological disease. Ann. Neurol. 36, 814–822 10.1002/ana.410360604 [DOI] [PubMed] [Google Scholar]
- 403. Pearson C. E. (2003) Slipping while sleeping? Trinucleotide repeat expansions in germ cells. Trends Mol. Med. 9, 490–495 10.1016/j.molmed.2003.09.006 [DOI] [PubMed] [Google Scholar]
- 404. Savouret C., Garcia-Cordier C., Megret J., te Riele H., Junien C., and Gourdon G. (2004) MSH2-dependent germinal CTG repeat expansions are produced continuously in spermatogonia from DM1 transgenic mice. Mol. Cell. Biol. 24, 629–637 10.1128/MCB.24.2.629-637.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Genovese M., Westhovens R., Meuleners L., Van der Aa A., Harrison P., Tasset C., and Kavanaugh A. (2018) Effect of filgotinib, a selective JAK 1 inhibitor, with and without methotrexate in patients with rheumatoid arthritis: patient-reported outcomes. Arthritis Res. Ther. 20, 57 10.1186/s13075-018-1541-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Pollard L. M., Bourn R. L., and Bidichandani S. I. (2008) Repair of DNA double-strand breaks within the (GAA*TTC)n sequence results in frequent deletion of the triplet-repeat sequence. Nucleic Acids Res. 36, 489–500 10.1093/nar/gkm1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Kim J. C., Harris S. T., Dinter T., Shah K. A., and Mirkin S. M. (2017) The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat. Struct. Mol. Biol. 24, 55–60 10.1038/nsmb.3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Kaushal S., and Freudenreich C. H. (2019) The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 58, 270–283 10.1002/gcc.22721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Benet A., and Azorín F. (1999) The formation of triple-stranded DNA prevents spontaneous branch-migration. J. Mol. Biol. 294, 851–857 10.1006/jmbi.1999.3295 [DOI] [PubMed] [Google Scholar]
- 410. Chang H. H. Y., Pannunzio N. R., Adachi N., and Lieber M. R. (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18, 495–506 10.1038/nrm.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Su X. A., Dion V., Gasser S. M., and Freudenreich C. H. (2015) Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev. 29, 1006–1017 10.1101/gad.256404.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Gazy I., Hayward B., Potapova S., Zhao X., and Usdin K. (2019) Double-strand break repair plays a role in repeat instability in a fragile X mouse model. DNA Repair (Amst.) 74, 63–69 10.1016/j.dnarep.2018.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Daley J. M., Laan R. L., Suresh A., and Wilson T. E. (2005) DNA joint dependence of pol X family polymerase action in nonhomologous end joining. J. Biol. Chem. 280, 29030–29037 10.1074/jbc.M505277200 [DOI] [PubMed] [Google Scholar]
- 414. Crespan E., Czabany T., Maga G., and Hübscher U. (2012) Microhomology-mediated DNA strand annealing and elongation by human DNA polymerases lambda and beta on normal and repetitive DNA sequences. Nucleic Acids Res. 40, 5577–5590 10.1093/nar/gks186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Alt F. W., and Schwer B. (2018) DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair (Amst.) 71, 158–163 10.1016/j.dnarep.2018.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Her J., and Bunting S. F. (2018) How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 293, 10502–10511 10.1074/jbc.TM118.000371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Liu D., Keijzers G., and Rasmussen L. J. (2017) DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. 773, 174–187 10.1016/j.mrrev.2017.07.001 [DOI] [PubMed] [Google Scholar]
- 418. Nojadeh J. N., Behrouz Sharif S., and Sakhinia E. (2018) Microsatellite instability in colorectal cancer. EXCLI J. 17, 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Pinto R. M., Dragileva E., Kirby A., Lloret A., Lopez E., St. Claire J., Panigrahi G. B., Hou C., Holloway K., Gillis T., Guide J. R., Cohen P. E., Li G. M., Pearson C. E., Daly M. J., and Wheeler V. C. (2013) Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington's disease mice: genome-wide and candidate approaches. PLoS Genet. 9, e1003930 10.1371/journal.pgen.1003930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Owen B. A., Yang Z., Lai M., Gajec M., Badger J. D. 2nd, Hayes J. J., Edelmann W., Kucherlapati R., Wilson T. M., and McMurray C. T. (2005) (CAG)n-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat. Struct. Mol. Biol. 12, 663–670 10.1038/nsmb965 [DOI] [PubMed] [Google Scholar]
- 421. Manley K., Shirley T. L., Flaherty L., and Messer A. (1999) Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet. 23, 471–473 10.1038/70598 [DOI] [PubMed] [Google Scholar]
- 422. van den Broek W. J., Nelen M. R., Wansink D. G., Coerwinkel M. M., te Riele H., Groenen P. J., and Wieringa B. (2002) Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum. Mol. Genet. 11, 191–198 10.1093/hmg/11.2.191 [DOI] [PubMed] [Google Scholar]
- 423. Zhao X., Zhang Y., Wilkins K., Edelmann W., and Usdin K. (2018) MutLγ promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. 14, e1007719 10.1371/journal.pgen.1007719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Zhao X. N., Kumari D., Gupta S., Wu D., Evanitsky M., Yang W., and Usdin K. (2015) Mutsβ generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum. Mol. Genet. 24, 7087–7096 10.1093/hmg/ddv408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Lokanga R. A., Zhao X. N., and Usdin K. (2014) The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 35, 129–136 10.1002/humu.22464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Dragileva E., Hendricks A., Teed A., Gillis T., Lopez E. T., Friedberg E. C., Kucherlapati R., Edelmann W., Lunetta K. L., MacDonald M. E., and Wheeler V. C. (2009) Intergenerational and striatal CAG repeat instability in Huntington's disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 33, 37–47 10.1016/j.nbd.2008.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Kovalenko M., Dragileva E., St Claire J., Gillis T., Guide J. R., New J., Dong H., Kucherlapati R., Kucherlapati M. H., Ehrlich M. E., Lee J. M., and Wheeler V. C. (2012) Msh2 acts in medium-spiny striatal neurons as an enhancer of CAG instability and mutant huntingtin phenotypes in Huntington's disease knock-in mice. PLoS ONE 7, e44273 10.1371/journal.pone.0044273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Tomé S., Manley K., Simard J. P., Clark G. W., Slean M. M., Swami M., Shelbourne P. F., Tillier E. R., Monckton D. G., Messer A., and Pearson C. E. (2013) MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington's disease mice. PLoS Genet. 9, e1003280 10.1371/journal.pgen.1003280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Ament S. A., Pearl J. R., Grindeland A., St. Claire J., Earls J. C., Kovalenko M., Gillis T., Mysore J., Gusella J. F., Lee J. M., Kwak S., Howland D., Lee M. Y., Baxter D., Scherler K., et al. (2017) High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington's disease CAG knock-in mice across multiple genetic backgrounds. Hum. Mol. Genet. 26, 913–922 10.1093/hmg/ddx006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Zhao X. N., Lokanga R., Allette K., Gazy I., Wu D., and Usdin K. (2016) A MutSβ-dependent contribution of MutSα to repeat expansions in Fragile X premutation mice? PLoS Genet. 12, e1006190 10.1371/journal.pgen.1006190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Williams G. M., and Surtees J. A. (2015) MSH3 promotes dynamic behavior of trinucleotide repeat tracts in vivo. Genetics 200, 737–754 10.1534/genetics.115.177303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Keogh N., Chan K. Y., Li G. M., and Lahue R. S. (2017) MutSβ abundance and Msh3 ATP hydrolysis activity are important drivers of CTG*CAG repeat expansions. Nucleic Acids Res. 45, 10068–10078 10.1093/nar/gkx650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Richard G. F., Dujon B., and Haber J. E. (1999) Double-strand break repair can lead to high frequencies of deletions within short CAG/CTG trinucleotide repeats. Mol. Gen. Genet. 261, 871–882 10.1007/s004380050031 [DOI] [PubMed] [Google Scholar]
- 434. Halabi A., Fuselier K. T. B., and Grabczyk E. (2018) GAA*TTC repeat expansion in human cells is mediated by mismatch repair complex MutLγ and depends upon the endonuclease domain in MLH3 isoform one. Nucleic Acids Res. 46, 4022–4032 10.1093/nar/gky143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Ezzatizadeh V., Sandi C., Sandi M., Anjomani-Virmouni S., Al-Mahdawi S., and Pook M. A. (2014) MutLα heterodimers modify the molecular phenotype of Friedreich ataxia. PLoS ONE 9, e100523 10.1371/journal.pone.0100523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Du J., Campau E., Soragni E., Ku S., Puckett J. W., Dervan P. B., and Gottesfeld J. M. (2012) Role of mismatch repair enzymes in GAA.TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J. Biol. Chem. 287, 29861–29872 10.1074/jbc.M112.391961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Ezzatizadeh V., Pinto R. M., Sandi C., Sandi M., Al-Mahdawi S., Te Riele H., and Pook M. A. (2012) The mismatch repair system protects against intergenerational GAA repeat instability in a Friedreich ataxia mouse model. Neurobiol. Dis. 46, 165–171 10.1016/j.nbd.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Kim H. M., Narayanan V., Mieczkowski P. A., Petes T. D., Krasilnikova M. M., Mirkin S. M., and Lobachev K. S. (2008) Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J. 27, 2896–2906 10.1038/emboj.2008.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Halabi A., Ditch S., Wang J., and Grabczyk E. (2012) DNA mismatch repair complex MutSβ promotes GAA.TTC repeat expansion in human cells. J. Biol. Chem. 287, 29958–29967 10.1074/jbc.M112.356758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Flower M., Lomeikaite V., Ciosi M., Cumming S., Morales F., Lo K., Hensman Moss D., Jones L., Holmans P., Track-HD Investigators, OPTIMISTIC Consortium, Monckton D. G., and Tabrizi S. J. (2019) MSH3 modifies somatic instability and disease severity in Huntington's and myotonic dystrophy type 1. Brain 10.1093/brain/awz115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Bettencourt C., Hensman-Moss D., Flower M., Wiethoff S., Brice A., Goizet C., Stevanin G., Koutsis G., Karadima G., Panas M., Yescas-Gómez P., García-Velázquez L. E., Alonso-Vilatela M. E., Lima M., Raposo M., et al. (2016) DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol. 79, 983–990 10.1002/ana.24656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Panigrahi G. B., Slean M. M., Simard J. P., Gileadi O., and Pearson C. E. (2010) Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSβ, but clustered slip-outs are poorly repaired. Proc. Natl. Acad. Sci. U.S.A. 107, 12593–12598 10.1073/pnas.0909087107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Wallace S. S. (2014) Base excision repair: a critical player in many games. DNA Repair (Amst.) 19, 14–26 10.1016/j.dnarep.2014.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Møllersen L., Rowe A. D., Illuzzi J. L., Hildrestrand G. A., Gerhold K. J., Tveterås L., Bjølgerud A., Wilson D. M. 3rd, Bjørås M., and Klungland A. (2012) Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum. Mol. Genet. 21, 4939–4947 10.1093/hmg/dds337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Kovtun I. V., Liu Y., Bjoras M., Klungland A., Wilson S. H., and McMurray C. T. (2007) OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447, 447–452 10.1038/nature05778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Lokanga R. A., Senejani A. G., Sweasy J. B., and Usdin K. (2015) Heterozygosity for a hypomorphic Polβ mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders. PLoS Genet. 11, e1005181 10.1371/journal.pgen.1005181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Liu Y., Prasad R., Beard W. A., Hou E. W., Horton J. K., McMurray C. T., and Wilson S. H. (2009) Coordination between polymerase β and FEN1 can modulate CAG repeat expansion. J. Biol. Chem. 284, 28352–28366 10.1074/jbc.M109.050286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Goula A. V., Berquist B. R., Wilson D. M. 3rd, Wheeler V. C., Trottier Y., and Merienne K. (2009) Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington's disease transgenic mice. PLoS Genet. 5, e1000749 10.1371/journal.pgen.1000749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Guo J., Gu L., Leffak M., and Li G. M. (2016) MutSβ promotes trinucleotide repeat expansion by recruiting DNA polymerase β to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis. Cell Res. 26, 775–786 10.1038/cr.2016.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Lai Y., Weizmann Y., and Liu Y. (2018) The deoxyribose phosphate lyase of DNA polymerase β suppresses a processive DNA synthesis to prevent trinucleotide repeat instability. Nucleic Acids Res. 46, 8940–8952 10.1093/nar/gky700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Lai Y., Budworth H., Beaver J. M., Chan N. L., Zhang Z., McMurray C. T., and Liu Y. (2016) Crosstalk between MSH2-MSH3 and polβ promotes trinucleotide repeat expansion during base excision repair. Nat. Commun. 7, 12465 10.1038/ncomms12465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Kim J. C., and Mirkin S. M. (2015) Putting the brakes on Huntington disease in a mouse experimental model. PLoS Genet. 11, e1005409 10.1371/journal.pgen.1005409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453. Spivak G. (2015) Nucleotide excision repair in humans. DNA Repair (Amst.) 36, 13–18 10.1016/j.dnarep.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Concannon C., and Lahue R. S. (2014) Nucleotide excision repair and the 26S proteasome function together to promote trinucleotide repeat expansions. DNA Repair (Amst.) 13, 42–49 10.1016/j.dnarep.2013.11.004 [DOI] [PubMed] [Google Scholar]
- 455. Koch M. R., House N. C. M., Cosetta C. M., Jong R. M., Salomon C. G., Joyce C. E., Philips E. A., Su X. A., and Freudenreich C. H. (2018) The chromatin remodeler Isw1 prevents CAG repeat expansions during transcription in Saccharomyces cerevisiae. Genetics 208, 963–976 10.1534/genetics.117.300529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Lin Y., and Wilson J. H. (2007) Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol. Cell. Biol. 27, 6209–6217 10.1128/MCB.00739-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Hubert L. Jr., Lin Y., Dion V., and Wilson J. H. (2011) Xpa deficiency reduces CAG trinucleotide repeat instability in neuronal tissues in a mouse model of SCA1. Hum. Mol. Genet. 20, 4822–4830 10.1093/hmg/ddr421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458. Zhao X. N., and Usdin K. (2014) Gender and cell-type-specific effects of the transcription-coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the fragile X-related disorders. Hum. Mutat. 35, 341–349 10.1002/humu.22495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Kovtun I. V., Johnson K. O., and McMurray C. T. (2011) Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging 3, 509–514 10.18632/aging.100324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Chao M. J., Kim K. H., Shin J. W., Lucente D., Wheeler V. C., Li H., Roach J. C., Hood L., Wexler N. S., Jardim L. B., Holmans P., Jones L., Orth M., Kwak S., MacDonald M. E., et al. (2018) Population-specific genetic modification of Huntington's disease in Venezuela. PLoS Genet. 14, e1007274 10.1371/journal.pgen.1007274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461. Long J. D., Lee J. M., Aylward E. H., Gillis T., Mysore J. S., Abu Elneel K., Chao M. J., Paulsen J. S., MacDonald M. E., and Gusella J. F. (2018) Genetic modification of Huntington disease acts early in the prediagnosis phase. Am. J. Hum. Genet. 103, 349–357 10.1016/j.ajhg.2018.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462. Goold R., Flower M., Moss D. H., Medway C., Wood-Kaczmar A., Andre R., Farshim P., Bates G. P., Holmans P., Jones L., and Tabrizi S. J. (2019) FAN1 modifies Huntington's disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet. 28, 650–661 10.1093/hmg/ddy375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Zhao X. N., and Usdin K. (2018) FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair (Amst.) 69, 1–5 10.1016/j.dnarep.2018.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. Liu T., Ghosal G., Yuan J., Chen J., and Huang J. (2010) FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329, 693–696 10.1126/science.1192656 [DOI] [PubMed] [Google Scholar]
- 465. Jin H., and Cho Y. (2017) Structural and functional relationships of FAN1. DNA Repair (Amst.) 56, 135–143 10.1016/j.dnarep.2017.06.016 [DOI] [PubMed] [Google Scholar]
- 466. Desai A., and Gerson S. (2014) Exo1 independent DNA mismatch repair involves multiple compensatory nucleases. DNA Repair (Amst.) 21, 55–64 10.1016/j.dnarep.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. MacKay C., Déclais A. C., Lundin C., Agostinho A., Deans A. J., MacArtney T. J., Hofmann K., Gartner A., West S. C., Helleday T., Lilley D. M., and Rouse J. (2010) Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, 65–76 10.1016/j.cell.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Cannavo E., Gerrits B., Marra G., Schlapbach R., and Jiricny J. (2007) Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 282, 2976–2986 10.1074/jbc.M609989200 [DOI] [PubMed] [Google Scholar]
- 469. Evans-Galea M. V., Hannan A. J., Carrodus N., Delatycki M. B., and Saffery R. (2013) Epigenetic modifications in trinucleotide repeat diseases. Trends Mol. Med. 19, 655–663 10.1016/j.molmed.2013.07.007 [DOI] [PubMed] [Google Scholar]
- 470. van Kuilenburg A. B. P., Tarailo-Graovac M., Richmond P. A., Drögemöller B. I., Pouladi M. A., Leen R., Brand-Arzamendi K., Dobritzsch D., Dolzhenko E., Eberle M. A., Hayward B., Jones M. J., Karbassi F., Kobor M. S., Koster J., et al. (2019) Glutaminase deficiency caused by short tandem repeat expansion in GLS. N. Engl. J. Med. 380, 1433–1441 10.1056/NEJMoa1806627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Matan Sorek L. R. Z. C., Eran Meshorer (2019) Open chromatin structure in PolyQ disease-related genes: a potential mechanism for CAG repeat expansion in the normal human population. NAR Genom. Bioinform. 1, e3 10.1093/nargab/lqz003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Sinden R. R. (1994) DNA Structure and Function, pp. 333–343, Academic Press, Inc., San Diego [Google Scholar]
- 473. Supek F., and Lehner B. (2019) Scales and mechanisms of somatic mutation rate variation across the human genome. DNA Repair (Amst.) 81, 102647 10.1016/j.dnarep.2019.102647 [DOI] [PubMed] [Google Scholar]
- 474. Mulvihill D. J., Nichol Edamura K., Hagerman K. A., Pearson C. E., and Wang Y. H. (2005) Effect of CAT or AGG interruptions and CpG methylation on nucleosome assembly upon trinucleotide repeats on spinocerebellar ataxia, type 1 and fragile X syndrome. J. Biol. Chem. 280, 4498–4503 10.1074/jbc.M413239200 [DOI] [PubMed] [Google Scholar]
- 475. Hagerman K. A., Ruan H., Edamura K. N., Matsuura T., Pearson C. E., and Wang Y. H. (2009) The ATTCT repeats of spinocerebellar ataxia type 10 display strong nucleosome assembly which is enhanced by repeat interruptions. Gene 434, 29–34 10.1016/j.gene.2008.12.011 [DOI] [PubMed] [Google Scholar]
- 476. Wang Y. H., and Griffith J. (1995) Expanded CTG triplet blocks from the myotonic dystrophy gene create the strongest known natural nucleosome positioning elements. Genomics 25, 570–573 10.1016/0888-7543(95)80061-P [DOI] [PubMed] [Google Scholar]
- 477. Godde J. S., and Wolffe A. P. (1996) Nucleosome assembly on CTG triplet repeats. J. Biol. Chem. 271, 15222–15229 10.1074/jbc.271.25.15222 [DOI] [PubMed] [Google Scholar]
- 478. House N. C., Polleys E. J., Quasem I., De la Rosa Mejia M., Joyce C. E., Takacsi-Nagy O., Krebs J. E., Fuchs S. M., and Freudenreich C. H. (2019) Distinct roles for S. cerevisiae H2A copies in recombination and repeat stability, with a role for H2A.1 threonine 126. Elife 8, e53362 10.7554/eLife.53362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Libby R. T., Hagerman K. A., Pineda V. V., Lau R., Cho D. H., Baccam S. L., Axford M. M., Cleary J. D., Moore J. M., Sopher B. L., Tapscott S. J., Filippova G. N., Pearson C. E., and La Spada A. R. (2008) CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet. 4, e1000257 10.1371/journal.pgen.1000257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480. Sun J. H., Zhou L., Emerson D. J., Phyo S. A., Titus K. R., Gong W., Gilgenast T. G., Beagan J. A., Davidson B. L., Tassone F., and Phillips-Cremins J. E. (2018) Disease-associated short tandem repeats co-localize with chromatin domain boundaries. Cell 175, 224–238.e15 10.1016/j.cell.2018.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Lynch M., Sung W., Morris K., Coffey N., Landry C. R., Dopman E. B., Dickinson W. J., Okamoto K., Kulkarni S., Hartl D. L., and Thomas W. K. (2008) A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. U.S.A. 105, 9272–9277 10.1073/pnas.0803466105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Blackburn E. H., Epel E. S., and Lin J. (2015) Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science 350, 1193–1198 10.1126/science.aab3389 [DOI] [PubMed] [Google Scholar]
- 483. McKinley K. L., and Cheeseman I. M. (2016) The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell Biol. 17, 16–29 10.1038/nrm.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Janssen A., Colmenares S. U., and Karpen G. H. (2018) Heterochromatin: guardian of the genome. Annu. Rev. Cell Dev. Biol. 34, 265–288 10.1146/annurev-cellbio-100617-062653 [DOI] [PubMed] [Google Scholar]
- 485. Persi E., Wolf Y. I., and Koonin E. V. (2016) Positive and strongly relaxed purifying selection drive the evolution of repeats in proteins. Nat. Commun. 7, 13570 10.1038/ncomms13570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Buschiazzo E., and Gemmell N. J. (2010) Conservation of human microsatellites across 450 million years of evolution. Genome Biol. Evol. 2, 153–165 10.1093/gbe/evq007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Rockman M. V., and Wray G. A. (2002) Abundant raw material for cis-regulatory evolution in humans. Mol. Biol. Evol. 19, 1991–2004 10.1093/oxfordjournals.molbev.a004023 [DOI] [PubMed] [Google Scholar]
- 488. Quilez J., Guilmatre A., Garg P., Highnam G., Gymrek M., Erlich Y., Joshi R. S., Mittelman D., and Sharp A. J. (2016) Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res. 44, 3750–3762 10.1093/nar/gkw219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Vinces M. D., Legendre M., Caldara M., Hagihara M., and Verstrepen K. J. (2009) Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324, 1213–1216 10.1126/science.1170097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490. Kouzine F., Wojtowicz D., Baranello L., Yamane A., Nelson S., Resch W., Kieffer-Kwon K. R., Benham C. J., Casellas R., Przytycka T. M., and Levens D. (2017) Permanganate/S1 nuclease footprinting reveals non-B DNA structures with regulatory potential across a mammalian genome. Cell Syst. 4, 344–356.e7 10.1016/j.cels.2017.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Zhao X., Su L., Schaack S., Sadd B. M., and Sun C. (2018) Tandem repeats contribute to coding sequence variation in bumblebees (Hymenoptera: Apidae). Genome Biol. Evol. 10, 3176–3187 10.1093/gbe/evy244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Press M. O., and Queitsch C. (2017) Variability in a short tandem repeat mediates complex epistatic interactions in Arabidopsis thaliana. Genetics 205, 455–464 10.1534/genetics.116.193359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Fondon J. W. 3rd, and Garner H. R. (2004) Molecular origins of rapid and continuous morphological evolution. Proc. Natl. Acad. Sci. U.S.A. 101, 18058–18063 10.1073/pnas.0408118101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Persi E., Prandi D., Wolf Y. I., Pozniak Y., Barnabas G. D., Levanon K., Barshack I., Barbieri C., Gasperini P., Beltran H., Faltas B. M., Rubin M. A., Geiger T., Koonin E. V., Demichelis F., and Horn D. (2019) Proteomic and genomic signatures of repeat instability in cancer and adjacent normal tissues. Proc. Natl. Acad. Sci. U.S.A. 116, 16987–16996 10.1073/pnas.1908790116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495. Frenkel Z. M., and Trifonov E. N. (2012) Origin and evolution of genes and genomes: crucial role of triplet expansions. J. Biomol. Struct. Dyn. 30, 201–210 10.1080/07391102.2012.677771 [DOI] [PubMed] [Google Scholar]
- 496. McMurray C. T. (2010) Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 11, 786–799 10.1038/nrg2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497. Callahan J. L., Andrews K. J., Zakian V. A., and Freudenreich C. H. (2003) Mutations in yeast replication proteins that increase CAG/CTG expansions also increase repeat fragility. Mol. Cell. Biol. 23, 7849–7860 10.1128/MCB.23.21.7849-7860.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Du J., Campau E., Soragni E., Jespersen C., and Gottesfeld J. M. (2013) Length-dependent CTG.CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum. Mol. Genet 22, 5276–5287 10.1093/hmg/ddt386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. Bontekoe C. J., Bakker C. E., Nieuwenhuizen I. M., van der Linde H., Lans H., de Lange D., Hirst M. C., and Oostra B. A. (2001) Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum. Mol. Genet. 10, 1693–1699 10.1093/hmg/10.16.1693 [DOI] [PubMed] [Google Scholar]
- 500. Entezam A., Biacsi R., Orrison B., Saha T., Hoffman G. E., Grabczyk E., Nussbaum R. L., and Usdin K. (2007) Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene 395, 125–134 10.1016/j.gene.2007.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Calado R. T., and Dumitriu B. (2013) Telomere dynamics in mice and humans. Semin. Hematol. 50, 165–174 10.1053/j.seminhematol.2013.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502. Gerhardt J., Zaninovic N., Zhan Q., Madireddy A., Nolin S. L., Ersalesi N., Yan Z., Rosenwaks Z., and Schildkraut C. L. (2014) Cis-acting DNA sequence at a replication origin promotes repeat expansion to fragile X full mutation. J. Cell Biol. 206, 599–607 10.1083/jcb.201404157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Shen X., Kilikevicius A., O'Reilly D., Prakash T. P., Damha M. J., Rigo F., and Corey D. R. (2018) Activating frataxin expression by single-stranded siRNAs targeting the GAA repeat expansion. Bioorg. Med. Chem. Lett. 28, 2850–2855 10.1016/j.bmcl.2018.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504. Haenfler J. M., Skariah G., Rodriguez C. M., Monteiro da Rocha A., Parent J. M., Smith G. D., and Todd P. K. (2018) Targeted reactivation of FMR1 transcription in Fragile X syndrome embryonic stem cells. Front. Mol. Neurosci. 11, 282 10.3389/fnmol.2018.00282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. Liu X. S., Wu H., Krzisch M., Wu X., Graef J., Muffat J., Hnisz D., Li C. H., Yuan B., Xu C., Li Y., Vershkov D., Cacace A., Young R. A., and Jaenisch R. (2018) Rescue of Fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172, 979–992.e6 10.1016/j.cell.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Simone R., Balendra R., Moens T. G., Preza E., Wilson K. M., Heslegrave A., Woodling N. S., Niccoli T., Gilbert-Jaramillo J., Abdelkarim S., Clayton E. L., Clarke M., Konrad M. T., Nicoll A. J., Mitchell J. S., et al. (2018) G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol. Med. 10, 22–31 10.15252/emmm.201707850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Thornton C. A., Wang E., and Carrell E. M. (2017) Myotonic dystrophy: approach to therapy. Curr. Opin. Genet. Dev. 44, 135–140 10.1016/j.gde.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Zamiri B., Reddy K., Macgregor R. B. Jr., and Pearson C. E. (2014) TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J. Biol. Chem. 289, 4653–4659 10.1074/jbc.C113.502336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509. Green K. M., Sheth U. J., Flores B. N., Wright S. E., Sutter A. B., Kearse M. G., Barmada S. J., Ivanova M. I., and Todd P. K. (2019) High-throughput screening yields several small-molecule inhibitors of repeat-associated non-AUG translation. J. Biol. Chem. 294, 18624–18638 10.1074/jbc.RA119.009951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510. Bargiela A., Sabater-Arcis M., Espinosa-Espinosa J., Zulaica M., Lopez de Munain A., and Artero R. (2019) Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models. Proc. Natl. Acad. Sci. U.S.A. 116, 25203–25213 10.1073/pnas.1820297116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Reddy K., Jenquin J. R., McConnell O. L., Cleary J. D., Richardson J. I., Pinto B. S., Haerle M. C., Delgado E., Planco L., Nakamori M., Wang E. T., and Berglund J. A. (2019) A CTG repeat-selective chemical screen identifies microtubule inhibitors as selective modulators of toxic CUG RNA levels. Proc. Natl. Acad. Sci. U.S.A. 116, 20991–21000 10.1073/pnas.1901893116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Verma A. K., Khan E., Bhagwat S. R., and Kumar A. (2020) Exploring the potential of small molecule-based therapeutic approaches for targeting trinucleotide repeat disorders. Mol. Neurobiol. 57, 566–584 10.1007/s12035-019-01724-4 [DOI] [PubMed] [Google Scholar]
- 513. Khan E., Biswas S., Mishra S. K., Mishra R., Samanta S., Mishra A., Tawani A., and Kumar A. (2019) Rationally designed small molecules targeting toxic CAG repeat RNA that causes Huntington's disease (HD) and spinocerebellar ataxia (SCAs). Biochimie 163, 21–32 10.1016/j.biochi.2019.05.001 [DOI] [PubMed] [Google Scholar]
- 514. Angelbello A. J., Rzuczek S. G., Mckee K. K., Chen J. L., Olafson H., Cameron M. D., Moss W. N., Wang E. T., and Disney M. D. (2019) Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U.S.A. 116, 7799–7804 10.1073/pnas.1901484116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Cheng W., Wang S., Zhang Z., Morgens D. W., Hayes L. R., Lee S., Portz B., Xie Y., Nguyen B. V., Haney M. S., Yan S., Dong D., Coyne A. N., Yang J., Xian F., et al. (2019) CRISPR-Cas9 screens identify the RNA helicase DDX3X as a repressor of C9ORF72 (GGGGCC)n repeat-associated non-AUG translation. Neuron 104, 885–898.e8 10.1016/j.neuron.2019.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Richard G. F., Viterbo D., Khanna V., Mosbach V., Castelain L., and Dujon B. (2014) Highly specific contractions of a single CAG/CTG trinucleotide repeat by TALEN in yeast. PLoS ONE 9, e95611 10.1371/journal.pone.0095611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Marcadier J. L., and Pearson C. E. (2003) Fidelity of primate cell repair of a double-strand break within a (CTG).(CAG) tract: effect of slipped DNA structures. J. Biol. Chem. 278, 33848–33856 10.1074/jbc.M304284200 [DOI] [PubMed] [Google Scholar]
- 518. van Agtmaal E. L., André L. M., Willemse M., Cumming S. A., van Kessel I. D. G., van den Broek W. J. A. A., Gourdon G., Furling D., Mouly V., Monckton D. G., Wansink D. G., and Wieringa B. (2017) CRISPR/Cas9-induced (CTGCAG)n repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol. Ther. 25, 24–43 10.1016/j.ymthe.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. Cinesi C., Aeschbach L., Yang B., and Dion V. (2016) Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat. Commun. 7, 13272 10.1038/ncomms13272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520. Dastidar S., Ardui S., Singh K., Majumdar D., Nair N., Fu Y., Reyon D., Samara E., Gerli M. F. M., Klein A. F., De Schrijver W., Tipanee J., Seneca S., Tulalamba W., Wang H., et al. (2018) Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 46, 8275–8298 10.1093/nar/gky548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521. Ouellet D. L., Cherif K., Rousseau J., and Tremblay J. P. (2017) Deletion of the GAA repeats from the human frataxin gene using the CRISPR-Cas9 system in YG8R-derived cells and mouse models of Friedreich ataxia. Gene Ther. 24, 265–274 10.1038/gt.2016.89 [DOI] [PubMed] [Google Scholar]
- 522. Park C. Y., Halevy T., Lee D. R., Sung J. J., Lee J. S., Yanuka O., Benvenisty N., and Kim D. W. (2015) Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 13, 234–241 10.1016/j.celrep.2015.08.084 [DOI] [PubMed] [Google Scholar]
- 523. Lo Scrudato M., Poulard K., Sourd C., Tomé S., Klein A. F., Corre G., Huguet A., Furling D., Gourdon G., and Buj-Bello A. (2019) Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in the muscle of DM1 mice. Mol. Ther. 27, 1372–1388 10.1016/j.ymthe.2019.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. André L. M., van Cruchten R. T. P., Willemse M., Bezstarosti K., Demmers J. A. A., van Agtmaal E. L., Wansink D. G., and Wieringa B. (2019) Recovery in the myogenic program of congenital myotonic dystrophy myoblasts after excision of the expanded (CTG)n repeat. Int. J. Mol. Sci. 20, E5685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Mosbach V., Poggi L., and Richard G. F. (2019) Trinucleotide repeat instability during double-strand break repair: from mechanisms to gene therapy. Curr. Genet. 65, 17–28 10.1007/s00294-018-0865-1 [DOI] [PubMed] [Google Scholar]
- 526. Ekman F. K., Ojala D. S., Adil M. M., Lopez P. A., Schaffer D. V., and Gaj T. (2019) CRISPR-Cas9-mediated genome editing increases lifespan and improves motor deficits in a Huntington's disease mouse model. Mol. Ther. Nucleic Acids 17, 829–839 10.1016/j.omtn.2019.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527. Pratte A., Prevost C., Puymirat J., and Mathieu J. (2015) Anticipation in myotonic dystrophy type 1 parents with small CTG expansions. Am. J. Med. Genet. A 167A, 708–714 10.1002/ajmg.a.36950 [DOI] [PubMed] [Google Scholar]
- 528. Chow J. F., Yeung W. S., Lau E. Y., Lam S. T., Tong T., Ng E. H., and Ho P. C. (2009) Singleton birth after preimplantation genetic diagnosis for Huntington disease using whole genome amplification. Fertil. Steril. 92, 828.e7–10 10.1016/j.fertnstert.2009.05.007 [DOI] [PubMed] [Google Scholar]
- 529. Rajan-Babu I. S., Lian M., Cheah F. S. H., Chen M., Tan A. S. C., Prasath E. B., Loh S. F., and Chong S. S. (2017) FMR1 CGG repeat expansion mutation detection and linked haplotype analysis for reliable and accurate preimplantation genetic diagnosis of fragile X syndrome. Expert Rev. Mol. Med. 19, e10 10.1017/erm.2017.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Perminov D., Voložonoka L., Korņejeva L., Jokste-Pţmane E., Blumberga A., Krasucka S., Seimuškina N., Kovaļova I., and Fodina V. (2017) First preimplantation genetic testing case for monogenic disease in Latvia. Gynecol. Endocrinol. 33, 47–49 10.1080/09513590.2017.1404239 [DOI] [PubMed] [Google Scholar]
- 531. Lian M., Lee C. G., and Chong S. S. (2019) Robust preimplantation genetic testing strategy for myotonic dystrophy type 1 by bidirectional triplet-primed polymerase chain reaction combined with multi-microsatellite haplotyping following whole-genome amplification. Front. Genet. 10, 589 10.3389/fgene.2019.00589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Zhao M., Cheah F. S. H., Tan A. S. C., Lian M., Phang G. P., Agarwal A., and Chong S. S. (2019) Robust preimplantation genetic testing of Huntington disease by combined triplet-primed PCR analysis of the HTT CAG repeat and multi-microsatellite haplotyping. Sci. Rep. 9, 16481 10.1038/s41598-019-52769-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. European Community Huntington's Disease Collaborative Study Group (1993) Ethical and social issues in presymptomatic testing for Huntington's disease: a European Community collaborative study. J. Med. Genet. 30, 1028–1035 10.1136/jmg.30.12.1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534. Tassicker R., Savulescu J., Skene L., Marshall P., Fitzgerald L., and Delatycki M. B. (2003) Prenatal diagnosis requests for Huntington's disease when the father is at risk and does not want to know his genetic status: clinical, legal, and ethical viewpoints. BMJ 326, 331–333 10.1136/bmj.326.7384.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535. Matsuura T., Yamagata T., Burgess D. L., Rasmussen A., Grewal R. P., Watase K., Khajavi M., McCall A. E., Davis C. F., Zu L., Achari M., Pulst S. M., Alonso E., Noebels J. L., Nelson D. L., et al. (2000) Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 26, 191–194 10.1038/79911 [DOI] [PubMed] [Google Scholar]
- 536. Orr H. T., Chung M. Y., Banfi S., Kwiatkowski T. J. Jr., Servadio A., Beaudet A. L., McCall A. E., Duvick L. A., Ranum L. P., and Zoghbi H. Y. (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 4, 221–226 10.1038/ng0793-221 [DOI] [PubMed] [Google Scholar]
- 537. Pulst S. M., Nechiporuk A., Nechiporuk T., Gispert S., Chen X. N., Lopes-Cendes I., Pearlman S., Starkman S., Orozco-Diaz G., Lunkes A., DeJong P., Rouleau G. A., Auburger G., Korenberg J. R., Figueroa C., and Sahba S. (1996) Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14, 269–276 10.1038/ng1196-269 [DOI] [PubMed] [Google Scholar]
- 538. Zhuchenko O., Bailey J., Bonnen P., Ashizawa T., Stockton D. W., Amos C., Dobyns W. B., Subramony S. H., Zoghbi H. Y., and Lee C. C. (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α 1A-voltage-dependent calcium channel. Nat. Genet. 15, 62–69 10.1038/ng0197-62 [DOI] [PubMed] [Google Scholar]
- 539. Nakamura K., Jeong S. Y., Uchihara T., Anno M., Nagashima K., Nagashima T., Ikeda S., Tsuji S., and Kanazawa I. (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum. Mol. Genet. 10, 1441–1448 10.1093/hmg/10.14.1441 [DOI] [PubMed] [Google Scholar]
- 540. Holmes S. E., O'Hearn E. E., McInnis M. G., Gorelick-Feldman D. A., Kleiderlein J. J., Callahan C., Kwak N. G., Ingersoll-Ashworth R. G., Sherr M., Sumner A. J., Sharp A. H., Ananth U., Seltzer W. K., Boss M. A., Vieria-Saecker A. M., Epplen J. T., Riess O., Ross C. A., and Margolis R. L. (1999) Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat. Genet. 23, 391–392 10.1038/70493 [DOI] [PubMed] [Google Scholar]
- 541. Lalioti M. D., Scott H. S., Buresi C., Rossier C., Bottani A., Morris M. A., Malafosse A., and Antonarakis S. E. (1997) Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 386, 847–851 10.1038/386847a0 [DOI] [PubMed] [Google Scholar]
- 542. Holmes S. E., O'Hearn E., Rosenblatt A., Callahan C., Hwang H. S., Ingersoll-Ashworth R. G., Fleisher A., Stevanin G., Brice A., Potter N. T., Ross C. A., and Margolis R. L. (2001) A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat. Genet. 29, 377–378 10.1038/ng760 [DOI] [PubMed] [Google Scholar]
- 543. Wieben E. D., Aleff R. A., Tosakulwong N., Butz M. L., Highsmith W. E., Edwards A. O., and Baratz K. H. (2012) A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2–2) gene predicts Fuchs corneal dystrophy. PLoS ONE 7, e49083 10.1371/journal.pone.0049083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. LaCroix A. J., Stabley D., Sahraoui R., Adam M. P., Mehaffey M., Kernan K., Myers C. T., Fagerstrom C., Anadiotis G., Akkari Y. M., Robbins K. M., Gripp K. W., Baratela W. A. R., Bober M. B., Duker A. L., et al. (2019) GGC repeat expansion and exon 1 methylation of XYLT1 is a common pathogenic variant in Baratela-Scott syndrome. Am. J. Hum. Genet. 104, 35–44 10.1016/j.ajhg.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Knight S. J., Flannery A. V., Hirst M. C., Campbell L., Christodoulou Z., Phelps S. R., Pointon J., Middleton-Price H. R., Barnicoat A., and Pembrey M. E. (1993) Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 74, 127–134 10.1016/0092-8674(93)90300-F [DOI] [PubMed] [Google Scholar]
- 546. Winnepenninckx B., Debacker K., Ramsay J., Smeets D., Smits A., FitzPatrick D. R., and Kooy R. F. (2007) CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am. J. Hum. Genet. 80, 221–231 10.1086/510800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Michaelis R. C., Velagaleti G. V., Jones C., Pivnick E. K., Phelan M. C., Boyd E., Tarleton J., Wilroy R. S., Tunnacliffe A., and Tharapel A. T. (1998) Most Jacobsen syndrome deletion breakpoints occur distal to FRA11B. Am. J. Med. Genet. 76, 222–228 10.1002/(SICI)1096-8628(19980319)76:3<222::AID-AJMG5>3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- 548. Sato N., Amino T., Kobayashi K., Asakawa S., Ishiguro T., Tsunemi T., Takahashi M., Matsuura T., Flanigan K. M., Iwasaki S., Ishino F., Saito Y., Murayama S., Yoshida M., Hashizume Y., et al. (2009) Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 85, 544–557 10.1016/j.ajhg.2009.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. Nakamori M., Panigrahi G.B., Lanni S., Gall-Duncan T., Hayakawa H., Tanaka H., Luo J., Otabe T., Li J., Sakata A., Caron M.C., Joshi N., Prasolava T., Chiang K., Masson J.Y., et al., (2020) A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat Genet. 52, 146–159 10.1038/s41588-019-0575-8 [DOI] [PMC free article] [PubMed] [Google Scholar]