

Abstract
Angiotensin-converting enzyme 2 (ACE2) counterbalances with ACE and functions as a negative regulator of the renin–angiotensin system (RAS). The importance of RAS in acute respiratory distress syndrome (ARDS) has recently re-emerged owing to the identification of ACE2 as a receptor for the SARS-coronavirus. Recent studies have demonstrated that ACE2 protects mice from acute lung injury as well as SARS-mediated lung injury. We review the role of the RAS, in particular ACE2, in the pathogenesis of ARDS.
Section editors:
Terry Delovitch – The John P. Robarts Research Institute, London, Ont., Canada
David Scott – University of Maryland School of Medicine, Baltimore, MD, USA
Introduction
Angiotensin-converting enzyme (ACE) and ACE2 share homology in their catalytic domain and provide different key functions in the renin–angiotensin system (RAS). ACE cleaves angiotensin I (Ang I) to generate angiotensin II (Ang II) [1, 2], whereas ACE2 reduces angiotensin II levels [3, 4] and is a negative regulator of the system. The importance of the RAS in acute respiratory distress syndrome (ARDS) has recently re-emerged owing to the identification of ACE2 as a receptor for the severe acute respiratory syndrome-coronavirus (SARS-CoV) [5]. In 2003, SARS spread throughout the world causing more than 800 deaths due to ARDS [6]. Our studies demonstrated that ACE2 protects mice from acute lung injury [7] as well as SARS-mediated lung injury [8]. In this article, we review the role of the RAS, in particular ACE2, in the pathogenesis of ARDS. The possible applications of modulating the RAS for the treatment of ARDS are also discussed.
Genetic susceptibilities to ARDS
ARDS is the most severe form of acute lung injury. Multiple predisposing factors for ARDS have been reported and include sepsis, acid aspiration, trauma or infections with viral pathogens such as SARS-CoV. Moreover, very recently H5N1 avian influenza virus has caused high lethality due to ARDS [9]. Despite recent progress, the mortality associated with ARDS remains very high, and there is no pharmacological therapy that has been shown in large-scale trials to impact mortality in ARDS [10].
Factors predicting the onset or severity of ARDS are poorly understood, but the low incidence of ARDS in the relatively large group of patients at risk has been shown. Also, there are large differences in plasma ACE concentrations between individuals but such concentrations tend to be similar within families [11]. These suggest the involvement of genetic components in the pathogenesis of ARDS. The human ACE gene (dcp1) is located on chromosome 17q23 and contains a restriction fragment length polymorphism defined by the presence (insertion, I) or absence (deletion, D) of a 287-bp alu repeat sequence in intron 16. Nearly 50% of the variance in plasma ACE activity can be accounted for by this ACE insertion/deletion (I/D) polymorphism, the D allele being associated with higher ACE activity [12]. Importantly, recent cohort studies of ARDS showed a significant association between an ACE I/D polymorphism and the susceptibility and mortality of ARDS [13]. The D/D genotype frequency was increased in patients with ARDS compared with the control cohort. Ninety-six patients fulfilling the criteria for ARDS were genotyped for the ACE polymorphism together with individuals from three comparison groups: 88 patients with non-ARDS respiratory failure ventilated in the Intensive Care Unit (ICU), 174 ICU patients undergoing coronary artery bypass grafting and 1906 individuals from a general population group. DD genotype frequency was increased in the patients with ARDS compared with the ICU (P = 0.00008), coronary artery bypass grafting (P = 0.0009) and the general population group (P = 0.00004). In addition, the ACE D/D allele significantly correlated with mortality in the ARDS group. Another study showed that patients carrying the ACE I/I genotype have a significantly increased survival rate [14]. In that study, the 28-day mortality rates were significantly different in the three ACE genotypes (42%, 65% and 75% for I/I, I/D and D/D, respectively; P = 0.036) in the ARDS group. Taken together, these data suggest a potential role for RAS in the pathogenesis of ARDS and implicate genetic factors in susceptibility and progression of this syndrome.
Clinical correlations between the RAS and ARDS
In addition to genetic correlation studies, substantial clinical studies suggest the possible activation of RAS in patients with ARDS. For instance, in patients with ARDS, an elevation of ACE levels in bronchoalveolar lavage fluid has been observed despite a reduction in circulating ACE [15]. Reduced circulating ACE levels may reflect loss of enzyme release from a damaged pulmonary vascular endothelium and may not be representative of activity in the lung compartment. Also, it has been reported that the pulmonary capillary endothelium-bound (PCEB) ACE activity correlates with the severity of lung injury in the patients with acute lung injury [16]. These data suggest the possible activation of RAS in patients with ARDS. Interestingly, recent retrospective epidemiological studies demonstrated that the prior outpatient use of an ACE inhibitor was associated with decreased mortality in patients hospitalized with community-acquired pneumonia, although further studies are needed to examine whether ACE inhibitors are protective when used in an inpatient settings for patients lacking traditional indications for the use of these medications [17].
Protective role of ACE2 in ARDS
In 2000, a novel homologue of ACE was cloned, termed ACE2 [3, 4]. ACE and ACE2 share homology in their catalytic domain and provide different key functions in the RAS. ACE cleaves angiotensin I to generate angiotensin II [1, 2], whereas ACE2 reduces angiotensin II levels [3, 4]. The biological effects of Ang II are mediated through two specific receptors, Ang II receptor type 1 (AT1R) and Ang II receptor type 2 (AT2R) [1, 2]. The targeted disruption of murine ACE2 resulted in increased Ang II levels, impaired cardiac contractility in aged mice, upregulation of hypoxia-induced genes in the heart [18] and worsened heart failure following aortic banding [19]. Loss of ACE on an ACE2 background or pharmacological inhibition of the RAS can at least partially reverse the cardiac phenotypes [18]. These genetic data demonstrate that ACE2 counterbalances the function of ACE and negatively regulates Ang II levels within the RAS (Fig. 1 ).
Figure 1.
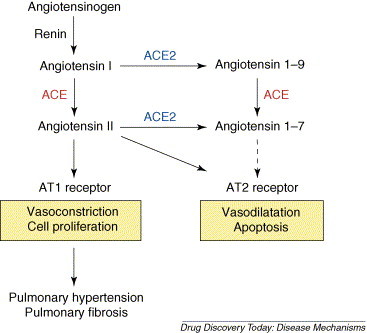
Current view of the renin–angiotensin system. Angiotensin I serves as a substrate for both ACE and ACE2. Angiotensin II is known to act as vasoconstrictor as well as mitogen for smooth muscle cells or fibroblasts, mainly through the angiotensin II type 1 receptor. The function of angiotensin 1–9 is not well understood. Both ACE and ACE2 are involved in the production of the vasodilator peptide angiotensin 1–7.
Our group has investigated the role of ACE2 in ARDS using ace2 knockout mice [7]. In acid aspiration-induced ARDS, endotoxin-induced ARDS, as well as peritoneal sepsis-induced ARDS, ace2 knockout mice exhibited very severe disease compared with control mice that express ACE2. Loss of ACE2 expression in mutant mice also resulted in enhanced vascular permeability, increased lung edema, neutrophil accumulation and worsened lung function. Of note, these pathologic manifestations occurred without apparent changes in heart contractility or pulmonary vascular tone among the experimental groups. Mechanistically, the negative regulation of Ang II levels by ACE2 accounts, in part, for the protective function of ACE2 in ARDS. For example, AT1 inhibitor treatment or additional ace gene deficiency on an ace2 knockout background rescues the severe phenotype of ace2 single mutant mice in acute lung injury. In addition, ace knockout mice and AT1R knockout mice, but not AT2R knockout mice, showed improved symptoms of acute lung injury. Therefore, in acute lung injury, ACE, Ang II and AT1R promote acute lung injury, whereas ACE2 and the AT2R protect from lung injury [7]. Importantly, the treatment with catalytically active, but not enzymatically inactive, recombinant ACE2 protein improved the symptoms of acute lung injury in wild type mice as well as in ace2 knockout mice, suggesting ACE2 protein as a possible novel therapeutic target for ARDS (Fig. 2 ).
Figure 2.
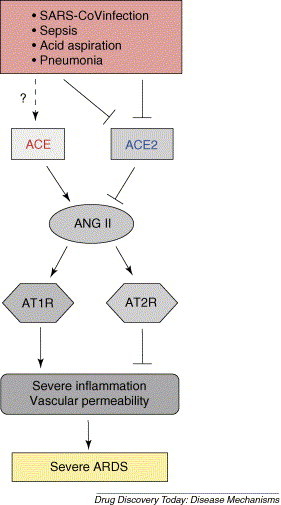
Schematic diagram of the proposed role of the renin–angiotensin system in development of ARDS. In ARDS induced by SARS-CoV infections, acid aspiration, pneumonias, sepsis or other pathogenic conditions, the generation of Ang II from Ang I is mediated by ACE. Ang II contributes to acute lung failure through stimulation of the angiotensin II type 1 receptor (AT1R), whereas ACE2 and angiotensin II type 2 receptor (AT2R) negatively regulate this pathway and protect from acute lung failure. However, additional ACE2-regulated, but Ang II-independent pathways seem to also contribute to ARDS.
Role of ACE2 in SARS infections and SARS-mediated lung injury
Within a few months following the publication of the SARS-CoV genome, ACE2 was identified as a potential receptor in cell line studies in vitro [5]. ACE2 binds SARS-CoV spike and supports ‘syncytia formation’, the fusion of spike-protein expressing cells into large multinucleated cells that can be also seen in ‘real’ SARS infections. Using a mouse SARS infection model with ace2 knockout mice, our group then provided evidence that ACE2 is indeed essential for SARS infections in vivo [8]. When ace2 knockout mice are infected with the SARS coronavirus, they were resistant to virus infection. Virus titers from the lung tissues of infected ace2 knockout mice were 105-fold lower than that isolated from the lungs of SARS-CoV-infected wild type mice. Thus, without excluding a second functional SARS-CoV receptor, ACE2 is an essential receptor for SARS infections in vivo.
Despite many studies on SARS-CoV, one mystery of SARS-CoV is why, in contrast to the other coronaviruses, infections with the SARS-CoV trigger severe lung disease with such high mortality. Accumulating evidence further indicates that severe SARS infections are dependent on the burden of viral replication as well as on the immunopathologic consequences of the host response. Our studies have implicated the involvement of the RAS in SARS pathogenesis. Intriguingly, wild type mice infected with SARS-CoV showed markedly downregulated ACE2 expression in lungs. Similarly, treatment with recombinant SARS-spike protein, in the absence of any other virus components, downregulates ACE2 expression in vitro and in vivo. Thus, SARS-CoV-infected or spike protein-treated wild type mice resemble ace2 knockout mice, and, similar to ace2 mutant mice, spike-treated wild type mice show markedly more severe pathology in acute lung injury. In addition, in spike-treated mice, Ang II peptide levels were increased and the worsened ARDS symptoms could be partially reversed by AT1R blocker treatment. Thus, the downregulation of ACE2 expression in SARS-CoV infections might play a causal role in SARS pathogenesis, especially in disease progression to ARDS.
RAS and pulmonary edema
Pulmonary circulation is a potentially important target for the RAS activation in the lung. For instance, Ang II via its type 1 receptor (AT1R) induces pulmonary vasoconstriction in response to hypoxia, suggesting important roles of AT1R in elevating pulmonary vascular tone that can result in pulmonary edema [20]. Also, infusion of Ang I [21] or Ang II [22] can produce pulmonary edema independent of catecholamine release. These data suggest an important role for the RAS in regulating pulmonary vascular tone, which might also contribute to the pathogenesis of ARDS.
In addition to increased vascular tone and a subsequent hydostatic edema formation, accumulating data suggest that Ang II also increases vascular permeability via AT1R, whereas stimulation of AT2 receptors exerts an opposite effect [23]. Several mediators have been implicated in Ang II regulated vascular permeability changes, including eicosanoids (i.e. leukotriene C4, prostaglandin E2 and I2), and vascular permeability factor. However, relatively few studies have addressed whether the RAS is involved in the increased vascular permeability observed in ARDS. Our study demonstrated that loss of ace2 results in increased vascular permeability using Evans Blue dye injections as an in vivo indicator of albumin leakage in mice. This vascular permeability was significantly attenuated in the lungs of AT1R mutant mice. These data indicate that loss of ACE2 expression and increased Ang II levels can trigger leaky pulmonary blood vessels through AT1R stimulation in ARDS [7].
Additional ACE and ACE2 targets in ARDS
Both ACE and ACE2 are unspecific proteases and can cleave additional substrates that might also play important roles in ACE/ACE2-regulated ARDS independent of Ang II. One of the ACE and ACE2 targets is bradykinin and its peptide metabolites. Bradykinin is the key effecter in the kallikrein–kinin system and also functions as a major proinflammatory mediator. Bradykinin is degraded by two main kinases, which are ACE (also known as kinase II in this system) and neutral endopeptidase. The biological effects of bradykinin are mediated by B1 receptor (B1R) and B2 receptor (B2R). B2R mediate most the known effects of bradykinin, including antiproliferative, antioxidant and antithrombotic effects [24]. The in vivo effects mediated via B1R or B2R are still poorly characterized. A recent study suggests that expression of B1R is induced by cytokines during inflammation in rats [25], albeit the role of bradykinin in Ang II-induced inflammation remains unclear.
ACE2 can remove in in vitro assays the C-terminal residue from apelin and other vasoactive peptides such as neurotensin and neurotensin-related peptide kinetensin. Moreover, the opioid peptides dynorphin A (1–13) and β-casamorphin are also substrates of ACE2. In contrast to ACE, ACE2 does not metabolize bradykinin, but catalytically inactivates both [des-Arg9]-bradykinin and lys[des-Arg9]-bradykinin [3, 26]. Thus, although many ACE2 functions have been attributed to the regulation of Ang II levels, Ang II itself is probably only part of the ACE2 story and other ACE2 substrates might play a major role in understanding ACE2 functions. Importantly, our own preliminary results suggest that inhibition of the bradykinin receptor can also alleviate ARDS (Y.I. and J.M.P., unpublished).
Perspectives
Recently, the critical importance of RAS in the pathogenesis of ARDS has been established in genetic animal models. ACE2 protects murine lungs from acute lung injury induced by acid aspiration, endotoxin shock and peritoneal sepsis [7]. In addition, ace knockout mice and AT1R knockout mice, but not AT2R knockout mice, showed improved symptoms of acute lung injury [7], consistent with other experimental studies [17, 27, 28]. Also, ACE2 has been identified as a key SARS-CoV receptor [5, 8] and importantly, ACE2 plays a protective role in SARS-CoV-mediated lung injury [8]. Therefore, in acute lung injury, ACE, Ang II and AT1R promote acute lung injury, whereas ACE2 and the AT2R protect from lung injury. These findings might provide the opportunity to develop recombinant ACE2 as a novel drug in ARDS along with AT1R inhibitor or ACE inhibitor to possibly treat emerging infectious lung diseases such as avian influenza A (H5N1) and other diseases that affect lung function (Table 1 ). In addition, as ACE2 is an unspecific protease, it would be also interesting to investigate the role of ACE2 and its metabolites including angiotensin-(1–7), des-Arg(9)-bradykinin, apelin or dynorphin in ARDS. We look forward to the use of angiotensin system-modulating agents/molecules, in particular ACE2, as novel therapeutic agents to treat severe acute lung failure, a syndrome that affects millions of people without any yet effective drug treatments.
Table 1.
Targets of modulating the renin–angiotensin system for the treatment of ARDS
Acknowledgements
We thank Chengyu Jiang, Shuan Rao and many others for their contributions. Supported by grants from The National Bank of Austria, The Austrian Ministry of Science and Education, Institute of Molecular Biotechnology in Austrian Academy of Sciences (IMBA) and EUGeneHeart to J.M.P. K.K. is supported by a Marie Curie Fellowship from the EU.
References
- 1.Skeggs L.T. The biochemistry of the renin–angiotensin system. Adv. Exp. Med. Biol. 1980;130:1–27. doi: 10.1007/978-1-4615-9173-3_1. [DOI] [PubMed] [Google Scholar]
- 2.Corvol P. Peptidyl dipeptidase A: angiotensin I-converting enzyme. Methods Enzymol. 1995;248:283–305. doi: 10.1016/0076-6879(95)48020-x. [DOI] [PubMed] [Google Scholar]
- 3.Donoghue M. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 4.Tipnis S.R. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 5.Li W. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peiris J.S. Severe acute respiratory syndrome. Nat. Med. 2004;10(12 Suppl.):S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imai Y. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuba K. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grose C., Chokephaibulkit K. Avian influenza virus infection of children in Vietnam and Thailand. Pediatr. Infect. Dis. J. 2004;23:793–794. doi: 10.1097/00006454-200408000-00024. [DOI] [PubMed] [Google Scholar]
- 10.Ware L.B., Matthay M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 11.Cambien F. Familial resemblance of plasma angiotensin-converting enzyme level: the Nancy Study. Am. J. Hum. Genet. 1988;43:774–780. [PMC free article] [PubMed] [Google Scholar]
- 12.Rigat B. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Invest. 1990;86:1343–1346. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marshall R.P. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2002;166:646–650. doi: 10.1164/rccm.2108086. [DOI] [PubMed] [Google Scholar]
- 14.Jerng J.S. Polymorphism of the angiotensin-converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit. Care Med. 2006;34:1001–1006. doi: 10.1097/01.CCM.0000206107.92476.39. [DOI] [PubMed] [Google Scholar]
- 15.Idell S. Angiotensin converting enzyme in bronchoalveolar lavage in ARDS. Chest. 1987;91:52–56. doi: 10.1378/chest.91.1.52. [DOI] [PubMed] [Google Scholar]
- 16.Orfanos S.E. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in acute lung injury. Circulation. 2000;102:2011–2018. doi: 10.1161/01.cir.102.16.2011. [DOI] [PubMed] [Google Scholar]
- 17.Mortensen E.M. The impact of prior outpatient ACE inhibitor use on 30-day mortality for patients hospitalized with community-acquired pneumonia. BMC Pulm. Med. 2005;5:12. doi: 10.1186/1471-2466-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crackower M.A. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto K. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension. 2006;47:718–726. doi: 10.1161/01.HYP.0000205833.89478.5b. [DOI] [PubMed] [Google Scholar]
- 20.Kiely D.G. Haemodynamic and endocrine effects of type 1 angiotensin II receptor blockade in patients with hypoxaemic cor pulmonale. Cardiovasc. Res. 1997;33:201–208. doi: 10.1016/s0008-6363(96)00180-0. [DOI] [PubMed] [Google Scholar]
- 21.Xu Z.H. Pulmonary edema induced by angiotensin I in rats. Jpn. J. Pharmacol. 1998;76:51–56. doi: 10.1254/jjp.76.51. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto T. Angiotensin II-induced pulmonary edema in a rabbit model. Jpn. J. Pharmacol. 1997;73:33–40. doi: 10.1254/jjp.73.33. [DOI] [PubMed] [Google Scholar]
- 23.Victorino G.P. Effect of angiotensin II on microvascular permeability. J. Surg. Res. 2002;104:77–81. doi: 10.1006/jsre.2002.6412. [DOI] [PubMed] [Google Scholar]
- 24.Marceau F., Regoli D. Bradykinin receptor ligands: therapeutic perspectives. Nat. Rev. Drug Discov. 2004;3:845–852. doi: 10.1038/nrd1522. [DOI] [PubMed] [Google Scholar]
- 25.Schanstra J.P. Bradykinin B(1) receptor-mediated changes in renal hemodynamics during endotoxin-induced inflammation. J. Am. Soc. Nephrol. 2000;11:1208–1215. doi: 10.1681/ASN.V1171208. [DOI] [PubMed] [Google Scholar]
- 26.Vickers C. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 27.Raiden S. Losartan, a selective inhibitor of subtype AT1 receptors for angiotensin II, inhibits neutrophil recruitment in the lung triggered by fMLP. J. Leukoc. Biol. 2000;68:700–706. [PubMed] [Google Scholar]
- 28.Lukkarinen H. Angiotensin II receptor blockade inhibits pneumocyte apoptosis in experimental meconium aspiration. Pediatr. Res. 2004;55:326–333. doi: 10.1203/01.PDR.0000100901.88697.66. [DOI] [PubMed] [Google Scholar]