

Abstract
Recent clinical investigations indicate that anthracycline-based chemotherapies induce early decline in heart mass in cancer patients. Heart mass decline may be caused by a decrease in cardiac cell number because of increased cell death or by a reduction in cell size because of atrophy. We previously reported that an anthracycline, doxorubicin (DOX), induces apoptotic death of cardiomyocytes by activating cyclin-dependent kinase 2 (CDK2). However, the signaling pathway downstream of CDK2 remains to be characterized, and it is also unclear whether the same pathway mediates cardiac atrophy. Here we demonstrate that DOX exposure induces CDK2-dependent phosphorylation of the transcription factor forkhead box O1 (FOXO1) at Ser-249, leading to transcription of its proapoptotic target gene, Bcl-2–interacting mediator of cell death (Bim). In cultured cardiomyocytes, treatment with the FOXO1 inhibitor AS1842856 or transfection with FOXO1-specific siRNAs protected against DOX-induced apoptosis and mitochondrial damage. Oral administration of AS1842856 in mice abrogated apoptosis and prevented DOX-induced cardiac dysfunction. Intriguingly, pharmacological FOXO1 inhibition also attenuated DOX-induced cardiac atrophy, likely because of repression of muscle RING finger 1 (MuRF1), a proatrophic FOXO1 target gene. In conclusion, DOX exposure induces CDK2-dependent FOXO1 activation, resulting in cardiomyocyte apoptosis and atrophy. Our results identify FOXO1 as a promising drug target for managing DOX-induced cardiotoxicity. We propose that FOXO1 inhibitors may have potential as cardioprotective therapeutic agents during cancer chemotherapy.
Keywords: anticancer drug, cardiovascular disease, cardiomyopathy, heart failure, cardiac muscle, cell cycle, Adriamycin
Introduction
Anthracyclines are currently the cornerstone of many modern chemotherapy regimens. Doxorubicin (DOX,2 trade name Adriamycin), a prototype anthracycline chemotherapeutic agent, is frequently used to treat hematological malignancies and solid tumors in children and adults (1). Unfortunately, administration of DOX also causes cumulative dose-dependent cardiotoxicity that manifests as cardiomyopathy, which can eventually lead to heart failure and death (2). These undesired cardiovascular side effects severely limit clinical use of anthracyclines. Recent evidence suggests that DOX exposure causes a decrease in myocardial mass in cancer patients as early as 1 month after treatment initiation (3, 4). In particular, early decline in left ventricular mass is associated with worsening heart failure scores, suggesting that a reduction in ventricular mass may serve as a cardiotoxicity biomarker for therapeutic intervention (3). The reduction in heart mass following DOX treatment can potentially be attributed to myocardial cell loss via apoptosis (5, 6) and/or atrophy of cardiomyocytes (4).
DOX induces cardiomyocyte apoptosis through multiple mechanisms, including introduction of DNA damage by targeting topoisomerase II-β (5) and generation of reactive oxygen species via mitochondrial iron accumulation (6). Despite these advances in knowledge, a widely adopted preventive strategy for DOX cardiotoxicity is currently unavailable (1). Therefore, further investigation of the cardiotoxic mechanisms and identification of novel drug targets are urgently needed. We have reported previously that the Cip/Kip-family cyclin-dependent kinase (CDK) inhibitor p21 protects cardiomyocytes against DOX-induced apoptosis (7). Our follow-up studies further identify CDK2 as an essential driver of cardiomyocyte apoptosis in response to DOX challenge (8). Activation of CDK2 enhances transcription of the proapoptotic gene Bim, which is required for execution of apoptosis upon DOX exposure (8). However, the specific transcription factor responsible for CDK2-dependent Bim expression remains to be identified.
The Bim promoter contains two forkhead response elements (5′-GTAAACAA-3′), which are recognized by the forkhead box family of transcription factors (9, 10). Interestingly, the forkhead box O (FOXO) subfamily protein FOXO1 is phosphorylated by CDK2 at Ser-249 (11), a site essential for its transcriptional activity in multiple cell types (11, 12). FOXO1 plays a critical role in cardiac development and pathophysiology and is expressed abundantly in the adult heart (13–16). To date, the pathophysiological significance of FOXO1 Ser-249 phosphorylation in the cardiac settings has not been examined. In this study, we used genetic and pharmacologic approaches to investigate the role of FOXO1 in DOX-induced cardiotoxicity. Our results suggest that phosphorylation of FOXO1 at Ser-249 by CDK2 mediates DOX-induced Bim expression and cardiomyocyte apoptosis.
In addition to apoptosis, FOXO transcription factors also regulate skeletal muscle atrophy through transcriptional activation of atrophy-related target genes such as atrogin-1 and muscle RING finger 1 (MuRF1) (17). Elevated FOXO levels in the heart, by adenovirus-mediated gene delivery (18) or heart-specific transgenic expression (19, 20), result in decreases in heart mass and cardiomyocyte size. However, it remains unclear whether FOXO family proteins mediate DOX-induced cardiac atrophy. By taking advantage of the selective FOXO1 inhibitor AS1842856 (21), we show that inhibition of FOXO1 maintains heart weight and preserves cardiac performance following challenge with DOX. Collectively, our study suggests that FOXO1 mediates DOX-induced cardiomyocyte apoptosis and cardiac atrophy downstream of CDK2.
Results
DOX exposure induced FOXO1 phosphorylation at Ser-249 in the heart and cardiomyocytes
We have reported previously that DOX exposure induces apoptotic death of cardiomyocytes through activation of CDK2 (8), which is known to phosphorylate FOXO1 at Ser-249 (11). To determine whether administration of DOX results in FOXO1 phosphorylation in the heart, adult male C57BL/6 mice received a single injection of DOX (5 mg/kg, i.p.) and heart samples were collected at 24 h, a time point showing elevated cardiac CDK2 activity (8). Western blotting revealed that DOX injection significantly increased the level of phospho-FOXO1 (Ser-249, Fig. S1A). Because cardiotoxicity correlates with the cumulative dose of DOX (2), mice were also challenged with a single DOX injection at the cumulative dose (5 mg/kg × 4 = 20 mg/kg) based on our established protocol (8). Compared with DOX (5 mg/kg), DOX (20 mg/kg) induced more robust FOXO1 Ser-249 phosphorylation on day 1, followed by a decline on day 5 (Fig. 1A). Two weeks after completion of DOX injections (4 weekly doses of 5 mg/kg), phospho-FOXO1 (Ser-249) levels were similar in DOX- and saline-injected hearts (Fig. S1B). These results suggested that administration of DOX induced cardiac FOXO1 phosphorylation at Ser-249 in a dose- and time-dependent manner. In response to DOX treatment, cultured adult mouse cardiomyocytes (AMCMs) exhibited a dramatic increase in phospho-FOXO1 (Ser-249) signal intensity (Fig. 1B), suggesting that DOX exposure induced FOXO1 phosphorylation through a cardiomyocyte-autonomous mechanism. To further corroborate these findings, neonatal rat cardiomyocytes (NRCMs) were challenged with DOX. As expected, the protein level of phospho-FOXO1 (Ser-249) was significantly increased as early as 2 h after DOX treatment (Fig. 1C) and remained elevated at 24 h before returning to baseline at 48 h (Fig. S1C). Moreover, DOX treatment also markedly increased the fluorescence intensity of phospho-FOXO1 (Ser-249) in NRCMs (Fig. 1D). In agreement with previous observations (13), FOXO1 predominantly localized in the nuclei of cardiomyocytes, and DOX-induced phosphorylation of FOXO1 at Ser-249 failed to alter its subcellular localization (Fig. S1D).
Figure 1.

DOX exposure induced FOXO1 phosphorylation at Ser-249 in the heart and cardiomyocytes. A, adult C57BL/6 mice received a single injection of DOX (20 mg/kg, i.p.) and were euthanized on days 0, 1, or 5 (n = 3/time point). Heart protein levels were measured by Western blotting. Values are mean ± S.E. and were analyzed using one-way ANOVA with Tukey post hoc test. *, p < 0.05 versus day 0. B, AMCMs were treated with DOX (1 μm) or vehicle for 4 h and subjected to immunofluorescence staining for phospho-FOXO1 (Ser-249, green, indicated by arrowheads), cardiac troponin T (cTnT, red), and nuclei (DAPI, blue). The boxed areas are shown at higher magnification on the right. Scale bar = 100 μm. C, NRCMs were treated with DOX (1 μm) for 0, 2, or 4 h. Cellular protein levels were measured by Western blotting. One-way ANOVA with Tukey post hoc test. **, p < 0.01 versus time 0. D, NRCMs were treated with DOX (1 μm) or vehicle for 4 h and subjected to immunofluorescence staining for phospho-FOXO1 (Ser-249, green), cTnT (red), and nuclei (DAPI, blue). Scale bar = 10 μm.
Stimulation with DOX enhanced FOXO1 phosphorylation by CDK2
In cancer cells, phosphorylation of FOXO1 at Ser-249 is mediated by CDK2 (11). However, whether FOXO1 is a CDK2 substrate in cardiomyocytes remains unknown. In response to DOX treatment, phospho-FOXO1 (Ser-249) was detected in CDK2 immunoprecipitants (Fig. 2A), indicating a potential role of CDK2 in DOX-induced cardiac FOXO1 phosphorylation. Moreover, DOX administration augmented interaction between FOXO1 and CDK2 in the mouse heart and markedly induced FOXO1 phosphorylation at the CDK target site (Fig. 2B), as assessed by immunoprecipitation with anti-FOXO1 followed by Western blotting for the phospho-CDK substrate motif ((K/H)pSP). A protein sequence analysis revealed that FOXO1 contains only one such motif, which is conserved between major mammalian species and is located around Ser-249 in humans (Ser-246 in mice and Ser-243 in rats, Fig. 2C). These results are in agreement with previous findings that FOXO1 Ser-249 is the primary site phosphorylated by CDKs (11, 12). Interestingly, inhibition of CDK2 with roscovitine completely blocked DOX-induced FOXO1 phosphorylation (Fig. 2D).
Figure 2.

Stimulation with DOX enhanced FOXO1 phosphorylation by CDK2. A, NRCMs were treated with DOX (1 μm) or vehicle for 4 h. Protein lysates were immunoprecipitated (IP) with anti-CDK2 antibody followed by Western blotting with the indicated antibodies. B, adult C57BL/6 mice were injected with DOX (5 mg/kg, i.p.) or saline and euthanized at 24 h. Protein lysates were immunoprecipitated with anti-FOXO1 antibody followed by Western blotting. Asterisk, IgG light chain. C, protein sequence analysis revealed that only one phospho-CDK substrate motif, (K/H)pSP, exists in FOXO1. D, NRCMs were pretreated with the CDK inhibitor roscovitine (50 μm) for 16 h prior to incubation with DOX (1 μm) for 4 h. Protein lysates were immunoprecipitated with anti-FOXO1 antibody followed by Western blotting. E, NRCMs were transfected with EGFP or HA-CDK2, and protein levels were measured by Western blotting. Two-tailed Student's t test. *, p < 0.05 versus EGFP. F, NRCMs transfected with HA-CDK2 were subjected to immunofluorescence staining for phospho-FOXO1 (Ser-249, green), HA tag (red), and nuclei (DAPI, blue). An intense phospho-FOXO1 (Ser-249) signal was observed in HA-positive cells (arrowhead) but not in HA-negative cells (arrows). Scale bar = 20 μm.
We previously demonstrated that transfection of NRCMs with CDK2 aggravated DOX-induced apoptosis but did not cause apoptosis in the absence of DOX (8). To further determine whether CDK2 is sufficient for FOXO1 phosphorylation at Ser-249, NRCMs were transfected with a CDK2 construct. As shown in Fig. 2E, overexpression of CDK2 significantly increased the protein level of phospho-FOXO1 (Ser-249). The fluorescence intensity of phospho-FOXO1 (Ser-249) was also dramatically higher in cells positive for exogenous CDK2 than in control cardiomyocytes (Fig. 2F). Together, these results suggested that CDK2 mediated DOX-induced FOXO1 phosphorylation at Ser-249.
Activation of FOXO1 was necessary for DOX-induced Bim transcription
Depending on cell type, phosphorylation of FOXO1 at Ser-249 has been shown to result in transcriptional inhibition (11) or activation (12). In the mouse heart and cultured cardiomyocytes, DOX-induced FOXO1 Ser-249 phosphorylation (Fig. 1) was associated with up-regulation of Bim (8), a FOXO1 target gene (22). To determine whether FOXO1 phosphorylation at Ser-249 contributes to Bim transcription, NRCMs were incubated with DOX in the presence of AS1842856, a selective FOXO1 inhibitor with no activity toward other forkhead proteins, including FOXO3a (21). Pretreatment with AS1842856 abolished DOX-induced phosphorylation of FOXO1 at Ser-249 (Fig. 3A) and suppressed expression of Bim at the protein and transcript levels (Fig. 3, B and C). To test the hypothesis that FOXO1 mediates DOX-induced Bim expression, NRCMs were transfected with FOXO1 siRNAs prior to incubation with DOX. Knockdown of FOXO1 significantly reduced the Bim mRNA level following DOX treatment (Fig. 3D). To further determine whether FOXO1 mediates DOX-induced Bim transcription, H9c2 myoblasts were transfected with the Bim-LUC reporter or a double mutant, Bim-LUC(dm) with mutations in the FOXO1-binding sites (9) prior to incubation with DOX. H9c2 myoblasts derived from rat heart were used in this experiment because the transfection efficiency of reporter constructs is relatively higher in these cells than in primary cardiomyocytes. Treatment with DOX increased Bim-LUC luciferase activity (Fig. 3E), indicating that DOX induced Bim transcription. Importantly, DOX exposure failed to activate the Bim-LUC(dm) reporter construct (Fig. 3E), suggesting that FOXO1-binding sites in the Bim promoter were necessary for DOX-induced Bim transcription.
Figure 3.

Activation of FOXO1 was necessary for DOX-induced Bim transcription. A–C, NRCMs were treated with DOX (1 μm) in the presence of the FOXO1 inhibitor AS1842856 (1 μm) or vehicle for 4 h. A and B, protein levels were measured by Western blotting. C, mRNA levels were analyzed by quantitative RT-PCR. Two-way ANOVA with Sidak test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, NRCMs were transfected with control (siControl) or FOXO1 siRNA (siFOXO1) prior to treatment with DOX (1 μm) for 4 h. Bim mRNA levels were analyzed by quantitative RT-PCR. Two-way ANOVA with Sidak test. **, p < 0.01. E, H9c2 cells were transfected with the Bim promoter construct Bim-LUC or Bim-LUC(dm) with mutations in the FOXO1-binding sites. Cells were then treated with DOX (1 μm) for various periods of time, and luminescence was measured. One-way ANOVA with Tukey test. **, p < 0.01; ***, p < 0.001 versus time 0.
Disruption of FOXO1 diminished DOX-induced cardiomyocyte apoptosis
Our data so far suggested that DOX induced FOXO1 activation, resulting in Bim transcription. Based on our recent report showing that Bim is necessary for DOX-induced apoptosis (8), we hypothesized that FOXO1 mediates DOX-induced cardiomyocyte apoptosis. Indeed, silencing of FOXO1 significantly reduced the levels of cleaved poly(ADP-ribose) polymerase (PARP) and cleaved caspase-3, two widely used markers of apoptosis, after challenge with DOX (Fig. 4A). Knockdown of FOXO1 also decreased the percentage of cardiomyocytes positive for TUNEL, a marker of DNA fragmentation in late-stage apoptosis (Fig. 4B). Consistently, inhibition of FOXO1 with AS1842856 suppressed DOX-induced cleavage of PARP and caspase-3 (Fig. 4C) and TUNEL labeling (Fig. 4D). These data suggested that FOXO1 activation was necessary for DOX-induced apoptotic death of cardiomyocytes.
Figure 4.

Disruption of FOXO1 diminished DOX-induced cardiomyocyte apoptosis. A and B, NRCMs were transfected with siControl or siFOXO1 prior to treatment with DOX (1 μm) for 24 h. A, protein levels were measured by Western blotting. B, apoptosis was evaluated by staining with TUNEL (green), cTnT (red), and nuclei (DAPI, blue). Scale bar = 20 μm. Two-tailed Student's t test. **, p < 0.01 versus siControl. C and D, NRCMs were treated with DOX (1 μm) in the presence of the FOXO1 inhibitor AS1842856 (1 μm) or vehicle for 24 h. C, protein levels were measured by Western blotting. D, apoptosis was evaluated by TUNEL staining. Two-tailed Student's t test. **, p < 0.01 versus vehicle.
Pretreatment with the FOXO1 inhibitor preserved mitochondrial integrity following DOX challenge
We showed previously that DOX-induced cardiomyocyte apoptosis is accompanied by loss of mitochondrial membrane potential (7, 8), which causes mitochondrial matrix remodeling and facilitates cytochrome c release into the cytosol during apoptosis (23). To determine whether FOXO1 inhibition preserves mitochondrial integrity, NRCMs were pretreated with AS1842856 prior to incubation with DOX. As expected, DOX-induced mitochondrial depolarization was significantly attenuated by AS1842856 (Fig. 5), suggesting that FOXO1 inhibition protected against DOX-induced mitochondrial damage.
Figure 5.
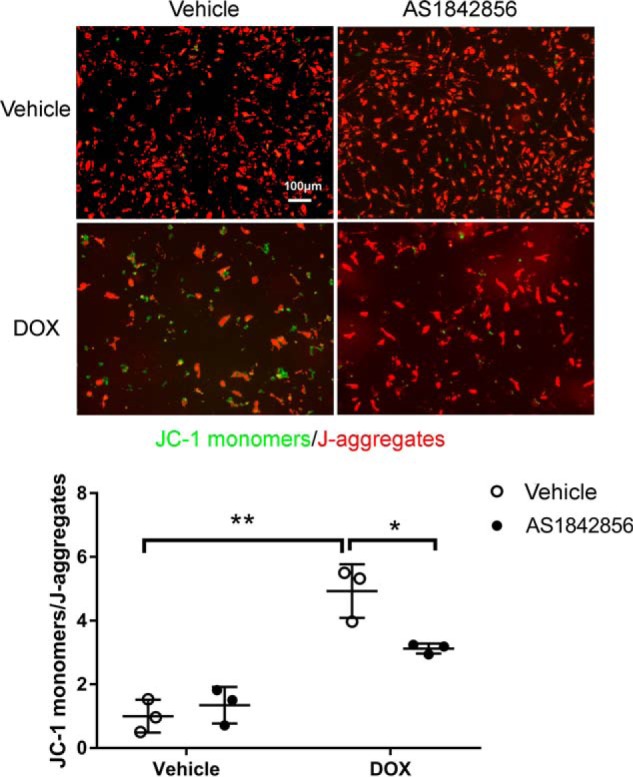
Pretreatment with the FOXO1 inhibitor preserved mitochondrial integrity following DOX challenge. NRCMs were treated with DOX (1 μm) in the presence of the FOXO1 inhibitor AS1842856 (1 μm) or vehicle for 48 h. Mitochondrial membrane potential was then assessed by JC-1 staining. JC-1 monomers (green) indicate damaged mitochondria, whereas J-aggregates (red) indicate healthy mitochondria. Two-way ANOVA with Sidak test. *, p < 0.05; **, p < 0.01. Scale bar = 100 μm.
Pharmacologic inhibition of FOXO1 with AS1842856 attenuated DOX-induced contractile dysfunction and fibrotic remodeling
Apoptotic death of cardiomyocytes has been suggested as a major mechanism underlying anthracycline-related cardiotoxicity (5, 6). Based on our results that DOX induced apoptosis through activation of FOXO1, we hypothesized that FOXO1 could serve as a novel drug target in anthracycline-induced cardiomyopathy. To test this hypothesis, we first determined whether the FOXO1 inhibitor AS1842856 inhibits cardiac FOXO1 activity in vivo. As shown in Fig. 6A, oral administration of AS1842856 significantly reduced the protein levels of phospho-FOXO1 (Ser-249) and Bim in the mouse heart, indicating effective FOXO1 inhibition. To further investigate whether FOXO1 inhibition preserves heart function after DOX exposure, animals were injected with DOX (5 mg/kg/week for 4 weeks, i.p.) to induce cardiomyopathy in the presence of AS1842856 (100 mg/kg, twice daily per week for 4 weeks, oral, Fig. 6B) or vehicle controls. Administration of AS1842856 almost completely prevented the DOX-induced decline in ejection fraction (Fig. 6C) and fractional shortening (Fig. 6D). It is noteworthy that administration of AS1842856 alone did not alter heart function in healthy animals. DOX-induced systolic dysfunction was associated with significant increases in left ventricular end-systolic volume and left ventricular end-systolic internal diameter; both were restored by AS1842856 (Fig. 6, E and F). In contrast, left ventricular diastolic function was not significantly altered by DOX or AS1842856, as indicated by comparable left ventricular end-diastolic volume (Fig. S2A) and left ventricular end-diastolic internal dimension (Fig. S2B). Moreover, left ventricular posterior wall thickness at end-systole (Fig. S2C) or end-diastole (Fig. S2D) and interventricular septal thickness at end-systole (Fig. S2E) or end-diastole (Fig. S2F) were similar in all groups.
Figure 6.

Pharmacologic inhibition of FOXO1 with AS1842856 attenuated DOX-induced contractile dysfunction and fibrotic remodeling. A, adult C57BL/6 mice received a single injection of DOX (5 mg/kg, i.p.) and were immediately fed the FOXO1 inhibitor AS1842856 (100 mg/kg, twice daily) or carrier solution control (n = 4/group). Hearts were collected 24 h after DOX injection. Two-tailed Student's t test. *, p < 0.05; ***, p < 0.001 versus carrier. B–H, assessment of DOX cardiotoxicity. B, experimental protocol. Adult C57BL/6 mice received weekly injections of DOX (5 mg/kg, i.p.) or saline for 4 weeks. Each DOX or saline injection was immediately followed by oral administration of AS1842856 (100 mg/kg, twice daily for 2 days) or carrier solution control. Animals were randomized into four groups: saline + carrier (n = 4), saline + AS1842856 (n = 4), DOX + carrier (n = 6), and DOX + AS1842856 (n = 6). C–F, left ventricular systolic function was evaluated by echocardiography before the first DOX injection and 2 weeks after the last DOX injection. C, Ejection fraction (EF). D, fractional shortening (FS). E, left ventricular end-systolic volume (LVVs). F, left ventricular end-systolic internal dimension (LVIDs). Two-way ANOVA with Sidak test. *, p < 0.05; NS, not significant. G, cardiomyocyte apoptosis was measured by staining for TUNEL (green), cTnT (red), and DAPI (blue). H, myocardial fibrosis was evaluated by Masson's trichrome staining. Collagen fibers were stained blue (arrows), and cardiac muscle was stained red. Two-tailed Student's t test. *, p < 0.05; **, p < 0.01 versus carrier.
To determine whether diminished cardiomyocyte apoptosis accounts for AS1842856-mediated cardioprotection, heart sections were subjected to TUNEL staining. As expected, the number of TUNEL-positive cardiomyocytes in DOX-challenged hearts was significantly reduced by AS1842856 treatment (Fig. 6G), suggesting that inhibition of FOXO1 protected against DOX-induced cardiomyocyte apoptosis. In addition, Masson's trichrome staining revealed that treatment with AS1842856 attenuated DOX-induced myocardial fibrosis (Fig. 6H).
DOX-induced cardiac atrophy was prevented by administration of the FOXO1 inhibitor
It has been reported recently that subacute DOX cardiotoxicity in humans is characterized by ventricular atrophy (3, 4). Indeed, echocardiographic measurement revealed that left ventricular mass was significantly decreased 2 weeks after the last DOX injection (Fig. 7A). Interestingly, inhibition of FOXO1 with AS1842856 prevented the DOX-induced decrease in left ventricular mass (Fig. 7A). DOX injection also significantly reduced the heart weight/tibia length ratio, which was preserved by administration of AS1842856 (Fig. 7B). Moreover, DOX-induced reduction in cardiomyocyte cross-sectional area was attenuated by AS1842856 (Fig. 7C), further supporting a critical role of FOXO1 in DOX-induced cardiac atrophy. Administration of AS1842856 alone did not significantly alter heart mass and cardiomyocyte size in mice (Fig. 7). Interestingly, DOX-induced body weight loss was also alleviated by AS1842856 (Fig. S3). Together, these results suggested that FOXO1 inhibition protected against DOX-induced cardiac atrophy.
Figure 7.

DOX-induced cardiac atrophy was prevented by administration of the FOXO1 inhibitor. A, left ventricular (LV) mass was assessed by echocardiography before the first DOX injection and 2 weeks after the last DOX injection. B, heart weight to tibia length ratio measured 2 weeks after the last DOX injection. C, cardiomyocyte cross-sectional area was analyzed by staining with wheat germ agglutinin (WGA, red) and cTnT (green). Scale bar = 20 μm. Two-way ANOVA with Sidak test. *, p < 0.05; **, p < 0.01.
Inhibition of FOXO1 blocked DOX-induced expression of the proatrophic gene MuRF1
Because the muscle-specific E3 ubiquitin ligase MuRF1 is necessary for DOX-induced subacute cardiac atrophy (4), we next measured MuRF1 expression in heart lysates. A single DOX injection at the cumulative dose (20 mg/kg) significantly up-regulated MuRF1 protein at 24 h, followed by a decline at day 5 (Fig. 8A). However, a single DOX injection at the lower dose (5 mg/kg) failed to increase the MuRF1 level at 24 h (data not shown). Because anthracycline cardiotoxicity highly depends on the cumulative dose (2), it is possible that DOX exposure induces cardiac MuRF1 expression only when the cumulative dose reaches or exceeds a threshold around 20 mg/kg in mice. In cultured NRCMs, stimulation with DOX also significantly increased the MuRF1 protein level (Fig. 8B). Interestingly, treatment with AS1842856 abrogated DOX-induced MuRF1 expression, suggesting that FOXO1-mediated transcription of MuRF1 likely mediated DOX-induced cardiac atrophy.
Figure 8.

Inhibition of FOXO1 blocked DOX-induced expression of the proatrophic gene MuRF1. A, Adult C57BL/6 mice received a single injection of DOX (20 mg/kg, i.p.) and were euthanized on day 0, 1, or 5 (n = 3/time point). Heart protein levels were measured by Western blotting. One-way ANOVA with Tukey post hoc test. *, p < 0.05 versus day 0. B, NRCMs were treated with DOX (1 μm) in the presence of the FOXO1 inhibitor AS1842856 (1 μm) or vehicle for 4 h. Protein levels were measured by Western blotting. **, p < 0.01; ***, p < 0.001. C, schematic of the role of FOXO1 in DOX-induced cardiotoxicity. DOX treatment induces CDK2-dependent activation of FOXO1, which mediates expression of the proapoptotic gene Bim and the proatrophic gene MuRF1, resulting in cardiomyocyte apoptosis and cardiac atrophy. Pharmacological inhibition of FOXO1 attenuates DOX-induced cardiotoxicity.
Discussion
The anthracycline compounds, widely used in current cancer chemotherapy, can cause irreversible, dose-dependent cardiac injury, including cardiomyocyte apoptosis (5, 6) and cardiac atrophy (3, 4). In this study, we demonstrated that DOX exposure induced CDK2-dependent FOXO1 activation, which was necessary for apoptosis and atrophy (Fig. 8C). Moreover, we identified FOXO1 as a transcription factor for the proapoptotic gene Bim and the proatrophic gene MuRF1. Using a small-molecule FOXO1 inhibitor, AS1842856, we showed that pharmacological inhibition of FOXO1 attenuated DOX-induced systolic dysfunction, cardiac atrophy, and ventricular remodeling. Our findings, for the first time, establish FOXO1 as a critical mediator of DOX-induced cardiotoxicity.
This study suggests that DOX exposure induces phosphorylation of FOXO1 at Ser-249 in a CDK activity–dependent manner. Ser-249 of FOXO1 (KS*PRR; S* indicates Ser-249) falls within the consensus CDK phosphorylation motif (K/R)(S/T)PX(K/R) and has been shown to be phosphorylated by CDK1 (12, 24), CDK2 (11), CDK4 (25), and CDK5 (26). However, the role of CDK4 in FOXO1 phosphorylation has been questioned, as Ser-249 phosphorylation appears to be independent of CDK4 in B cells (27). Because we have shown previously that DOX induces cardiac CDK2 activation (8), we determined whether CDK2 is involved in DOX-induced FOXO1 phosphorylation at Ser-249. Interestingly, DOX exposure enhanced interaction between FOXO1 and CDK2, and phospho-FOXO1 (Ser-249) was detected in CDK2 immunoprecipitants. Moreover, overexpression of CDK2 markedly increased phospho-FOXO1 (Ser-249) level in cardiomyocytes. These results suggest that CDK2 mediates FOXO1 Ser-249 phosphorylation in response to DOX exposure. However, it cannot be excluded that phosphorylation of FOXO1 at Ser-249 may also be mediated by additional CDK family member(s).
The effect of Ser-249 phosphorylation on FOXO1 transcriptional activity has been controversial. It was initially reported that Ser-249 phosphorylation results in FOXO1 nuclear export and inactivation in prostate cancer cells (11, 24, 28), glioma cells (29), and primary cortical neurons (26). Other studies, however, revealed that Ser-249 phosphorylation promotes FOXO1 nuclear accumulation and activation by inhibiting interaction with 14-3-3 in granule neurons (12), NIH 3T3 fibroblast cells (12), breast cancer cells (30), as well as muscle sarcoma cells (25). In agreement with the latter, our findings suggest that DOX-induced FOXO1 Ser-249 phosphorylation is associated with FOXO1 activation, resulting in elevated transcription of the downstream target genes Bim and MuRF1. Therefore, FOXO1 activity seems to be modulated by Ser-249 phosphorylation in a cell type–specific manner. These conflicting results may be caused by distinct confounding factors in different cell types, such as kinase and phosphatase expression patterns and cell cycle profiles. For example, subcellular localization of FOXO1 is regulated by phosphorylation at multiple sites by many kinases, such as phosphorylation at Thr-24, Ser-256, and Ser-319 by Akt and phosphorylation at Ser-212 by mammalian sterile 20–like kinase 1 (31). It is possible that Ser-256 phosphorylation may interfere with Ser-249 phosphorylation, considering the relative proximity of these two sites. In addition, FOXO1 localization and activity are also regulated by kinases expressed at specific phases of the cell cycle (32). Therefore, the final outcome of Ser-249 phosphorylation may also depend on the cell cycle status. Further investigations are warranted to clarify these issues.
Although FOXO1 is essential for myocardial development and homeostasis (13–16), its role in chemotherapy-related heart damage remains largely unknown. Our study revealed that activation of FOXO1 by DOX augmented expression of the BH3-only protein Bim and exaggerated apoptosis in cardiomyocytes. Consequently, administration of the FOXO1 inhibitor AS1842856 repressed Bim, attenuated myocyte apoptosis, and preserved cardiac function in mice challenged with DOX. Our results are consistent with recent findings showing that depletion of FOXO1 in lamin A/C–deficient mouse hearts suppresses apoptosis and prolongs survival (33). Along similar lines, activation of FOXO1 aggravates myocardial ischemia/reperfusion injury in diabetic hearts via induction of nitrosative stress and endoplasmic reticulum stress (34). Moreover, sustained activation of cardiac FOXO1 or FOXO3 results in diabetic cardiomyopathy (35, 36) and heart failure (37). Interestingly, FOXO1 and FOXO3 may also promote cardiomyocyte survival through transcription of genes encoding antioxidant enzymes (catalase and manganese superoxide dismutase 2) and autophagy-related proteins (LC3 and Gabarapl1) (15). This discrepancy might be due to time-dependent regulation of FOXO function, as activation of FOXO3 is initially protective through up-regulation of manganese superoxide dismutase 2 and repression of oxidative stress but eventually causes cell death through transcription of Bim and FasL (38). Collectively, these studies suggest that FOXO transcription factors play critical roles in the regulation of cardiomyocyte apoptosis in pathological conditions.
A common manifestation of subacute DOX cardiotoxicity in cancer patients and rodent models is a reduction in heart mass (3, 4). In this study, we showed that DOX exposure reduced heart mass and cardiomyocyte size through activation of FOXO1. Intriguingly, DOX injection induces transcription of FOXO1 target genes involved in atrophy, including atrogin-1, MuRF1, and Bnip3 (39). Most recently, it has been shown that MuRF1 null mice are protected from DOX-induced cardiac atrophy and contractile dysfunction (4). Based on these findings, we speculate that DOX induces cardiac atrophy through FOXO1-mediated transcription of MuRF1. Indeed, inhibition of FOXO1 suppressed DOX-induced MuRF1 expression (Fig. 8, A and B). FOXO1 may also reduce cardiomyocyte size through enhancing autophagy via transcription of the autophagy pathway genes LC3, Gabarapl1, and Atg12 (16). In line with the above findings, activation of FOXO1 blunts cardiomyocyte hypertrophy induced by multiple agonists, including angiotensin II, isoproterenol, and phenylephrine (14). Overexpression of FOXO3 also induces transcription of atrogin-1 and MuRF1, resulting in a decrease in cardiomyocyte size (18). Conversely, FOXO3-deficient mice exhibit increased heart weight (14). It is noteworthy that heart-specific deletion of FOXO1, somewhat surprisingly, fails to increase cardiomyocyte size (36). In agreement with this finding, we showed that administration of the FOXO1 inhibitor AS1842856 alone did not significantly increase cardiomyocyte size in mice in the absence of DOX (Fig. 7). Collectively, these findings suggest that inhibition of FOXO1 attenuates DOX-induced cardiac atrophy without causing apparent morphological changes in the healthy adult heart.
DOX cardiotoxicity is also characterized by myofibrillar disarray (40). Interestingly, the FOXO target gene MuRF1 is a muscle-specific E3 ubiquitin ligase involved in proteasomal degradation of sarcomeric proteins, and activation of the FOXO-MuRF1 signaling axis has been shown to cause myofibril disarray in cardiomyocytes (41). Therefore, cardioprotection conferred by FOXO1 inhibition may also include preserving myofibril integrity following DOX administration.
In conclusion, this study reveals that DOX induces CDK2-dependent phosphorylation and activation of FOXO1, resulting in cardiomyocyte apoptosis and cardiac atrophy. Our study identifies FOXO1 as a potential drug target in anthracycline cardiotoxicity. Small-molecule FOXO1 inhibitors, which are currently in clinical development (42), could be promising therapeutic agents for cardiomyopathy and heart failure caused by cancer chemotherapy.
Experimental procedures
Animals
Animals used in this study, including C57BL/6 mice and Sprague-Dawley rats, were obtained from Envigo and were housed in the Washington State University Spokane vivarium, which is accredited by the American Association for Accreditation of Laboratory Animal Care. All procedures were approved by the Institutional Animal Care and Use Committee at Washington State University and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (2011).
Cell culture
AMCMs were isolated from adult C57BL/6 mice as described previously (43). In brief, the heart was digested with collagenase 2 (0.5 mg/ml), collagenase 4 (0.5 mg/ml), and protease XIV (0.05 mg/ml). Ventricular cardiac myocytes were then dissociated using forceps, separated by gentle trituration and gravity sedimentation, and plated in chamber slides coated with laminin (5 μg/ml). NRCMs were isolated from Sprague-Dawley rat pups using a neonatal cardiomyocyte isolation system (Worthington Biochemical Corp.), as described previously (8). Briefly, hearts were harvested and digested in trypsin (50 μg/ml) at 4 °C overnight and then in collagenase 2 (100 units/ml) at 37 °C for 45 min. NRCMs were plated in dishes coated with 0.2% gelatin in medium 199 supplemented with 15% fetal bovine serum for 24 h and then cultured in serum-free medium 199. H9c2 cells (CRL-1446, ATCC) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum.
Transfection and luciferase reporter assay
NRCMs were transfected with siRNAs using HiPerfect transfection reagent (Qiagen). The siRNA sequences used were as follows: FOXO1 siRNA, GAGGAUUGAACCAGUAUAACU; control siRNA, UAAGGCUAUGAAGAGAUAC[dT][dT]. Lipofectamine 3000 reagent (Thermo Fisher Scientific) was used for transfection of plasmids, including EGFP, HA-CDK2 (1884, Addgene), Bim-LUC, or Bim-LUC(dm) luciferase reporters (kindly provided by Dr. Jonathan Ham, University College London) (9). The luciferase assay was performed using Glo Lysis buffer (E2661, Promega) and the Steady-Glo luciferase assay system (E2520, Promega) according to the manufacturer's protocol.
Immunoprecipitation and immunoblotting
Protein lysates from tissue or cultured cells were immunoprecipitated with mouse anti-CDK2 (sc-6248, Santa Cruz Biotechnology) or rabbit anti-FOXO1 (2880, Cell Signaling Technology) using Dynabeads protein G (Thermo) according to the manufacturer's protocol. Western blotting was performed as described previously (8) with the following antibodies: rabbit anti-phospho-FOXO1 (Ser-249) (PA5-64676, Thermo Fisher Scientific, 1:1000), rabbit anti-PARP (9542, Cell Signaling Technology, 1:1000), rabbit anti-FOXO1 (2880, Cell Signaling Technology, 1:1000), rabbit anti-Bim (2933, Cell Signaling Technology, 1:1000), goat-anti-MuRF1 (AF5366, R&D Systems, 1:1000), rabbit anti-GAPDH (sc-25778, Santa Cruz Biotechnology, 1:1000), and rabbit anti-histone H3 (4499, Cell Signaling Technology, 1:1000).
Immunostaining
Immunofluorescence staining was carried out as described previously (8), with the following antibodies: rabbit anti-phospho-FOXO1 (Ser-249) (AF8271, Affinity Bioscience, 1:50), mouse anti-cardiac Troponin T (MS-295-P, Thermo Scientific, 1:100), and rabbit anti-HA (3724, Cell Signaling Technology, 1:50). Apoptosis was assessed by TUNEL staining using the In Situ Cell Death Detection Kit (Roche Applied Science) according to the manufacturer's instructions.
Quantitative RT-PCR
Quantitative RT-PCR was performed as described previously (8), using the iScript cDNA Synthesis Kit (Bio-Rad) for reverse transcription and Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific) for quantitative PCR. The following primers were used: Bim, 5′-CCATGAGTTGTGACAAGTCAACAC-3′ (forward) and 5′-GATCTTCAGGTTCCTCCTGAGACTG-3′ (reverse); 18S rRNA, 5′-TGACTCAACACGGGAAACCTCAC-3′ (forward) and 5′-ATCGCTCCACCAACTAAGAACGG-3′ (reverse).
Subcellular fractionation
NRCMs were homogenized in isolation buffer (190 mm d-mannitol, 70 mm sucrose, 20 mm HEPES, and 0.2 mm EDTA) on ice. Nuclear and cytosolic fractions were separated by centrifugation at 600 × g for 10 min and 20,000 × g for 60 min, respectively.
JC-1 staining
Mitochondrial membrane potential was evaluated with the JC-1 Mitochondrial Membrane Potential Assay Kit (Cayman Chemical) as described previously (8). Briefly, NRCMs were incubated with JC-1 at 37 °C for 30 min. JC-1 forms J-aggregates (red fluorescence) in healthy cells and monomers (green fluorescence) in apoptotic cells. The fluorescence intensity ratio of JC-1 monomers to J-aggregates was used as an indicator of mitochondrial depolarization.
In vivo DOX cardiotoxicity studies
Male C57BL/6 mice (8–12 weeks old) received DOX injections (dissolved in saline, 5 mg/kg/week for 4 weeks, i.p., LC Laboratories) to induce cardiotoxicity as described previously (8). The FOXO1 inhibitor AS1842856 (HY-100596, MedChemExpress) was dissolved in a carrier solution (6% (2-hydroxypropyl)-β-cyclodextrin, H107, Sigma) and sonicated for 5 s according to a published protocol (21). Immediately following each DOX or saline injection, mice were treated with AS1842856 (100 mg/kg, oral gavage) or the carrier solution twice daily for 2 days (21). Animals were randomly divided into four groups: saline + carrier, saline + AS1842856, DOX + carrier, and DOX + AS1842856. Heart function was assessed by echocardiography using the VisualSonics VEVO 2100 imaging system before the first DOX injection and 2 weeks after the last DOX injection. Animals were then euthanized for heart tissue collection and analysis. Cardiac atrophy was examined by staining with wheat germ agglutinin, and cardiomyocyte cross-sectional area was analyzed using ImageJ software (National Institutes of Health) as described previously (44). Myocardial fibrosis was evaluated by Masson's trichrome staining (HT10516, Sigma) according to the manufacturer's protocol. A separate cohort of animals was sacrificed 24 h after DOX injection for biochemical and molecular analyses.
Statistics
Statistical analyses were conducted using the GraphPad Prism 7.02 software. Results are presented as mean ± S.E. Statistical differences between two groups were determined using two-tailed Student's t test. For multiple comparisons, one-way analysis of variance (ANOVA) followed by Tukey post hoc test or two-way ANOVA followed by Sidak test were used, as appropriate. Differences were considered significant at p < 0.05.
Author contributions
P. X. data curation; P. X., J. C., Y. L., and M. F. formal analysis; P. X. validation; P. X., J. C., Y. L., M. F., and Z. C. investigation; P. X. visualization; P. X., J. C., and Y. L. methodology; P. X. and Z. C. writing-original draft; B. C. J. and Z. C. supervision; B. C. J. and Z. C. writing-review and editing; Z. C. conceptualization; Z. C. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Jonathan Ham (University College London) for providing the Bim-LUC and Bim-LUC(dm) constructs and Megan Chastain (Microscopy Core, Washington State University) for technical assistance.
This work was supported by NHLBI, National Institutes of Health Grants R00HL119605 and R56HL145034 (to Z. C.) and R01HL140067 (to B. C. J.); the ASPET Summer Undergraduate Research Program (to M. F.); and the Washington State University College of Pharmacy and Pharmaceutical Sciences. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- DOX
- doxorubicin
- CDK
- cyclin-dependent kinase
- FOXO
- forkhead box O
- AMCM
- adult mouse cardiomyocyte
- NRCM
- neonatal rat cardiomyocyte
- LUC
- luciferase
- dm
- double mutant
- PARP
- poly(ADP-ribose) polymerase
- EGFP
- enhanced GFP
- ANOVA
- analysis of variance
- cTnT
- cardiac troponin T.
References
- 1. Curigliano G., Cardinale D., Dent S., Criscitiello C., Aseyev O., Lenihan D., and Cipolla C. M. (2016) Cardiotoxicity of anticancer treatments: epidemiology, detection, and management. CA Cancer J. Clin. 66, 309–325 10.3322/caac.21341 [DOI] [PubMed] [Google Scholar]
- 2. Cardinale D., Colombo A., Bacchiani G., Tedeschi I., Meroni C. A., Veglia F., Civelli M., Lamantia G., Colombo N., Curigliano G., Fiorentini C., and Cipolla C. M. (2015) Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 131, 1981–1988 10.1161/CIRCULATIONAHA.114.013777 [DOI] [PubMed] [Google Scholar]
- 3. Jordan J. H., Castellino S. M., Meléndez G. C., Klepin H. D., Ellis L. R., Lamar Z., Vasu S., Kitzman D. W., Ntim W. O., Brubaker P. H., Reichek N., D'Agostino R. B. Jr., and Hundley W. G. (2018) Left ventricular mass change after anthracycline chemotherapy. Circ. Heart Fail. 11, e004560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Willis M. S., Parry T. L., Brown D. I., Mota R. I., Huang W., Beak J. Y., Sola M., Zhou C., Hicks S. T., Caughey M. C., D'Agostino R. B. Jr., Jordan J., Hundley W. G., and Jensen B. C. (2019) Doxorubicin exposure causes subacute cardiac atrophy dependent on the striated muscle-specific ubiquitin ligase MuRF1. Circ. Heart Fail. 12, e005234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang S., Liu X., Bawa-Khalfe T., Lu L. S., Lyu Y. L., Liu L. F., and Yeh E. T. (2012) Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 18, 1639–1642 10.1038/nm.2919 [DOI] [PubMed] [Google Scholar]
- 6. Ichikawa Y., Ghanefar M., Bayeva M., Wu R., Khechaduri A., Naga Prasad S. V., Mutharasan R. K., Naik T. J., and Ardehali H. (2014) Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 124, 617–630 10.1172/JCI72931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng Z., DiMichele L. A., Rojas M., Vaziri C., Mack C. P., and Taylor J. M. (2014) Focal adhesion kinase antagonizes doxorubicin cardiotoxicity via p21(Cip1.). J. Mol. Cell Cardiol. 67, 1–11 10.1016/j.yjmcc.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xia P., Liu Y., Chen J., Coates S., Liu D. X., and Cheng Z. (2018) Inhibition of cyclin-dependent kinase 2 protects against doxorubicin-induced cardiomyocyte apoptosis and cardiomyopathy. J. Biol. Chem. 293, 19672–19685 10.1074/jbc.RA118.004673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilley J., Coffer P. J., and Ham J. (2003) FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162, 613–622 10.1083/jcb.200303026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chi H. C., Chen S. L., Cheng Y. H., Lin T. K., Tsai C. Y., Tsai M. M., Lin Y. H., Huang Y. H., and Lin K. H. (2016) Chemotherapy resistance and metastasis-promoting effects of thyroid hormone in hepatocarcinoma cells are mediated by suppression of FoxO1 and Bim pathway. Cell Death Dis. 7, e2324 10.1038/cddis.2016.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang H., Regan K. M., Lou Z., Chen J., and Tindall D. J. (2006) CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 314, 294–297 10.1126/science.1130512 [DOI] [PubMed] [Google Scholar]
- 12. Yuan Z., Becker E. B., Merlo P., Yamada T., DiBacco S., Konishi Y., Schaefer E. M., and Bonni A. (2008) Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science 319, 1665–1668 10.1126/science.1152337 [DOI] [PubMed] [Google Scholar]
- 13. Evans-Anderson H. J., Alfieri C. M., and Yutzey K. E. (2008) Regulation of cardiomyocyte proliferation and myocardial growth during development by FOXO transcription factors. Circ. Res. 102, 686–694 10.1161/CIRCRESAHA.107.163428 [DOI] [PubMed] [Google Scholar]
- 14. Ni Y. G., Berenji K., Wang N., Oh M., Sachan N., Dey A., Cheng J., Lu G., Morris D. J., Castrillon D. H., Gerard R. D., Rothermel B. A., and Hill J. A. (2006) Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114, 1159–1168 10.1161/CIRCULATIONAHA.106.637124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sengupta A., Molkentin J. D., Paik J. H., DePinho R. A., and Yutzey K. E. (2011) FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 286, 7468–7478 10.1074/jbc.M110.179242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sengupta A., Molkentin J. D., and Yutzey K. E. (2009) FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 284, 28319–28331 10.1074/jbc.M109.024406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S. H., and Goldberg A. L. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412 10.1016/S0092-8674(04)00400-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skurk C., Izumiya Y., Maatz H., Razeghi P., Shiojima I., Sandri M., Sato K., Zeng L., Schiekofer S., Pimentel D., Lecker S., Taegtmeyer H., Goldberg A. L., and Walsh K. (2005) The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 280, 20814–20823 10.1074/jbc.M500528200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schips T. G., Wietelmann A., Höhn K., Schimanski S., Walther P., Braun T., Wirth T., and Maier H. J. (2011) FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc. Res. 91, 587–597 10.1093/cvr/cvr144 [DOI] [PubMed] [Google Scholar]
- 20. Cao D. J., Jiang N., Blagg A., Johnstone J. L., Gondalia R., Oh M., Luo X., Yang K. C., Shelton J. M., Rothermel B. A., Gillette T. G., Dorn G. W., and Hill J. A. (2013) Mechanical unloading activates FoxO3 to trigger Bnip3-dependent cardiomyocyte atrophy. J. Am. Heart Assoc. 2, e000016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagashima T., Shigematsu N., Maruki R., Urano Y., Tanaka H., Shimaya A., Shimokawa T., and Shibasaki M. (2010) Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Mol. Pharmacol. 78, 961–970 10.1124/mol.110.065714 [DOI] [PubMed] [Google Scholar]
- 22. Lam E. W., Brosens J. J., Gomes A. R., and Koo C. Y. (2013) Forkhead box proteins: tuning forks for transcriptional harmony. Nat. Rev. Cancer 13, 482–495 10.1038/nrc3539 [DOI] [PubMed] [Google Scholar]
- 23. Gottlieb E., Armour S. M., Harris M. H., and Thompson C. B. (2003) Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 10, 709–717 10.1038/sj.cdd.4401231 [DOI] [PubMed] [Google Scholar]
- 24. Liu P., Kao T. P., and Huang H. (2008) CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene 27, 4733–4744 10.1038/onc.2008.104 [DOI] [PubMed] [Google Scholar]
- 25. Liu L., Wu J., Ong S. S., and Chen T. (2013) Cyclin-dependent kinase 4 phosphorylates and positively regulates PAX3-FOXO1 in human alveolar rhabdomyosarcoma cells. PLoS ONE 8, e58193 10.1371/journal.pone.0058193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou J., Li H., Li X., Zhang G., Niu Y., Yuan Z., Herrup K., Zhang Y. W., Bu G., Xu H., and Zhang J. (2015) The roles of Cdk5-mediated subcellular localization of FOXO1 in neuronal death. J. Neurosci. 35, 2624–2635 10.1523/JNEUROSCI.3051-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu Y., Wu Y., Feng X., Shen R., Wang J. H., Fallahi M., Li W., Yang C., Hankey W., Zhao W., Ganju R. K., Li M. O., Cleveland J. L., and Zou X. (2014) CDK4 deficiency promotes genomic instability and enhances Myc-driven lymphomagenesis. J. Clin. Invest. 124, 1672–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu H., Liu P., Pan Y., and Huang H. (2011) Inhibition of cyclin-dependent kinase phosphorylation of FOXO1 and prostate cancer cell growth by a peptide derived from FOXO1. Neoplasia 13, 854–863 10.1593/neo.11594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lau C. J., Koty Z., and Nalbantoglu J. (2009) Differential response of glioma cells to FOXO1-directed therapy. Cancer Res. 69, 5433–5440 10.1158/0008-5472.CAN-08-4540 [DOI] [PubMed] [Google Scholar]
- 30. Feng X., Wu Z., Wu Y., Hankey W., Prior T. W., Li L., Ganju R. K., Shen R., and Zou X. (2011) Cdc25A regulates matrix metalloprotease 1 through Foxo1 and mediates metastasis of breast cancer cells. Mol. Cell. Biol. 31, 3457–3471 10.1128/MCB.05523-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao Y., Wang Y., and Zhu W. G. (2011) Applications of post-translational modifications of FoxO family proteins in biological functions. J Mol. Cell. Biol. 3, 276–282 10.1093/jmcb/mjr013 [DOI] [PubMed] [Google Scholar]
- 32. Yuan C., Wang L., Zhou L., and Fu Z. (2014) The function of FOXO1 in the late phases of the cell cycle is suppressed by PLK1-mediated phosphorylation. Cell Cycle 13, 807–819 10.4161/cc.27727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Auguste G., Gurha P., Lombardi R., Coarfa C., Willerson J. T., and Marian A. J. (2018) Suppression of activated FOXO transcription factors in the heart prolongs survival in a mouse model of laminopathies. Circ. Res. 122, 678–692 10.1161/CIRCRESAHA.117.312052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo W., Jiang T., Lian C., Wang H., Zheng Q., and Ma H. (2014) QKI deficiency promotes FoxO1 mediated nitrosative stress and endoplasmic reticulum stress contributing to increased vulnerability to ischemic injury in diabetic heart. J. Mol. Cell Cardiol. 75, 131–140 10.1016/j.yjmcc.2014.07.010 [DOI] [PubMed] [Google Scholar]
- 35. Ni Y. G., Wang N., Cao D. J., Sachan N., Morris D. J., Gerard R. D., Kuro-O M., Rothermel B. A., and Hill J. A. (2007) FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc. Natl. Acad. Sci. U.S.A. 104, 20517–20522 10.1073/pnas.0610290104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Battiprolu P. K., Hojayev B., Jiang N., Wang Z. V., Luo X., Iglewski M., Shelton J. M., Gerard R. D., Rothermel B. A., Gillette T. G., Lavandero S., and Hill J. A. (2012) Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Invest. 122, 1109–1118 10.1172/JCI60329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qi Y., Zhu Q., Zhang K., Thomas C., Wu Y., Kumar R., Baker K. M., Xu Z., Chen S., and Guo S. (2015) Activation of Foxo1 by insulin resistance promotes cardiac dysfunction and β-myosin heavy chain gene expression. Circ. Heart Fail. 8, 198–208 10.1161/CIRCHEARTFAILURE.114.001457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi C., Viccaro K., Lee H. G., and Shah K. (2016) Cdk5-Foxo3 axis: initially neuroprotective, eventually neurodegenerative in Alzheimer's disease models. J. Cell Sci. 129, 1815–1830 10.1242/jcs.185009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kavazis A. N., Smuder A. J., and Powers S. K. (2014) Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. Physiol. 117, 223–230 10.1152/japplphysiol.00210.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sawyer D. B., Zuppinger C., Miller T. A., Eppenberger H. M., and Suter T. M. (2002) Modulation of anthracycline-induced myofibrillar disarray in rat ventricular myocytes by neuregulin-1beta and anti-erbB2: potential mechanism for trastuzumab-induced cardiotoxicity. Circulation 105, 1551–1554 10.1161/01.CIR.0000013839.41224.1C [DOI] [PubMed] [Google Scholar]
- 41. Shimizu H., Langenbacher A. D., Huang J., Wang K., Otto G., Geisler R., Wang Y., and Chen J. N. (2017) The Calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes. Elife 6, e27955 10.7554/eLife.27955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hornsveld M., Dansen T. B., Derksen P. W., and Burgering B. M. T. (2018) Re-evaluating the role of FOXOs in cancer. Semin. Cancer Biol. 50, 90–100 10.1016/j.semcancer.2017.11.017 [DOI] [PubMed] [Google Scholar]
- 43. Ackers-Johnson M., Li P. Y., Holmes A. P., O'Brien S. M., Pavlovic D., and Foo R. S. (2016) A simplified, Langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ. Res. 119, 909–920 10.1161/CIRCRESAHA.116.309202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng Z., Zhu Q., Dee R., Opheim Z., Mack C. P., Cyr D. M., and Taylor J. M. (2017) Focal adhesion kinase-mediated phosphorylation of Beclin1 protein suppresses cardiomyocyte autophagy and initiates hypertrophic growth. J. Biol. Chem. 292, 2065–2079 10.1074/jbc.M116.758268 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.