

Abstract
There are a number of antivirals as well as antiviral strategies that could be envisaged to prevent or treat severe acute respiratory syndrome (SARS) (or similar) coronavirus (CoV) infections. Targets for the prophylactic or therapeutic interventions include interaction of the spike (S) glycoprotein (S1 domain) with the host cell receptor, fusion of the S2 domain with the host cell membrane, processing of the replicase polyproteins by the virus-encoded proteases (3C-like cysteine protease [3CLpro] and papain-like cysteine protease) and other virus-encoded enzymes such as the NTPase/helicase and RNA-dependent RNA polymerase. Human monoclonal antibody blocking S1 may play an important role in the immunoprophylaxis of SARS. Fusion inhibitors reminiscent of enfuvirtide in the case of HIV may also be developed for SARS-CoV. Various peptidomimetic and nonpeptidic inhibitors of 3CLpro have been described, the best ones inhibiting SARS-CoV replication with a selectivity index greater than 1000. Human interferons, in particular α- and β-interferon, as well as short interfering RNAs could further be pursued for the control of SARS. Various other compounds, often with an ill-defined mode of action but selectivity indexes up to 100, have been reported to exhibit in vitro activity against SARS-CoV: valinomycin, glycopeptide antibiotics, plant lectins, hesperetin, glycyrrhizin, aurintricarboxylic acid, chloroquine, niclosamide, nelfinavir and calpain inhibitors.
Keywords: coronavirus, interferon, monoclonal antibody, protease inhibitors, SARS, SARS-CoV inhibitors, siRNA, S protein
Severe acute respiratory syndrome (SARS) is a new infectious disease that is mainly characterized by influenza-like symptoms, high fever, myalgia, dyspnea, lymphopenia and lung infiltrates (pneumonia) leading to acute breathing problems, with an overall mortality rate of approximately 10% (in the elderly as high as 50%). The disease appeared in the Guandong province of southern China at the end of 2002 from where it swept into 29 countries. When the epidemic finally waned after more than 100 days, the WHO had counted a cumulative number of more than 8000 probable SARS cases with almost 800 deaths. The etiological agent of SARS has been unequivocally identified as the SARS-associated coronavirus (SARS-CoV) [1–6].
SARS-CoV was, following the human CoVs 229E and OC43, the third CoV to be identified in humans. Subsequently, a fourth human CoV, NL63, was isolated from individuals suffering from respiratory illness [7], and it is likely that additional human CoVs may be uncovered in the future. This underscores the importance of the search for inhibitors of the SARS-CoV, and given the multitude of targets that could be envisaged for chemotherapeutic intervention [8], numerous approaches have already been proposed to inhibit SARS-CoV replication and spread [9,10].
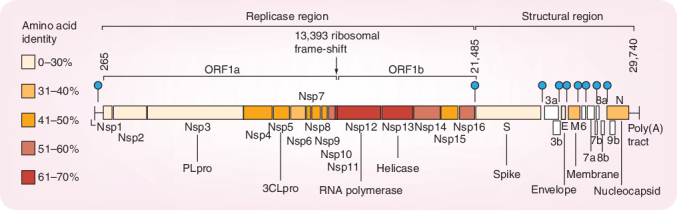
The organization of the SARS-CoV genome (29,740 bases long plus-stranded RNA) is similar to that of other CoVs (Figure 1). There are essentially two regions, the replicase region and the structural region. The replicase region encompasses two overlapping open reading frames (ORFs 1a and 1b). A translational read-through by a -1 position ribosomal frameshift allows the translation of the overlapping reading frames into a single polyprotein. Virus-encoded proteases, namely the papain-like cysteine protease (PLpro) and the picornavirus 3C-like cysteine protease (3CLpro) cleave the polyprotein into the individual polypeptides required for replication and transcription [8]. The remaining one third of the genome encodes for at least four structural proteins: spike protein (S), envelope protein (E), membrane glycoprotein (M) and nucleocapsid protein (N). Several additional genes encoding additional nonstructural proteins are known as ‘accessory genes’.
Figure 1. Genome structure of severe acute respiratory syndrome coronavirus.
Reprinted with permission from [8]. 3CLpro: 3C-like cysteine protease; Nsp: Nonstructural protein: ORF: Open reading frame; PLpro: Papain-like cysteine protease.
This review will evaluate the different gene products encoded by the SARS-CoV genome as possible points of attack for chemotherapeutic (or prophylactic) agents, thereby reviewing the various strategies that have already been proposed to curb (treat or prevent) a potential SARS-CoV infection. These approaches have to be viewed in the framework of antivirals and antiviral strategies against virus infections at large [11].
Virus entry into the host cell
The metallopeptidase angiotensin-converting enzyme 2 (ACE2) has been identified as a functional receptor for the SARS-CoV [12]. ACE2 interacts with the S1 domain of the viral S glycoprotein, and this interaction and the subsequent infection can be blocked by both a soluble form of ACE2 and their anti-ACE2 antibody [12]. ACE2 expression in cell lines correlates with their susceptibility to SARS-CoV S-driven infection, suggesting that ACE2 must be a major receptor for SARS-CoV. This, in turn, suggests that the SARS-CoV S protein may be considered as an attractive target for therapeutic intervention [13]. Interestingly, ACE2 expression positively correlated with the differentiation state of human airway epithelia; undifferentiated cells expressing little ACE2 were poorly infected with SARS-CoV, while well differentiated cells expressing more ACE2 were readily infected [14].
A 193-amino acid fragment of the S protein (corresponding to residues 318–510) binds to ACE2 more efficiently than the full S1 domain, and, in fact, the 193-residue fragment blocks S protein-mediated infection with an inhibitory concentration of 50% (IC50) of less than 10 nM (the IC50 of the full S1 domain being ∼50 nM) [15]. Also, human monoclonal antibodies to the S1 protein domain block the association of SARS-CoV with ACE2, indicating that the ACE2 binding site of S1 could be a target for drug development [16]. A small-molecular-weight inhibitor that was found to interact with the ACE2 active catalytic site, (S,S)-2-(1-carboxy-2-[3-[3,5-dichlorobenzyl]-3H-imidazol-4-yl]-ethylamino)-4-methylpentanoic acid (MLN-4760) has been described [17]. Whether MLN-4760 inhibits SARS-CoV infection has not, as yet, been demonstrated.
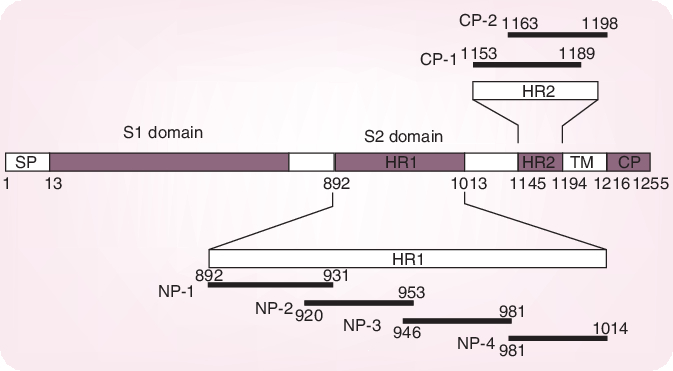
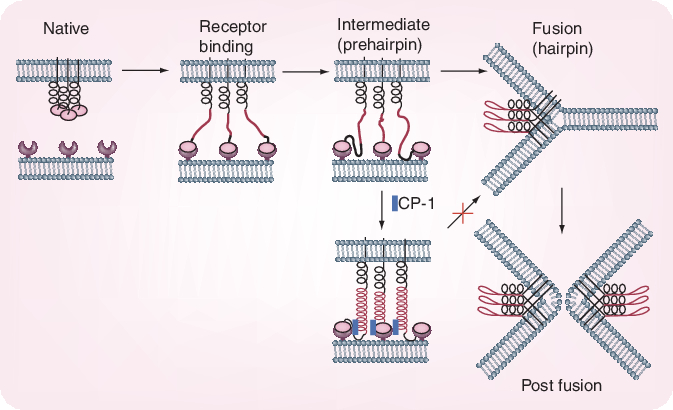
Whereas the S1 domain of the S glycoprotein determines virus attachment to the host cells, the subsequent virus–cell fusion process is governed by conformational changes of the two heptad regions (HRs)-N (or HR1) and HR-C (or HR2) within the S2 domain, resulting in the formation of a 6-helix bundle (trimer of dimers). Systemic peptide mapping has shown that the site of interaction between the HR1 and HR2 regions is between residues 916 and 950 of HR1 and residues 1151–1185 of HR2 [18]. It has also been shown that a peptide, CP-1, derived from the HR2 region, inhibits SARS-CoV infection in the micromolar range: CP-1 bound with high affinity to a peptide from the HR1 region, NP-1 (Figure 2)[19]. CP-1 could bind to the HR1 region, thereby interfering with the conformational changes leading to the 6-helix bundle formation (Figure 3) and the therewith associated virus–cell fusion process. Obviously, the HR1–HR2 interaction may be viewed as an attractive target for the design of potent peptide-type SARS-CoV entry inhibitors [20,21], reminiscent of the HIV-1 gp41 HR2-derived peptide T20 (enfuvirtide) which has been developed as a HIV-1 fusion inhibitor [22]. The latter could inhibit the fusion of SARS-CoV with target cells, but apparently with too low efficiency to be therapeutically meaningful.
Figure 2. Severe acute respiratory syndrome coronavirus (SARS-CoV) spike protein.
Residue numbers of each region correspond to their positions in the spike protein of SARS-CoV. Six peptides corresponding to the sequences of HR1 and HR2 regions are also shown. Reprinted with permission from [19]. CP: Cytoplasmic domain; HR: Heptad region; SP: Signal peptide; TM: Transmembrane domain.
Figure 3. Conformational changes of severe acute respiratory syndrome coronavirus spike protein during the process of fusion between the virus and target cell membranes.
Reprinted with permission from [19].
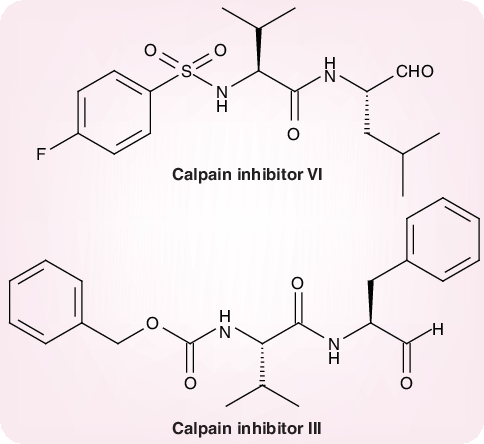
Recently, Simmons and colleagues suggested that following receptor binding and induced conformational changes in the S glycoprotein, a third step would be involved in the viral entry process, namely cathepsin-L proteolysis within endosomes [23]. They demonstrated that a cathepsin-L-specific inhibitor, MDL 28170 (also known as calpain inhibitor III or Z-Val–Phe[CHO]), at the same time inhibited cathepsin-L activity and S protein-mediated infection (at an IC50 of 2.5 nM and 0.1 µM, respectively). In addition to calpain inhibitor III, some other calpain inhibitors have been described as inhibitors of SARS-CoV replication, the most selective (selectivity index >100) being calpain inhibitor VI (4-fluorophenylsulfonyl Val–Leu[CHO]) (Figure 4)[24].
Figure 4. Calpain inhibitors.
Calpain inhibitor VI: 4-flurophenylsulfonyl-Val-Leu(CHO). Calpain inhibitor III: Z-Val-Phe(CHO).
Virus-encoded proteases
Following entry of SARS-CoV into the host cell, the genomic plus-stranded RNA is translated to produce two large overlapping replicase polyproteins, which are further processed to functional polypeptides through extensive proteolytic cleavage, mainly by the 3CLpro. The SARS 3C-like protease, also called main protease (Mpro) cleaves the replicase polyproteins at as many as 11 conserved sites, so as to generate the functional proteins necessary for virus replication. Mpro was quickly recognized as an attractive target for the development of anti-SARS-CoV agents [25]. It was proposed that compounds such as AG-7088, which had proven to be active against the rhinovirus 3C protease, could be modified in order to make them active against CoVs such as SARS-CoV [25]. As a first modification of AG-7088, the methylene group of the p-fluorophenylalanine residue was removed, and the resulting KZ7088 was modeled into the structure of the SARS-CoV Mpro [26].
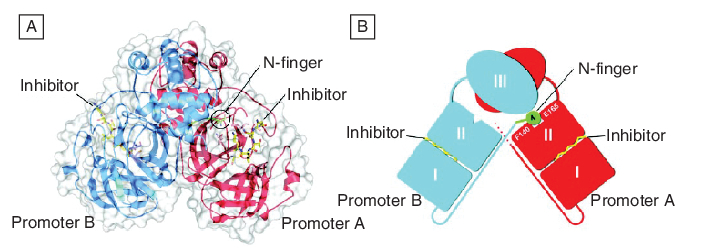
The crystal structures of the SARS-CoV Mpro complexed with various substrate analogs, such as a hexapeptidyl chloromethyl ketone (Figure 5)[27] or aza-peptide epoxide [28] have been determined, and the recombinant SARS-CoV Mpro has been successfully cloned and expressed [29]. The information thus gathered on the mode of inhibitor binding and enzyme catalysis should help to provide a structural basis for rational drug design.
Figure 5. The severe acute respiratory syndrome coronavirus (SARS-CoV) main protease (Mpro) dimer structure complexed with a substrate–analog hexapeptidyl chloromethyl ketone inhibitor.
(A) The SARS-CoV Mpro dimer structure is presented as ribbons, and inhibitor molecules are shown as ball-and-stick models. Promoter A (the catalytically competent enzyme) is red, promoter B (the inactive enzyme) is blue and the inhibitor molecules are yellow. The N-finger residues of promoter B are green. The molecular surface of the dimer is superimposed. (B) A cartoon diagram illustrating the important role of the N-finger in both dimerization and maintenance of the active form of the enzyme. Reprinted with permission from [27].
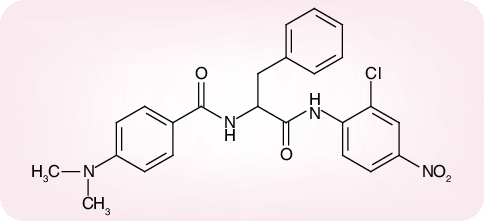
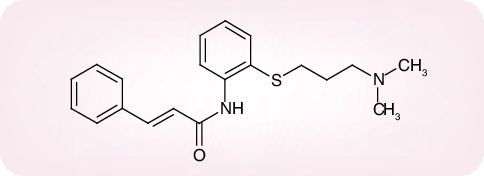
Of a number of peptidomimetic compounds (aziridinyl peptides [30], keto-glutamine analogs [31], chymotrypsin-like protease inhibitors [32] and peptide anilides [33]) that have been reported as inhibitors of the SARS-CoV Mpro, the niclosamide anilide (Figure 6), with a Ki = 0.03 µM (IC50 = 0.06 µM), proved to the most potent (competitive) inhibitor [33]. Also, several nonpeptidic compounds have been described as inhibitors of the SARS-CoV Mpro: that is, etacrynic acid derivatives such as etacrynic acid amide (Ki = 35.3 µM) [34], isatin derivatives (IC50 values ranging from 0.95 to 17.50 µM) [35], hexachlorophene derivatives (IC50 values ranging from 7.6 to 84.5 µM) [36] and natural products from teas such as theaflavin-3,3´-digallate (IC50 = 9.5 µM) [37]. However, in none of these cases [30–37] was it ascertained whether the compounds were also effective in inhibiting SARS-CoV infection in cell culture, except for two of the chymotrypsin-like protease inhibitors [32] which were found to inhibit SARS-CoV in cell culture at a relatively high concentration (45 and 70 µM, respectively) [32].
Figure 6. Niclosamide anilide (JMF 1507).
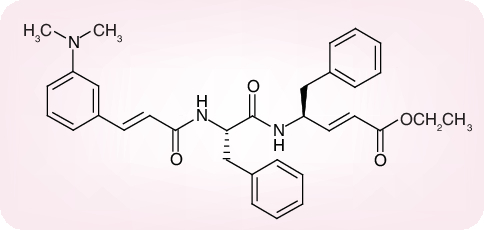
There are only a few cases where the 3CL protease inhibitors were shown to inhibit both the SARS-CoV protease activity and virus replication in cell culture. The Phe–Phe dipeptide inhibitor shown in Figure 7 was found to inhibit the 3CL protease at an IC50 of 1 µM (Ki = 0.52) and inhibited virus replication in Vero cells at an effective concentration of 50% (EC50) of 0.18 µM, while not being toxic to the host cells at a concentration of 200 µM (selectivity index: >1000) [38]. Cinanserin (SQ 10,643, a well characterized serotonin antagonist) Figure 8 is another example of an inhibitor of SARS-CoV replication which may act via inhibition of the 3CL protease (IC50 for the enzyme = 5 µM; EC50 values for reduction of viral RNA and infectious particles ranging from 19 to 34 µM) [39]. Finally, an octapeptide, designed for the SARS-CoV Mpro, namely AVLQSGFR, was reported to inhibit SARS-CoV replication in Vero cells at an EC50 of 0.027 µg/ml, while not being cytotoxic at 100 µg/ml, thus establishing a selectivity index of greater than 3700 [40]. Whether this highly selective antiviral effect was actually mediated by an inhibition of the SARS-CoV Mpro was not ascertained in this study [40].
Figure 7. Phe-Phe dipeptide.
Figure 8. Cinanserin.
In addition to 3CLpro, a PLpro is encoded by the SARS-CoV genome. SARS-CoV PLpro processes the replicase polyprotein at three conserved cleavage sites, thus generating the nonstructural proteins NSP1, NSP2 and NSP3. It was recently demonstrated that SARS-CoV PLpro also had deubiquitinating activity [41,42]. The possibility that SARS-CoV PLpro could deubiquitinate host or viral proteins has added a higher level of functional complexity to this enzyme, and should, in principle, elevate the value of SARS-CoV PLpro as a potential target for therapeutic intervention.
Virus-encoded helicase, polymerase & endonuclease
SARS-CoV encodes for an helicase which must unwind the double-stranded (±)RNA helices during the viral replication cycle. This helicase, akin to the herpesviral DNA helicase, also possesses NTPase activity, and may therefore be termed an NTPase/helicase. The SARS-CoV NTPase/helicase has been considered a potential target for the development of anti-SARS-CoV agents [43]. These agents could, in theory, be targeted at any of the three major domains of the enzyme, the N-terminal metal-binding domain, the hinge domain and the NTPase/helicase domain. Bananin (Figure 9) and three of its derivatives (iodobananin, vanillinbananin and eubananin) were shown to inhibit both the ATPase and helicase activity of the SARS-CoV NTPase/helicase, with IC50 values (for the ATPase activity) in the range of 0.5–3 µM [44]. Bananin was also found to inhibit SARS-CoV replication in fetal rhesus kidney (FRhK)-4 cells at an EC50 of less than 10 µM and a 50% cytotoxicity concentration (CC50) of over 300 µM, thus exhibiting a selectivity index of over 30 [44]. Whether the antiviral effect obtained in cell culture was causally linked to the inhibition of the NTPase/helicase was not ascertained.
Figure 9. Bananin.
The SARS-CoV RNA-dependent RNA polymerase (RdRp), due to its pivotal role in viral replication, represents another potential target for anti-SARS therapy. This enzyme (Figure 10) does not contain a hydrophobic pocket for non-nucleoside inhibitors similar to those that have proven effective against the hepatits C virus (HCV) polymerase or HIV-1 reverse transcriptase [45]. In fact, non-nucleoside HIV-1 reverse transcriptase inhibitors were shown to have no evident inhibitory effect on SARS-CoV RdRp activity [46]. It is intriguing that during purification, the full-length SARS-CoV RdRp was unstable and was hydrolytically cleaved into three fragments, a N-terminal p12 fragment, a middle p30 fragment and a C-terminal p64 fragment comprising the catalytic domain. The cause of the cleavage is unclear. Nor is it clear whether this cleavage also occurs in the viral life cycle [46]. At present, few, if any, nucleoside analogs have been recognized as specific inhibitors of the SARS-CoV RdRp. There is N4-hydroxycytidine, which has been accredited with both anti-HCV and anti-SARS-CoV effects. Against SARS-CoV it proved active at an EC50 of 10 µM (selectivity index ≥ 10) [24]. However, whether this antiviral effect was mediated by an inhibition of the viral RdRp was not ascertained.
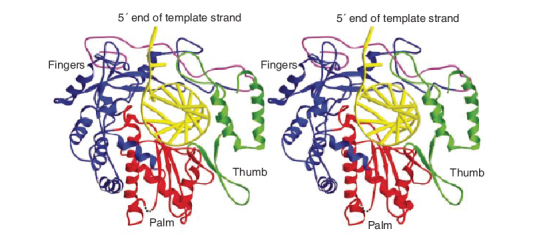
Figure 10. Ribbon diagram of the homology model of severe acute respiratory syndrome coronavirus RNA-dependent RNA polymerase with a docked RNA template primer.
α-helices are shown as spirals and β-strands as arrows. The subdomains of the catalytic domain are colored as the N-terminal portion of the fingers subdomain (376–424) in magenta, the base of the fingers (residues 425–584 and 626–679) in blue, palm (residues 585–625 and 680–807) in red, and thumb (residues 808–932) in green. Reprinted with permission from [45].
Given the large genome of SARS-CoV (and other CoVs), and the need for discontinuous transcription to generate subgenomic transcripts, they could be expected to encode novel RNA-processing functions. Indeed, a number of such proteins have been identified: NSP14, a putative exonuclease; NSP15, an endoribonuclease and NSP16, a putative RNA methyltransferase. RNA endonuclease activity is unusual among positive-strand RNA viruses, suggesting that, because it specifically occurs during SARS-CoV replication, it could be considered a target for antiviral drug development. This pertains, in particular, NSP15, to an Mn2+-dependent endoribonuclease that specifically cleaves RNA at unpaired uridylate residues. The role of NSP15, which consists of six subunits (arranged as a dimer of trimers) [47] in the CoV infection process, still remains to be elucidated.
Human monoclonal antibody
As mentioned above, human monoclonal antibody (mAb) to the S1 domain of the S protein of SARS-CoV blocks its association with the host cell ACE2 receptor [16]. The mAb concerned, 80R immunoglobulin (Ig)G1, was further evaluated for its immunoprophylactic efficacy in vivo in a mouse model [48]. When 80R IgG1 was given prophylactically to mice at doses therapeutically achievable in humans, viral replication was reduced by more than four orders of magnitude to below assay limits. The results demonstrated that the vast majority of SARS-CoVs isolated thus far remain sensitive to 80R. In an outbreak setting, early and rapid genotyping of the S1 gene fragment encoding the 80R epitope should provide an accurate guide for installment of immunoprophylaxis with 80R [48].
Data generated with human mAb (CR304) against SARS-CoV in ferrets also point to the feasibility of immunoprophylaxis with human mAb for the control of human SARS-CoV infections [49]. Similar observations in a mouse model demonstrating that primary infection with SARS-CoV provides protection from reinfection and that antibody alone can protect against viral replication, suggest that vaccines that induce neutralizing antibodies and strategies for immunoprophylaxis or, perhaps, immunotherapy are likely to be effective against SARS [50].
Most of the SARS-CoV-infected patients spontaneously recover, and recovered patients have higher and sustainable levels of both N and S glycoprotein-specific antibody responses, suggesting that antibody responses are likely to play an important role in determining the ultimate disease outcome of SARS-CoV-infected patients [51]. It would, therefore, not be unreasonable to expect that antibody to SARS-CoV, as present in convalescent plasma [52], may favorably influence the course of SARS.
As an interesting hypothesis, derived from the experience gathered with HIV [53], it may be proposed that antibody towards carbohydrate-hidden immunogenic epitopes on the SARS-CoV envelope may confer an immunoprophylactic, as well as an immunotherapeutic approach towards CoV and various other enveloped virus infections. To enable this approach, carbohydrate-binding agents, such as the Hippeastrum hybrid (amaryllis), Galanthus nivalis (snowdrop) and Urtica dioica mannose- or N-acetylglucosamine-binding lectins could be used to cause deletions, upon repeated exposure (passages), of the glycosylation sites in the SARS-CoV envelope in order to expose the carbohydrate-hidden epitopes to both active (vaccine) and passive (antibody) immune responses.
Human interferon
Shortly after SARS-CoV had been identified as the causative agent of SARS, Cinatl and colleagues [54] were the first to note that interferons (IFNs) inhibited the replication of SARS-CoV in cell culture in vitro; IFN-β being more potent than either IFN-α or -γ. These observations were subsequently confirmed in several other studies [55–58]. IFN-β exhibited potent antiviral activity at doses that had been shown to have acceptable safety profiles [55]. Also, IFN-α showed an in vitro inhibitory effect starting at concentrations of 1000 IU/ml [56]. In contrast with type I IFNs (α, β), type II IFN (γ) had little, if any, inhibitory effect on SARS-CoV replication [57]. The human MxA protein is one of the most prominent proteins induced by IFN-β. Nevertheless, no interference with SARS-CoV replication was observed in Vero cells stably expressing MxA, which implies that other IFN-induced proteins must be responsible for the strong inhibitory activity of IFN-β against SARS-CoV [58].
IFN-β, in conjunction with IFN-γ, was found to synergistically inhibit the replication of SARS-CoV in Vero cells [59], and a preliminary uncontrolled study with the IFN alfacon-1 (a synthetic IFN-α designed to represent a consensus IFN-α) suggested that this type of IFN, in combination with corticosteroids might be effective in vivo in the treatment of SARS [60]. Furthermore, a CpG oligodeoxynucleotide, which is able to induce human peripheral blood mononuclear cells (PBMCs) to produce high levels of IFN-α/β, has been accredited with strong activity against SARS-CoV in vitro[61].
Being a prophylactic rather than therapeutic agent, IFN(s) may have their highest utility in the prophylaxis or early postexposure management of SARS. Pegylated (PEG)-IFN-α has been shown to reduce viral replication and excretion, viral antigen expression by type 1 pneumocytes and the attendant pulmonary damage in cynomolgous macaques that were infected experimentally with SARS-CoV [62]. PEG-IFN-α is commercially available for the treatment of HCV (where it is generally used in combination with ribavirin) and hepatitis B. PEG-IFN-α as well as the other commercially available IFNs (e.g., IFN-β and alfacon-1) could be considered for prevention and/or early postexposure treatment of SARS should it re-emerge.
RNA interference
RNA interference (RNAi) can be defined as silencing of gene expression through degradation of (the target) RNA [63]. RNAi can be broken down into two main phases. In the first phase, long double-stranded RNA (dsRNA) is processed by Dicer, an RNAse III enzyme into duplexes of short interfering RNA (siRNA) of 21–24 nucleotides in length. Exogenous synthetic siRNAs can also be incorporated into the RNA-induced silencing complex (RISC), thereby bypassing the requirement for dsRNA processing by Dicer. In the second phase, a helicase present in RISC unwinds the duplex siRNA, which then pairs by means of its unwound antisense strand to its target messenger RNA (mRNA) that bears a high degree of sequence complementarity to the siRNA. An RNAse (Slicer) within RISC then proceeds to degrade the target mRNA at sites not bound by the siRNA, that is, 10 nucleotides upstream of the 5´-most residue of the siRNA-target mRNA duplex [63].
Thus, siRNAs have been developed that target the replicase [64] and S [65] genes of the SARS-CoV genome, thereby silencing their expression in cell culture. Potent siRNA inhibitors of SARS-CoV in vitro (i.e., the siRNA duplexes siSC2 [forward sequence: 5´-GCUCCUAAUUACACUCAACdtdt-3´] and siSC5 [forward sequence: 5´-GGAUGAGGAAGGCAAUUUAdtdt-3´], targeting the SARS-CoV genomeat S protein- and NSP12-coding regions, respectively) were further evaluated for their efficacy in a rhesus macaque SARS model [66], and were found to provide relief from SARS-CoV infection-induced fever, diminish SARS-CoV levels and reduce acute diffuse alveolar damage. No sign of toxicity was observed with the siRNA concerned [66]. Whether SARS can be conquered by the siRNA approach remains to be proven, however [67]. Therefore, the siRNA delivery forms should be further optimized, and the most effective target within the SARS-CoV genome should be identified. Since siRNAs, at least in their second stage of action, obey the antisense principle, antisense strategies such as those based on peptide-conjugated antisense phosphorodiamidate morpholino oligomers (P-PMOs) also deserve closer attention [68].
Miscellaneous compounds
A growing number of compounds have been identified as SARS-CoV replication inhibitors exhibiting mechanisms of action which are both diverse and largely unexplored. Of greater than 10,000 agents tested against SARS-CoV in Vero cells, approximately 50 compounds were found active at less than or equal to 10 µM [69]; as the most potent inhibitor, with an EC50 of 0.85 µM (selectivity index = 80), emerged valinomycin, a peptidic insecticide acting as a potassium ion transporter (Figure 11). Inhibitory effects on SARS-CoV replication, with selectivity indexes of up to 100, and EC50 values as low as 1 µg/ml, have been observed for a variety of compounds including the vancomycin, eremomycin and teicoplanin aglycon derivatives [70], and the mannose-specific plant lectins derived from G. nivalis, Hippeastrum hybrid[71] and Allium porrum (leek) [72]. The mode of action of these compounds has not been assessed, but it is tempting to speculate that they interfere with the binding of the S glycoprotein to the host cells.
Figure 11. Valinomycin.
Isatis indigotica root and phenolic Chinese herbs were frequently used for the prevention of SARS during the SARS outbreaks in China, Hong Kong and Taiwan. I. indigotica root (Radix isatidis) is native to China. From the I. indigotica root extracts several compounds, that is, indigo, sinigrin, aloe-emodin and hesperetin, were isolated that inhibited the cell-free and cell-based cleavage activity of the SARS Mpro (3CLpro) at IC50 values ranging from 10 to 1000 µM [73]. The inhibitory effects on SARS-CoV replication in cell culture (i.e., Vero cells) were not determined in this study. The cytotoxicity was determined, however, and, based on the ratio of the CC50 to the IC50 (cell-based cleavage), hesperetin appeared to be the most selective (selectivity index: ∼300) [73].
Glycyrrhizin, another plant product that has been isolated from the licorice root (Glycyrrhiza radix), is known as an anti-inflammatory substance in Chinese medicine. Glycyrrhizin consists of one molecule of glycyrrhetinic acid linked to two molecules of glucuronic acid (Figure 12). It has long since been recognized as an antiviral substance, active in vitro against both DNA and RNA viruses, including HIV [74,75]. Glycyrrhizin has also been shown to inhibit the replication of SARS-CoV, but only at a concentration (EC50: 300 µg/ml or ∼365 µM) that would be difficult to achieve in vivo[76]. Through the introduction of certain chemical modifications it proved possible to increase the antiviral potency of glycyrrhizin, but, as these modifications also increased the cytotoxicity, the selectivity index of the glycyrrhizin derivatives was reduced as compared with that of glycyrrhizin (selectivity index: ≥65) [77].
Figure 12. Glycyrrhizin.
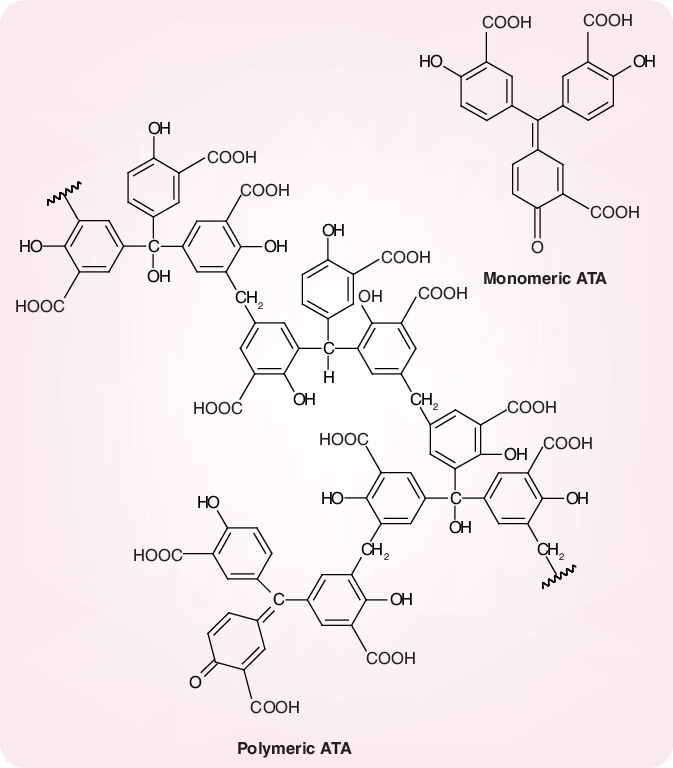
Potent and selective inhibition of SARS-CoV replication has also been shown for aurintricarboxylic acid (ATA) [78]; reported values for its EC50 and CC50 (in Vero cells) were 0.2 and 37.5 mg/ml, respectively. Thus, the selectivity index of ATA was estimated to be 187 [78]. The anti-SARS-CoV activity of ATA was tentatively attributed to an inhibitory effect on the viral RdRp [79]. It should be recognized, however, that ATA, which is commonly represented in its monomeric structure but actually occurs as a heterogenous mixture of polymers (Figure 13), is able to bind to, and inhibit, a variety of proteins and cellular processes [80,81]. ATA has since long been known as an inhibitor of HIV replication [82], and its anti-HIV activity is at least partially mediated by a specific interaction with the HIV cell receptor CD4 [83].
Figure 13. Monomeric and polymeric structures of aurintricarboxylic acid, according to [81].
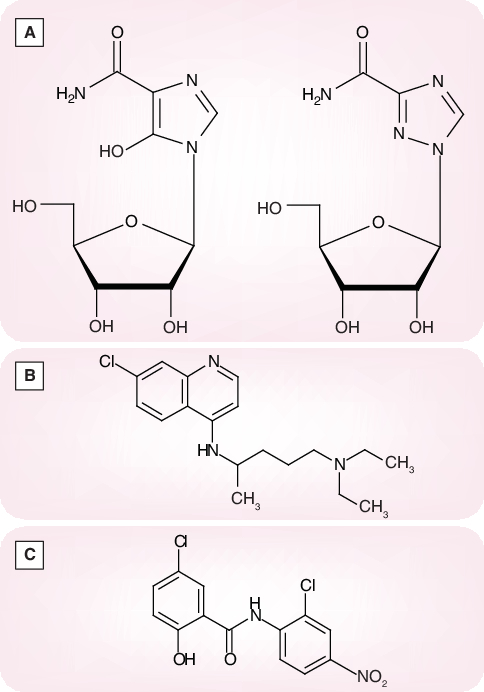
Ribavirin, another compound that has long since been known as a broad-spectrum antiviral agent [84], targeted at inosine monophosphate (IMP) dehydrogenase (a key enzyme involved in the de novo biosynthesis of GTP) [85], did not show meaningful activity against SARS-CoV replication in cell culture [56,76]. Its EC50, determined by virus yield reduction in Vero cells, was 40 µg/ml (CC50 > 200 µg/ml; selectivity index >5) [86]. In comparison, mizoribine, which is also assumed to act in its monophosphorylated form, as an inhibitor of the IMP dehydrogenase, exhibited an EC50 of 10 µg/ml (CC50 > 200 µg/ml; selectivity index > 20) [86]. Although these findings do not legitimate the use of ribavirin or mizoribine (Figure 14A) as single agents in the treatment of SARS, they point to IMP dehydrogenase as a potential target for the development of more potent anti-SARS-CoV agents.
Figure 14. (A) Mozoribine and ribavirin, (B) chloroquine and (C) niclosamide.
The 4-aminoquinoline chloroquine, another ‘old’ compound best known for its antimalarial effects, but also accredited with antiviral and anti-inflammatory effects, has been recommended for its potential use, preferably in combination with other antivirals, in the treatment of AIDS as well as SARS [87]. Chloroquine (Figure 14B) was found to inhibit SARS-CoV replication in Vero cells at an EC50 of 8.8 µM (CC50 = 261 µM; selectivity index = 30) [88]. These inhibitory effects were observed when the cells were treated with the drug either before or after exposure to the virus [89]. The EC50 of chloroquine for inhibition of SARS-CoV in vitro approximates the plasma concentrations of chloroquine reached during treatment of acute malaria [88].
Inhibition of SARS-CoV infection in vitro has also been reported for a nitric oxide (NO) generating compound, S-nitroso-N-acetylpenicillamine (SNAP), but only at an EC50 as high as 222 µM and a selectivity index as low as 2.6 [90]. Niclosamide (Figure 14C), an existing antihelminthic drug, was able to inhibit the replication of SARS-CoV in Vero cells at an EC50 of 2 µM, while its CC50 was 250 µM; its selectivity could therefore be estimated at 125 [91].
Then there are the HIV protease inhibitors nelfinavir [92] and lopinavir [93] which have been reported to inhibit the replication of SARS-CoV in Vero cells and FRhK-4 cells, respectively. In vitro activity against SARS-CoV was demonstrated for lopinavir at a concentration of 4 µg/ml (in comparison with 50 µg/ml for ribavirin) [93]. Yamamoto and colleagues were not able to find a selective antiviral effect with lopinavir, but for nelfinavir they found an EC50 of 0.048 µM (CC50 = 14.5 µM; selectivity index = 300) [91]. Preliminary clinical trials in the treatment of SARS with lopinavir (boosted by ritonavir) point to an apparent favorable clinical response [93,94].
Expert commentary
Detailed knowledge on the molecular structure and functioning of most of the SARS-encoded proteins, except for the Mpro (3CLpro) is lacking, which means that thus far, few, if any, successful attempts have been made to rationally design anti-SARS-CoV compounds. Most of the antiviral compounds which have thus far been found effective against SARS-CoV, were evaluated because they had previously been shown (or were analogous to compounds that had been shown previously) to be effective against viruses other than SARS-CoV, in particular HIV.
It could be postulated that to become a potential anti-SARS candidate drug, the SARS-CoV inhibitor should possess a selectivity index of greater than 100, equally importantly an EC50 of not (much) higher than 1–3 µM, so as to be able to achieve sufficiently high plasma (and tissue) drug levels upon systemic administration. Thus, qualifying as potential anti-SARS drug candidates are some of the calpain inhibitors [23,24] and 3CLpro inhibitors, for example, the Phe–Phe dipeptide inhibitor shown in Figure 7[38], which exhibited an EC50 of 0.18 µM and a selectivity index greater than 1000. Also, the octapeptide, AVLQSGFR, with an EC50 of 0.027 µg/ml (selectivity index >3700) [40] seems quite promising following the proposed criteria, and thus would be a number of the miscellaneous compounds such as valinomycin [69], niclosamide [90] and nelfinavir [91].
Of equal, if not greater, importance is proof of efficacy against SARS in the in vivo setting, which remains to be provided with the aforementioned compounds. Such proof of principle exists for human mAbs [48,49], IFN-α [62] and siRNA [66], which means that, should SARS-CoV, or a similar CoV, re-emerge, attempts could be immediately undertaken to prevent and/or treat these infections with human mAbs, IFN and/or siRNA. Being commercially available at present, IFNs (PEG-IFN-α, IFN-β and alfacon-1 etc.) should possibly be considered the best choice to curb SARS, should it strike again.
Finally, as has thus far been applied in several viral (i.e., HIV and HCV) infections, and could be recommended for others (i.e., avian influenza), drug combination therapy certainly represents a valuable option for the management of SARS-CoV infection. As for any drug combination approach, this should allow the individual drugs to show a synergistic antiviral effect, thereby reducing the likelihood for drug resistance development (which, admittedly, has so far not been recognized as a problem in the treatment of SARS).
Five-year view
It is hard to predict whether SARS-CoV, or a similar CoV, would strike again in the future. This makes it impossible to speculate on how the field will evolve over the next 5 years. However, as for avian influenza H5N1, we ought to be better prepared. Therefore, attempts should continue to develop the appropriate means to prevent and/or treat SARS-CoV infections. The development of an adequate vaccine against SARS (not discussed here) remains, of course, mandatory, but, in addition, other prophylactic/therapeutic options should be duly explored, and these include, besides human mAbs, IFNs and siRNAs, also low-molecular-weight SARS-CoV inhibitors targeted at any of the specific processes involved in the viral replication cycle (i.e., viral entry into the cells, proteolytic cleavage, RNA replication and transcription).
-
•
Severe acute respiratory syndrome (SARS) is a new epidemic disease that emerged at the end of 2002, but subsequently subsided during the course of 2003.
-
•
Measures should be undertaken to prevent or treat SARS should the SARS coronavirus (CoV) re-emerge.
-
•
A first target for prophylactic or therapeutic intervention is the spike (S) glycoprotein involved in viral entry into the cells. This process can be hit by monoclonal antibody and fusion inhibitors.
-
•
A second target is the processing of the replicase polyproteins by virus-encoded proteases. A number of protease inhibitors have been shown to interact at this level.
-
•
Additional virus-encoded enzymes have been identified, among which are the NTP/helicase and RNA-dependent RNA polymerase, as potential targets for chemotherapeutic intervention.
-
•
Human interferons (α and β) seem be indicated for the prophylaxis and early treatment of SARS-CoV infection.
-
•
A possible approach to treat SARS may be based on RNA interference, using short interfering RNA duplexes.
-
•
Other compounds that have been shown to inhibit SARS-CoV replication in cell culture include valinomycin, glycopeptide antibiotics, plant lectins, hesperetin, glycyrrhizin, aurintricarboxylic acid, chloroquine, niclosamide, nelfinavir and some of the calpain inhibitors.
References
Papers of special note have been highlighted as: • of interest •• of considerable interest
- 1.Peiris JS, Lai ST, Poon LL et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361, 1319–1325 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee N, Hui D, Wu A et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 348, 1986–1994 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek TG, Erdman D, Goldsmith CS et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Drosten C, Gunther S, Preiser W et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Kuiken T, Fouchier RA, Schutten M et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362, 263–270 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fouchier RA, Kuiken T, Schutten M et al. Aetiology: Koch’s postulates fulfilled for SARS virus. Nature 423, 240 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Hoek L, Pyrc K, Jebbink MF et al. Identification of a new human coronavirus. Nature Med. 10, 368–373 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stadler K, Masignani V, Eickmann M et al. SARS – beginning to understand a new virus. Nature Rev. Microbiol. 1, 209–218 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides an excellent insight into the organization of the severe acute respiratory syndrome coronavirus (SARS-CoV) genome and the targets for chemotherapeutic intervention.
- 9.Oxford JS, Balasingam S, Chan C, Catchpole A, Lambkin R. New antiviral drugs, vaccines and classic public health interventions against SARS coronavirus. Antiviral Chem. Chemother. 16, 13–21 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Shigeta S, Yamase T. Current status of anti-SARS agents. Antiviral Chem. Chemother. 16, 23–32 (2005). [DOI] [PubMed] [Google Scholar]
- 11.De Clercq E. Antivirals and antiviral strategies. Nature Rev. Microbiol. 2, 704–720 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides a comprehensive overview of the different antiviral strategies that could be used against virus infections at large. These strategies could, mutatis mutandis, also extend towards the SARS-CoV.
- 12.Li W, Moore MJ, Vasilleva N et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofmann H, Geier M, Marzi A et al. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem. Biophys. Res. Commun. 319, 1216–1221 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia HP, Look DC, Shi L et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 79, 14614–14621 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 279, 3197–3201 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sui J, Li W, Murakami A et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl Acad. Sci. USA 101, 2536–2541 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Towler P, Staker B, Prasad SG et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 279, 17996–18007 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tripet B, Howard MW, Jobling M, Holmes RK, Holmes KV, Hodges RS. Structural characterization of the SARS-coronavirus spike S fusion protein core. J. Biol. Chem. 279, 20836–20849 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Xiao G, Chen Y et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 363, 938–947 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Presents an ingenious approach towards blocking the so-called 6-helix bundle formation that leads to virus–cell fusion.
- 20.Zhu J, Xiao G, Xu Y et al. Following the rule: formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochem. Biophys. Res. Commun. 319, 283–288 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ni L, Zhu J, Zhang J, Yan M, Gao GF, Tien P. Design of recombinant protein-based SARS-CoV entry inhibitors targeting the heptad-repeat regions of the spike protein S2 domain. Biochem. Biophys. Res. Commun. 330, 39–45 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veiga S, Yuan Y, Li X et al. Why are HIV-1 fusion inhibitors not effective against SARS-CoV? Biophysical evaluation of molecular interactions. Biochim. Biophys. Acta 1760, 55–61 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl Acad. Sci. USA 102, 11876–11881 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnard DL, Hubbard VD, Burton J et al. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and β-d-N4-hydroxycytidine. Antiviral Chem. Chemother. 15, 15–22 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Chou KC, Wei DQ, Zhong WZ. Binding mechanisms of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 308, 148–151 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Yang M, Ding Y et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA 100, 13190–13195 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Insight into the crystal structure of the SARS 3C-like cysteine protease complexed with a substrate analog should help to provide a structural basis for rational drug design.
- 28.Lee T-W, Cherney MM, Huitema C et al. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol. 353, 1137–1151 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan K, Wei P, Feng Q et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 279, 1637–1642 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martina E, Stiefl N, Degel B et al. Screening of electrophilic compounds yields an aziridinyl peptide as new active-site directed SARS-CoV main protease inhibitor. Bioorg. Med. Chem. Lett. 15, 5365–5369 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain RP, Pettersson HI, Zhang J et al. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J. Med. Chem. 47, 6113–6116 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Ghosh AK, Xi K, Ratia K et al. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem. 48, 6767–6771 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Shie J-J, Fang J-M, Kuo C-J et al. Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease. J. Med. Chem. 48, 4469–4473 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Kaeppler U, Stiefl N, Schiller M et al. A new lead for nonpeptidic active-site-directed inhibitors of the severe acute respiratory syndrome coronavirus main protease discovered by a combination of screening and docking methods. J. Med. Chem. 48, 6832–6842 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Chen L-R, Wang Y-C, Lin YW et al. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg. Med. Chem. Lett. 15, 3058–3062 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y-C, Huang V, Chao T-C et al. Screening of drugs by FRET analysis identifies inhibitors of SARS-CoV 3CL protease. Biochem. Biophys. Res. Commun. 333, 194–199 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C-N, Lin CPC, Huang K-K et al. Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3,3´-digallate (TF3). Evid. Based Complement Alternat. Med. 2, 209–215 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shie J-J, Fang J-M, Kuo T-H et al. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α,β-unsaturated esters. Bioorg. Med. Chem. 13, 5240–5252 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes one of the most potent and selective SARS-CoV inhibitors targeted at the 3C-like protease.
- 39.Chen L, Gui C, Luo X et al. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro J. Virol. 79, 7095–7103 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gan Y-R, Huang H, Huang Y-D et al. Synthesis and activity of an octapeptide inhibitor designed for SARS coronavirus main proteinase. Peptides (2005) (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79, 15189–15198 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 79, 15199–15208 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanner JA, Watt RM, Chai YB et al. The severe acute respiratory syndrome (SARS) coronavirus NTPase/helicase belongs to a distinct class of 5´ to 3´ viral helicases. J. Biol. Chem. 278, 39578–39582 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanner JA, Zheng B-J, Zhou J et al. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chem. Biol. 12, 303–311 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu X, Liu Y, Weiss S, Arnold E, Sarafianos SG, Ding J. Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 31, 7117–7130 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng A, Zhang W, Xie Y et al. Expression, purification, and characterization of SARS coronavirus RNA polymerase. Virology 335, 165–176 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guarino LA, Bhardwaj K, Dong W, Sun J, Holzenburg A, Kao C. Mutational analysis of the SARS virus Nsp15 endoribonuclease: identification of residues affecting hexamer formation. J. Mol. Biol. 353, 1106–1117 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sui J, Li W, Roberts A et al. Evaluation of human monoclonal antibody 80R for immunoprophylaxis of severe acute respiratory syndrome by an animal study, epitope mapping, and analysis of spike variants. J. Virol. 79, 5900–5906 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.ter Meulen J, Bakker ABH, van den Brink EN et al. Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet 363, 2139–2141 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Experiments reported in references 48 and 49 demonstrate the feasibility of immunoprophylaxis with human monoclonal antibody for the control of SARS-CoV infections.
- 50.Subbarao K, McAuliffe J, Vogel L et al. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J. Virol. 78, 3572–3577 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, Zhang F, Yu W et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J. Med. Virol. 78, 1–8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong VWS, Dai D, Wu AKL, Sung JJY. Treatment of severe acute respiratory syndrome with convalescent plasma. Hong Kong Med. J. 9, 199–201 (2003). [PubMed] [Google Scholar]
- 53.Balzarini J, Van Laethem K, Hatse S et al. Carbohydrate-binding agents cause deletions of highly conserved glycosylation sites in HIV gp120. A new therapeutic concept to hit the Achilles heel of HIV. J. Biol. Chem. 280, 41005–41014 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW. Treatment of SARS with human interferons. Lancet 362, 293–294 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hensley LE, Fritz EA, Jahrling PB, Karp CL, Huggins JW, Geisberg TW. Interferon-β 1a and SARS coronavirus replication. Emerg. Infect. Dis. 10, 317–319 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ströher U, DiCaro A, Li Y et al. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-α. J. Infect. Dis. 189, 1164–1167 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng B, He M-L, Wong K-L et al. Potent inhibition of SARS-associated coronavirus (SCoV) infection and replication by type I interferons (IFN-α/β) but not by type II interferon (IFN-γ). J. Interferon Cytokine Res. 24, 388–390 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Spiegel M, Pichlmair A, Mühlberger E, Haller O, Weber F. The antiviral effect of interferon-beta against SARS-coronavirus is not mediated by MxA protein. J. Clin. Virol. 30, 211–213 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sainz B Jr, Mossel EC, Peters CJ, Garry RF. Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV). Virology 329, 11–17 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loutfy MR, Blatt LM, Siminovitch KA et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome. JAMA 290,3222–3228 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Bao M, Zhang Y, Wan M et al. Anti-SARS-CoV immunity induced by a novel CpG oligodeoxynucleotide. Clin. Immunol. 118(2–3), 180–187 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haagmans BL, Kuiken T, Martina BE et al. Pegylated interferon-α protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nature Med. 10, 290–293 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates, for the first time, that interferon may be useful, in vivo, in reducing SARS-CoV replication and the attendant pulmonary damage.
- 63.Stevenson M. Therapeutic potential of RNA interference. N. Engl. J. Med. 351, 1772–1777 (2004). [DOI] [PubMed] [Google Scholar]
- 64.He M-L, Zheng B, Peng Y et al. Inhibition of SARS-associated coronavirus infection and replication by RNA interference. JAMA 290,2665–2666 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Li T, Fu L et al. Silencing SARS-CoV spike protein expression in cultured cells by RNA interference. FEBS Lett. 560, 141–146 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Li B-j, Tang Q, Cheng D et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in rhesus macaque. Nature Med. 11, 944–951 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates that the principle of RNA interference, a revolutionary approach for silencing gene expression, also holds for SARS.
- 67.Chang Z, Hu J. RNAi therapeutics: can siRNAs conquer SARS? Gene Ther. (2005) (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neuman BW, Stein DA, Kroeker AD et al. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J. Virol. 79, 9665–9676 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu C-Y, Jan J-T, Ma S-H et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl Acad. Sci. USA 101, 10012–10017 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balzarini J, Keyaerts E, Vijgen L, De Clercq E, Preobrazhenskaya M, Van Ranst M. Inhibitory activity of vancomycin, eremomycin and teicoplanin aglycon derivatives against feline and human coronaviruses (i.e. SARS). 17th International Conference on Antiviral Research. AZ, USA, 2–6 May 2004. Antiviral Res. 62(A59), 78 (2004). [Google Scholar]
- 71.Balzarini J, Vijgen L, Keyaerts E et al. Mannose-specific plant lectins are potent inhibitors of coronavirus infection including the virus causing SARS. 17th International Conference on Antiviral Research. AZ, USA, 2–6 May 2004. Antiviral Res. 62(A76), 122 (2004). [Google Scholar]
- 72.Vijgen L, Keyaerts E, Van Damme E et al. Antiviral effect of plant compounds of the Alliaceae family against the SARS coronavirus. 17th International Conference on Antiviral Research. AZ, USA, 2–6 May 2004. Antiviral Res. 62(A76), 123 (2004). [Google Scholar]
- 73.Lin C-W, Tsai F-J, Tsai C-H et al. Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antiviral Res. 68, 36–42 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ito M, Nakashima H, Baba M et al. Inhibitory effect of glycyrrhizin on the in vitro infectivity and cytopathic activity of the human immunodeficiency virus [HIV (HTLV-III/LAV)]. Antiviral Res. 7, 127–137 (1987). [DOI] [PubMed] [Google Scholar]
- 75.Ito M, Sato A, Hirabayashi K et al. Mechanism of inhibitory effect of glycyrrhizin on replication of human immunodeficiency virus (HIV). Antiviral Res. 10, 289–298 (1988). [DOI] [PubMed] [Google Scholar]
- 76.Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 361, 2045–2046 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoever G, Baltina L, Michaelis M et al. Antiviral activity of glycyrrhizic acid derivatives against SARS-coronavirus. J. Med. Chem. 48, 1256–1259 (2005). [DOI] [PubMed] [Google Scholar]
- 78.He R, Adonov A, Traykova-Adonova M et al. Potent and selective inhibition of SARS coronavirus replicatoin by aurintricarboxylic acid. Biochem. Biophys. Res. Commun. 320, 1199–1203 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ya YL, Zhang XW, Andonov A, He RT. Structural analysis of inhibition mechanisms of aurintricarboxylic acid on SARS-CoV polymerase and other proteins. Comp. Biol. Chem. 29, 212–219 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cushman M, Wang P, Chang SH et al. Preparation and anti-HIV activities of aurintricarboxylic acid fractions and analogues: direct correlation of antiviral potency with molecular weight. J. Med. Chem. 34, 329–337 (1991). [DOI] [PubMed] [Google Scholar]
- 81.Cushman M, Kanamathareddy S, De Clercq E, Schols D, Goldman ME, Bowen JA. Synthesis and anti-HIV activities of low molecular weight aurintricarboxylic acid fragments and related compounds. J. Med. Chem. 34, 337–342 (1991). [DOI] [PubMed] [Google Scholar]
- 82.Balzarini J, Mitsuya H, De Clercq E, Broder S. Aurintricarboxylic acid and Evans blue represent two different classes of anionic compounds which selectively inhibit the cytopathogenicity of human T-cell lymphotropic virus type III/lymphadenopathy-associated virus. Biochem. Biophys. Res. Commun. 136, 64–71 (1986). [DOI] [PubMed] [Google Scholar]
- 83.Schols D, Baba M, Pauwels R, Desmyter J, De Clercq E. Specific interaction of aurintricarboxylic acid with the human immunodeficiency virus/CD4 cell receptor. Proc. Natl Acad. Sci. USA 86, 3322–3326 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowski JT, Robins RK. Broad-spectrum antiviral activity of virazole: 1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 177, 705–706 (1972). [DOI] [PubMed] [Google Scholar]
- 85.Streeter DG, Witkowski JT, Khare GP et al. Mechanism of action of 1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide (virazole), a new broad-spectrum antiviral agent. Proc. Natl Acad. Sci. USA 70, 1174–1178 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saijo M, Morikawa S, Fukushi S et al. Inhibitory effect of mizoribine and ribavirin on the replication of severe acute respiratory syndrome (SARS)-associated coronavirus. Antiviral Res. 66, 159–163 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect. Dis. 3, 722–727 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem. Biophys. Res. Commun. 323, 264–268 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vincent MJ, Bergeron E, Benjannet S et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2, 69 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Keyaerts E, Vijgen L, Chen L, Maes P, Hedenstierna G, Van Ranst M. Inhibition of SARS-coronavirus infection in vitro by S-nitroso-N-acetylpenicillamine, a nitric oxide donor compound. Int. J. Infect. Dis. 8, 223–226 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu C-J, Jan J-T, Chen C-M et al. Inhibition of severe acute respiratory syndrome coronavirus replication by niclosamide. Antimicrob. Agents Chemother. 48, 2693–2696 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamamoto N, Yang R, Yoshinaka Y et al. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 318, 719–725 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chu CM, Cheng VCC, Hung IFN et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax 59, 252–256 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chan KS, Lai ST, Chu CM et al. Treatment of severe acute respiratory syndrome with lopinavir/ritonavir: a multicentre retrospective matched cohort study. Hong Kong Med. J. 9, 399–406 (2003). [PubMed] [Google Scholar]