

Abstract
Rhinovirus (RV) infection is involved in acute exacerbations of asthma and chronic obstructive pulmonary disease (COPD). RV primarily infects upper and lower airway epithelium. Immunoproteasomes (IP) are proteolytic machineries with multiple functions including the regulation of MHC class I antigen processing during viral infection. However, the role of IP in RV infection has not been explored. We sought to investigate the expression and function of IP during airway RV infection. Primary human tracheobronchial epithelial (HTBE) cells were cultured at air–liquid interface (ALI) and treated with RV16, RV1B, or interferon (IFN)-λ in the absence or presence of an IP inhibitor (ONX-0914). IP gene (i.e. LMP2) deficient mouse tracheal epithelial cells (mTECs) were cultured for the mechanistic studies. LMP2-deficient mouse model was used to define the in vivo role of IP in RV infection. IP subunits LMP2 and LMP7, antiviral genes MX1 and OAS1 and viral load were measured. Both RV16 and RV1B significantly increased the expression of LMP2 and LMP7 mRNA and proteins, and IFN-λ mRNA in HTBE cells. ONX-0914 down-regulated MX1 and OAS1, and increased RV16 load in HTBE cells. LMP2-deficient mTECs showed a significant increase in RV1B load compared with the wild-type (WT) cells. LMP2-deficient (compared with WT) mice increased viral load and neutrophils in bronchoalveolar lavage (BAL) fluid after 24 h of RV1B infection. Mechanistically, IFN-λ induction by RV infection contributed to LMP2 and LMP7 up-regulation in HTBE cells. Our data suggest that IP are induced during airway RV infection, which in turn may serve as an antiviral and anti-inflammatory mechanism.
Introduction
Rhinovirus (RV) infection is involved in the pathogenesis of multiple respiratory diseases, from the common cold to life-threatening acute exacerbations of asthma and chronic obstructive pulmonary disease (COPD) [1,2]. RV infects both upper and lower respiratory tracts, leading to airway inflammation and bronchial constriction [3,4]. RV-induced asthma exacerbations are a major cause of disease morbidity and mortality, and increase healthcare costs [5,6], but there is no effective therapy for RV infection. Therefore, it is imperative to elucidate novel mechanisms against RV infection in the airways.
Proteasomes are proteolytic machineries that contribute to protein quality control in eukaryotic cells[7]. They have a complex structure consisting of the 20S central proteolytic core, which contains the standard subunits β1, β2, and β5 [8]. Binding of one or two 19S multisubunit regulatory caps to the 20S core forms the 26S proteasome, while the PA28 regulatory complex binds to either the 26S proteasome or to the 20S core [8,9]. The three proteolytically active subunits β1, β2, and β5 in the constitutive proteasomes are replaced by immunoproteasome (IP) subunits LMP2 (β1i), MECL-1 (β2i), and LMP7 (β5i) upon exposure to pro-inflammatory stimuli such as interferon (IFN)-γ or tumor necrosis factor α (TNF-α) [10,11]. The known functions of IP include facilitating antigen presentation, splicing antigenic peptides, modulating pro-inflammatory cytokines, and maintaining protein homeostasis after oxidative stress [12–15].
Research on IP in the lungs is getting more attention due to their emerging role as a potential therapeutic target. IP may be involved in the pathophysiology of lung diseases particularly COPD and idiopathic pulmonary fibrosis. Kammerl et al. [16] demonstrated reduced levels of IP subunits in bronchoalveolar lavage (BAL) cells from patients with COPD. Moreover, cigarette smoke exposure inhibited IP expression by alveolar macrophages of COPD patients. However, conflicting results exist as Fujino et al. [17] reported increased LMP2 and LMP7 expression in primary alveolar type II cells from patients with early stages of COPD.
It has been proposed that IP dysfunction might result in impaired antiviral immune responses and increased disease severity [16], but this has not been firmly established. IP has been shown to participate in the cleavage of viral proteins into pathogen-derived peptides that enhance MHC I antigen presentation for effective pathogen clearance by the cytotoxic CD8+ T cells [16,18–20]. Keller et al. [21] observed an increase in IP mRNA and protein expression during lung murine gammaherpesvirus-68 (MHV-68) infection in mice. Their data suggest that IP is involved in ubiquitin-dependent and -independent degradation of proteins during infection. However, Robek et al. [22] found that IP were not essential for the antiviral effect of IFN against hepatitis B virus in vivo.
In the current study, we sought to determine the role of IP in airway defense against RV infection. We hypothesized that RV infection induces IP, which in turn serves as a novel mechanism to defend viral infection in airway epithelium, the primary site of RV infection. As RV infection induces IFN (e.g. IFN-λ/IL-28) responses in airway epithelium [23–26], we further hypothesized that IP up-regulation is dependent on IFN-λ induction by RV. A better understanding on the mechanism of airway defense against RV infection may lead to the development of novel and effective therapy for patients experiencing exacerbations associated with viral infection.
Materials and methods
RV16 and RV1B propagation
RV16 and RV1B (American Type Culture Collection, Manassas, VA) were propagated in H1-Hela cells (CRL-1958, ATCC), purified and titrated to plaque-forming unit (PFU) as described previously [27].
Human tracheobronchial epithelial cell culture and treatments
Human lungs from de-identified adult organ donors without smoking history were obtained through the National Disease Research Interchange (Philadelphia, PA), the International Institute for the Advancement of Medicine (Edison, NJ) or Donor Alliance of Colorado. The collected lungs were not suitable for transplantation and were donated for medical research. The Institutional Review Board (IRB) at National Jewish Health approved the study. Donors were selected based on lung function with >225 ratio of a PaO2/FIO2, no history of clinical lung disease, a chest radiograph that indicated no infection, and a time on the ventilator of <5 days. Tracheal and main bronchial tissue were digested with 0.1% protease derived from Streptomyces griseus (P5147–56, Sigma–Aldrich, St. Louis, MO) in DMEM (GE Life Sciences, Logan, Utah) overnight at 4°C, and processed as previously reported [28].
Air–liquid interface (ALI) culture of human tracheobronchial epithelial (HTBE) cells was performed as previously described [29,30]. Briefly, HTBE cells at passage 1 were expanded onto collagen-coated 60-mm tissue culture dishes containing BEGM™ with supplements (Lonza, Walkersville, MD, U.S.A.). Cells (passage 2) were then transferred on to collagen-coated 12-well transwell plates (Transwell 2460, Corning Incorporated, Corning, NY, U.S.A.) with PneumaCult™-ALI medium (StemCell, Vancouver, BC, Canada). After 7 days of submerged culture, cells were shifted to ALI for the next 14 days to induce mucociliary differentiation. On day 14, cells were infected with RV in the presence or absence of an IP inhibitor ONX-0914 (APExBIO, Houston, TX, U.S.A.). For ONX-0914 treatment, cells were pre-treated with 100 nM of ONX-0914 in both apical and basolateral media for 2 h prior to RV infection. To infect the cells, the apical surface was washed with PBS to remove mucus, followed by incubation with 0.25% BSA-PBS (control), RV at 106 PFU/well, or 10 ng/ml IFN-λ (PeproTech, Rocky Hill, NJ, U.S.A.) at 37°C, 5% CO2 for 2 h. Thereafter, viruses at the apical surface were removed and the ALI cultures continued for up to 48 h. Basolateral supernatants were then collected. Cells were lysed with RLT buffer for RNA extraction and with RIPA buffer for Western blot analysis.
IFN-λ Receptor 1 small interfering RNA (siRNA) transfection
To define the role of type III IFN (IFN-λ) in RV-induced IP, we performed IFN-λ Receptor 1 (IFN-λR1) siRNA transfection to knockdown gene expression. We chose the submerged cell culture system as siRNA-mediated gene knockdown is transient, and thus works best in the submerged (short-term) but not in the ALI (long-term) system. HTBE cells under submerged culture in 24-well plates (105 cells/well) were transfected with 10 nM IFN-λR1 siRNA (ID: 1102892; Thermo Fisher Scientific, Waltham, MA, U.S.A.) or with Scrambled (Scr) control siRNA using Lipofetamine RNAiMax transfection reagent (Thermo Fisher Scientific, Waltham, MA, U.S.A.) according to manufacturer’s protocol. After 48 h of transfection, cells were incubated with 0.25% BSA-PBS (uninfected) or infected with RV16 at MOI of 10 at 37°C, 5% CO2 for 2 h, and then washed with PBS to remove the viruses from the cell surface. Cells were further incubated for 24 h until harvest. Cells were lysed with RLT buffer for RNA extraction and with RIPA buffer for Western blot analysis.
Mouse model of intranasal RV1B infection
Wild-type (WT) C57BL/6 and LMP2 knockout (KO) mice on a C57BL/6 background were kindly provided by Dr. Deborah A. Ferrington at the University of Minnesota. Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Minnesota. Age-matched female and male mice (8–12 weeks) anesthetized by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (10 mg/kg) were intranasally inoculated with 50 μl of PBS (control) or RV1B at 107 PFU/mouse for 24 h. At the time of killing, mouse lungs were lavaged with 1 ml sterile saline and BAL fluid was collected for leukocyte counts. Cytospins of BAL cells were stained with Diff-Quick kit (IMEB Inc., San Marcos, CA) and cell differentials were determined as the percentage of 500 counted leukocytes. BAL fluid (cell free) was utilized for RNA extraction and quantitation of viral load by quantitative real-time reverse-transcription PCR (RT-PCR). In addition, the left lung lobe was placed in RNAlater (Thermo Fisher Scientific, Waltham, MA, U.S.A.) for RNA extraction by using the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, U.S.A.).
Mouse tracheal epithelial cells’ isolation and culture
Mouse tracheas were excised from WT and LMP2 KO mice and digested in DMEM with 0.1% protease (Sigma–Aldrich, St. Louis, MO) and 50 μg/ml amphotericin B (Sigma–Aldrich, St. Louis, MO) at 4°C for 4 h. Cells were washed, collected, and resuspended in F media with a Rho kinase inhibitor Y-27632 at 10 μM and 125 ng/ml of mouse epidermal growth factor (mEGF) before expansion onto irradiated 3T3 fibroblast feeder cells [31,32]. Expanded mouse tracheal epithelial cells (mTECs) were then trypsinized and seeded on to collagen-coated 12-well transwell plates with PneumaCult™-ALI medium (StemCell, Vancouver, BC, Canada) and 15 ng/ml mEGF. Cells were grown for 7 days in submerged conditions and then shifted to ALI for 14 days. Thereafter, cells were treated apically with 106 PFU RV1B per transwell or 0.25% BSA-PBS (uninfected) at 37°C, 5% CO2 for 2 h. Viral particles were removed and the ALI cultures were placed back in the incubator for 24 h. Basolateral supernatants were collected and cells were lysed with RLT buffer for RNA extraction and with RIPA buffer for Western blot analysis.
Quantitative real-time reverse-transcription PCR
mRNA levels of human LMP2, LMP7, MX1, and OAS1, and mouse LMP2, LMP7, and MX1 were measured using TaqMan gene expression assays (Thermo Fisher Scientific, Waltham, MA, U.S.A.). Quantitative real-time PCR was performed on the CFX96™ real-time PCR Detection System (Bio-Rad Laboratories Inc., Hercules, CA, U.S.A.). RV (forward: 5′-CCT CCG GCC CCT GAA T-3′; reverse: 5′-GGT CCC ATC CCG CAA TT-3′, probe: 5′-CTA ACC TTA AAC CTG CAG CCA-3′) and human IFN-λ1 (forward: 5′-GGG AAC CTG TGT CTG AGA ACG T-3′; reverse: 5′-GAG TAG GGC TCA GCG CAT AAA TA-3′; probe: 5′-CTG AGT CCA CCT GAC ACC CCA CAC C-3′) were purchased from Integrated DNA Technologies (Coralville, IA, U.S.A.).
Target gene expression was expressed as relative mRNA levels. ΔCT was calculated as the difference in the threshold cycle (CT) between the target gene and the reference (housekeeping) genes GAPDH and 18S rRNA for human and mouse samples, respectively. Then, the mean of the ΔCT from the control (e.g. non-infected) samples was calculated. To obtain the ΔΔCT, the ΔCT of a treatment condition (e.g. RV infection) was subtracted from the mean ΔCT of the control samples. The 2−ΔΔCT method was applied to calculate the changes (relative levels) in gene expression under various treatments [33].
Viral load in cell-free mouse BAL fluid was assessed by extracting total RNA from 50 μl of BAL fluid, synthesizing cDNA, and performing quantitative PCR using RV specific PCR primers and a probe as described before [34,35]. The viral load was then calculated by using the 2−ΔΔCT method with the mean CT value from the control mice (WT C57BL/6) as the reference sample.
Western blot
Cells were lysed in Pierce™ RIPA Buffer with Halt™ protease and phosphatase inhibitor cocktail (1:100) (catalog number: 78440, Thermo Fisher Scientific, Waltham, MA, U.S.A.). Equal amount of proteins were loaded and separated on 12% Mini-Protean protein gels (ten wells, Bio-Rad, Hercules, CA, U.S.A.) and transferred onto a PVDF membrane (Bio-Rad, Hercules, CA, U.S.A.). Membranes were incubated with primary antibodies: rabbit anti-LMP2, rabbit anti-LMP7, rabbit anti-IFN-λR1 (Abcam, Cambridge, MA, U.S.A.), mouse anti-GAPDH (Santa Cruz Biotechnology Inc., Dallas, TX, U.S.A). Membranes were then probed with a horseradish peroxidase-conjugated secondary anti-rabbit or anti-mouse antibody (GE Healthcare Bio-Sciences, Pittsburgh, PA, U.S.A.). Membranes were stripped using Restore™ PLUS stripping buffer to re-probe with another primary antibody (Thermo Fisher Scientific, Waltham, MA, U.S.A.). Target proteins were visualized using Amersham™ ECL™ Western blotting substrate (GE Healthcare Bio-Sciences, Pittsburgh, PA, U.S.A.).
Densitometry was performed using the NIH’s ImageJ software to obtain the density data for the target protein band as well as the loading control protein (GAPDH) band in all the samples across different blots. Thereafter, the ratio of target proteins (e.g. LMP2, LMP7, and IFN-λR1) compared with GAPDH was calculated in each sample. Finally, by giving the control (e.g. no infection) densitometric data a value of 1, the fold change of the target protein under different treatment conditions (e.g. RV infection) over the control was calculated from each donor.
Statistical analysis
Data were analyzed and graphs were made using Prism7 software (GraphPad). For parametric data, two-group comparisons were made using the Student’s t test, and multiple comparisons were performed using one-way ANOVA with Tukey’s post hoc test. Correlation was assessed using the Pearson coefficient. For non-parametric data, two-group comparisons were done using the Mann–Whitney test, and multiple comparisons were made using the Kruskal–Wallis test. A p value <0.05 was considered statistically significant.
Results
RV infection induces IP expression in primary human airway epithelial cells
To determine if RV induces IP in airway epithelium, HTBE cells cultured at ALI were infected with RV1B and RV16. As shown in Figure 1A and B, mRNA levels of IP subunits LMP2 and LMP7 were significantly up-regulated after 24 h of RV16 and RV1B infection as compared with the uninfected control. Similar data were obtained at 48 h post-infection (p.i.) (data not shown). At the protein level, there was a marginal increase in LMP2 and LMP7 at 24 h p.i. (data not shown), but LMP2 and LMP7 proteins were significantly increased after 48 h of RV1B or RV16 infection (Figure 1C).
Figure 1. Human tracheobronchial epithelial (HTBE) cells infected with rhinovirus increase immunoproteasome subunit expression.
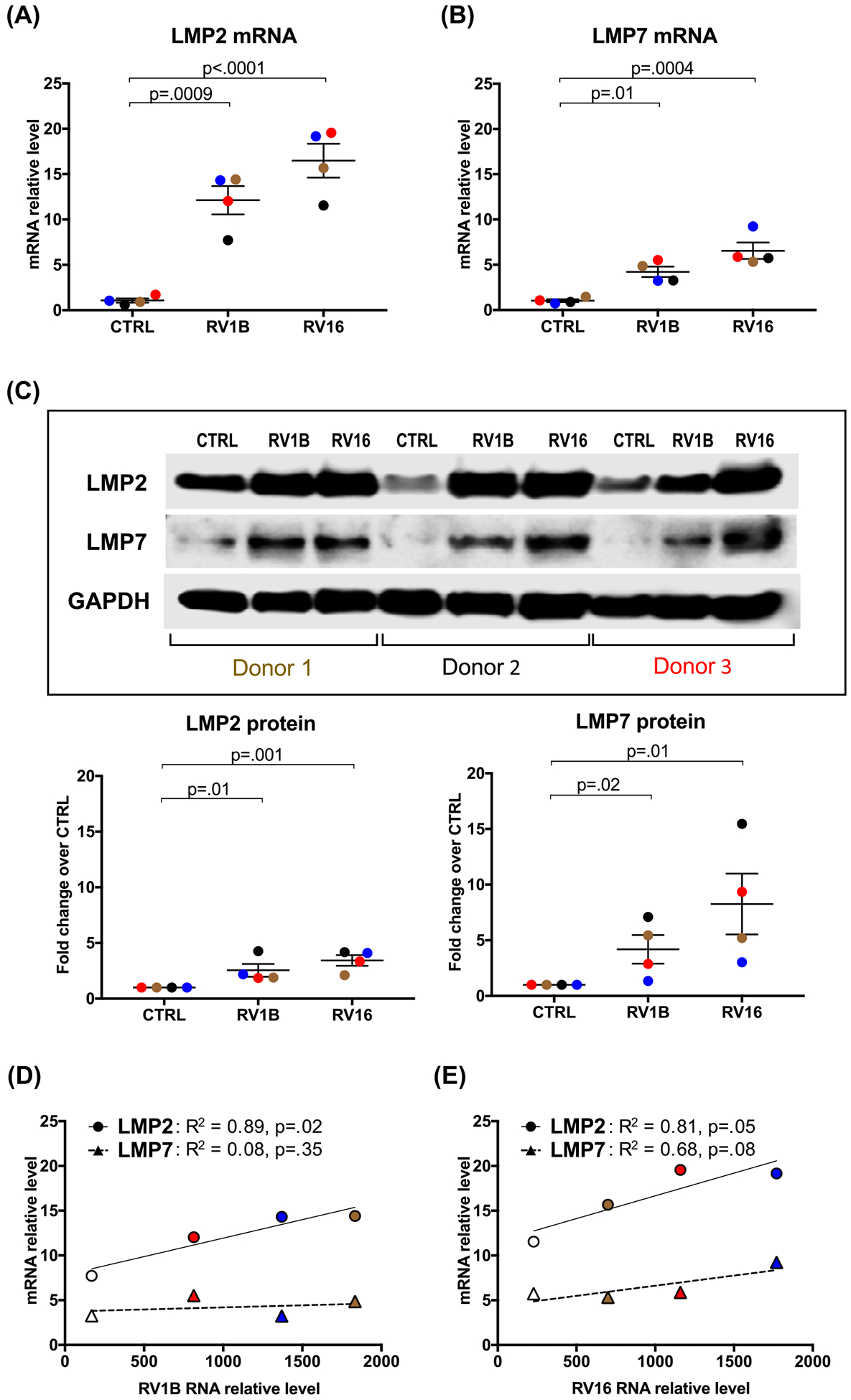
HTBE cells (n=4 subjects, as represented by different color symbols) grown at ALI were infected with RV1B and RV16 or treated with 0.25% BSA-PBS (control, CTRL) for 24 and 48 h. mRNA levels of IP subunits LMP2 (A) and LMP7 (B) were quantitated by RT-PCR (24 h) and were normalized to GAPDH gene. (C) Representative Western blot images and densitometry of LMP2 and LMP7 proteins at 48 h post RV infection. Correlations between RV1B (D) and RV16 (E) load and the levels of LMP2 and LMP7 mRNA. Data were presented as means ± S.E.M. and analyzed using ANOVA.
To further demonstrate the role of RV infection in the induction of IP subunits, we correlated the levels of RV RNA with those of LMP2 and LMP7 mRNA. There was a significant positive correlation between RV1B or RV16 load and LMP2, but not LMP7 mRNA expression (Figure 1D and E). This may suggest that the LMP2 subunit might be more involved in the induction of IP during RV infection compared with LMP7.
IP subunit inhibition increases viral load
To investigate the role of IP in regulating airway epithelial antiviral genes and viral load, we used a pharmacological inhibitor ONX-0914, a highly selective inhibitor of LMP7 [14,36,37]. Application of ONX-0914 in RV-infected HTBE cells cultured at ALI significantly reduced antiviral genes MX1 and OAS1 (Figure 2A and B), and increased the viral load (Figure 2C). Interestingly, ONX-0914 in the absence of RV infection did not significantly change the expression of LMP2 and LMP7 proteins (Figure 2D). However, in the presence of RV16 infection, ONX-0914 significantly inhibited the induction of LMP2 and LMP7. Collectively, these data support our hypothesis that RV-induced IP serves as a mechanism against viral infection.
Figure 2. Inhibition of immunoproteasomes down-regulates antiviral genes and increases viral load in human tracheobronchial epithelial (HTBE) cells.

HTBE cells (n=4 subjects, as represented by different color symbols) grown at ALI were pre-treated with ONX-0914 for 2 h prior to RV16 infection or 0.25% BSA-PBS treatment (control, CTRL). Cells were harvested after 48 h of infection for measurement of antiviral genes MX1 (A) and OAS1 (B) and RV load (C) by RT-PCR and were normalized to GAPDH gene. (D) Densitometry of LMP2 and LMP7 proteins and representative Western blot images from two subjects. Data were presented as means ± S.E.M. and analyzed using ANOVA.
IFN-λ promotes the induction of IP in primary human airway epithelial cells
To determine how RV infection induces IP expression, we examined the role of type III IFN (IFN-λ) due to the increasing reports on its protective role against viral infection in epithelial cells [23–26]. We found that IFN-λ was significantly up-regulated during RV infection in HTBE cells (Figure 3A). Also, IFN-λ-treated cells increased LMP2 and LMP7 mRNA levels (Figure 3B and C).
Figure 3. Interferon (IFN)-λ induces immunoproteasome expression in primary human airway epithelial cells.

HTBE cells (n=4 subjects, as represented by different color symbols) grown at ALI were treated with RV16, recombinant human IFN-λ protein (10 ng/ml) or 0.25% BSA-PBS (control, CTRL) for 48 h to measure IFN-λ (A), LMP2 (B), and LMP7 (C) by RT-PCR and were normalized to GAPDH gene. Data were presented as means ± S.E.M. and analyzed using ANOVA.
To determine the molecular mechanisms underlying IP induction by IFN-λ during RV infection, we carried out the gene knockdown experiment of IFN-λ receptor 1 (IFN-λR1) using siRNA in submerged culture of airway epithelial cells. IFN-λR1 siRNA reduced its mRNA and protein expression by approximately 40% (P=0.03; Figure 4A and B). IFN-λR1 protein expression was also decreased in IFN-λR1 siRNA-transfected cells infected with RV16 (Figure 4B). In Scr siRNA-transfected cells, RV16 infection resulted in a significant increase in LMP2 and LMP7 subunits, which was consistent with our ALI culture results of IP induction by RV. IFN-λR1 siRNA in RV-infected cells significantly decreased LMP2 and LMP7 protein expression (Figure 4B), and increased the viral load (Figure 4C). This suggests that RV-induced IFN-λ signaling is in part responsible for IP up-regulation during RV infection.
Figure 4. Small interfering RNA (siRNA)-mediated knockdown of IFN-λR1 decreases immunoproteasome subunit expression in rhinovirus-infected human tracheobronchial epithelial (HTBE) cells.

HTBE cells in submerged culture (n=5 subjects, as represented by different color symbols) were transfected with IFN-λR1 siRNA or Scr siRNA control, and then treated with or without RV16 at MOI 10. Cells were harvested after 24 h of infection to verify knockdown of IFN-λR1 mRNA (A) and protein (B). (B) – LMP2 and LMP7 protein densitometric data and representative Western blot images. (C) – RV load was measured by RT-PCR and was normalized to GAPDH gene. Data were presented as means ± S.E.M. and analyzed using ANOVA.
Involvement of IP subunit LMP2 in airway epithelial RV infection
Having explicitly demonstrated the role of LMP7 in RV-infected human airway epithelial cells by using the LMP7 inhibitor, we next aimed to assess the specific role of LMP2 subunit against viral infection by leveraging our ALI culture system of LMP2-deficient mTECs. Like HTBE cells, WT mTECs infected with RV1B showed a significant increase in LMP2 mRNA expression (Figure 5A). As expected, LMP2 KO mTECs did not express LMP2 in the absence or presence of RV infection. LMP2 deficiency resulted in a significant increase in RV levels compared with the WT cells (Figure 5B), which was coupled with up-regulation of antiviral gene MX1 in LMP2 KO cells infected with RV1B (Figure 5C).
Figure 5. LMP2-deficient (LMP2 KO) mouse tracheal epithelial cells (mTECs) increase viral load.

WT and LMP2 KO mTECs grown at ALI were infected with RV1B for 24 h. The IP subunit LMP2 (A), RV load (B), and MX1 (C) were measured by RT-PCR (n=9 from three independent experiments) and was normalized to 18S gene. Data were presented as medians and analyzed by the Kruskal–Wallis test (A,C) or the Mann–Whitney test (B).
Higher levels of RV and neutrophils in BAL fluid of RV1B-infected LMP2 KO mice
To establish the in vivo role of IP against viral infection, we used a model of RV infection in LMP2-deficient mice. After 24 h of intranasal inoculation of RV1B, WT mice increased LMP2 mRNA level in the lungs (Figure 6A), which is in line with our in vitro results (Figures 1 and 4). Importantly, viral load in the BAL fluid of LMP2 KO mice was increased compared with the C57 WT mice (Figure 6B). LMP2 deficiency (compared with sufficiency) also resulted in increased total number and percentage of neutrophils in the BAL fluid following RV1B infection (Figure 6C and D). Of note, no difference in the number of other types of leukocytes such as macrophages and lymphocytes was observed between the two groups (data not shown). Collectively, this suggests that inhibition of LMP2 subunit enhances viral infection and inflammation in vivo.
Figure 6. LMP2 knockout (KO) mice are more susceptible to RV infection than the WT C57BL/6 (C57 WT) mice.

Mice were intranasally inoculated with PBS or RV1B at 107 PFU/mouse for 24 h. (A) – LMP2 mRNA levels were measured in the left lung lobe by RT-PCR and was normalized to 18S gene. (B) – RV load was measured in BAL fluid by RT-PCR. Total (C) and percentage (D) of neutrophils were counted in BAL. n=7–15 mice per group. Data were presented as medians and analyzed by the Kruskal–Wallis test (A,C,D) or the Mann–Whitney test (B).
Discussion
The present study demonstrates the first evidence for a role of IP in respiratory RV infection. Specifically, we demonstrated that RV-induced IP is critical to airway defense against viral infection in cultured human and mouse airway epithelial cells as well as in mice. We have also, for the first time, shown the contribution of IFN-λ to IP induction during RV infection in human airway epithelium.
Although several groups have examined the role of IP in animal models of viral infection [19,21], little is known regarding the expression and function of IP in the human airways exposed to respiratory viruses. The classical role of IP in immune responses has been attributed to its regulation of the antigen-processing machinery [19,21]. In the current study, we investigated a non-classical role of IP in airway epithelium. First, we found that RV infection in well-differentiated human primary airway epithelial cells strongly and consistently induced IP expression. Second, by using several complementary approaches such as inhibition or deletion of IP activity or expression, we have revealed the in vitro and in vivo antiviral functions of IP subunits LMP2 and LMP7 during airway RV infection.
How IP is regulated during airway RV infection is poorly understood. Type II IFN-γ is one of the main pro-inflammatory cytokines that induce the formation of IP. However, IFN-γ is secreted by immune cells but not by airway epithelial cells. Recent studies demonstrated that IFN-λ contributes to the defense against viruses including RV [24,38]. Our data, for the first time, suggest IFN-λ as an upstream regulator of RV-induced IP. We found increased IFN-λ in airway epithelial cells infected with RV as well as induction of LMP2 and LMP7 by IFN-λ. We further established IFN-λ as an enhancer in RV-induced IP using siRNA-mediated knockdown of IFN-λR1, an essential receptor involved in the function of type III IFNs in airway epithelial cells [26]. IFN-λR1 knockdown in RV-infected human airway epithelial cells decreased LMP2 and LMP7 expression, which was paralleled with increased viral load. Over-all, our findings demonstrate a critical role of the IFN-λ/IFN-λR1 axis in RV-induced IP expression and antiviral function. However, how the IFN-λ/IFN-λR1 axis up-regulates LMP2 and LMP7 expression warrants further investigation. IFN-λ, like IFN-γ, is known to activate STAT1 signaling [39,40], and promoters of LMP2 and LMP7 have putative binding sites of STAT1 [41,42]. Thus, it is possible that STAT1 activation following IFN-λ stimulation during RV infection would increase LMP2 or LMP7, but this hypothesis needs to be tested in our future experiments. Although we examined the role of IFN-λ in IP induction and function, we are aware that type I IFN signaling may also contribute to IP induction during RV infection as IFN-α and IFN-β can be up-regulated by RV infection [43] and they do activate STAT1. Nonetheless, revealing the role of IFN-λ as a new mechanism in IP induction and antiviral function will further improve our understanding of airway defense against RV infection.
The classical function of IP is on the adaptive immune response by generating peptides for antigen presentation [8,9]. In the current study, we focused on the innate immune functions of IP in RV-infected airway epithelium, a non-classical immune cell type that has not been well-studied for its utilization of the IP mechanism. Our findings from the in vitro and in vivo studies using the LMP2 or LMP7 deficiency or inhibition approach collectively support the antiviral function of IP during RV infection. By using a highly selective LMP7 inhibitor, we demonstrated less induction of antiviral genes but increased viral load in RV-infected human primary airway epithelial cells. Interestingly, results from cultured LMP2-deficient (compared with sufficient) mTECs showed increased viral load as well as antiviral genes. Our results suggest that LMP7 may act primarily as an enhancer of antiviral gene expression with sub-sequent reduction in viral infection. On the other hand, LMP2 may directly inhibit viral infection (e.g. replication). Although the exact mechanisms underlying the antiviral function of IP remain unclear, it is possible that IP-mediated antiviral gene expression and other mechanisms such as degradation of viral proteins may be involved. Also, as LMP2 deficiency likely changes the IP structure, the effect of LMP2 deficiency on antiviral gene expression may not depend on LMP2 activity as previous studies show the distinct role of the structure compared with activity of IP subunits in IP functions [44,45]. Likewise, the effect of LMP7 inhibition by ONX-0914 may be independent of the IP structure. Whether this mechanistic difference explains the varying effect of LMP2 deficiency and LMP7 inhibition on antiviral gene expression during viral infection deserves further investigation.
Importantly, by using the RV infection mouse model, we have shown the in vivo functions of IP. The increased viral load in LMP2-deficient mouse airway lumen coupled with high levels of neutrophils suggests an anti-inflammatory function of IP in the lung. Our finding is in agreement with studies from a mouse model of Candida albicans infection treated with ONX-0914 [36] as well as from proteasome dysfunction-associated autoimmune diseases showing massive infiltration of neutrophils [46,47]. Excessive neutrophil accumulation in the airways is a pathological feature of patients with RV-associated acute asthma exacerbations [48–50]. Whether IP deficiency exists in those asthma patients is unclear. Our data regarding the antiviral and anti-inflammatory functions of IP will encourage clinicians and scientists to further determine IP regulation in patients and to explore the possibility to enhance IP expression and function for the treatment of RV infection.
There are several interesting findings from the current study that need further investigation. First, we found that LMP2, but not LMP7, significantly correlated with the viral load in cultured human airway epithelial cells. This could suggest that different subunits of IP may not be equally regulated by the same RV infection or they exert varying functions during viral infection. Second, the LMP7 inhibitor ONX-0914 was found to inhibit LMP7 and LMP2 expression in RV-infected human airway epithelial cells. At the concentration used (100 nM), ONX-0914 is highly selective to inhibit LMP7 enzymatic activity, but is not expected to directly reduce RV-induced IP expression. We propose that the decrease in RV-induced LMP7 expression following ONX-0914 treatment may be explained by the fact that the covalent modification of LMP7 with ONX-0914 may reduce its recognition by the anti-LMP7 antibody [51,52]. The reduction in RV-induced LMP2 expression following ONX-0914 may be secondary to the changes of antiviral genes as we observed the decrease in MX1 and OAS1 in ONX-0914-treated cells. Additionally, we found a trend of decreased IFN-λ in RV-infected cells with ONX-0914 treatment (data not shown). Third, we found that ONX-0914 led to the upper shift of LMP2 protein on the gel. While we do not know the exact reason for this upper shift of LMP2 protein, it likely reflects an accumulation of the LMP2 precursor protein including N-terminal amino acids that could not be cleaved by the inhibited LMP7 during proteasome maturation [9,53].
One limitation of the present study is that we focussed on IP protein expression, but not its activity in response to RV infection. Future experiments will be planned to correlate IP protein expression with its activity. Another limitation is that we examined the regulation and function of IP during infection without inclusion of asthma or other lung disease conditions. Future studies are warranted to define how asthma or other lung diseases impact IP levels and functions.
In conclusion, we found that RV infection induces IP both in vitro and in vivo, which in turn enhances the host antiviral mechanism. Discovery of IP as a key component of airway defense against RV infection would provide novel insight into the development of new strategies to eliminate airway viruses from patients suffering from various respiratory diseases.
Clinical perspectives.
Respiratory rhinovirus infection is a common, but significant clinical problem in patients suffering from various lung diseases such as asthma. However, no effective therapy is available for this viral infection. Immunoproteasomes may be involved in the pathophysiology of lung diseases, but their role in respiratory RV infection has not been explored.
We have provided the first evidence that rhinovirus induces immunoproteasomes in human primary airway epithelial cell culture and mouse models, and uncovered the antiviral function of immunoproteasomes during acute rhinovirus infection. Mechanistically, we found that virus-induced IFN-λ contributes to immunoproteasome induction.
Our basic research findings will provide rationale for future clinical studies looking into immunoproteasomes in airways of patients with various lung diseases, and designing new therapeutic approaches to enhance immunoproteasome expression and function in patients who have deficiency in immunoproteasomes and experience exacerbations associated with viral infection.
Funding
This work was supported by the National Institutes of Health [grant numbers R01 HL122321, R01 AI106287, R01 HL125128, U19 AI125357].
Abbreviations
- ALI
air–liquid interface
- BAL
bronchoalveolar lavage
- COPD
chronic obstructive pulmonary disease
- CT
threshold cycle
- HTBE
human tracheobronchial epithelial
- IFN
interferon
- IFN-λR1
IFN-λ Receptor 1
- IP
immunoproteasome
- KO
knockout
- mEGF
mouse epidermal growth factor
- mTECs
mouse tracheal epithelial cells
- PFU
plaque-forming unit
- p.i.
post-infection
- RV
rhinovirus
- Scr
scrambled
- siRNA
small interfering RNA
- WT
wild-type
Footnotes
Competing interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Bartlett NW, Singanayagam A and Johnston SL (2015) Mouse models of rhinovirus infection and airways disease. Methods Mol. Biol 1221,181–188, [DOI] [PubMed] [Google Scholar]
- 2.Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T et al. (2008) Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc. Natl. Acad. Sci. U.S.A 105, 13562–13567, 10.1073/pnas.0804181105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai TR, Vonk JM, Postma DS and Boezen HM (2007) Severe exacerbations predict excess lung function decline in asthma. Eur. Respir. J 30, 452–456, 10.1183/09031936.00165106 [DOI] [PubMed] [Google Scholar]
- 4.Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V et al. (2005) Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med 201, 937–947, 10.1084/jem.20041901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castillo JR, Peters SP and Busse WW (2017) Asthma exacerbations: pathogenesis, prevention, and treatment. J. Allergy Clin. Immunol. Pract 5, 918–927, 10.1016/j.jaip.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinke JW and Borish L (2016) Immune responses in rhinovirus-induced asthma exacerbations. Curr. Allergy Asthma Rep 16, 78, 10.1007/s11882-016-0661-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker TA, Bach HH, Gamelli RL, Love RB and Majetschak M (2014) Proteasomes in lungs from organ donors and patients with end-stage pulmonary diseases. Physiol. Res 63, 311–319 [DOI] [PubMed] [Google Scholar]
- 8.McCarthy MK and Weinberg JB (2015) The immunoproteasome and viral infection: a complex regulator of inflammation. Front. Microbiol 6, 21, 10.3389/fmicb.2015.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrington DA and Gregerson DS (2012) Immunoproteasomes: structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci 109, 75–112, 10.1016/B978-0-12-397863-9.00003-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niewerth D, Kaspers GJ, Assaraf YG, van Meerloo J, Kirk CJ, Anderl J et al. (2014) Interferon-gamma-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. J. Hematol. Oncol 7, 7, 10.1186/1756-8722-7-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aki M, Shimbara N, Takashina M, Akiyama K, Kagawa S, Tamura T et al. (1994) Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J. Biochem 115, 257–269, 10.1093/oxfordjournals.jbchem.a124327 [DOI] [PubMed] [Google Scholar]
- 12.Verbrugge SE, Scheper RJ, Lems WF, de Gruijl TD and Jansen G (2015) Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. Ther 17, 17, 10.1186/s13075-015-0529-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basler M, Kirk CJ and Groettrup M (2013) The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol 25, 74–80, 10.1016/j.coi.2012.11.004 [DOI] [PubMed] [Google Scholar]
- 14.Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C et al. (2009) A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med 15, 781–787, 10.1038/nm.1978 [DOI] [PubMed] [Google Scholar]
- 15.Lundh M, Bugliani M, Dahlby T, Chou DH, Wagner B, Ghiasi SM et al. (2017) The immunoproteasome is induced by cytokines and regulates apoptosis in human islets. J. Endocrinol 233, 369–379, 10.1530/JOE-17-0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kammerl IE, Dann A, Mossina A, Brech D, Lukas C, Vosyka O et al. (2016) Impairment of immunoproteasome function by cigarette smoke and in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med 193, 1230–1241, 10.1164/rccm.201506-1122OC [DOI] [PubMed] [Google Scholar]
- 17.Fujino N, Ota C, Takahashi T, Suzuki T, Suzuki S, Yamada M et al. (2012) Gene expression profiles of alveolar type II cells of chronic obstructive pulmonary disease: a case-control study. BMJ Open 2, 10.1136/bmjopen-2012-001553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basler M, Beck U, Kirk CJ and Groettrup M (2011) The antiviral immune response in mice devoid of immunoproteasome activity. J. Immunol 187, 5548–5557, 10.4049/jimmunol.1101064 [DOI] [PubMed] [Google Scholar]
- 19.Shin EC, Seifert U, Kato T, Rice CM, Feinstone SM, Kloetzel PM et al. (2006) Virus-induced type I IFN stimulates generation of immunoproteasomes at the site of infection. J. Clin. Invest 116, 3006–3014, 10.1172/JCI29832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kammerl IE and Meiners S (2016) Proteasome function shapes innate and adaptive immune responses. Am. J. Physiol. Lung Cell. Mol. Physiol 311, L328–L336 [DOI] [PubMed] [Google Scholar]
- 21.Keller IE, Vosyka O, Takenaka S, Kloβ A, Dahlmann B, Willems LI et al. (2015) Regulation of Immunoproteasome function in the lung. Sci. Rep 5, 10230, 10.1038/srep10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robek MD, Garcia ML, Boyd BS and Chisari FV (2007) Role of immunoproteasome catalytic subunits in the immune response to hepatitis B virus. J. Virol 81, 483–491, 10.1128/JVI.01779-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulraiz F, Bellinghausen C, Dentener MA, Reynaert NL, Gaajetaan GR, Beuken EV et al. (2014) Efficacy of IFN-lambda1 to protect human airway epithelial cells against human rhinovirus 1B infection. PLoS ONE 9, e95134, 10.1371/journal.pone.0095134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V et al. (2010) Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol 84, 5670–5677, 10.1128/JVI.00272-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S and Baldridge MT (2017) Interferon-lambda: a potent regulator of intestinal viral infections. Front. Immunol 8, 749, 10.3389/fimmu.2017.00749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW et al. (2006) Role of deficient type III interferon-lambda production in asthma exacerbations. Nat. Med 12, 1023–1026, 10.1038/nm1462 [DOI] [PubMed] [Google Scholar]
- 27.Wu Q, van Dyk LF, Jiang D, Dakhama A, Li L, White SR et al. (2013) Interleukin-1 receptor-associated kinase M (IRAK-M) promotes human rhinovirus infection in lung epithelial cells via the autophagic pathway. Virology 446, 199–206, 10.1016/j.virol.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Q, Jiang D, Smith S, Thaikoottathil J, Martin RJ, Bowler RP et al. (2012) IL-13 dampens human airway epithelial innate immunity through induction of IL-1 receptor-associated kinase M. J. Allergy Clin. Immunol 129, 825e2–833e2, 10.1016/j.jaci.2011.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berman R, Jiang D, Wu Q and Chu HW (2016) alpha1-Antitrypsin reduces rhinovirus infection in primary human airway epithelial cells exposed to cigarette smoke. Int. J. Chron. Obstruct. Pulmon. Dis 11, 1279–1286, 10.2147/COPD.S105717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gross CA, Bowler RP, Green RM, Weinberger AR, Schnell C and Chu HW (2010) beta2-agonists promote host defense against bacterial infection in primary human bronchial epithelial cells. BMC Pulm. Med 10, 30, 10.1186/1471-2466-10-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu HW, Rios C, Huang C, Wesolowska-Andersen A, Burchard EG, O’Connor BP et al. (2015) CRISPR-Cas9-mediated gene knockout in primary human airway epithelial cells reveals a proinflammatory role for MUC18. Gene Ther 22, 822–829, 10.1038/gt.2015.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stevenson C, Jiang D, Schaefer N, Ito Y, Berman R, Sanchez A et al. (2017) MUC18 regulates IL-13-mediated airway inflammatory response. Inflamm. Res 66, 691–700, 10.1007/s00011-017-1050-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wenzel SE, Trudeau JB, Barnes S, Zhou X, Cundall M, Westcott JY et al. (2002) TGF-beta and IL-13 synergistically increase eotaxin-1 production in human airway fibroblasts. J. Immunol 169, 4613–4619, 10.4049/jimmunol.169.8.4613 [DOI] [PubMed] [Google Scholar]
- 34.Loens K, van Loon AM, Coenjaerts F, van Aarle Y, Goossens H, Wallace P et al. (2012) Performance of different mono- and multiplex nucleic acid amplification tests on a multipathogen external quality assessment panel. J. Clin. Microbiol 50, 977–987, 10.1128/JCM.00200-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Rijn AL, Claas EC, von dem Borne PA, Kroes ACM and de Vries JJC (2017) Rhinovirus viremia in adult patients with high viral load in bronchoalveolar lavages. J. Clin. Virol 96, 105–109, 10.1016/j.jcv.2017.10.007 [DOI] [PubMed] [Google Scholar]
- 36.Mundt S, Basler M, Buerger S, Engler H and Groettrup M (2016) Inhibiting the immunoproteasome exacerbates the pathogenesis of systemic Candida albicans infection in mice. Sci. Rep 6, 19434, 10.1038/srep19434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Althof N, Goetzke CC, Kespohl M, Voss K, Heuser A, Pinkert S et al. (2018) The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. EMBO Mol. Med 10, 200–218, 10.15252/emmm.201708089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR and Paludan SR (2006) Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol 80, 4501–4509, 10.1128/JVI.80.9.4501-4509.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamauchi S, Takeuchi K, Chihara K, Honjoh C, Kato Y, Yoshiki H et al. (2016) STAT1 is essential for the inhibition of hepatitis C virus replication by interferon-lambda but not by interferon-alpha. Sci. Rep 6, 38336, 10.1038/srep38336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dickensheets H, Sheikh F, Park O, Gao B and Donnelly RP (2013) Interferon-lambda (IFN-lambda) induces signal transduction and gene expression in human hepatocytes, but not in lymphocytes or monocytes. J. Leukoc. Biol 93, 377–385, 10.1189/jlb.0812395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatterjee-Kishore M, Wright KL, Ting JP and Stark GR (2000) How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 19, 4111–4122, 10.1093/emboj/19.15.4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foss GS and Prydz H (1999) Interferon regulatory factor 1 mediates the interferon-gamma induction of the human immunoproteasome subunit multicatalytic endopeptidase complex-like 1. J. Biol. Chem 274, 35196–35202, 10.1074/jbc.274.49.35196 [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Hamati E, Lee PK, Lee WM, Wachi S, Schnurr D et al. (2006) Rhinovirus induces airway epithelial gene expression through double-stranded RNA and IFN-dependent pathways. Am. J. Respir. Cell Mol. Biol 34, 192–203, 10.1165/rcmb.2004-0417OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basler M, Lauer C, Moebius J, Weber R, Przybylski M, Kisselev AF et al. (2012) Why the structure but not the activity of the immunoproteasome subunit low molecular mass polypeptide 2 rescues antigen presentation. J. Immunol 189, 1868–1877, 10.4049/jimmunol.1103592 [DOI] [PubMed] [Google Scholar]
- 45.Gileadi U, Moins-Teisserenc HT, Correa I, Booth BL Jr, Dunbar PR, Sewell AK et al. (1999) Generation of an immunodominant CTL epitope is affected by proteasome subunit composition and stability of the antigenic protein. J. Immunol 163, 6045–6052 [PubMed] [Google Scholar]
- 46.Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M et al. (2011) A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Invest 121, 4150–4160, 10.1172/JCI58414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S et al. (2012) Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 64, 895–907, 10.1002/art.33368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fahy JV (2009) Eosinophilic and neutrophilic inflammation in asthma: insights from clinical studies. Proc. Am. Thorac. Soc 6, 256–259, 10.1513/pats.200808-087RM [DOI] [PubMed] [Google Scholar]
- 49.Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF and Barnes PJ (1999) Neutrophilic inflammation in severe persistent asthma. Am. J. Respir. Crit. Care Med 160, 1532–1539, 10.1164/ajrccm.160.5.9806170 [DOI] [PubMed] [Google Scholar]
- 50.Ray A and Kolls JK (2017) Neutrophilic inflammation in asthma and association with disease severity. Trends Immunol. 38, 942–954, 10.1016/j.it.2017.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M et al. (2012) Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148, 727–738, 10.1016/j.cell.2011.12.030 [DOI] [PubMed] [Google Scholar]
- 52.Federspiel JD, Codreanu SG, Goyal S, Albertolle ME, Lowe E, Teague J et al. (2016) Specificity of protein covalent modification by the electrophilic proteasome inhibitor carfilzomib in human cells. Mol. Cell. Proteomics 15, 3233–3242, 10.1074/mcp.M116.059709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Griffin TA, Nandi D, Cruz M, Fehling HJ, Kaer LV, Monaco JJ et al. (1998) Immunoproteasome assembly: cooperative incorporation of interferon gamma (IFN-gamma)-inducible subunits. J. Exp. Med 187, 97–104, 10.1084/jem.187.1.97 [DOI] [PMC free article] [PubMed] [Google Scholar]