

Abstract
Mitochondrial dysfunction has recently been implicated as an underlying factor to several common neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s and amyotrophic lateral sclerosis (ALS). Valosin containing protein (VCP)-associated multisystem proteinopathy is a new hereditary disorder associated with inclusion body myopathy, Paget disease of bone (PDB), frontotemporal dementia (FTD) and ALS. VCP has been implicated in several transduction pathways including autophagy, apoptosis and the PINK1/Parkin cascade of mitophagy. In this report, we characterized VCP patient and mouse fibroblasts/myoblasts to examine their mitochondrial dynamics and bioenergetics. Using the Seahorse XF-24 technology, we discovered decreased spare respiratory capacity (measurement of extra ATP that can be produced by oxidative phosphorylation in stressful conditions) and increased ECAR levels (measurement of glycolysis), and proton leak in VCP human fibroblasts compared with age- and sex-matched unaffected first degree relatives. We found decreased levels of ATP and membrane potential, but higher mitochondrial enzyme complexes II + III and complex IV activities in the patient VCP myoblasts when compared to the values of the control cell lines. These results suggest that mutations in VCP affect the mitochondria’s ability to produce ATP, thereby resulting in a compensatory increase in the cells’ mitochondrial complex activity levels. Thus, this novel in vitro model may be useful in understanding the pathophysiology and discovering new drug targets of mitochondrial dynamics and physiology to modify the clinical phenotype in VCP and related multisystem proteinopathies (MSP).
Keywords: Inclusion body myopathy, Paget’s disease of the bone, Frontotemporal dementia, ALS, Valosin containing protein, Mitochondrial bioenergetics
1. Introduction
Hereditary inclusion body myopathy, Paget’s disease of bone (PDB), and frontotemporal dementia (IBMPFD), more recently termed multisystem proteinopathy (MSP), caused by mutations in the valosin containing protein (VCP) gene are associated with weakness and atrophy of skeletal, pelvic and shoulder girdle muscles in 90% of individuals (Kimonis et al., 2000; Kovach et al., 2001; Watts et al., 2003). Affected individuals exhibit scapular winging, progressive muscle weakness in a limb girdle muscular dystrophy (LGMD) distribution and die from cardiac and respiratory failure (Kimonis et al., 2000, 2008b). Histologically, patients display rimmed vacuoles and ubiquitinated TAR DNA binding protein-43 (TDP-43)-positive inclusion bodies in muscles (Kimonis et al., 2000, 2008a, 2008b; Watts et al., 2004), the heart and brain. Affected individuals also suffer from Paget’s disease of bone (PDB) in 50%, frontotemporal dementia (FTD) in 33%, and amyotrophic lateral sclerosis (ALS) in approximately 15% of cases.
Our previous analyses of IBMPFD myoblasts with VCP mutations showed enlarged vacuoles, increased apoptosis and a defective maturation process to myotubes (Vesa et al., 2009). VCP is involved in a plethora of cellular functions including the autophagy and apoptosis signaling transduction pathways (Nalbandian et al., 2011). Autophagy is a catabolic process whereby cytoplasmic components are sequestered in autophagosomes and brought to lysosomes for degradation and recycling. Accumulation of lysosome-associated membrane proteins (LAMP-1 and LAMP-2) and light chain 3 (LC3-I) was observed in the vacuolar membranes, suggestive of disturbed final fusion steps between autophagosome and lysosome, thus, leading to immature autophagosomes (Tresse et al., 2010). More recently, exploring the function of VCP has demonstrated that its impairment by pathogenic mutations cannot facilitate the PINK1/Parkin signaling cascade, thereby promoting mitophagy (Kim et al., 2013). Current studies are investigating the role of mitophagy as one of the pathogenic manifestations underlying VCP disease.
Mitochondrial dysfunction has recently been implicated as an underlying factor to several neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease and ALS. With its multi-faceted phenotype, VCP MSP was considered a good candidate for a disease resulting from disrupted mitochondrial function. Thus, in this report, we have examined the mitochondrial dynamics and bioenergetics in VCP human fibroblasts using Seahorse XF-24 flux analyzer technology and discovered decreased spare respiratory capacity (a measurement of extra ATP that can be produced by oxidative phosphorylation in stressful conditions); increased ECAR levels, (a measurement of glycolysis); decreased ATP content; and proton leak in VCP human fibroblasts compared with age- and sex-matched unaffected first degree relatives. This new interplay between mitochondrial dynamics and cell biology may provide a deeper understanding of the cellular and molecular mechanism(s) underlying VCP pathophysiology and may be of critical importance in discovering new drug targets for VCP and related multisystem diseases.
2. Materials and methods
2.1. Patient myoblast cultures
Control and patient primary myoblasts (with the R155H mutation) were obtained from Muscle Tissue Culture Collection (MTCC)/EuroBioBank (München, Germany). Cells were maintained in Skeletal Muscle Cell Growth Medium (PromoCell, Heidelberg, Germany) supplemented with the supplement mix, 10% FBS, gentamicin (Life Technologies, Carlsbad, CA) and GlutaMAX-1 in 5% CO2 at 37 °C. Cultured patient myoblasts were plated in 35 mm dishes with a ConA-laminin coated cover glasses (MatTek Corp, Ashland, MA).
2.2. VCP murine myoblast cultures
Murine myoblasts (3-week old) from WT, VCPR155H/+, and VCPR155H/R155H were harvested and cultured. Briefly, myoblasts were cultured in DMEM with 5% FBS supplemented with antibiotics in a 37 °C with 5% CO2. Trypsin/EDTA (Life Technologies, Carlsbad, CA) was used for trypsinization purposes. All cell lines were of the same genetic background.
2.3. Immunocytochemical (ICC) analysis
Patient fibroblasts and myoblasts were incubated for 24 h at 37 °C in 5% CO2. Briefly, myoblasts were cultured in DMEM with 5% FBS supplemented with antibiotics in a 37 °C with 5% CO2. Immunocytochemical (ICC) staining were performed using routine methods (Badadani et al., 2010). Patient fibroblasts and myoblasts were stained with TDP-43, ubiquitin, VCP, LC3-I/II, p62/SQSTM1, and JC-1 (Life Technologies, Carlsbad, CA) -specific antibodies. All primary antibodies were purchased from Abcam (Cambridge, MA) and analyzed by fluorescence microscopy (Carl Zeiss, Thornwood, NY) as described previously (Nalbandian et al., 2013).
2.4. Mitochondrial isolation and OXPHOS enzyme analysis
Mitochondria were isolated from cultured human myoblasts by homogenization and differential centrifugation. Mitochondrial enzyme complex activities were determined by oxidative phosphorylation (OXPHOS) enzyme assays using a previously described standard protocol (Kokoszka et al., 2004; Trounce et al., 1996). In brief, the complexes II + III analyses were performed as follows: 550 μl dH2O, 400 μl 100 mM potassium phosphate buffer (pH 7.4), 20 μl 1 M succinate, 1 μl 0.5 M EDTA, 1 μl 2 M KCN and 10 μl 5 mg/ml mitochondria were added to a 1 ml cuvette. Subsequently, 30 μl 1 mM cytochrome c was added and the cuvette and samples were read for 1 min at 550 nm. For complex IV analysis, reduced cytochrome c was prepared by adding 20 μl 0.1 M DTT in 1 ml of 1 mM cytochrome c, and incubating for 15 min at room temperature after which A550/A565 ratio was determined. To determine the complex IV activity: 850 μl dH2O, 100 μl 100 mM potassium phosphate buffer (pH 7.4), 50 μl 1 mM reduced cytochrome c and 2 μl 5 mg/ml mitochondria were pipetted into a 1 ml cuvette and samples were read for 1 min at 550 nm. For the citrate synthase analysis: 740 μl dH2O, 100 μl 1 M Tris–HCl (pH 8.0), 50 μl 6 mM Acetyl–CoA, 10 μl 10 mM DTNB and 50 μl 1 mg/ml mitochondria were added into a 1 ml cuvette and incubated at 30 °C for 5 min. Thereafter, 50 μl 10 mM oxaloacetate was added and samples were read for 1 min at 412 nm. Data was acquired with a Spectra Max plus (Molecular Devices, Sunny-vale, CA) and SoftMax Pro software, according to the manufacturer’s protocol.
2.5. Seahorse XF-24 metabolic flux analysis
For the Seahorse in vitro mitochondrial respiration studies, 25,000 human fibroblast cells/well were grown in 24-well plates (Seahorse Bioscience, Billerica, MA) with 5% CO2 in a 37 °C incubator in DMEM supplemented with 10% fetal bovine serum (FBS). The assay was performed over time-resolved measurements of a single population of cells over a period of hours as previously described (Ledbetter et al., 1981). Mitochondrial function parameters were evaluated by basal respiration, ATP turnover rate, proton leak and maximal and spare respiratory capacity measurements.
The cellular bioenergetic profiles were measured by serial injections of oligomycin (1 μM final concentration, which blocks ATP synthase to assess respiration required for ATP turnover), FCCP (carbonyl cyanide 4-trifluoromethoxy-phenylhydrazone, 1 μM final concentration, a proton ionophore which induces chemical uncoupling and maximal respiration), and rotenone plus antimycin A (1 μM final concentration of each, which completely inhibits electron transport to measure non-mitochondrial respiration). The data were normalized to the total protein in each well. The OCR and ECAR values were determined from 3 wells per sample and experiments were replicated.
2.6. Bioenergetics: Mitochondrial membrane potential
Cultured patient myoblasts were plated in 35 mm dishes with ConA-laminin coated cover glasses (MatTek Corp, Ashland, MA) and were rinsed with a HEPES-buffered salt solution (HBSS) containing 120 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 15 mM glucose, 20 mM HEPES, 10 mM NaOH, and 0.001% phenol red, pH 7.2–7.4. Cells were incubated with the mitochondrial membrane potential (ΔΨm) sensitive probe rhodamine 123 for 30 min in the dark at room temperature. Fluorescent illumination was provided by a 150-W xenon arc lamp, and band-specific filters were used for excitation (ex: 490 nm, diachronic: 580 nm, em: 530 nm). In order to produce maximal loss of ΔΨm, the cultures were first incubated with rhodamine 123 for 30 min and thereafter challenged with 5 μM FCCP. Images were acquired with a 12-bit digital CCD camera (Photometrics, Tucson, AZ). Pseudocolor representations were generated digitally using Metafluor 4.0 imaging software (Universal Imaging, West Chester, PA). Data were collected at 5–10 s intervals. Total cells analyzed included: 30 control A cells, 31 R155H cells, 64 control B cells, and 47 R155S cells.
To determine mitochondrial membrane potential by immunohistochemistry, cultured live human myoblasts were stained with JC-1 antibody (Life Technologies, Carlsbad, CA). Briefly, cells were seeded onto chamber slides and stained with the mitochondrial membrane potential probe JC-1 (a cationic carbocyanine mitochondrial dye) for 45 min at 37 °C. Cells were then washed with warm DMEM and mounted with DAPI hard set mounting medium (Vector Labs, Burlingame, CA). Slides were analyzed by fluorescence microscopy (Carl Zeiss Microscope).
2.7. Determination of cellular energy state ATP levels
Levels of ATP were determined in VCP patient myoblasts and fibroblasts and mouse myoblasts versus controls as per manufacturer’s instructions (Life Technologies, Carlsbad, CA). Briefly, standard reaction solutions were prepared (100 ul) for each sample reaction (emission maximum ~560 nm at pH 7.8). A standard curve was generated for a series of ATP concentrations and used in calculating the amount of ATP in each experimental sample.
2.8. Statistical analyses
Means were used as summary statistics for all experiments. We compared the above experiments – including immunocytochemical, Western blot and in vitro studies – in patient fibroblasts, myoblasts, and WT, VCPR155H/+, and VCPR155H/R155H mice myoblasts with mixed model analysis of variance and pair-wise t-tests. For Seahorse XF-24 metabolic and ATP content analyses, results are expressed as means ± SEM and significance was determined using two-tailed Student’s t-test or two-way ANOVA with Bonferroni post-test. A probability of p < 0.05 was considered to be significant.
3. Results
3.1. VCP human fibroblasts illustrate increased pathology and decreased ATP
Upon examination of the VCPR155H/+ and VCPR155H/R155H mouse fibroblasts, we subsequently explored patient VCP fibroblasts to understand the pathology of VCP disease in vitro. The autophagy pathway is dysregulated in VCP disease, hence, we performed immunocytochemistry with LC3-I/II (Fig. 1A), p62/SQSTM1 (Fig. 1B), ubiquitin (Fig. 1C), and TDP-43-specific (Fig. 1D) antibodies to evaluate any protein expression differences in VCP patient fibroblasts. We discovered that the following markers were increased: LC3, p62/SQSTM1, and ubiquitin in VCP patient fibroblasts compared to age- and disease-matched control fibroblasts (Fig. 1A–C). Interestingly, we also observed translocation of the TDP-43 into the cytoplasmic of patient fibroblasts, this being considered the hallmark of pathology in VCP and related neurodegenerative disorders (Fig. 1D). Additionally, we stained the patient VCP fibroblasts with JC-1, a mitochondrial membrane marker, and found a decreased membrane potential (Fig. 1E). Western blot for protein determination illustrated increased expression levels for LC3-I/II, p62/SQSTM1, ubiquitin and TDP-43 in VCP patient versus control fibroblasts (Fig. 1F). Interestingly, we detected significantly decreased ATP levels in our VCP patient versus control fibroblasts (Fig. 1G).
Fig. 1.
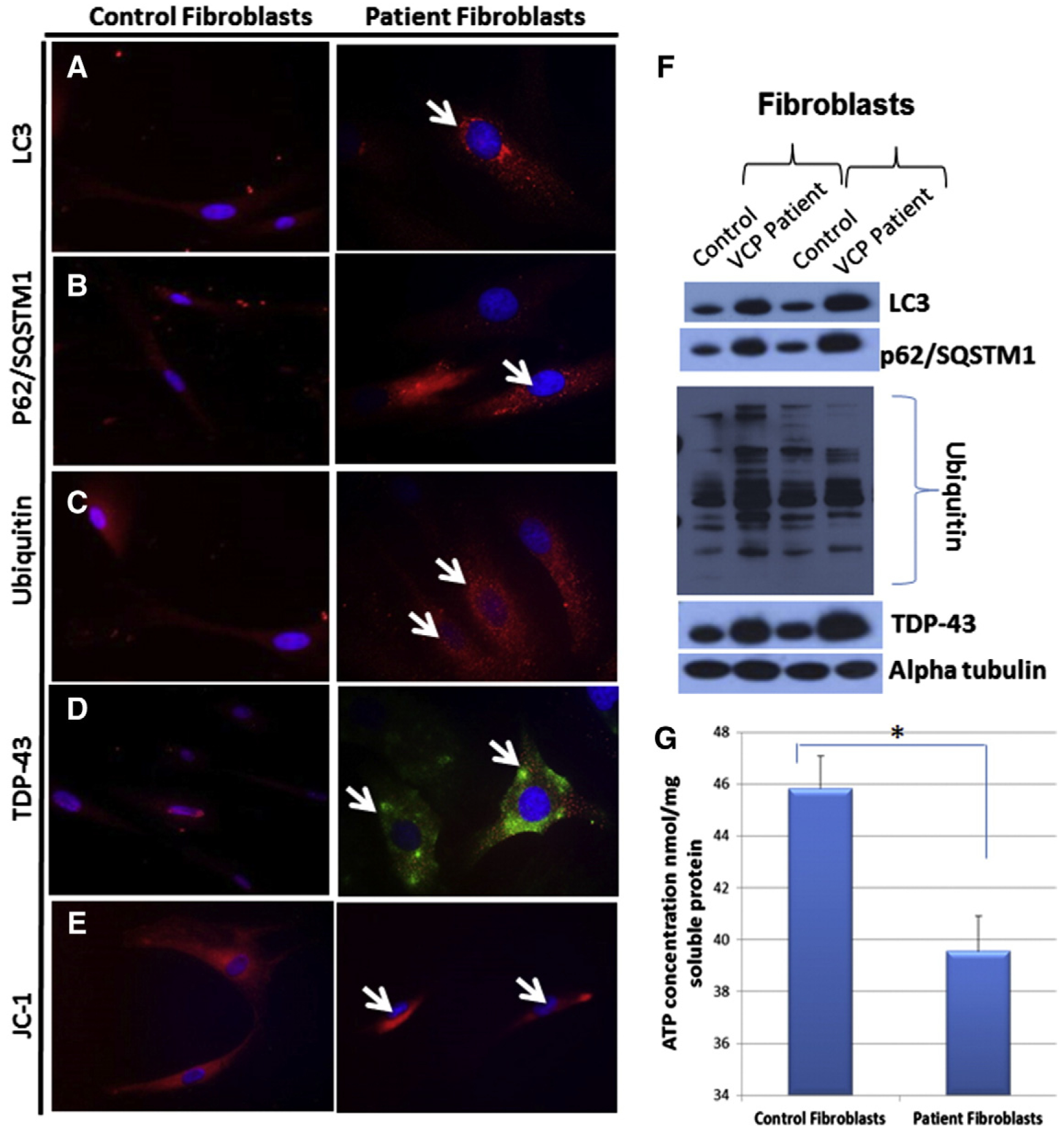
Immunocytochemical analyses of autophagy markers in human fibroblasts affected with VCP disease and controls. Immunocytochemical staining of control and patient VCP fibroblasts with (A) LC3-I/II, (B) p62/SQSTM1, (C) Ubiquitin and (D) TDP-43 showing increased staining suggestive of disrupted autophagy pathways. (E) Staining of human fibroblasts with JC-1, a mitochondrial membrane potential marker. (Magnification 630×) White arrows pointing to increased protein expression levels of LC3, p62/SQSTM1, ubiquitin and TDP-43 and decreased expression levels of JC-1 in patient fibroblasts. (F) Western blot protein expression analysis of LC3, p62/SQSTM1, ubiquitin and TDP-43 antibodies in patient fibroblasts. Alpha tubulin was used as a loading control. (G) Levels of ATP (nmol/mg soluble protein) were determined in the control and patient fibroblasts. Statistical significance is denoted by *p ≤ 0.05.
3.2. VCP human fibroblasts show decreased spare respiratory capacity
Recent data indicate that mitochondrial dysfunction is an initial event underlying the development and progression of several neurodegenerative disorders. Seahorse technology has been used to assess mitochondrial dysfunction in several diseases (Ho et al., 2012; Jarrett et al., 2013) and was performed as previously described (Ledbetter et al., 1981). The rate of oxygen consumption (OCR), an indicator of mitochondrial respiration and the extracellular acidification rate (ECAR), a measure of lactic acid formed during glycolytic energy metabolism, were both measured with a Seahorse XF-24 flux analyzer (Fig. 2A). The OCR and ECAR measurements in cells are related to the flux through catabolic pathways utilized in ATP synthesis rates that occur in response to changes in ATP consumption. Our bioenergetic data in VCP human fibroblasts indicates decreased spare respiratory capacity (Fig. 2B,D), a measurement of additional ATP that can be produced by oxidative phosphorylation in stressful conditions; and increased ECAR levels (Fig. 2C), which represent glycolysis, and elevated proton leak (Fig. 2E) compared with age- and sex-matched unaffected first degree relatives. These results suggest that mutations in VCP affect the mitochondria’s ability to produce ATP, thereby increasing the cells’ glycolysis levels.
Fig. 2.

Seahorse XF-24 metabolic flux analyses in fibroblasts from patients affected with VCP disease and controls. (A) Seahorse Bioscience analysis of cellular bioenergetics assay approach (measurements of glycolysis and mitochondrial function). The Seahorse Bioscience analysis comprised of measuring (B) % oxygen consumption rate (OCR), (C) % extracellular acidification rate (ECAR), (D) spare respiratory capacity and (E) proton leak in controls 1 and 2 and VCP patient fibroblast samples 1–4.
3.3. Mitochondrial membrane potential is unchanged, but mitochondrial enzyme proteins and activities are upregulated in patient VCP myoblasts
Defective mitochondrial functions are commonly observed in several disorders including human muscle diseases. First, we stained our VCP patient myoblast (421/07) lines with autophagy markers to assess pathology and observed co-localization with ubiquitin and p62/SQSTM1 (Fig. 3A), VCP and LC3-I/II (Fig. 3B), and VCP and TDP-43 (Fig. 3C). Additionally, we stained the patient VCP myoblasts with JC-1, a mitochondrial membrane marker, and found a decreased membrane potential (Fig. 3D). Protein expressions for LC3, p62/SQSTM1, TDP-43, were examined and showed increased levels in VCP patient versus control myoblasts (Fig. 3E). Interestingly, the protein expression levels of complexes I–V were also significantly increased in the VCP patient versus control myoblasts (Fig. 3E).
Fig. 3.

Measurements of mitochondrial membrane potential, enzyme complex activities, and ATP levels in VCP patient myoblasts. Immunocytochemical staining of control myoblasts versus patient myoblasts (421/07) stained with (A) ubiquitin and p62/SQSTM1, (B) VCP and LC3, (C) VCP and TDP-43 (magnification 400×), and (D) JC-1 (mitochondrial membrane potential marker) (magnification 630×). White arrows pointing to increased protein expression levels of ubiquitin and p62/SQSTM1, VCP and LC3, VCP and TDP-43, and decreased expression levels of JC-1 in patient myoblasts. (E) Western blot protein expression analyses of LC3, p62/SQSTM1, TDP-43, and complexes I–V in VCP patient and control myoblasts. Alpha tubulin was used as a loading control. (F) Complex I activity levels in VCP patient and control myoblasts. (G) Comparison of mitochondrial ΔΨm in the control and patient myoblasts after the maximal loss of the mitochondrial ΔΨm produced by FCCP. The peak/basal (Fx/Fo) ratio of rhodamine 123 fluorescence was measured at baseline (before adding FCCP) and peak (upon addition of FCCP). Results are presented as relative values compared to the values of control A. Data are normalized by the citrate synthase activity. (H) Levels of ATP (nmol/mg soluble protein) were determined in the control and VCP patient myoblasts. Statistical significance is denoted by *p ≤ 0.05.
Next, we evaluated whether mitochondrial functions were affected in cultured VCP patient myoblasts by determining the mitochondrial membrane potential (ΔΨm) using rhodamine 123. This positively charged probe partitions across membranes as a function of potential differences. To induce rapid mitochondrial depolarization, we used carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP). When the cultured myoblasts were incubated with rhodamine 123 and exposed to 5 μM FCCP, an abrupt increase in cytoplasmic fluorescence was observed in both control and mutant cell lines indicating the abruption and full loss of ΔΨm. To analyze if there is a difference between VCP and control cells in responding to FCCP-induced mitochondrial depolarization, we measured the peak ratios (after adding FCCP) and baseline (before adding FCCP) of rhodamine 123 fluorescence. Our results obtained from these analyses showed no significant difference in the mitochondrial ΔΨm between two VCP patients and two control cell lines studied (Fig. 3G).
Although the mitochondrial ΔΨm was not significantly different between patient and control myoblasts, we could not rule out possible differences in the enzyme activities of mitochondrial complexes. For this purpose, we measured the activities of complexes II + III and IV. They were readily measured by following the oxidation of reduced cytochrome c at 550 minus 540 nm (extinction coefficient 19.0 mM−1 cm−1). Specific rates can be calculated either from the initial quasi-liner phase of the reaction or by estimation of first-order rate constants. The assay of citrate synthase follows the reduction of 5,5′-dithiobis (2-nitrobenzoic acid, DTNB) at 412 minus 360 nm (extinction coefficient 13.6 mM−1 cm−1), coupled to the reduction of CoA by the citrate synthase reaction in the presence of oxaloacetate. The complex activities were normalized with the activities of the citrate synthase. Interestingly, the complex II + III activity was 4.4 times higher and complex IV 1.7 times higher in the patient cell line when compared to the values of the control cell line (Fig. 3G). There were no significant differences observed in complex I activity levels between the controls versus VCP patient myoblasts (Fig. 3F).
3.4. ATP levels are decreased in patient VCP myoblasts
To further analyze the basal cellular ATP levels, determined by the rates of ATP production (oxidative phosphorylation and glycolysis) and consumption, we monitored ATP levels in our VCP patient versus control myoblasts (Fig. 3H). ATP levels were significantly lower in VCP patient versus control myoblasts, possibly suggesting that VCP deficient cells generate less ATP.
3.5. Characterization of VCP mouse myoblast shows pathological phenotype
Since the autophagy pathway is dysregulated in VCP disease, we performed immunocytochemistry on VCP myoblasts using autophagy markers including LC3-I/II and p62/SQSTM1 to evaluate any protein expression differences (Fig. 4B,C). In comparison to the age-matched controls, we observed increased levels of autophagy markers LC3-I/II and p62/SQSTM1 in the VCPR155H/+ and VCPR155H/R155H animals as compared to WT littermates. Interestingly, TDP-43, a transcriptional factor used in assessing pathology when translocated to the cytoplasm was not found to be translocated in the 3-week old VCPR155H/+ and VCPR155H/R155H fibroblasts (Fig. 4D). We detected decreased levels of JC-1 mitochondrial membrane potential in our VCP heterozygote and homozygotes myoblasts as compared to the Wild Type myoblasts (Fig. 4E). Protein expressions for LC3, p62/SQSTM1, TDP-43 were examined and demonstrated increased levels in VCP mouse (heterozygote and homozygote) versus Wild Type myoblasts (Fig. 4F). Interestingly, the protein expression levels of complexes I-–V were also significantly increased in the VCP heterozygote and homozygote myoblasts as compared to Wild Type (Fig. 4F). Lastly, we discovered decreased levels of ATP in the VCP mouse myoblasts (both heterozygous and homozygous) versus Wild Type myoblasts (Fig. 4G).
Fig. 4.

DIC images and immunocytochemical analyses of autophagy markers in Wild Type, VCPR155H/+, and VCPR155H/R155H myoblasts. (A) DIC images of myoblasts from WT, VCPR155H/+ and VCPR155H/R155H mice. Immunocytochemical staining of WT, VCPR155H/+ and VCPR155H/R155H fibroblasts with (B) LC3-I/II, (C) p62/SQSTM1, (D) TDP-43, and (E) JC-1 mitochondrial membrane potential marker (magnification 630×). White arrows pointing to increased protein expression levels of LC3-I/II, p62/SQSTM1, ubiquitin and TDP-43, and decreased expression levels of JC-1 in mouse myoblasts. (F) Western blot expression analyses of mouse myoblasts with LC3, p62/SQSTM1, ubiquitin and TDP-43 antibodies. Alpha tubulin was used as a loading control. (G) Levels of ATP (nmol/mg soluble protein) were determined in the control and VCP mouse myoblasts.
4. Discussion
VCP multisystem proteinopathy (MSP) is caused by mutations in the VCP gene and results in a variety of clinical symptoms including primarily proximal myopathy, Paget disease of the bone, and Frontotemporal Dementia (FTD) (Watts et al., 2004). To date, several disease mutations have been reported, all of which are missense mutations causing an amino acid change in the N-terminal part of the polypeptide. The high evolutionary conservation suggests an important role for the VCP protein in the normal cellular functions. This is further supported by the finding that homozygous knock-out mice lacking both VCP alleles die in early embryogenesis (Muller et al., 2007). Also the transgenic mice overexpressing the R155H mutation show muscle weakness and muscle tissue pathology typical of patients with VCP disease (Weihl et al., 2007).
We have generated a novel neomycin cassette-free heterozygous knock-in (KI) mouse model expressing the common disease-related R155H VCP mutation (VCPR155H/+). This mouse model recapitulates the human VCP-associated myopathy including progressive muscle, bone, spinal cord and brain pathologies (Nalbandian et al., 2013; Yin et al., 2012), manifesting increased TDP-43, ubiquitin, autophagy and mitochondrial pathologies. The R155H (VCPR155H/R155H) mouse model expressing two mutant alleles exhibits progressive weakness and accelerated pathology in skeletal muscle, spinal cord, brain, and heart prior to their early demise at 21 days of age. These mice are a useful model providing us with a high-throughput screening platform with which to test potential therapies. Within a short period of time, we can obtain valuable in vivo data about whether a treatment would be effective for VCP-associated diseases. These mice are also ideal for examining key cellular and molecular interactions as well as signaling transduction pathways underlying VCP-related diseases.
The pathological mechanisms resulting in the clinical and cellular features in the muscle tissues of human and mouse are still to be resolved. Therefore, we have started analyzing human and mouse fibroblast and myoblast cell lines expressing the mutant VCP at an endogenous level. By analyzing these cells that express the protein of interest at a physiological level we can exclude the possibility of overexpression artifacts and the influence of transfection treatments.
Impairment of the autophagy cascade for protein degradation and mitochondrial dysfunction have been implicated in VCP-associated disease (Ju et al., 2008, 2009; Kim et al., 2013; Tresse et al., 2010; Vesa et al., 2009), as well as other neurodegenerative diseases such as Parkinson’s disease, Huntington’s, Alzheimer’s and ALS (Batlevi and La Spada, 2011; Choi et al., 2013; Komatsu et al., 2006a, 2006b; Levine and Kroemer, 2008; Malicdan and Nishino, 2012; Malicdan et al., 2007; Masiero et al., 2009, 2012; Raben et al., 2007; Rabinowitz and White, 2010; Schiaffino et al., 2008; Son et al., 2012; Vesa et al., 2009; Wang et al., 2012). The dysfunction of autophagy in VCP-associated disease is a failure of the autophagosomes to fuse with the lysosome, thus causing autophagosome accumulation (Ju et al., 2009). VCP participates in the formation of ubiquitinated endocytic autophagosomes and this failure leads to a buildup of vacuoles in the muscle tissue of VCP-associated disease patients (Ju and Weihl, 2010; Weihl et al., 2006). In our study, we detected increased levels of TDP-43, ubiquitin, LC3, and p62/SQSTM1, indicative of defective autophagy. Mitophagy is a form of autophagy that selectively targets damaged mitochondria causing mitochondria dysfunction, damage and production of toxic reactive oxygen species (ROS) (Sentelle et al., 2012; Wallace, 2005). We have previously shown that the VCPR155H/R155H homozygote mouse muscle have mitochondrial proliferation and architecture with megaconia and disrupted cristae and mitochondrial respiratory chain dysfunction (Nalbandian et al., 2012). We are currently investigating this process in VCP disease to assess its biophysical and functional role.
Mitochondrial membrane potential is an indicator of cellular mitochondrial health and function. Our findings showed no significant difference in the mitochondrial ΔΨm between two VCP patients and two control cell lines studied, however, showed a decrease in JC-1 immunohistochemical staining of patient fibroblasts and myoblasts. The mitochondrial respiratory chain (MRC) is composed of five protein complexes (I–V) and two small electron carriers ubiquinol and cytochrome C. Complexes I, III and IV produce an electrochemical proton gradient across the mitochondrial inner membrane (MIM) driving the ATP synthesis by complex V. Interestingly, in our study, we found increased complexes I–V protein expression levels, possibly suggestive of a block in the mitochondrial respiratory chain. Remarkably, this corroborates with the low levels of ATP production in the VCP patient myoblasts and fibroblasts versus controls. Similar results were also observed in the VCP heterozygote and homozygote myoblasts versus Wild Type controls. These results suggest that mutations in VCP affect the mitochondria’s ability to produce ATP, thereby increasing the cells’ glycolysis levels. Our findings are in accordance with a previously reported study by Bartolome et al. (2013) illustrating low membrane potential and ATP levels in VCP deficient cells (Bartolome et al., 2013).
This paper investigates dysregulated mitochondrial bioenergetics and proposes a mechanism by which pathogenic VCP mutations lead to VCP disease. These studies may provide an understanding of the physiopathology and the promise of new pharmacologic avenues for interventions and treatments for VCP and related neurodegenerative diseases. Future physiological and biochemical experiments will be necessary to examine the crosstalk between mitochondrial dysfunction and autophagosome formation, autophagy and mitophagy pathways in VCP disease.
5. Conclusions
Despite intense investigations, the discovery of effective therapies and the disease mechanisms underlying VCP-associated myopathies and neurodegenerative disorders remains elusive. To understand the pathological mechanisms underlying the myopathy of VCP disease, we studied cellular consequences of VCP mutations in patient fibroblast and mouse primary myoblast cell lines and found changes in their mitochondrial dynamics and bioenergetics. These findings highlight pathophysiological events that may occur in VCP disease and may ultimately pave the way towards promising therapeutics in VCP and related multisystem proteinopathies.
Acknowledgments
We thank the families and their physicians as well as our many collaborators for their contributions to this work. Human fibroblasts and myoblast cultures were obtained from patients under IRB Protocol# 2007-5832. Myoblast cultures were also obtained from the Muscle Tissue Culture Collection, a partner of Eurobiobank (www.eurobiobank.org) at the Friedrich-Baur-Institute (Department of Neurology, Ludwig-Maximilians-University, Munich, Germany) (MD-NET, service structure S1, 01GM0601) funded by the German ministry of education and research (BMBF, Bonn, Germany).
Funding
This work was supported by National Institutes of Health (NIH) grants R01, R21 and Muscular Dystrophy Association (MDA) 175682 to VEK. Clinical evaluations were performed in the GCRC at Boston Children’s Hospital (Boston, MA), and at the ICTS at University of California, Irvine (Irvine, CA) and University of Kentucky (Lexington, KY).
Abbreviations:
- MSP
multisystem proteinopathy
- VCP
valosin containing protein
- IBMPFD
inclusion body myopathy associated with Paget’s disease of the bone and frontotemporal dementia
- ALS
amyotrophic lateral sclerosis
- ETC
mitochondrial electron transport chain
- OCR
oxygen consumption rate
- ECAR
extracellular acidification rate
- ATP
adenosine triphosphate
References
- Badadani M, Nalbandian A, Watts GD, Vesa J, Kitazawa M, Su H, Tanaja J, Dec E, Wallace DC, Mukherjee J, Caiozzo V, Warman M, Kimonis VE, 2010. VCP associated inclusion body myopathy and Paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS ONE 5 (pii: e13183). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome F, Wu HC, Burchell VS, Preza E, Wray S, Mahoney CJ, Fox NC, Calvo A, Canosa A, Moglia C, Mandrioli J, Chio A, Orrell RW, Houlden H, Hardy J, Abramov AY, Plun-Favreau H, 2013. Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlevi Y, La Spada AR, 2011. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol. Dis 43, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B, 2013. Autophagy in human health and disease. N. Engl. J. Med 368, 651–662. [DOI] [PubMed] [Google Scholar]
- Ho J, de Moura MB, Lin Y, Vincent G, Thorne S, Duncan LM, Hui-Min L, Kirkwood JM, Becker D, Van Houten B, Moschos SJ, 2012. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol. Cancer 11, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Rohrer B, Perron NR, Beeson C, Boulton ME, 2013. Assessment of mitochondrial damage in retinal cells and tissues using quantitative polymerase chain reaction for mitochondrial DNA damage and extracellular flux assay for mitochondrial respiration activity. Methods Mol. Biol 935, 227–243. [DOI] [PubMed] [Google Scholar]
- Ju JS, Weihl CC, 2010. p97/VCP at the intersection of the autophagy and the ubiquitin proteasome system. Autophagy 6, 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Miller SE, Hanson PI, Weihl CC, 2008. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J. Biol. Chem 283, 30289–30299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, Weihl CC, 2009. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol 187, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP, 2013. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimonis VE, Kovach MJ, Waggoner B, Leal S, Salam A, Rimer L, Davis K, Khardori R, Gelber D, 2000. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet. Med 2, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimonis VE, Fulchiero E, Vesa J, Watts G, 2008a. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: Review of a unique disorder. Biochim. Biophys. Acta 12, 744–748. [DOI] [PubMed] [Google Scholar]
- Kimonis VE, Mehta SG, Fulchiero EC, Thomasova D, Pasquali M, Boycott K, Neilan EG, Kartashov A, Forman MS, Tucker S, Kimonis K, Mumm S, Whyte MP, Smith CD, Watts GD, 2008b. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am. J. Med. Genet 146, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC, 2004. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kominami E, Tanaka K, 2006a. Autophagy and neurodegeneration. Autophagy 2, 315–317. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K, 2006b. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. [DOI] [PubMed] [Google Scholar]
- Kovach MJ, Ruiz J, Kimonis K, Mueed S, Sinha S, Higgins C, Elble S, Elble R, Kimonis VE, 2001. Genetic heterogeneity in autosomal dominant essential tremor. Genet. Med 3, 197–199. [DOI] [PubMed] [Google Scholar]
- Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD, 1981. Deletions of chromosome 15 as a cause of the Prader–Willi syndrome. N. Engl. J. Med 304, 325–329. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G, 2008. Autophagy in the pathogenesis of disease. Cell 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicdan MC, Nishino I, 2012. Autophagy in lysosomal myopathies. Brain Pathol. 22, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicdan MC, Noguchi S, Nishino I, 2007. Autophagy in a mouse model of distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Autophagy 3, 396–398. [DOI] [PubMed] [Google Scholar]
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M, 2009. Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515. [DOI] [PubMed] [Google Scholar]
- Moresi V, Carrer M, Grueter CE, Rifki OF, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN, 2012. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl. Acad. Sci. U. S. A 109, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JM, Deinhardt K, Rosewell I, Warren G, Shima DT, 2007. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem. Biophys. Res. Commun 354, 459–465. [DOI] [PubMed] [Google Scholar]
- Nalbandian A, Donkervoort S, Dec E, Badadani M, Katheria V, Rana P, Nguyen C, Mukherjee J, Caiozzo V, Martin B, Watts GD, Vesa J, Smith C, Kimonis VE, 2011. The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J. Mol. Neurosci 45, 522–531. [DOI] [PubMed] [Google Scholar]
- Nalbandian A, Llewellyn KJ, Kitazawa M, Yin HZ, Badadani M, Khanlou N, Edwards R, Nguyen C, Mukherjee J, Mozaffar T, Watts G, Weiss J, Kimonis VE, 2012. The homozygote VCP(R(1)(5)(5)H/R(1)(5)(5)H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS ONE 7, e46308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian A, Llewellyn KJ, Badadani M, Yin HZ, Nguyen C, Katheria V, Watts G, Mukherjee J, Vesa J, Caiozzo V, Mozaffar T, Weiss JH, Kimonis VE, 2013. A progressive translational mouse model of human valosin-containing protein disease: the VCP(R155H/+) mouse. Muscle Nerve 47, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Roberts A, Plotz PH, 2007. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 26, 45–48. [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E, 2010. Autophagy and metabolism. Science (New York, N.Y.) 330, 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S, Mammucari C, Sandri M, 2008. The role of autophagy in neonatal tissues: just a response to amino acid starvation? Autophagy 4, 727–730. [DOI] [PubMed] [Google Scholar]
- Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, Bielawski J, Ogretmen B, 2012. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol 8, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JH, Shim JH, Kim KH, Ha JY, Han JY, 2012. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med 44, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP, 2010. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, Wallace DC, 1996. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 264, 484–509. [DOI] [PubMed] [Google Scholar]
- Vesa J, Su H, Watts GD, Krause S, Walter MC, Martin B, Smith C, Wallace DC, Kimonis VE, 2009. Valosin containing protein associated inclusion body myopathy: abnormal vacuolization, autophagy and cell fusion in myoblasts. Neuromuscul. Disord 19, 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CK, 2012. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. U. S. A 109, 15024–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GD, Thorne M, Kovach MJ, Pestronk A, Kimonis VE, 2003. Clinical and genetic heterogeneity in chromosome 9p associated hereditary inclusion body myopathy: exclusion of GNE and three other candidate genes. Neuromuscul. Disord 13, 559–567. [DOI] [PubMed] [Google Scholar]
- Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE, 2004. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet 36, 377–381. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Dalal S, Pestronk A, Hanson PI, 2006. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet 15, 189–199. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Miller SE, Hanson PI, Pestronk A, 2007. Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum. Mol. Genet 16, 919–928. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Nalbandian A, Hsu CI, Li S, Llewellyn KJ, Mozaffar T, Kimonis VE, Weiss JH, 2012. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 3, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]