

Abstract
Organophosphate (OP) intoxications from nerve agent and OP pesticide exposures are managed with pyridinium aldoxime–based therapies whose success rates are currently limited. The pyridinium cation hampers uptake of OPs into the central nervous system (CNS). Furthermore, it frequently binds to aromatic residues of OP-inhibited acetylcholinesterase (AChE) in orientations that are nonproductive for AChE reactivation, and the structural diversity of OPs impedes efficient reactivation. Improvements of OP antidotes need to include much better access of AChE reactivators to the CNS and optimized orientation of the antidotes' nucleophile within the AChE active-center gorge. On the basis of X-ray structures of a CNS-penetrating reactivator, monoxime RS194B, reversibly bound to native and venomous agent X (VX)–inhibited human AChE, here we created seven uncharged acetamido bis-oximes as candidate antidotes. Both oxime groups in these bis-oximes were attached to the same central, saturated heterocyclic core. Diverse protonation of the heterocyclic amines and oxime groups of the bis-oximes resulted in equilibration among up to 16 distinct ionization forms, including uncharged forms capable of diffusing into the CNS and multiple zwitterionic forms optimal for reactivation reactions. Conformationally diverse zwitterions that could act as structural antidote variants significantly improved in vitro reactivation of diverse OP-human AChE conjugates. Oxime group reorientation of one of the bis-oximes, forcing it to point into the active center for reactivation, was confirmed by X-ray structural analysis. Our findings provide detailed structure-activity properties of several CNS-directed, uncharged aliphatic bis-oximes holding promise for use as protonation-dependent, conformationally adaptive, “smart” accelerated antidotes against OP toxicity.
Keywords: acetylcholinesterase (AChE), crystal structure, molecular modeling, enzyme structure, drug design, structure-function, aliphatic bis-oxime, organophosphate intoxication, oxime reactivation, uncharged oxime antidote
Introduction
Nucleophilic oximes reacting with organophosphate (OP)4-inhibited acetylcholinesterase (AChE; EC 3.1.1.7) are the only true reactivating antidotes against OP intoxication in nerve agent or OP pesticide exposure. Based on the structure of the pyridinium monoxime 2PAM (1), structures and in vitro reactivation efficacies of pyridinium aldoxime-based antidotes have evolved during 70 years of research to yield more efficient bis-pyridinium bis-oximes, such as ortho-7, obidoxime, or MMB4 (2–5). Nevertheless, those cationic antidotes are ineffective in vivo, for reactivation of brain AChE or for reactivation of AChE in any tissue when administered orally (6), due to the inability of pyridinium cations to cross biological membranes (7). Within the past decade, it has become increasingly clear that for effective and complete recovery from an OP intoxication, antidotal action is needed in both the peripheral and central nervous systems (8, 9). A series of centrally active oximes have been recently designed and are currently at different stages of in vitro and in vivo testing (7, 10–14). One mechanism for antidotes to traverse biological membranes and reach the central nervous system (CNS) is by diffusion, which is most effective for uncharged molecular species when they are formed by fast equilibration with ionized species. An acetamido oxime, RS194B (7) (Fig. 1), is an example of an uncharged but ionizable antidote with demonstrated capacity to cross biological membranes, including the blood-brain barrier (BBB), and effectively recover activity of OP-inhibited AChE, both in vitro (6, 7) and in vivo (7, 12). Protonation states of its two ionizable groups (oxime and heterocyclic amine; both with pKa values of ∼9 (7)) will equilibrate in an aqueous environment between four ionization species, cationic, anionic, zwitterionic, and uncharged (Fig. 1A). At physiological pH, their relative abundance is calculated as 93% cationic, 0.51% zwitterionic, 6.4% uncharged, and a negligible fraction in the anionic form. Good BBB penetration of RS194B and its high bioavailability (7) suggest that even 6.4% of the uncharged species, with constant rapid re-equilibration, can be sufficient for an efficient diffusion across biological membranes. Reactivation of OP-inhibited human AChE (OP-hAChE), on the other hand, relies primarily on the small percentage of RS194B in the zwitterionic form that combines a cationic center critical for AChE binding with an anionic oximate that can initiate a nucleophilic SN2 attack. The kinetics of RS194B reactivation revealed relatively weak affinity for productive binding of this monoxime to OP-hAChE, as reflected by the relatively large, millimolar values of Kox constants (7). Those could be a consequence of competition between its less abundant, reactive zwitterionic and much less reactive, abundant cationic forms, but also due to a nonproductive orientation of the RS194B zwitterion when bound to the OP-hAChE conjugate. Either increasing the fraction of zwitterion or enhancing the binding to OP-hAChE in a productive orientation should reduce the Kox constant, leading to enhanced antidotal efficacy of the compound. An appropriate modification of the molecular structure of the antidote has the potential of enhancing both properties.
Figure 1.

A and B, ionization equilibria and abundance of individual ionization forms at pH 7.4, for monoxime RS194B (all forms) (A) and for bis-oxime LG-703 (4 most abundant of 16 possible forms) (B). C, general reactivation scheme of OP-conjugated AChE. Shown is VX-hAChE conjugate in reaction with one of the zwitterionic forms of the oxime antidote LG-703. Indicated are individual rate constants that describe formation (the second-order association constant k1) and disappearance (the first-order dissociation constant k−1 and the first-order reactivation reaction constant k2, representing the maximal rate of reactivation) of the reversible {oxime*OP-hAChE} complex. The Michaelis–Menten type constant for the reactivation reaction Ka (Ka = (k2 + k−1)/k1 and the overall second-order reactivation rate constant kr (kr = k2/Ka) are composite constants dependent on k1, k−1, and k2.
Here, we present a structure-based reactivator design of a new class of oxime antidotes with enhanced properties for reactivation of OP-hAChE conjugates. Starting with the RS194B oxime, we have analyzed its molecular interactions with both native and VX-inhibited hAChE by resolving X-ray structures of their respective complexes at 2.4 and 2.25 Å resolutions. Surprisingly, the nucleophilic aldoxime moiety of RS194B was found 9 and 16 Å distant, respectively, and pointing away from the active and OP-conjugated catalytic Ser-203, the target of the intended reactivation reaction. This is farther than distances between 5 and 9 Å typically observed in reversible complexes of pyridinium aldoximes with AChE (15). To solve the oxime orientation problem, we have used RS194B structures as molecular templates to design and test, first in silico, a small library of bis-oximes with an aldoxime on both ends of the molecule. Unlike structures of typical bis-pyridinium bis-oximes, where each oxime group is directly attached to a separate pyridinium ring, our bis-oximes were formed around a single, uncharged, saturated heterocycle. Inclusion of additional ionizable groups, an aldoxime and a heterocyclic amine, resulted in the possible formation of 8–16 different ionization species per compound, instead of only four for monoxime RS194B. As a consequence, higher concentrations of nucleophilically reactive zwitterions at physiological pH values were observed (Fig. 1). Furthermore, our X-ray structures suggest that due to discrete flexibility of acetamido-alkyl arms bearing oxime groups, a single zwitterionic bis-oxime could form multiple conformationally diverse zwitterions. Those can effectively act as structural variants of the compound in interaction with diverse OP-hAChE conjugates and thus adjust to specific structural requirements of the reactivation reaction. For seven of these “smart” bis-oximes, selected by computational docking analysis and synthesized in 20–150-mg quantities, we demonstrate here good in vitro reactivation efficacy. In reactivation of sarin-, cyclosarin-, VX-, and paraoxon-inhibited hAChE, the designed piperidine, piperazine, and homopiperazine derivatives exceeded the efficacy of RS194B. For one of the representative bis-oximes, LG-703, we also confirmed, by solving the X-ray structure, that one of its oxime groups bound 5 Å close and pointing toward Ser-203, in an orientation productive for reactivation. We present here the design, synthesis, and functional in vitro characterization of seven uncharged heterocyclic bis-oxime reactivators with a conceptually novel scaffold as promising prototypes for the creation of a next generation of adaptable, accelerated CNS-active antidotes against OP intoxication.
Results and discussion
X-ray structures as templates
To develop a structural template for the design of accelerated uncharged antidotes of OP-conjugated hAChE, we have solved X-ray crystal structures of RS194B, one of the currently most promising uncharged oxime reactivators (7, 12), in complex with apo-hAChE and with VX-hAChE conjugate (Table 1).
Table 1.
X-ray crystallographic data collection and refinement statistics
| LT |
|||
|---|---|---|---|
| hAChE*RS194B | VX-hAChE*RS194B | hAChE*LG-703 | |
| Data collection | |||
| Space group | P31 | ||
| Cell dimensions a, b, c (Å) | 125.0, 125.0, 129.8 | 124.4, 124.4, 129.1 | 125.4, 125.4, 129.8 |
| Resolution (Å) | 40.00–2.40 (2.49–2.40)a | 40.00–2.25 (2.33–2.25) | 40.00–3.00 (3.11–3.00) |
| Rmerge | 0.034 (0.522) | 0.063 (0.594) | 0.160 (0.561) |
| I/σI | 11.3 (1.5) | 9.6 (1.2) | 4.3 (1.3) |
| Completeness (%) | 94.1 (94.1) | 92.2 (91.4) | 96.7 (94.4) |
| Redundancy | 1.8 (1.7) | 1.9 (1.8) | 2.6 (2.4) |
| CC1/2 | 0.975 (0.522) | 0.983 (0.465) | 0.944 (0.582) |
| Refinement | |||
| No. of reflections | 76164 | 88333 | 38127 |
| Resolution (Å) | 39.02–2.41 | 38.82–2.26 | 39.14–3.00 |
| Rwork/Rfree | 0.206/0.229 | 0.207/0.229 | 0.205/0.253 |
| No. of atoms (non-H) | 8686 | 8830 | 8473 |
| Water | 242 | 366 | 70 |
| Root mean square deviation | |||
| Bonds (Å) | 0.002 | 0.002 | 0.002 |
| Bond angles (degrees) | 0.521 | 0.475 | 0.484 |
| Ramachandran favored | 96.56 | 96.00 | 95.07 |
| Ramachandran allowed | 3.35 | 4.00 | 4.83 |
| Ramachandran outliers | 0.09 | 0.00 | 0.09 |
| Average B factors | |||
| Protein | 41.0 | 37.2 | 61.3 |
| Ligand | 46.8 | 56.9 | 85.6 |
| PDB ID | 6U34 | 6U37 | 6U3P |
a Values in parentheses are for the highest-resolution shell.
Given that an oxime reactivator represents a substrate for the OP-hAChE conjugate, functional evaluation of relevant X-ray structural data in addition to general analysis of binding site interactions needs to consider the proximity of the reactive aldoxime group to the conjugated phosphorus, as well as the ionization state of the aldoxime group that is critical for the reactivation reaction. Out of the four ionization states of RS194B (Fig. 1), the cationic state, is the most abundant and the anionic state the least abundant at pH 7.4, whereas the zwitterionic state would represent a chemically reactive state of primary interest. Although the X-ray diffraction data in principle cannot reveal positions of protons, thus being unable to unambiguously determine the ionization states of RS194B in the resulting structures, some suggestions can be made based on the likely hydrogen-bonding patterns with surrounding water molecules and hAChE residues.
RS194B binds to the active site of apo-hAChE in a nonproductive pose
In the 2.4 Å X-ray structure of the binary, co-crystalized complex with unmodified (apo-) hAChE, RS194B binds inside the active-site gorge in an obvious nonproductive orientation (Figs. 2A and 3A). Its nucleophilic aldoxime points away from the active Ser-203, where inhibitory OPs covalently bind. The electron density of the RS194B azepane ring is clearly defined at the choline-binding site positioned against the Trp-86 indole ring through C-H…π interactions.
Figure 2.

A and B, X-ray structures of monoxime RS194B bound in the active-center gorge of apo-hAChE (A) and VX-hAChE conjugate (B). C, location of the active center gorge in the hAChE monomer. A rectangular portion of the green water-accessible Connolly surface was removed to reveal the gorge.
Figure 3.
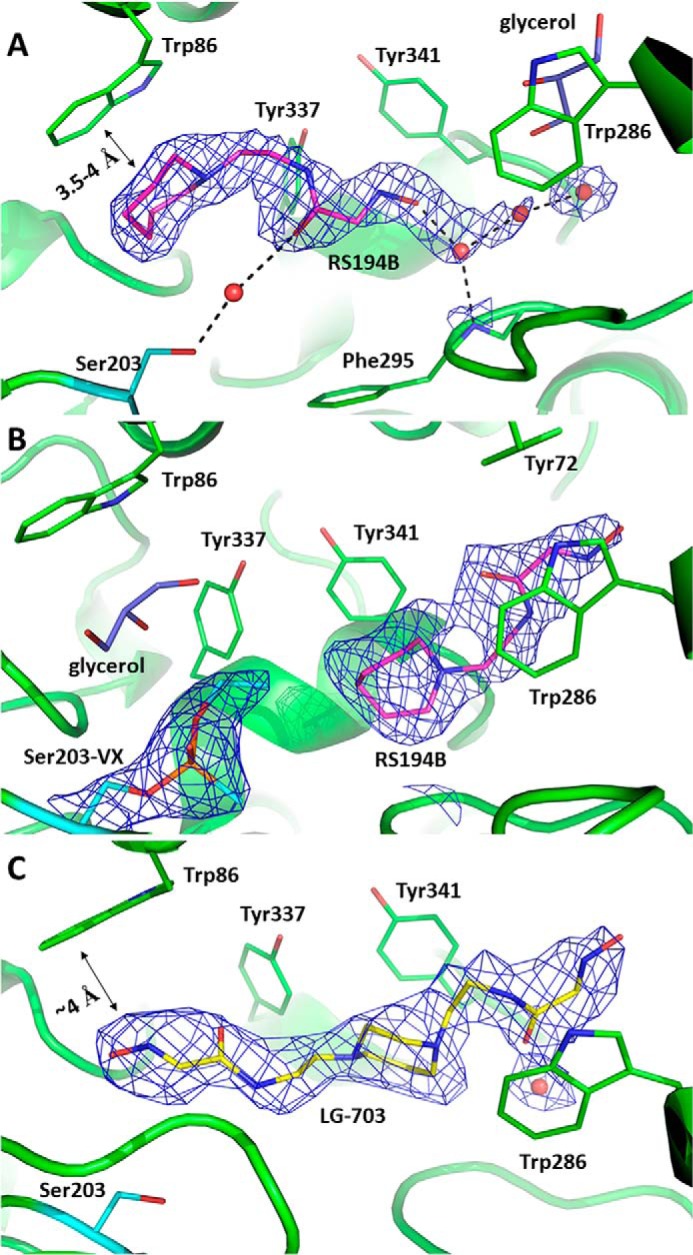
Binding modes of the oxime reactivators in complexes with apo- (A and C) and VX-inhibited (B) hAChE. Shown are views of the active-site gorge with nonproductively bound RS194B at the choline-binding site of apo-AChE (A), nonproductively bound RS194B at the peripheral site of VX-hAChE (B), or bound LG-703 (C). For all panels, black dashed lines represent possible hydrogen-bonding interactions; the 2Fo − Fc electron density maps are shown as blue mesh and contoured at 1.5σ in A and 1.0σ levels in B and C; ligands are shown in stick models with carbon atoms in magenta for RS194B, yellow for LG-703, and purple for glycerol. Red spheres represent water molecules; catalytic Ser-203 is colored with cyan carbon atoms, and the rest of the enzyme residues are shown in green.
The short ∼3.5-4-Å distances between the two heterocyclic rings strongly indicate the existence of cation-π interactions, suggesting that the RS194B azepane amine is protonated (Fig. 3A).
The choline-binding site's evolved preference for cationic species, such as the positively charged quaternary amino group of the physiological substrate acetylcholine (seen in 2HA4, with mouse AChE), or the protonated heterocyclic nitrogen of 9-aminoacridine (in 6O4X, with hAChE) or tacrine (in 1ACJ, with Torpedo californica AChE) is consistent with protonation of the bound azepane ring nitrogen in the most abundant ionization state of RS194B. The entire molecule of RS194B is submerged deep into the lower part of the active center gorge, beyond the narrow choke point defined by the side chains of tyrosines 124 and 337, escaping interactions with the peripheral anionic site (PAS) Trp-286. Lodging of the azepane moiety into the choline-binding site results in a slight but noticeable displacement of the Tyr-337 side chain, which in our recently published hAChE structures (16, 17) assumes a variety of conformers, consistent with the known conformational diversity observed in all other AChEs (18). The acetamide carbonyl group of RS194B is water-bridged to the side chain of Ser-203, whereas the oxygen of the reactive aldoxime group makes a water-mediated interaction with the main-chain amide of Phe-295 (Figs. 2A and 3A and Fig. S1). The chain of water molecules then tailgates the aldoxime group at the active center gorge opening. The observed water-mediated network indicates significant hydration of the reactivator inside the active-site gorge, which may suggest that RS194B enters the active-site gorge with its aldoxime group being deprotonated. If that is the case, our structure indicates that the zwitterionic form of this reactivator binds to the active center of hAChE more than 2 orders of magnitude better than the 180 times more abundant cationic form (Fig. 1A). The abundance of the less reactive cationic form could, in part, explain the large Kox values of RS194B. Obviously, here we did not capture a “productive” nucleophilically reactive zwitterion orientation, where oxime group should be pointing into the active center gorge, possibly due to its lower abundance and to interference of the bound cryoprotectant glycerol (Figs. 2A and 3A). Nevertheless, efficient in vitro reactivation kinetics of RS194B unequivocally confirms the existence of such a “productive” complex, yet at a low abundance not detected by X-ray diffraction.
Effect of VX inhibition on RS194B binding
Covalent binding of the ethylmethylphosphonate to Ser-203 in the crystal of the binary hAChE*RS194B complex, upon brief VX analogue soak, displaces the RS194B molecule from the choline-binding site (Figs. 2B and 3B and Fig. S1). RS194B shifts upward, above the choke point, and toward the PAS at the opening of the gorge, replacing a glycerol molecule observed in the binary complex by the acetamido-aldoxime moiety. RS194B is kept in the same generally nonproductive orientation, with the azepane ring directed toward the VX-Ser-203 conjugate. Because the 185-Å3 volume of RS194B fills most of ∼300-Å3 large lower gorge area in the RS194B*hAChE binary complex, it is unlikely that covalent binding of the large VX analog (with a volume of ∼250 Å3) can occur in the crystal without complete dissociation of the RS194B molecule from the complex, followed by its reassociation with the VX-hAChE conjugate once the leaving group of the analog has left the gorge. It appears, therefore, that the nonproductive RS194B orientation is dictated by the geometry of the VX-conjugated gorge and is not a remnant of the RS194B orientation in the binary complex. At this location, the RS194B molecule is sandwiched between the side chains of Tyr-341 and Trp-286. The binding is stabilized through hydrophobic contacts of the azepane ring with the Tyr-341 side chain and π-π interactions between the acetamido-aldoxime–conjugated π-system and the Trp-286 indole ring of the PAS. Whereas protonation of the heterocycle seems most likely, there is no structural evidence regarding the ionization state of the aldoxime group. The RS194B electron density is less well-defined at this site relative to that observed in the binary complex, which is characteristic of a weaker, more dynamic binding. The weaker stabilization of RS194B at the PAS of VX-hAChE is also consistent with the millimolar Kox value determined from the kinetics of VX-hAChE reactivation by RS194B (7), which is about an order of magnitude higher than the Ki constant of 157 μm determined for the reversible inhibition of hAChE by RS194B (data not shown).
The formation of the VX-hAChE conjugate also affects the conformation of the bound RS194B reactivator, which is different between the two complexes. The main difference comes from how the planes of the azepane ring and the acetamido-aldoxime group are oriented in space relative to each other (Fig. S2). In the binary complex, the two planes are nearly perpendicular to each other (dihedral angle of 82°), whereas in the complex with VX conjugate, they are essentially co-planar (dihedral angle of 150°). Both conformers could sterically fit into the PAS area of the VX-hAChE conjugate while maintaining the azepane-Tyr-341 hydrophobic interaction, but the π-π stacking interaction of acetamido-aldoxime with Trp-286 preferably stabilizes the flattened conformation. However, in the choline-binding site of native hAChE, only the perpendicular conformation of RS194B could fit while maintaining the strong stabilizing anchor of the azepane ring with Trp-86. It is additionally well-stabilized by hydrogen-bonding to Ser-203 via adjacent waters. Thus, the nucleophile-bearing arm of RS194B can assume multiple distinct conformations to adapt to different binding site environments, contributing to the diversity of reactive geometries of the reactivating RS194B molecule.
In silico design and screening of accelerated lead reactivator: Uncharged bis-oxime library
A possible remedy for the nonproductive binding orientation in reactivation is introduction of an additional reactive oxime group on the opposite side of the central RS194B heterocycle. Molecules of such architecture would be able to span the entire length of the active-site gorge, utilizing weak interactions at the PAS to guide the attacking oxime group toward the OP-modified serine residue. More importantly, two aldoxime-bearing flexible linkers could assume a range of well-defined yet distinct conformations (as seen in RS194B structures) and place reactive nucleophiles in a variety of positions relative to the protonated heterocyclic amines that serve as an anchor. The resulting enhanced diversity of productive reactivation orientations and geometries would exceed that of bis-pyridinium bis-oximes (5, 15) when those are compared with the corresponding pyridinium monoximes 2-PAM and HI6 (15, 19). Often distracting for reactivation reaction, interactions of two aromatic pyridinium cations with aromatic residues lining the AChE gorge would be eliminated as well. Thus, heterocyclic bis-oximes created around a single heterocycle represent a significantly different class of bis-oximes from those obtained by combining pyridinium monoximes into bis-pyridinium bis-oximes with the capacity to produce more efficient, uncharged, centrally active reactivators. This is due to the unique conformational control stemming from dynamically controlled intramolecular charge distribution of distinct protonated heterocyclic amines influencing conformations of alkyl arms bearing critical reactive nucleophiles.
Consequently, we built 17 uncharged heterocyclic bis-oximes in silico (Table S1), generated the bis-oxime*hAChE models by replacing RS194B from the X-ray structure of VX-hAChE conjugate with a bis-oxime, and then computationally docked bis-oximes within the VX-hAChE structure. A composite docking score (combination of the raw docking score, the resulting distance between the closer oximate and the conjugated phosphorus, and the oxime attack angle) revealed homopiperazine compound S5 as the best lead (Table S1) for chemical synthesis and further in vitro evaluation.
Chemical synthesis and physico-chemical properties of the uncharged bis-oxime library
Based on compound S5 (Table S1), we synthesized seven novel uncharged bis-oximes by N-alkylation of the central homopiperazine, piperazine, or piperidine heterocycles (Table 2). Detailed synthetic procedures and confirmation of their structures are provided in the supporting information.
Table 2.
Acidity, lipophilicity, and CNS MPO analyses of synthesized bis-oximes
Experimentally determined acidity (pKa), distribution coefficient (logD7.4), and partition coefficient (logPneutral) between water and n-octanol were obtained via potentiometric titrations (SiriusT3, Pion, Inc.); calculated values (in italics), including topological polar surface area (TPSA in Å2) and number of hydrogen bond donors (HBD), were determined using MarvinView 5.4.1.2 (ChemAxon Ltd). The CNS MPO analysis was conducted as described by Wager et al. (20). MPO, multiparameter optimization.

Synthesis of 20–150-mg quantities of pure material additionally allowed for potentiometric determination of pKa values and logD7.4 and logPneutral values for all bis-oximes and their comparison with RS194B. Potentiometric titrations over pH ranges of 2–12 revealed either three or four ionizable groups per bis-oxime (Table 2), two of which were attributed to aldoxime groups (Acid 1 and 2), and the other one or two to heterocyclic amines (Base 1 and 2). Values of pKa for RS194B (8.56 and 9.66 in Table 2) were comparable with those previously determined (7). 13 of 14 bis-oxime oxime groups (Acids 1 and 2, Table 2) had pKa values lower than the oxime group in RS194B, by up to 0.94 pH units, allowing for their better nucleophilic reactivity. Values of pKa for oxime groups were also confirmed by monitoring bis-oxime nucleophilic reactivity (for oximolysis of ATCh) in the pH range 5–11 (Table S2), showing good agreement with values obtained by potentiometric titration. Values of pKa for disubstituted heterocyclic amines were, as expected, lower than that of monosubstituted RS194B, indicating their lower general protonation. Moreover, the larger difference between Acid 2 and Base 2 pKa values in all bis-oximes, compared with RS194B, will result in formation of a larger fraction of uncharged forms in their ionization equilibria, which is important for enhancement of bis-oxime diffusion across biological membranes.
In the all-heterocyclic diamines, the second of the two ring nitrogens had a significantly lower pKa value, suggesting that the ring will be singly protonated over a wide pH range. Bis-oximes of otherwise symmetric structure (LG-703, LG-804, LG-700, and LG-747) will therefore assume asymmetric configurations. In the symmetric environment of aqueous solution, fast exchange of protonation states between heterocyclic amines will likely result in a compound of dynamically symmetric conformation. Nevertheless, in an asymmetric environment, such as the active center gorge of hAChE, compound symmetry will be lost. Consequently, a single heterocyclic bis-oxime could have the unique ability to instantaneously adjust (or select) its ionization state and thus its conformation to fit the geometry of the enzyme-binding site. With the structural diversity of oxime-binding sites in different OP-hAChE conjugates in mind, this conformationally “adaptive” property could be valuable for broadening the spectrum of its reactivation efficiency.
For small drug candidate molecules with intended CNS activity, partition (P) and distribution (D) coefficients are used to predict the ability to cross biological membranes, including the BBB. Values of logD7.4 determined for bis-oximes LG-700 and LG-823 compare generally well with RS194B (Table 2). To characterize CNS-applicable druglike properties of the bis-oximes, we tested one of the commonly used multiparameter scoring approaches (20) and compared it with RS194B (Table 2). Scoring was based on compound molecular weight, topological polar surface area (TPSA), number of hydrogen bond donors (HBD) and pKa. For the bis-oximes, the calculated CNS MPO (multiparameter optimization) desirability scores were found to be at or near 4.0 (Table 2), indicating good likelihood of CNS penetration.
pKa values of the individual ionizable groups and the pH of the medium will dictate distribution of different ionization forms and availability of uncharged and zwitterionic forms. Optimal pH for formation of both uncharged and zwitterionic forms of RS194B is calculated to be pH 9, where 58% of the compound will exist in the uncharged and 6% in the zwitterionic form (Fig. 4 and Fig. S3), dictating that at physiological pH 7.4, both fractions would be reduced to one-tenth of those values. For all bis-oximes, the pH optima for both uncharged and zwitterionic forms are shifted to lower pH, by up to two pH units (Fig. 5 and Fig. S4), resulting in a significant increase in their concentrations at physiological pH.
Figure 4.

Calculated pH-dependent abundance of ionization species for selected (8 of 16) species of LG-703 (A) compared with all four ionization species of RS194B (B). Curves were calculated using pKa values from Table 2 and equations given in Figs. S3 and S4. Dotted, uncharged; blue, cationic; red, anionic (for LG-703, it is anionic−2); black, zwitterionic (for LG-703, it is zwitterionic 1). Species found only for LG-703 are zwitterionic 2 (light green) and anionic−1 (orange).
Figure 5.

pH-dependent abundance of ionization species for LG bis-oximes compared with RS194B. A, uncharged oxime species; B, zwitterionic oxime species. Curves were calculated using pKa values from Table 2 and equations given in the supporting materials.
For RS194B, even the relatively small fractional concentration of the uncharged form present at pH 7.4 was demonstrated to effectively allow the drug to reach the CNS and reactivate OP-hAChE (7, 12), and further improvement of compound efficacy by optimization of its corresponding pKa values seems reasonable. Due to the larger number of ionizable groups, the bis-oximes equilibrate between eight (heterocyclic monoamines, LG-823, and LG-829) and 16 (heterocyclic diamines, LG-703, LG-804, LG-700, LG-750, and LG-747) ionization forms (Fig. S4). For example, two oxime groups and two heterocyclic amines of LG-703 rapidly equilibrate between an uncharged form, four zwitterions, one cationic+2 form, two cationic+1 forms, one anionic−2 form, two anionic−1 forms, two triple-ionic+1−2 and two triple-ionic+2−1 forms, and one quadruple-ionic+2−2 form. Most of those forms are present only at high or low extremes of pH. At the physiological pH of 7.4, the most abundant ionization states are one of the cationic+1 form (75%), the uncharged form (21%), and two zwitterionic forms (3 and 0.7%). The different distributions of positive and negative charges in the two zwitterions, where the protonated amine “anchors” are at different distances from the corresponding reactive anionic oximates, allow them to preferentially bind to the different geometries of the various OP-hAChE conjugates. The solution of bis-oxime LG-703 at pH 7.4 contains two conformationally and electrostatically different nucleophilic oxime reactivator zwitterions, one at 3% and the other at 0.7% of its total concentration. This diversity of nucleophilically reactive zwitterions has the potential to be explored for the design of a broad range of nucleophilic antidotes effective against OP-hAChEs of diverse structures. In addition, the total fraction of zwitterionic species is severalfold higher at pH 7.4 in bis-oximes (2–4%) compared with RS194B (0.5%) (Fig. 5), which should translate into their better reactivity. The fraction of uncharged forms in bis-oximes at 6–60% appears higher than the one observed in RS194B (Fig. 5A), suggesting predisposition for more effective diffusion through biological membranes and CNS penetration than that observed for RS194B (7).
In vitro efficacy of novel bis-oximes for reactivation of OP-hAChE conjugates
Three kinetic constants, kr, k2, and Kox, obtained from the analysis of concentration dependence in reactivation (Fig. S5) of four OP-hAChE conjugates by each LG bis-oxime and the monoxime RS194B (Fig. 6; listed in Table 3) reflect generally good in vitro reactivation efficacy of the bis-oximes. The second-order reactivation rate constant kr (equal to the ratio of the maximal reactivation rate constant k2 and Michaelis–Menten type constant Kox) is frequently used as a “key metric” in evaluation of in vitro reactivation efficacy of oxime antidotes. However, a thorough analysis requires that all three constants be considered. The same large numeric value of kr could come either from a ratio of a large value of k2 and an intermediate value of Kox or from a ratio of a small value of k2 and an even smaller value of Kox, the former being more favored.
Figure 6.

Concentration dependence of oxime reactivation of sarin- (A), paraoxon- (B), cyclosarin- (C), and VX-inhibited (conjugated) (D) hAChE. Shown is dependence for the LG bis-oximes LG-703 (black circles), LG-804 (black triangles), LG-700 (white circles), LG-750 (white triangles), LG-747 (white squares), LG-823 (gray inverted triangles), LG-829 (gray diamonds) compared with RS194B (stars and dashed curves). Curves for piperidine-based bis-oximes LG-823 and LG-829 are gray. All measurements (n ≥ 3; error bars show individual kobs spread) were at 37 °C in 0.10 m phosphate buffer, pH 7.4, and with up to 1% DMSO. Curves were calculated by nonlinear regression from the relationship kobs = k2/(1 + Kox/[oxime]) as described previously (7), and resulting constants are listed in Table 3.
Table 3.
Kinetic constants for in vitro reactivation by LG bis-oximes and RS194B monoxime
OP-hAChE conjugates were obtained by inhibition with paraoxon and analogues of sarin, cyclosarin, and VX. Maximal reactivation rate constant k2(min−1), Michaelis–Menten type constant Kox (mm), and the overall second-order reactivation rate constant kr(m−1 min−1) were calculated by non-linear regression from curves in Fig. 6 as described previously (7). S.E. values of constants k2 and kr obtained from regression were typically smaller than 30% of the reported values. Constants for 2PAM (in the absence of DMSO (7)) are listed for comparison (gray shading). Different shading for LG bis-oximes indicates difference in the central heterocycle (yellow, homopiperazines; white, piperazines; blue, piperidines). Due to 1% DMSO in the reactivation assay, values of constants for RS194B do not correspond exactly to our previously published ones (7), which were obtained at lower DMSO concentration.

Overall, the kinetics of bis-oxime reactivation was more efficient than that by monoxime RS194B. Importantly, more than half of the novel bis-oximes showed noticeably larger rate constants, both kr (Fig. S6) and k2, for nearly all tested OPs.
Based on the values of the k2 constants, the most efficient reactivators for all four OP conjugates appeared to be two homopiperazine bis-oximes, LG-703 and LG-804, followed by two piperidine bis-oximes, LG-829 and the one-methylene-group-shorter LG-823. The two piperazines LG-747 and LG-700 were generally similar to RS194B or slower. Nevertheless, their Kox constants were the lowest of all oximes for all OP-hAChE conjugates, reflecting their enhanced affinity to form reversible complexes. Generally, the bis-oxime Kox constants were lower than those for the RS194B for sarin, cyclosarin, and VX conjugates, as expected, indicating that the nonproductive complex formation observed in the RS194B*VX-hAChE X-ray structure was prevented or reduced.
Overall, our in vitro analysis of the reactivation potency of seven novel bis-oximes reveals a slight dominance in potency of homopiperazines (LG-703 and LG-804) over piperidines (LG-823 and LG-829) due to their better ability to assume transition state geometries (larger k2 constant). Piperazine-based bis-oximes form the highest-affinity reversible complexes with OP-hAChE conjugates (lowest Kox constants) but have difficulty forming proper transition state geometry (lowest k2 constants) for the nucleophilic reaction likely due to their suboptimal charge distribution (compared with isosteric piperidines). One methylene group longer linkers (three versus two methylenes) generally improved reactivation efficiencies either by increasing k2 (piperidines and piperazines) or by reducing Kox (homopiperazines). Most bis-oximes were able to form better transition state geometries than monoxime RS194B (larger k2 constants), whereas piperazine- and piperidine-based bis-oximes also formed better reversible complexes with OP-hAChEs (lower Kox constants). Their in vitro reactivation potencies are comparable with the BBB-impermeable 2PAM (Table 3). Thus, all seven of our new LG bis-oximes can be considered as promising antidotes worth investigating further in ex vivo and in vivo experimental settings.
Structure of LG703 bound to the active site of hAChE
The improved in vitro reactivation efficacy of the uncharged bis-oximes compared with the monoxime RS194B is indeed a likely consequence of the productive binding orientation of their second oxime group, as seen in the X-ray crystal structure of the apo-hAChE in complex with LG-703, a bis-oxime structurally closest to RS194B (Table 2). The LG-703 binds the entire length of the active-site gorge, spanning its oxime groups between Trp-86 at the gorge base and Trp-286 at the gorge opening (Figs. 3C and 7). At the active site, the oxime-bearing arm extends all the way to the choline-binding site to occupy a position similar to that of the RS194B azepane ring in the apo-hAChE complex, slightly overshooting Ser-203 (Figs. 3C, 7, and 8).
Figure 7.

X-ray structure of homopiperazine-based bis-oxime LG-703 bound in the active-center gorge of Apo-hAChE. Orange/green water-accessible Connolly surface of hAChE is removed in part to reveal the gorge interior. Catalytic triad side chains (Ser-203, His-447, and Glu-334) are rendered in yellow sticks. The choke point narrow is defined by Tyr-124 and Tyr-337.
Figure 8.
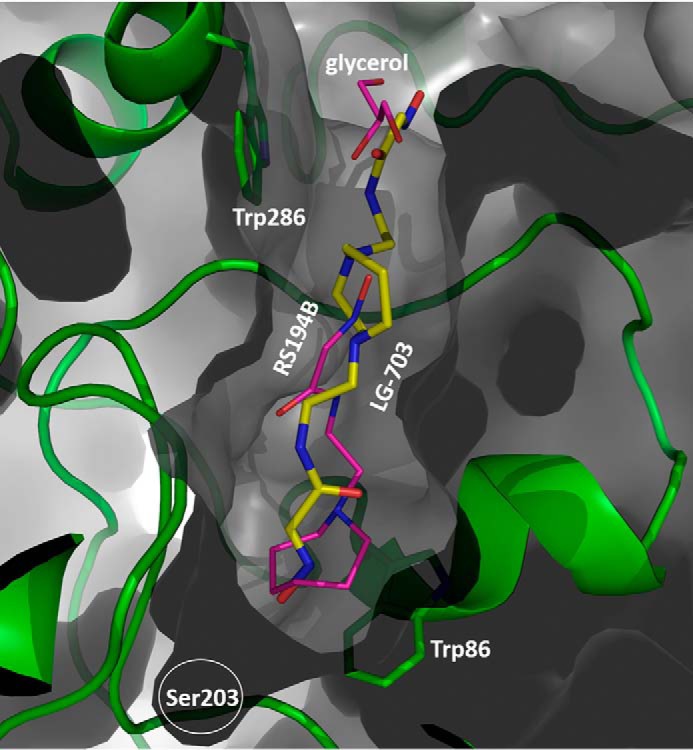
LG-703 (stick model with yellow carbons) traverses the entire length of the active-site gorge. Magenta, model of RS194B in hAChE*RS194B X-ray structure superimposed on hAChE*LG-703.
In this conformation, the aldoxime N-O group is positioned nearly equidistant between Ser-203 and Trp-86 and participates in n→π* interactions with the indole ring of Trp-86. The reactive hydroxyl group is additionally stabilized by a weak (3.4 Å) hydrogen-bonding interaction with the Glu-202 carboxylate. The combination of the LG-703 length and its deep penetration into the gorge results in ∼1-Å displacement of the choke point residues, Tyr-124 and Tyr-337, and rotation of the Tyr-341 side chain relative to the position in the binary complex of RS194B with apo-hAChE. Although the alignment of the aldoxime group at the active site can be considered a productive one because of its extended orientation toward the catalytic Ser-203, the reactivator will not be able to perform the nucleophilic attack on OP-Ser-203 from the position it occupies in this structure. Nevertheless, the overall “reorientation” of the reactive aldoxime group from the position 9–16 Å distant from the Ser-203 in our two RS194B structures (Figs. 2 (A and B) and 3 (A and B)) to the position 5 Å close to it (Fig. 7) appears structurally and functionally much more significant than the corresponding “reorientations” observed in X-ray structures of any of the bis-pyridinium bis-oximes (5–8 Å) (15) compared with typically nonproductive orientations of the pyridinium monoximes 2-PAM or HI6 (5–9 Å) (15, 21). Apparently, the intensity of interactions of individual pyridinium rings with aromatic rings of the AChE active center gorge “locks” their orientation in either mono- or bis- pyridinium oximes.
Protonation of the two oxime groups and the two cyclic amines of LG-703, essential for the bis-oxime binding within the hAChE gorge and for its nucleophilic reactivity, result at pH 7.4 in formation of cationic+1 (75%), uncharged (21%), and two zwitterionic forms (3 and 0.7%). Three of those major forms should have one of the two cyclic amines protonated, and the X-ray structure suggests that it would be the one facing the gorge entry. The axial position of the acetamidoethylene substituent on that cyclic nitrogen would place a proton in the sterically more favorable equatorial position that is also open to cation-π stabilization by the 3.7-Å distant Trp-286, 4.7-Å distant Tyr-124, and 4.6-Å distant Tyr-341 near the hAChE gorge entry. Furthermore, hydrogen bonding of one LG-703 oxime group to Glu-202 strongly indicates that the oxime is protonated. The other oxime group is stabilized by n→π* interaction with Trp-286 at a distance (∼4 Å) similar to that between Trp-86 and the first oxime, possibly hinting at similar ionization states of the two oxime groups. It is thus possible that the cationic+1 form of LG-703 is bound in the gorge, but it could also be one of its two zwitterions.
In this X-ray structure, the two acetamido-alkyl substituents of LG-703 adopt very different conformations not only with respect to the relative positions of the planes containing atoms of the specific functional groups, but also with respect to the imino and carbonyl double bonds of each acetamido-oxime moiety being cis-trans isomers (Fig. S2C). At the peripheral site, the external acetamido-oxime substituent adopts a flattened conformation relative to the homopiperazine core. Similar to RS194B binding in the VX-hAChE*RS194B complex, this conformation is stabilized through weak C-H…π interactions of the piperazine ring with Tyr-341 and n→π* of the acetamido-oxime group with Trp-286 (Fig. 7). The two double bonds of the external tail are trans-oriented with respect to each other. The same trans-positioning was observed for the imino and carbonyl double bonds of RS194B in both complexes. In contrast, these groups are cis-oriented in the internal tail of LG-703, and the plane of the acetamido-oxime group is almost perpendicular to the central heterocycle, resembling the conformation of RS194B in apo-hAChE. As a result, and critical for reactivation efficacy, distances between the heterocyclic amine and each of two oxime group oxygens appear consistently more than 1 Å longer in the internal tail. To quantify the energetics of cis and trans acetamido-oxime isomers and the interconversion between the two conformations, we performed molecular geometry optimization of LG-703 and torsion angle scan (0–180°) with quantum mechanics calculations in implicit water (Fig. S7). The calculations produced two stationary points corresponding to cis (torsion angle 0°) and trans (torsion angle 180°) orientations of almost equal stability. The two conformers differ only by 0.3 kcal/mol but are separated by an energy barrier of 5 kcal/mol. This suggests that three conformers of the LG-703 molecule can coexist in solution: cis-cis, cis-trans, or trans-trans (referring to the geometry of the two acetamido-aldoxime moieties). Furthermore, the calculations imply that these conformations are not imposed by the active-site gorge environment but rather are selectively stabilized by it. Nevertheless, likely protonation of the peripherally oriented cyclic amine in LG-703 seems to imply a substituent geometry similar to the one seen in both RS194B structures where the cyclic amine is protonated.
In our structure, LG-703 binds to only one of two hAChE monomers of the asymmetric unit homodimer, allowing for direct comparison of the complex and the apo-hAChE structures derived from the same crystal (described in the supporting information and Fig. S8).
Whereas relatively small differences are expected for the overall conformational sterics of the cationic+1 and the two zwitterionic ionization forms of LG-703 (Fig. 1), different distances and interaction patterns between protonated amine and deprotonated aldoxime groups (∼6 Å in zwitterion 1 versus ∼11 Å in zwitterion 2; Fig. 1B) could have substantial influence on their reactivation efficacy against OP-hAChE conjugates having different structures. The protonated amine as a main anchoring point of the compound in the AChE gorge would thus position the attacking nucleophilic oximates quite differently, as already indicated by the asymmetric conformation (Figs. 7 and 8) of otherwise two-dimensionally symmetric (Fig. 1) LG-703 molecule, bound in the hAChE active-center gorge. The option of having both zwitterions in significant abundance in solution, in combination with conformational flexibility, could be advantageous for an antidote, could broaden its efficacy against a wider range of nerve agents, and could render it a more universal antidote, a previously unexplored aspect of improving antidote efficacy in the design of oxime reactivators. We thus consider this new class of uncharged bis-oximes to be “dynamically adaptable” or “smart” oxime reactivators, because in solution at physiological pH, one compound can assume a multitude of ionization forms, each with different reactivation reaction potential against single or diverse OP-hAChE conjugates.
Conclusions
We present here structure-assisted rational design, synthesis, and initial in vitro functional characterization of a directed library of seven novel uncharged bis-oxime reactivators (Table S3). Initial X-ray structural analysis of the prototypical uncharged acetamido monoxime RS194B revealed nonproductive orientations bound to either native or VX-inhibited hAChEs that we successfully corrected, first in silico by introduction of an additional reactive oxime group in a focused library of 17 bis-oximes. Subsequent computational rank-ordering led us to the selection and synthesis of seven novel compounds: two piperidine, three piperazine, and two homopiperazine doubly substituted alkyl acetamido oximes. 4–6 novel bis-oximes were faster in vitro reactivators of sarin-, VX-, cyclosarin-, and paraoxon-derived OP-hAChE conjugates, compared with the in vivo proven uncharged monoxime reactivator RS194B. Whereas reactivation rates may be slower than those reported for the best bis-pyridinium bis-oximes, one has to keep in mind that none of pyridinium oximes has the capacity to reactivate OP-conjugated AChE in the CNS. We thus compared reactivation efficacies of our novel bis-oximes with those of the confirmed centrally active antidote RS194B. By solving an X-ray structure of the bis-oxime LG-703, in complex with hAChE, we confirmed that improved reactivation efficiency was consistent with the productive orientation of one of the two nucleophilically reactive aldoxime groups. Reactive aldoxime-bearing “arms” of LG-703 assumed an asymmetric conformation in the complex, despite two-dimensional symmetry of the LG-703 molecule. In addition to the correct binding orientation, the dynamic charge distribution between the three or four ionizable groups of the novel heterocyclic bis-oximes leads to an increase in the diversity of the zwitterionic forms believed to be primarily responsible for the nucleophilic reactivation of OP-hAChEs. Fast equilibration between multiple zwitterions resulted in the formation of multiple, structurally diverse and asymmetric reactivation species for each bis-oxime. To the best of our knowledge, this is the first time that such dynamically adaptable “smart” oxime reactivators have been created, and we believe that this novel approach in optimizing reactivator efficacy and broadening specificity against structurally diverse OP-hAChE conjugates deserves to be further explored in subsequent studies.
These novel reactivators maintain a larger fraction of their uncharged forms at pH 7.4 compared with RS194B and should therefore readily pass across the BBB and reach the CNS. Additional ex vivo and in vivo testing of these bis-oximes is under way with the expectation that they could yield a new class of accelerated uncharged reactivator antidotes against OP intoxication.
Experimental procedures
Protein expression and purification
The monomeric form of human AChE truncated at the C-terminal amino acid position 547 was expressed in Gnt1− HEK293 cells and purified using N-terminal FLAG tag on the anti-FLAG affinity column by proteolytic elution, as described before (16).
Crystallization
In preparation for crystallization, samples of hAChE were dialyzed in 10 mm NaCl, 10 mm HEPES, pH 7, and concentrated to 6–10 mg/ml. About 1 h prior to crystallization, the solution of hAChE was combined with stock solutions of ligands in a molar ratio of 1:5 (LG-703) or 1:10 (RS194B) to obtain binary complexes. Crystals were grown by vapor diffusion at 10 °C in sitting-drop microbridges (Hampton Research, Aliso Viejo, CA). Well solutions containing 10–20 mm sodium citrate, 100 mm HEPES, pH 7, 8–8.5% PEG 6000 and 100 mm potassium nitrate, 100 mm HEPES, pH 7.5, 10.5% PEG 3350 were used in crystallization experiments with RS194B and LG-703. To obtain the RS194B complex with VX-hAChE conjugate, crystals of the binary complex, hAChE*RS194B, were soaked in a solution of nonhazardous analogue of VX (17). Low-toxicity, nonvolatile fluorescent methylphosphonate (Flu-MP) (22) analogue of nerve agent VX was used. The Flu-MPs differ from actual nerve agent OPs only by the structure of their respective leaving groups that are excluded from the resulting OP-hAChE structures. Inhibition of hAChE by Flu-MPs results in OP-hAChE covalent conjugates identical to the ones formed upon inhibition with the corresponding volatile OPs.
X-ray data collection
X-ray crystallographic data were collected from frozen crystals at 100 K. Prior to data collection, crystals were subjected to two very brief consecutive soaks in the cryoprotectant solutions, first in 12.5% glycerol followed by 25% glycerol, and then flash-cooled by plunging into liquid nitrogen. Diffraction data were collected on the ID19 (100 K) beamline at SBC-CAT using a Pilatus3 X 6M (ID19) detector at the Advanced Photon Source (APS). X-ray diffraction data were integrated and scaled using the HKL3000 software suite (23). The structures were solved by molecular replacement using the CCP4 suite (24). The structure of the apo-hAChE (PDB code 4EY4) (21) was used as a starting model with all waters and the N-linked glycosylated saccharides removed. Refinement was performed using the phenix.refine program in the PHENIX (25) suite, and the resulting structure was analyzed with MolProbity (26). The structures were built and manipulated with the program Coot (27). Each hAChE structure contained two independent enzyme molecules in the asymmetric unit. All ligands in the reported hAChE structures were refined with an occupancy of 1.0. Structures were also analyzed using Nanome, VR macromolecular visualization and modeling suite (Nanome Inc.). Figures were generated using Coot, Biovia Discovery Visualizer (Dassault Systems), and PyMOL molecular graphics software (Schrödinger LLC). A summary of the crystallographic data and refinement is given in Table 1. Crystallographic data have been deposited to the PDB under codes 6U34, 6U37, and 6U3P.
Computational molecular modeling
Seventeen hypothetical uncharged bis-oxime compounds were generated in silico and evaluated computationally as potential reactivators of the VX-hAChE conjugate using computational docking. The co-crystal structures of RS194B*hAChE and RS194B*VX-hAChE were used as the receptors for the docking calculations upon removal of RS194B. All 17 designed hAChE reactivators were sketched using maestro and minimized using the ligprep module with their possible protonation states generated by Epik. Optimized ligands were docked into the binding pocket of the native and VX-conjugated hAChE using the Ligand Docking protocol with the extra precision (XP) scoring mode of Glide. For each system, the receptor grid box for docking was generated and centered on the ligand in the binding pocket of hAChE with a box size of 8 × 8 × 8 Å using the Receptor Grid Generation protocol. Considering the mechanism of hAChE reactivation, the Ranking Score (Rscore) of each designed hAChE reactivator that accounts for both binding energy and binding pose is defined as follows: Rscore = 0.3*(l − 4.0)2 + 0.0025*(Φ − 150)2 + Edocking. In this scoring equation, l represents the closest distance between the OP phosphorus and the oxime group oxygen, and Φ stands for the angle between the oxime oxygen the phosphorus and the oxygen of Ser-203.
Synthetic procedures
RS194B was prepared as described before (7). The synthesis of all bis-oxime acetamides was conducted as highlighted in Scheme 1 and described below. Compounds LG-700, LG-703, and LG-804 were prepared starting with N-alkylation reactions of either homopiperazine 5 (for LG-703 and LG-804) or piperazine 4 (for LG-700), with the appropriate ω-halogenalkylcarbonitriles. Next, reduction of the nitrile moieties to the corresponding primary amines (17, 18, and 21, Scheme 1), followed by condensation reaction with ethyl (2E)-2-(hydroxyimino)acetate 2, provided the desired bis-oximes. It is noteworthy that compound 23 is commercially available, and its condensation with 2 afforded LG-747. The synthesis of bis-oxime, LG-750, which features carbon spacers of different length, called for two sequential N-alkylation reactions starting from N-Boc-protected piperazine 12, followed by the same reduction and condensation sequence. Piperidine derivatives, LG-823 and LG-829, were prepared in a similar fashion starting from N-Boc-4-cyanomethylpiperidine 13 (Scheme 1).
SCHEME 1.

General synthetic approaches: N-alkylation with appropriate ω-halogenalkylcarbonitrile (a), reduction of carbonitriles to amines (b), N-Boc deprotection (c), and condensation reaction with compound 2 (d). Detailed synthetic procedures and confirmation of their structures are provided in the supporting information.
Determination of physico-chemical properties
Determination of acidity (i.e. pKa) as well as lipophilicity (i.e. logD7.4 and logPneutral) of all bis-oximes and monoxime RS194B was conducted via potentiometric titrations using a SiriusT3 instrument (Pion Inc.) according to the manufacturer's instructions. Values of pKa for oxime groups were also determined by monitoring their nucleophilic reactivity (at 0.30 mm concentration) for oximolysis of ATCh (0.30 mm) in phosphate-pyrophosphate buffers at pH 5, 6, 7, 7.5, 8, 8.5, 9, 9.5, 10, 10.5, and 11, at 22 °C, as described earlier (7).
Quantitative distribution of mono- and bis-oxime ionization species
Individual abundance of cationic, anionic, zwitterionic, and uncharged oxime reactivator forms were calculated for each compound in the pH range between 6 and 12, using experimentally determined pKa values and equations derived for each of the species (Figs. S3 and S4), based on the Henderson–Haselbach equation.
Reactivation kinetics
Covalent conjugates of hAChE identical to those inhibited by nerve agent OPs VX, sarin, and cyclosarin were prepared using low-toxicity Flu-MP analogues of nerve agents. Inhibition of hAChE by Flu-MPs results in OP-hAChE covalent conjugates identical to the ones formed upon inhibition with the corresponding volatile OPs. 4–10-Fold stoichiometric excess of Flu-MPs or paraoxon was used to inhibit hAChE (∼20 μm) for several minutes at room temperature, resulting in >99% loss of catalytic activity, to ensure homogeneous inhibition by the more potent SP enantiomer of Flu-MPs. Excess unreacted Flu-MP was removed from the inhibition mixture by Sephadex G-50 separation on two consecutive spin columns (Bio-Rad, Bio-Spin 6 Tris columns). Time-dependent recovery of hAChE activity was monitored spectrophotometrically (28) in 0.1 m phosphate buffer, pH 7.4 (containing 0.01% BSA) upon the addition of LG bis-oximes at 37 °C. The first-order reactivation rate constant (kobs) for each (oxime + OP-hAChE conjugate) combination was calculated by nonlinear regression. The dependence of reactivation rates on oxime concentrations and determination of maximal reactivation rate constant k2, Michaelis–Menten type constant Kox, and the overall second-order reactivation rate constant kr were conducted as described previously (7).
Author contributions
L. G., O. G., X. C., D. K. B., P. T., C. B., A. K., and Z. R. conceptualization; X. C., D. K. B., P. T., C. B., A. K., and Z. R. resources; L. G., O. G., X. K., X. C., D. K. B., P. T., C. B., A. K., and Z. R. formal analysis; C. B., A. K., and Z. R. supervision; X. C., D. K. B., A. K., and Z. R. funding acquisition; L. G., O. G., X. K., X. C., D. K. B., P. T., C. B., A. K., and Z. R. validation; L. G., O. G., X. K., X. C., D. K. B., P. T., C. B., A. K., and Z. R. investigation; L. G., O. G., X. K., X. C., D. K. B., P. T., C. B., A. K., and Z. R. methodology; L. G., O. G., A. K., and Z. R. writing-original draft; A. K., C. B., and Z. R. project administration; L. G., O. G., X. K., X. C., D. K. B., P. T., C. B., A. K., and Z. R. writing-review and editing; O. G., X. K., X. C., C. B., A. K., and Z. R. data curation; O. G., X. K., X. C., A. K., and Z. R. software; O. G., X. K., X. C., A. K., and Z. R. visualization.
Supplementary Material
Acknowledgments
X-ray crystallographic data presented in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source, beamline ID19. Use of the Advanced Photon Source, an Office of Science User Facility operated for the United States Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the United States DOE under Contract DE-AC02-06CH11357. The Office of Biological and Environmental Research supported research at the Center for Structural Molecular Biology at Oak Ridge National Laboratory using facilities supported by the Scientific User Facilities Division, Office of Basic Energy Sciences, United States DOE. The assistance of Kwok-Yiu Ho and University of California San Diego undergraduate students Stephanie Luedtke, Yunshen Li, Celine Bojo, and Bianca Pomar in completion of oxime reactivation and oximolysis experiments is greatly appreciated.
This work was supported by CounterACT Program (National Institutes of Health (NIH) Office of the Director and NINDS, NIH) Grants U01 NS083451 and R21 NS098998 and by University of California San Diego, Academic Senate Grant BG084144. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S3 and Figs. S1–S8.
The atomic coordinates and structure factors (codes 6U34, 6U37, and 6U3P) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- OP
- organophosphate
- 2-PAM
- 2-pyridine aldoxime methiodide
- AChE
- acetylcholinesterase
- hAChE
- human AChE
- ATCh
- acetylcholine
- CNS
- central nervous system
- BBB
- blood-brain barrier
- OP-hAChE
- OP-inhibited human AChE
- PAS
- peripheral anionic site
- VX
- venomous agent X
- Flu-MP
- fluorescent methylphosphonate
- Boc
- t-butoxycarbonyl
- PDB
- Protein Data Bank.
References
- 1. Wilson I. B., Ginsburg B. (1955) A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim. Biophys. Acta 18, 168–170 10.1016/0006-3002(55)90040-8 [DOI] [PubMed] [Google Scholar]
- 2. Hobbiger F., and Sadler P. W. (1958) Protection by oximes of bis-pyridinium ions against lethal diisopropyl phosphonofluoridate poisoning. Nature 182, 1672–1673 10.1038/1821672a0 [DOI] [PubMed] [Google Scholar]
- 3. Heilbronn E., and Tolagen B. (1965) Toxogonin in sarin, soman and tabun poisoning. Biochem. Pharmacol. 14, 73–77 10.1016/0006-2952(65)90059-6 [DOI] [PubMed] [Google Scholar]
- 4. Pang Y. P., Kollmeyer T. M., Hong F., Lee J. C., Hammond P. I., Haugabouk S. P., and Brimijoin S. (2003) Rational design of alkylene-linked bis-pyridiniumaldoximes as improved acetylcholinesterase reactivators. Chem. Biol. 10, 491–502 10.1016/S1074-5521(03)00126-1 [DOI] [PubMed] [Google Scholar]
- 5. Ekström F., Pang Y. P., Boman M., Artursson E., Akfur C., and Börjegren S. (2006) Crystal structures of acetylcholinesterase in complex with HI-6, Ortho-7 and obidoxime: structural basis for differences in the ability to reactivate tabun conjugates. Biochem. Pharmacol. 72, 597–607 10.1016/j.bcp.2006.05.027 [DOI] [PubMed] [Google Scholar]
- 6. Sit R. K., Kovarik Z., Maček Hrvat N., Žunec S., Green C., Fokin V. V., Sharpless K. B., Radić Z., Taylor P. (2018) Pharmacology, pharmacokinetics, and tissue disposition of zwitterionic hydroxyiminoacetamido alkylamines as reactivating antidotes for organophosphate exposure. J. Pharmacol. Exp. Ther. 367, 363–372 10.1124/jpet.118.249383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Radić Z., Sit R. K., Kovarik Z., Berend S., Garcia E., Zhang L., Amitai G., Green C., Radić B., Fokin V. V., Sharpless K. B., and Taylor P. (2012) Refinement of structural leads for centrally acting oxime reactivators of phosphylated cholinesterases. J. Biol. Chem. 287, 11798–11809 10.1074/jbc.M111.333732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor P., Yan-Jye S., Momper J., Hou W., Camacho-Hernandez G. A., Radić Z., Rosenberg Y., Kovarik Z., Sit R., and Sharpless K. B. (2019) Assessment of ionizable, zwitterionic oximes as reactivating antidotal agents for organophosphate exposure. Chem. Biol. Interact. 308, 194–197 10.1016/j.cbi.2019.05.015 [DOI] [PubMed] [Google Scholar]
- 9. Kobrlova T., Korabecny J., and Soukup O. (2019) Current approaches to enhancing oxime reactivator delivery into the brain. Toxicology 423, 75–83 10.1016/j.tox.2019.05.006 [DOI] [PubMed] [Google Scholar]
- 10. Sit R. K., Radić Z., Gerardi V., Zhang L., Garcia E., Katalinić M., Amitai G., Kovarik Z., Fokin V. V., Sharpless K. B., and Taylor P. (2011) New structural scaffolds for centrally acting oxime reactivators of phosphylated cholinesterases. J. Biol. Chem. 286, 19422–19430 10.1074/jbc.M111.230656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalisiak J., Ralph E. C., and Cashman J. R. (2012) Nonquaternary reactivators for organophosphate-inhibited cholinesterases. J. Med. Chem. 55, 465–474 10.1021/jm201364d [DOI] [PubMed] [Google Scholar]
- 12. Rosenberg Y. J., Mao L., Jiang X., Lees J., Zhang L., Radić Z., and Taylor P. (2017) Post-exposure treatment with the oxime RS194B rapidly reverses early and advanced symptoms in macaques exposed to sarin vapor. Chem. Biol. Interact. 274, 50–57 10.1016/j.cbi.2017.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zorbaz T., Braïki A., Maraković N., Renou J., de la Mora E., Maček Hrvat N., Katalinić M., Silman I., Sussman J. L., Mercey G., Gomez C., Mougeot R., Pérez B., Baati R., Nachon F., et al. (2018) Potent 3-hydroxy-2-pyridine aldoxime reactivators of organophosphate-inhibited cholinesterases with predicted blood-brain barrier penetration. Chemistry 24, 9675–9691 10.1002/chem.201801394 [DOI] [PubMed] [Google Scholar]
- 14. Chambers J. E., and Meek E. C. (2020) Novel centrally active oxime reactivators of acetylcholinesterase inhibited by surrogates of sarin and VX. Neurobiol. Dis. 133, 104487 10.1016/j.nbd.2019.104487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kovalevsky A., Blumenthal D. K., Cheng X., Taylor P., and Radić Z. (2016) Limitations in current acetylcholinesterase structure-based design of oxime antidotes for organophosphate poisoning. Ann. N.Y. Acad. Sci. 1378, 41–49 10.1111/nyas.13128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gerlits O., Ho K. Y., Cheng X., Blumenthal D., Taylor P., Kovalevsky A., and Radić Z. (2019) A new crystal form of human acetylcholinesterase for exploratory room-temperature crystallography studies. Chem. Biol. Interact. 309, 108698 10.1016/j.cbi.2019.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gerlits O., Kong X., Cheng X., Wymore T., Blumenthal D. K., Taylor P., Radić Z., and Kovalevsky A. (2019) Productive reorientation of a bound oxime reactivator revealed in room temperature X-ray structures of native and VX-inhibited human acetylcholinesterase. J. Biol. Chem. 294, 10607–10618 10.1074/jbc.RA119.008725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Radić Z., and Taylor P. (2006) Structure and function of cholinesterases. In Toxicology of Organophosphate and Carbamate Compounds (Gupta R., ed) p. 161–186, Elsevier, Amsterdam [Google Scholar]
- 19. Franklin M. C., Rudolph M. J., Ginter C., Cassidy M. S., and Cheung J. (2016) Structures of paraoxon-inhibited human acetylcholinesterase reveal perturbations of the acyl loop and the dimer interface. Proteins 84, 1246–1256 10.1002/prot.25073 [DOI] [PubMed] [Google Scholar]
- 20. Wager T. T., Hou X., Verhoest P. R., and Villalobos A. (2010) Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 1, 435–449 10.1021/cn100008c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheung J., Rudolph M. J., Burshteyn F., Cassidy M. S., Gary E. N., Love J., Franklin M. C., and Height J. J. (2012) Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 55, 10282–10286 10.1021/jm300871x [DOI] [PubMed] [Google Scholar]
- 22. Amitai G., Adani R., Limanovich O., Teitlboim S., Yishay S., Tveria L., Yacov G., Meshulam H., and Raveh L. (2008) Characterization of asymmetric fluorogenic phosphonates as probes for developing organophosphorus hydrolases with broader stereoselectivity. Chem. Biol. Interact. 175, 249–254 10.1016/j.cbi.2008.05.036 [DOI] [PubMed] [Google Scholar]
- 23. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 10.1107/S0907444906019949 [DOI] [PubMed] [Google Scholar]
- 24. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ellman G. L., Courtney K. D., Andres V. Jr., and Featherstone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95 10.1016/0006-2952(61)90145-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.