

Abstract
Renpenning syndrome belongs to a group of X-linked intellectual disability disorders. The Renpenning syndrome–associated protein PQBP1 (polyglutamine-binding protein 1) is intrinsically disordered, associates with several splicing factors, and is involved in pre-mRNA splicing. PQBP1 uses its C-terminal YxxPxxVL motif for binding to the splicing factor TXNL4A (thioredoxin like 4A), but the biological function of this interaction has yet to be elucidated. In this study, using recombinant protein expression, in vitro binding assays, and immunofluorescence microscopy in HeLa cells, we found that a recently reported X-linked intellectual disability–associated missense mutation, resulting in the PQBP1-P244L variant, disrupts the interaction with TXNL4A. We further show that this interaction is critical for the subcellular location of TXNL4A. In combination with other PQBP1 variants lacking a functional nuclear localization signal required for recognition by the nuclear import receptor karyopherin β2, we demonstrate that PQBP1 facilitates the nuclear import of TXNL4A via a piggyback mechanism. These findings expand our understanding of the molecular basis of the PQBP1–TXNL4A interaction and of the etiology and pathogenesis of Renpenning syndrome and related disorders.
Keywords: nuclear transport, protein complex, protein import, protein-protein interaction, autism, neurodevelopment, PQBP1, Renpenning syndrome, splicing factor, TXNL4A, X-linked intellectual disability
Introduction
Renpenning syndrome, a group of X-linked intellectual disability, is caused by mutations in human PQBP1 (polyglutamine-binding protein 1, also known as Npw38) gene on Xp11.2 (1–3). PQBP1 gene encodes an intrinsically disordered protein predominantly expressed in the central nervous system during development and playing important roles in neurodevelopment and neuronal functions (4–7). PQBP1 is composed of three interacting domains: a WW domain interacting with the nucleocytoplasmic shuttling splicing factor WBP11 (also known as SIPP1/NPWBP/SNP70) (8–10); a polar amino acid–rich domain (PRD)2 interacting with polyglutamine tracts (11, 12); a proline–tyrosine nuclear localization signal (PY-NLS) recognized by nuclear import receptor karyopherin β2 (Kapβ2) (13); and an unstructured C-terminal domain (CTD) interacting with splicing factor TXNL4A (14–18). Despite its interaction with splicing factors, PQBP1 is also found present in the precatalytic B complex (19, 20), suggesting the involvement of PQBP1 in alternative pre-mRNA splicing. Wang et al. (21) reported that depletion of Pqbp1 in mouse primary cortical neurons changed alternative splicing of mRNAs enriched for neuron projection functions and resulted in reduced outgrowth and branching of dendrites. However, most mutations of PQBP1 found in Renpenning syndrome patients, except for the missense mutation Y65C in the WW domain (22), are frameshift mutations in the PRD or CTD region, resulting in premature termination (1–3). How different mutations affect PQBP1 function remains unclear. We previously discovered that PQBP1-Δ459–462 and Δ463–464 mutants encode a new epitope that specifically associates with FMRP (fragile X mental retardation protein) and promotes its degradation (23). It points out the fact that frameshift mutations may gain unexpected functions with the new sequences, instead of just losing the original ones. It makes the mechanistic study more complicated.
Human TXNL4A (also known as U5–15kD/snRNP15/hDim1) is an evolutionarily highly conserved component of the U5 and U4/U6.U5 small nuclear ribonucleoprotein particles (24). Its yeast ortholog Dim1/Dib1/Snu16 is essential for pre-mRNA splicing in vivo (25, 26). TXNL4A has a thioredoxin-like fold formed by four-stranded β-sheets flanked by three α-helices (24) and binds a continuous 23-residue segment of the C-terminal domain of PQBP1 (14, 16). The structure of the binary complex of TXNL4A and PQBP1 shows that the hydrophobic groove of TXNL4A formed by Val14, Ile18, Phe69, Met72, Tyr73, Met82, and Phe84 recognizes the 245YxxPxxVL motif of PQBP1 (17). This complex is mainly stabilized by these hydrophobic interactions as well as two hydrogen bonds (PQBP1-Y245: TXNL4A-E74; PQBP1-N255:TXNL4A-D68) (17). PQBP1 frameshift mutants that lack the C-terminal domain cannot interact with TXNL4A, which is believed to affect the function of PQBP1 in splicing (17). The interaction of PQBP1 and TXNL4A seems important for the localization of TXNL4A. Exogenously expressed TXNL4A is predominately diffused in the cells but will form large punctate nuclear structure if co-expressed with PQBP1 (15). Whether PQBP1 promotes the nuclear import of TXNL4A has yet to be elucidated.
Recently, Redin et al. (27) identified a new missense mutation of PQBP1 (c.731C>T, P244L) in a targeted high-throughput sequencing test in intellectual disability or autism patients. This mutation is right before the 245YxxPxxVL252 motif of PQBP1 for TXNL4A binding. In this study, we found that the PQBP1-P244L mutant disrupted the interaction with TXNL4A. In combination with other PQBP1 NLS mutants, we clearly showed that PQBP1 facilitates the nuclear import of TXNL4A in a piggyback mechanism.
Results
PQBP1-P244L mutant disrupts its interaction with TXNL4A
Previous studies show that TXNL4A recognizes a 245YxxPxxVL252 motif in the C-terminal domain of PQBP1, which is missing in PQBP1 frameshift mutants found in Renpenning syndrome patients. Our in vitro binding assay also demonstrated that GST–PQBP1-Δ461–462 and Δ575–576 lost their binding capacity to TXNL4A, whereas the missense mutation Y65C in the WW domain did not affect TXNL4A binding (Fig. 1, A–C), and the RPY mutant in the NLS region only slightly affected TXNL4A binding (Fig. 1, A–C). Redin et al. (27) reported a new missense mutation in the CTD of PQBP1 (c.731C>T) using targeted high-throughput sequencing, which changes residue 244 from proline to leucine (P244L). Pro244 is right before the 245YxxPxxVL252 motif (Fig. 1A) and was not identified in crystal structures as a “hot spot” for TXNL4A binding (17). We were curious about whether P244L would affect the binding to TXNL4A, so we generated the recombinant mutant protein GST–PQBP1-P244L for in vitro binding assay. The result showed a significant decrease in TXNL4A binding of GST–PQBP1-P244L (∼17.6% of GST–PQBP1–WT), which is comparable with that of GST–PQBP1-Δ575–576 (∼13.2% of GST–PQBP1–WT) (Fig. 1, B and C).
Figure 1.

The C-terminal mutations of PQBP1 disrupt its interaction with TXNL4A. A, schematic demonstration of protein domain structures of PQBP1–WT and mutants. The mutated regions are highlighted with red. Y65C, the missense mutant in the WW domain; Δ461–462, frameshift mutant c.461–462del, p.E154Afs*12; Δ575–576, frameshift mutant c.575–576del, p.K192Sfs*7; P244L, the missense mutation in C-terminal domain; RPY, tetra-point mutant (180RR181 and 186PY187 to alanines) in the nuclear localization signal. B, in vitro binding assays show the interactions of GST–PQBP1–WT and mutants with TXNL4A. Immobilized GST–PQBP1–WT and mutants were incubated with purified recombinant TXNL4A. Representative results from three independent experiments. C, densitometric analysis of B. The relative density of TXNL4A band against GST–PQBP1 band in each reaction was normalized to that in the reaction with GST–PQBP1–WT (100%). The data show as means ± S.D. from three independent experiments. ****, p < 0.0001. MW, molecular mass.
In the structure of PQBP1–CT43 (238–260 amino acids) in complex with TXNL4A (Protein Data Bank code 4BWQ), PQBP1 takes an L-shaped structure, and residues 238–247 are in an extended conformation followed by an α-helix formed by residues 248–259 (17). Pro244 sits in a small cavity formed by Lys88, His89, and Met91 of TXNL4A, and it is not involved in hydrophobic interactions or hydrogen bonds with TXNL4A (Fig. 2A). Indeed, P244A mutant only showed a fairly small change in TXNL4A binding, indicating that Pro244 is not a hot spot (Fig. 2, B and C). Mutagenesis analysis using PyMOL (28) showed P244L mutation introduced a long side chain in the small space, and the collision might disrupt the interface between PQBP1 and TXNL4A (Fig. 2A). To test this, we made an additional mutant P244V that also introduces a long side chain at that site and performed the binding assay. As we expected, both PQBP1-P244V and P244L mutant proteins showed remarkable reduction in TXNL4A binding (Fig. 2, B and C). We demonstrated that a single residue mutation P244L in the CTD of PQBP1 abolished its interaction with TXNL4A.
Figure 2.

The C-terminal mutations of PQBP1 disrupt its interaction with TXNL4A. A, structure mutagenesis analysis on the complex of PQBP1–CT43 and TXNL4A. The structure of PQBP1–WT and TXNL4A was adopted from Protein Data Bank structure 4BWQ. Green cartoon, PQBP1; gray surface, TXNL4A; yellow surface, the three residues Lys88, His89, and Met91 in TXNL4A that form the small cavity accommodating Pro244 in PQBP1. Mutagenesis analysis was done with PyMOL, and the side chains of mutated residues are shown as stick model and highlighted with red. B, in vitro binding assays show the interactions of GST–PQBP1–WT and P244 site mutants with TXNL4A. Immobilized GST–PQBP1–WT and mutants were incubated with purified recombinant TXNL4A. Representative results from three independent experiments. C, densitometric analysis of B. The relative density of TXNL4A band against GST–PQBP1 band in each reaction was normalized to that in the reaction with GST–PQBP1–WT (100%). The data are shown as means ± S.D. from three independent experiments. **, p < 0.01; ***, p < 0.001. MW, molecular mass.
PQBP1 contains a PY-NLS recognized by Kapβ2
PQBP1 is dominantly localized in nuclei (6, 8, 29, 30). Previous reports show that PQBP1 contains a PY-NLS and is a substrate of nuclear import receptor Kapβ2 (13). The PY-NLS of PQBP1 is within the residues 170–187 region right before the C-terminal domain (Fig. 3A). It is composed of a N-terminal basic residue-enriched epitope, a conserved Arg residue in the middle, and PY residues at the C terminus. To disrupt the interaction between PQBP1 and Kapβ2, we mutated 180RR181 and 186PY187 to alanines by site-directed mutagenesis (Fig. 3A) and tested its binding ability to Kapβ2. An in vitro binding assay showed that Kapβ2 bound to the GST–PQBP1–RPY mutant reduced to less than 10% of that bound to GST–PQBP1–WT (Fig. 3, B and C). Meanwhile, we compared the interactions of other PQBP1 pathogenic mutants with Kapβ2. The missense mutation in the WW domain Y65C did not affect the interaction between PQBP1 and Kapβ2, whereas the Δ461–462 frameshift mutant lacking the partial PRD, NLS, and the entire CTD lost its binding to Kapβ2 (Fig. 3, B and C). The frameshift mutant Δ575–576 that changes the sequence right after the NLS caused ∼50% loss of Kapβ2 binding, and the missense mutant in CTD P244L slightly affected Kapβ2 binding (Fig. 3, B and C). These data verify that the interaction between PQBP1 and nuclear import receptor Kapβ2 is mainly mediated by the PY-NLS within the region of residues 170–187.
Figure 3.

PQBP1 binds to the nuclear import receptor Kapβ2 through its PY-NLS. A, the sequences of PQBP1–WT and RPY mutants at PY-NLS. The three-epitope structure of PY-NLS consensus sequence is shown at the top. The mutated residues are highlighted with red. B, in vitro binding assays show the interactions of GST–PQBP1–WT and mutants with Kapβ2. Immobilized GST–PQBP1–WT and mutants were incubated with purified recombinant Kapβ2. Representative results from three independent experiments are shown here. C, densitometric analysis of B. The relative density of Kapβ2 band against GST–PQBP1 band in each reaction was normalized to that in the reaction with GST–PQBP1–WT (100%). The data are shown as means ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01; ****, p < 0.0001. MW, molecular mass.
PQBP1 can simultaneously bind Kapβ2 and TXNL4A
PQBP1 interacts with Kapβ2 and TXNL4A through different regions, which made us suspect that PQBP1 might bind Kapβ2 and TXNL4A simultaneously. We immobilized GST–PQBP1–WT proteins on GSH resins and incubated with a mixture of TXNL4A and Kapβ2. The results clearly showed that PQBP1 bound Kapβ2 and TXNL4A together (Fig. 4A), indicating that PQBP1 might function as a connector between Kapβ2 and TXNL4A. Furthermore, when we immobilized MBP–TXNL4A to test its interaction with soluble Kapβ2 in the presence or absence of different PQBP1 proteins, we found that MBP–TXNL4A bound Kapβ2 only in the presence of PQBP1–WT (Fig. 4B). In the presence of PQBP1-P244L, MBP–TXNL4A could not bind Kapβ2 because its interaction with PQBP1 was disrupted; whereas in the presence of PQBP1–RPY mutant, MBP–TXNL4A associated with PQBP1–RPY, but PQBP1–RPY could not bind Kapβ2 because of the loss of PY-NLS (Fig. 4B). Our data confirm that PQBP1 mediates the interaction between TXNL4A and nuclear import receptor Kapβ2.
Figure 4.
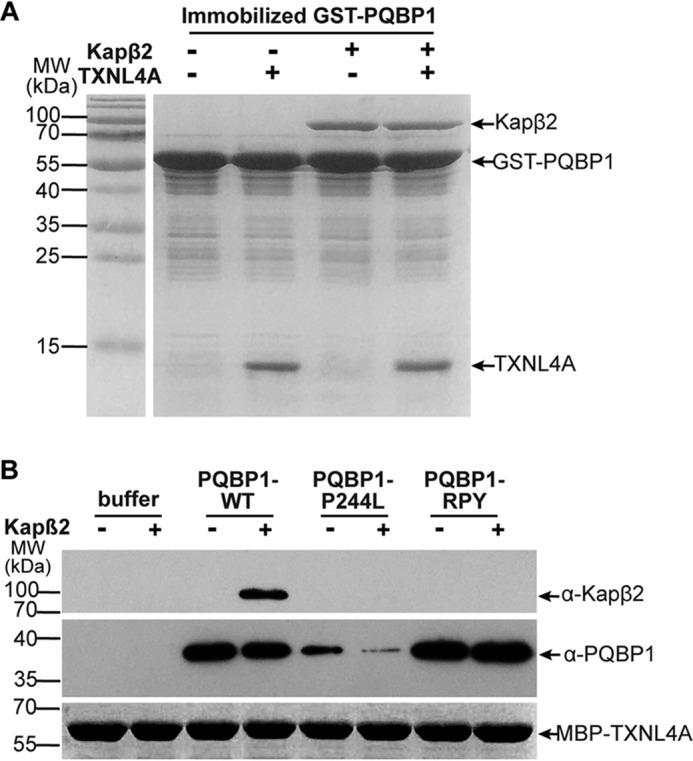
PQBP1 binds Kapβ2 and TXNL4A simultaneously. A, in vitro binding assays show the interactions of GST–PQBP1–WT with Kapβ2 and TXNL4A. Immobilized GST–PQBP1–WT was incubated with purified recombinant TXNL4A alone, Kapβ2 alone, or Kapβ2 with TXNL4A. Shown are representative results from three independent experiments. B, Western blots show the interactions of MBP–TXNL4A with PQBP1 and Kapβ2. Immobilized MBP–TXNL4A was incubated with purified recombinant Kapβ2 in the presence or absence of PQBP1–WT or mutants. Representative results from three independent experiments. MW, molecular mass.
The interaction with PQBP1 is critical for the nuclear import of TXNL4A
Next, we investigated whether Kapβ2-mediated nuclear import of PQBP1 also facilitates the import of TXNL4A. We co-transfected pEGFP–C1–TXNL4A with pFLAG–CMV2–PQBP1–WT or different mutants into HeLa cells and measured the cytoplasmic and nuclear fluorescence signals of EGFP and anti-FLAG, respectively. FLAG–PQBP1–WT, Y65C, Δ575–576, and P244L were mainly in the nucleus (N/C ratio, 4.69, 4.80, 4.68, and 5.74), but the other two mutants without the PY-NLS FLAG–PQBP1-Δ461–462 and RPY diffused throughout the cell (N/C ratio, 1.06 and 1.87) (Fig. 5A). It suggests that PY-NLS is critical for proper nuclear location of PQBP1. EGFP-TXNL4A by itself diffused in the cell (N/C ratio, 1.28), but when it was co-transfected with PQBP1–WT or Y65C, it was dominantly in the nucleus (N/C ratio, 2.90 or 2.63) (Fig. 5, A, and B). This PQBP1-facilitated nuclear import of EGFP-TXNL4A was abolished when the cells were co-transfected with FLAG–PQBP1-Δ461–462, Δ575–576, P244L, and RPY (N/C ratio, 1.38, 1.13, 1.80, and 1.42) (Fig. 5, A and B). Among these four mutants, FLAG–PQBP1-Δ461–462 and RPY lost their PY-NLS, whereas FLAG–PQBP1-Δ575–576 and P244L lost their interaction with TXNL4A. Collectively, both the PY-NLS and C-terminal TXNL4A-binding string are required for PQBP1-facilitated TXNL4A nuclear import (Fig. 5C).
Figure 5.

PQBP1 facilitates the nuclear import of TXNL4A. A, immunostaining shows the subcellular localizations of transfected PQBP1 and TXNL4A. HeLa cells were transfected with pFLAG–CMV2–PQBP1–WT or mutants in the presence of pEGFP–C1–TXNL4A. Scale bar, 20 μm. B, quantification of the nuclear/cytoplasmic fluorescent intensities of EGFP-TXNL4A signals. The data show the means ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01. C, the model of PQBP1-facilitated nuclear import of TXNL4A via the Kapβ2-mediated pathway. DAPI, 4′,6′-diamino-2-phenylindole.
Discussion
Both PQBP1 and TXNL4A are evolutionarily conserved molecules and components of spliceosome (15, 19, 20, 24, 31–33). The molecular interaction between them is well-established (14–18), but what the biological function of this interaction is remains unclear. TXNL4A is a member of the U5 spliceosomal complex and mainly functions in the nucleus. There is no obvious NLS in its sequence, and its molecular mass is only 15 kDa. So, it is easy to believe that it gets into the nucleus through passive diffusion by default. However, our study using exogenously expressed proteins demonstrates that receptor-mediated active transport is also involved in TXNL4A nuclear import. It is transported with PQBP1 through Kapβ2-mediated pathway in a piggyback mechanism. Interestingly, we noticed that TXNL4A-GFP was more accumulated in the nucleus (32) than EGFP-TXNL4A (15), indicating the EGFP tag position might affect the subcellular distribution of TXNL4A. Given that the N terminus of TXNL4A is responsible for PQBP1 binding (17), N-terminal tagging could affect its interaction with PQBP1 and reduce its import efficiency. Because of the very low expression level and inaccessibility to antibodies of endogenous TXNL4A, we have not been able to test the import of endogenous TXNL4A yet. Our data show that PQBP1 and TXNL4A are not just associated in the spliceosome, but they may be transported together. Llorian et al. (34) showed that splicing factor WBP11, another PQBP1 interactor, could be piggyback-transported into the nucleus through the interaction with the PQBP1 WW domain. It seems that the full function of PQBP1 in splicing factor transport requires both the WW domain (35) and the C-terminal domain to be intact, suggesting that PQBP1 is a “two-pronged” adaptor for the transport of splicing factors.
PQBP1 is an intrinsically disordered protein with a small folded WW domain and long disordered regions encompassing the PRD and CTD. The unstructured C-terminal region of PQBP1 undergoes disorder-to-order transition upon complex formation with TXNL4A, and the evolutionarily conserved 245YxxPxxVL252 motif in this region is critical for PQBP1–TXNL4A binding (17, 36). Residues Tyr245, Pro248, Val251, and Leu252 are involved in the hydrophobic interactions with TXNL4A to stabilize the complex. Residue Pro244 not involved in any hydrophobic interactions or hydrogen bonds with TXNL4A sits in a small cavity on the surface of TXNL4A that can only accommodate short side chains. This accommodation is critical for the proper position of Tyr245 in the hydrophobic groove of TXNL4A because P244V and P244L mutants do not bind TXNL4A, but P244A does. In the structure of a fusion protein of TXNL4A residues 4–137 and PQBP1–CT43, residues 223–243 and 260–265 of PQBP1 are missing because of conformational flexibility, whereas residue Pro244 is still present in the structure (17), which provides another line of evidence to support the importance of proper accommodation of residue 244.
Most mutations of PQBP1 found in Renpenning syndrome patients are frameshift mutations in the PRD and CTD regions, resulting in premature terminations (1–3). They share the common phenotypes like intellectual deficiency, microcephaly, short stature, and microrchidia (2, 37). PQBP1-P244L missense mutation was recently identified in intelligent disability patients using targeted high-throughput sequencing (27). These patients show moderate intellectual disability, poor autonomy, communication and social interaction disorders, learning difficulties, and obvious autistic behaviors (27). The quite different phenotypes in P244L patients indicate a distinct molecular pathogenesis. Our study shows that P244L mutation has a slight effect on its own subcellular localization and structure but significantly affects the subcellular distribution and potential function of its partner TXNL4A. It suggests a new pathogenic mechanism of Renpenning syndrome.
Experimental procedures
Plasmid construction
For protein expression, the cDNAs encoding full-length protein of PQBP1 WT or TXNL4A were inserted into pGEXTEV or pMALTEV vector, respectively. PQBP1-Y65C, Δ461–462, Δ575–576, P244A, P244V, P244L, and RPY mutants were generated by site-directed mutagenesis. For cell transfection, PQBP1–WT and mutants were subcloned into pFLAG–CMV2 vector and TXNL4A into pEGFP–C1 vector. pGEXTEV–Kapβ2 was obtained from Chook lab (University of Texas Southwestern Medical Center). All constructs were verified by DNA sequencing.
Recombinant protein expression
All GST or MBP fusion proteins were expressed in Escherichia coli BL21 cells. Bacteria were cultured in LB broth (Generay Biotech, Shanghai, China) and induced by 1 mm of isopropyl β-d-thiogalactopyranoside for 4 h at 25 °C or 12–16 h at 4 °C. The proteins were purified with GSH–Sepharose (G-Biosciences) and amylose resins (New England Biolabs, Ipswich, MA) as previous described (23).
In vitro binding assays
GST–PQBP1–WT and mutant fusion proteins immobilized to GSH resins were incubated with purified TXNL4A or Kapβ2 proteins at 4 °C for 30 min and extensively washed with TB buffer (20 mm HEPES, pH 7.3, 110 mm KOAc, 2 mm MgOAc, 2 mm DTT, 1 mm EGTA, 10% glycerol). Bound proteins were visualized using SDS-PAGE and Coomassie Blue staining. Approximately 5 μg of immobilized GST–PQBP1 proteins were incubated with ∼20 μg of purified TXNL4A or ∼20 μg of purified Kapβ2. Approximately 25% of the bound proteins was loaded for gel analysis.
MBP–TXNL4A proteins immobilized on amylose beads were incubated with Kapβ2 in the presence or absence of PQBP1–WT, PQBP1-P244L, and PQBP1–RPY mutants. Bound proteins were separated using SDS-PAGE and visualized with Coomassie Blue staining (MBP–TXNL4A) or Western blotting (Kapβ2 and PQBP1).
Cell culture, transfection, and immunofluorescence
HeLa cells were transfected with plasmids using X-treme GENE HP DNA transfection reagent (Roche, Basel, Switzerland) according to the manufacturer's protocol. After 16 h, the transfected cells were fixed with 4% paraformaldehyde in PBS for 20 min followed by permeabilization in 0.5% PBST for 10 min. After three washes, the cells were briefly blocked with 3% BSA in PBS, followed by incubation with anti-FLAG antibody (1:300, mouse, Abmart, Shanghai, China) at room temperature for 2 h. Then the samples were washed in 0.1% PBST and incubated with Alexa 555–labeled anti-mouse antibody (1:400, Abcam, Cambridge, UK) for 2 h at room temperature before mounting using the mounting medium with 4′,6′-diamino-2-phenylindole (Vector Laboratories Inc., Burlingame, CA). The cells were examined under a Leica DM5000 B (Leica, Germany). The fluorescence images were analyzed with ImageJ software (National Institutes of Health), and the data from three independent experiments were averaged. The quantitative data are means ± S.D.
Author contributions
X. L. and Z. C. Z. data curation; X. L., L.-X.D., and Z. C. Z. formal analysis; X. L. and L.-X.D. methodology; X. L., J. H., and Z. C. Z. writing-original draft; J. H. and Z. C. Z. funding acquisition; J. H. and Z. C. Z. project administration; Z. C. Z. conceptualization; Z. C. Z. supervision.
Acknowledgments
We thank Dr. Yuh Min Chook (University of Texas Southwestern Medical Center) for the construct and Yu-Qian Shen and Jian Zuo for technical help and members of the Han laboratory for critical comments on the manuscript.
This work was supported by National Natural Science Foundation of China Grant 31671045 (to Z. C. Z.), Jiangsu Natural Science Foundation Basic Research Program of Jiangsu Province Grant BK20170080 (to Z. C. Z.), and a National Natural Science Foundation of China Grant 81730034 (to J. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- PRD
- polar amino acid–rich domain
- CTD
- C-terminal domain
- PY-NLS
- proline–tyrosine nuclear localization signal
- Kapβ2
- karyopherin β2
- EGFP
- enhanced green fluorescence protein
- MBP
- maltose-binding protein
- GST
- glutathione S-transferase.
References
- 1. Kalscheuer V. M., Freude K., Musante L., Jensen L. R., Yntema H. G., Gécz J., Sefiani A., Hoffmann K., Moser B., Haas S., Gurok U., Haesler S., Aranda B., Nshedjan A., Tzschach A., et al. (2003) Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat. Genet. 35, 313–315 10.1038/ng1264 [DOI] [PubMed] [Google Scholar]
- 2. Stevenson R. E., Bennett C. W., Abidi F., Kleefstra T., Porteous M., Simensen R. J., Lubs H. A., Hamel B. C., and Schwartz C. E. (2005) Renpenning syndrome comes into focus. Am. J. Med. Genet. A 134, 415–421 [DOI] [PubMed] [Google Scholar]
- 3. Rejeb I., Ben Jemaa L., Abaied L., Kraoua L., Saillour Y., Maazoul F., Chelly J., and Chaabouni H. (2011) A novel frame shift mutation in the PQBP1 gene identified in a Tunisian family with X-linked mental retardation. Eur. J. Med. Genet. 54, 241–246 10.1016/j.ejmg.2011.01.010 [DOI] [PubMed] [Google Scholar]
- 4. Qi Y., Hoshino M., Wada Y., Marubuchi S., Yoshimura N., Kanazawa I., Shinomiya K., and Okazawa H. (2005) PQBP-1 is expressed predominantly in the central nervous system during development. Eur. J. Neurosci. 22, 1277–1286 10.1111/j.1460-9568.2005.04339.x [DOI] [PubMed] [Google Scholar]
- 5. Iwamoto K., Huang Y., and Ueda S. (2000) Genomic organization and alternative transcripts of the human PQBP-1 gene. Gene 259, 69–73 10.1016/S0378-1119(00)00437-6 [DOI] [PubMed] [Google Scholar]
- 6. Komuro A., Saeki M., and Kato S. (1999) Npw38, a novel nuclear protein possessing a WW domain capable of activating basal transcription. Nucleic Acids Res. 27, 1957–1965 10.1093/nar/27.9.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wan D., Zhang Z. C., Zhang X., Li Q., and Han J. (2015) X chromosome-linked intellectual disability protein PQBP1 associates with and regulates the translation of specific mRNAs. Hum. Mol. Genet. 24, 4599–4614 10.1093/hmg/ddv191 [DOI] [PubMed] [Google Scholar]
- 8. Komuro A., Saeki M., and Kato S. (1999) Association of two nuclear proteins, Npw38 and NpwBP, via the interaction between the WW domain and a novel proline-rich motif containing glycine and arginine. J. Biol. Chem. 274, 36513–36519 10.1074/jbc.274.51.36513 [DOI] [PubMed] [Google Scholar]
- 9. Bedford M. T., Sarbassova D., Xu J., Leder P., and Yaffe M. B. (2000) A novel pro-Arg motif recognized by WW domains. J. Biol. Chem. 275, 10359–10369 10.1074/jbc.275.14.10359 [DOI] [PubMed] [Google Scholar]
- 10. Llorian M., Beullens M., Andrés I., Ortiz J. M., and Bollen M. (2004) SIPP1, a novel pre-mRNA splicing factor and interactor of protein phosphatase-1. Biochem. J. 378, 229–238 10.1042/bj20030950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Imafuku I., Waragai M., Takeuchi S., Kanazawa I., Kawabata M., Mouradian M. M., and Okazawa H. (1998) Polar amino acid-rich sequences bind to polyglutamine tracts. Biochem. Biophys. Res. Commun. 253, 16–20 10.1006/bbrc.1998.9725 [DOI] [PubMed] [Google Scholar]
- 12. Okazawa H., Rich T., Chang A., Lin X., Waragai M., Kajikawa M., Enokido Y., Komuro A., Kato S., Shibata M., Hatanaka H., Mouradian M. M., Sudol M., and Kanazawa I. (2002) Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death. Neuron 34, 701–713 10.1016/S0896-6273(02)00697-9 [DOI] [PubMed] [Google Scholar]
- 13. Lee B. J., Cansizoglu A. E., Süel K. E., Louis T. H., Zhang Z., and Chook Y. M. (2006) Rules for nuclear localization sequence recognition by karyopherin β2. Cell 126, 543–558 10.1016/j.cell.2006.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Waragai M., Junn E., Kajikawa M., Takeuchi S., Kanazawa I., Shibata M., Mouradian M. M., and Okazawa H. (2000) PQBP-1/Npw38, a nuclear protein binding to the polyglutamine tract, interacts with U5–15kD/dim1p via the carboxyl-terminal domain. Biochem. Biophys. Res. Commun. 273, 592–595 10.1006/bbrc.2000.2992 [DOI] [PubMed] [Google Scholar]
- 15. Zhang Y., Lindblom T., Chang A., Sudol M., Sluder A. E., and Golemis E. A. (2000) Evidence that dim1 associates with proteins involved in pre-mRNA splicing, and delineation of residues essential for dim1 interactions with hnRNP F and Npw38/PQBP-1. Gene 257, 33–43 10.1016/S0378-1119(00)00372-3 [DOI] [PubMed] [Google Scholar]
- 16. Takahashi M., Mizuguchi M., Shinoda H., Aizawa T., Demura M., Okazawa H., and Kawano K. (2010) Polyglutamine tract-binding protein-1 binds to U5–15kD via a continuous 23-residue segment of the C-terminal domain. Biochim. Biophys. Acta 1804, 1500–1507 10.1016/j.bbapap.2010.03.007 [DOI] [PubMed] [Google Scholar]
- 17. Mizuguchi M., Obita T., Serita T., Kojima R., Nabeshima Y., and Okazawa H. (2014) Mutations in the PQBP1 gene prevent its interaction with the spliceosomal protein U5–15 kD. Nat. Commun. 5, 3822 10.1038/ncomms4822 [DOI] [PubMed] [Google Scholar]
- 18. Mizuguchi M., Obita T., Kajiyama A., Kozakai Y., Nakai T., Nabeshima Y., and Okazawa H. (2016) Allosteric modulation of the binding affinity between PQBP1 and the spliceosomal protein U5–15kD. FEBS Lett. 590, 2221–2231 10.1002/1873-3468.12256 [DOI] [PubMed] [Google Scholar]
- 19. Deckert J., Hartmuth K., Boehringer D., Behzadnia N., Will C. L., Kastner B., Stark H., Urlaub H., and Lührmann R. (2006) Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol. Cell. Biol. 26, 5528–5543 10.1128/MCB.00582-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Makarova O. V., Makarov E. M., Urlaub H., Will C. L., Gentzel M., Wilm M., and Lührmann R. (2004) A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J. 23, 2381–2391 10.1038/sj.emboj.7600241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Q., Moore M. J., Adelmant G., Marto J. A., and Silver P. A. (2013) PQBP1, a factor linked to intellectual disability, affects alternative splicing associated with neurite outgrowth. Genes Dev. 27, 615–626 10.1101/gad.212308.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lubs H., Abidi F. E., Echeverri R., Holloway L., Meindl A., Stevenson R. E., and Schwartz C. E. (2006) Golabi-Ito-Hall syndrome results from a missense mutation in the WW domain of the PQBP1 gene. J. Med. Genet. 43, e30 10.1136/jmg.2005.037556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang X. Y., Qi J., Shen Y. Q., Liu X., Liu A., Zhou Z., Han J., and Zhang Z. C. (2017) Mutations of PQBP1 in Renpenning syndrome promote ubiquitin-mediated degradation of FMRP and cause synaptic dysfunction. Hum. Mol. Genet. 26, 955–968 [DOI] [PubMed] [Google Scholar]
- 24. Reuter K., Nottrott S., Fabrizio P., Lührmann R., and Ficner R. (1999) Identification, characterization and crystal structure analysis of the human spliceosomal U5 snRNP-specific 15 kD protein. J. Mol. Biol. 294, 515–525 10.1006/jmbi.1999.3258 [DOI] [PubMed] [Google Scholar]
- 25. Carnahan R. H., Feoktistova A., Ren L., Niessen S., Yates J. R. 3rd, and Gould K. L. (2005) Dim1p is required for efficient splicing and export of mRNA encoding lid1p, a component of the fission yeast anaphase-promoting complex. Eukaryotic Cell 4, 577–587 10.1128/EC.4.3.577-587.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stevens S. W., and Abelson J. (1999) Purification of the yeast U4/U6.U5 small nuclear ribonucleoprotein particle and identification of its proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 7226–7231 10.1073/pnas.96.13.7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Redin C., Gérard B., Lauer J., Herenger Y., Muller J., Quartier A., Masurel-Paulet A., Willems M., Lesca G., El-Chehadeh S., Le Gras S., Vicaire S., Philipps M., Dumas M., Geoffroy V., et al. (2014) Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 51, 724–736 10.1136/jmedgenet-2014-102554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeLano W. L. (2015) The PyMOL Molecular Graphics System, version 1.8, Schroedinger, LLC, New York [Google Scholar]
- 29. Nicolaescu E., Beullens M., Lesage B., Keppens S., Himpens B., and Bollen M. (2008) Nature of the nuclear inclusions formed by PQBP1, a protein linked to neurodegenerative polyglutamine diseases. Eur. J. Cell Biol. 87, 817–829 10.1016/j.ejcb.2008.05.001 [DOI] [PubMed] [Google Scholar]
- 30. Okazawa H., Sudol M., and Rich T. (2001) PQBP-1 (Np/PQ): a polyglutamine tract-binding and nuclear inclusion-forming protein. Brain Res. Bull. 56, 273–280 10.1016/S0361-9230(01)00579-2 [DOI] [PubMed] [Google Scholar]
- 31. Nasu M., Mizuno F., and Ueda S. (2012) Comparative aspects of polyglutamine binding domain in PQBP-1 among Vertebrata. Gene 511, 243–247 10.1016/j.gene.2012.09.047 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y. Z., Gould K. L., Dunbrack R. L. Jr., Cheng H., Roder H., and Golemis E. A. (1999) The evolutionarily conserved Dim1 protein defines a novel branch of the thioredoxin fold superfamily. Physiol. Genomics 1, 109–118 10.1152/physiolgenomics.1999.1.3.109 [DOI] [PubMed] [Google Scholar]
- 33. Stevens S. W., Barta I., Ge H. Y., Moore R. E., Young M. K., Lee T. D., and Abelson J. (2001) Biochemical and genetic analyses of the U5, U6, and U4/U6 x U5 small nuclear ribonucleoproteins from Saccharomyces cerevisiae. RNA 7, 1543–1553 [PMC free article] [PubMed] [Google Scholar]
- 34. Llorian M., Beullens M., Lesage B., Nicolaescu E., Beke L., Landuyt W., Ortiz J. M., and Bollen M. (2005) Nucleocytoplasmic shuttling of the splicing factor SIPP1. J. Biol. Chem. 280, 38862–38869 10.1074/jbc.M509185200 [DOI] [PubMed] [Google Scholar]
- 35. Tapia V. E., Nicolaescu E., McDonald C. B., Musi V., Oka T., Inayoshi Y., Satteson A. C., Mazack V., Humbert J., Gaffney C. J., Beullens M., Schwartz C. E., Landgraf C., Volkmer R., Pastore A., et al. (2010) Y65C missense mutation in the WW domain of the Golabi–Ito–Hall syndrome protein PQBP1 affects its binding activity and deregulates pre-mRNA splicing. J. Biol. Chem. 285, 19391–19401 10.1074/jbc.M109.084525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sudol M., McDonald C. B., and Farooq A. (2012) Molecular insights into the WW domain of the Golabi-Ito-Hall syndrome protein PQBP1. FEBS Lett. 586, 2795–2799 10.1016/j.febslet.2012.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nizon M., Andrieux J., Rooryck C., de Blois M. C., Bourel-Ponchel E., Bourgois B., Boute O., David A., Delobel B., Duban-Bedu B., Giuliano F., Goldenberg A., Grotto S., Heron D., Karmous-Benailly H., et al. (2015) Phenotype-genotype correlations in 17 new patients with an Xp11.23p11.22 microduplication and review of the literature. Am. J. Med. Genet. A 167A, 111–122 10.1002/ajmg.a.36807 [DOI] [PubMed] [Google Scholar]