

Abstract
Although the term virtual screening as the in silico analog of high throughput screening has been coined only a decade ago, virtual screening is now a widespread lead identification method in the pharmaceutical industry. A myriad of different methods have been developed exploiting the growing library of target structures and assay data as a basis for finding new lead structures. Exploiting synergies between different methods best utilizes the information available and is at the center of recent developments.
Section editors:
Tudor Oprea – University of New Mexico, School of Medicine, Albuquerque, USA
Alex Tropsha – University of North Carolina, Chapel Hill, USA
Introduction
Virtual screening has become an integral part of the drug discovery process in recent years [1]. Related to the more general and long pursued concept of database searching [2, 3] the term ‘virtual screening’ (VS) is relatively young [4]. Walters et al. define virtual screening as ‘automatically evaluating very large libraries of compounds’ using computer programs [5]. As this definition suggests, VS has largely been a numbers game focusing on questions like how can we filter down the enormous chemical space of >1060 conceivable compounds [6] to a manageable number that can be synthesized, purchased and tested. Although filtering the entire chemical universe might be a fascinating question, more practical VS scenarios focus on designing/optimizing targeted combinatorial libraries and enriching libraries of available compounds from in-house compound repositories or vendor offerings.
The main goal of a virtual screen is to come up with hits of novel chemical structure that yield a unique pharmacological profile. Thus, success of a virtual screen is defined in terms of finding interesting new scaffolds rather than many hits. Interpretations of VS accuracy should therefore be considered with caution. Low hit rates of interesting scaffolds are clearly preferable over high hit rates of already known scaffolds. Box 1 lists some practical considerations for a VS set-up.
Box 1.
-
•
VS methods need to be able to process 10 million individual compounds in a few weeks time.
-
•
Given that typically hundreds of VS hits are biologically tested and given that hit rates known from HTS are typically in the order of ≤0.1 percent, VS methods should enrich active ligands in the VS hit set ≥10 fold over random to obtain a reasonable chance of finding a true hit. In cases of low hit rate targets, enrichment rates might have to be significantly higher.
-
•
VS hit sets should balance compounds ranked highly and being structurally diverse to increase the chance of finding novel chemotypes. Hits should then be followed up in a second iteration by analog testing.
VS has experienced increased attention in recent years (Fig. 1 ) due to the rise in available datasets, VS techniques and excitement created by successful screening studies. When integrated with high throughput screening (HTS), VS can aid in the rapid identification of novel ligands [7, 8]. VS includes target specific search criteria but also target independent considerations such as drug-likeness [9, 10]. The use of VS technologies has also aided the identification of bioactive molecules from natural products [11]. VS methods are often divided into structure-based VS (SBVS) [12, 13] and ligand-based VS (LBVS) [14, 15]. SBVS and LBVS have been reviewed frequently in the literature. Therefore, we focus here on possible synergies between SBVS and LDVS, some new interests in machine learning techniques for VS, and highlight recent success stories.
Figure 1.
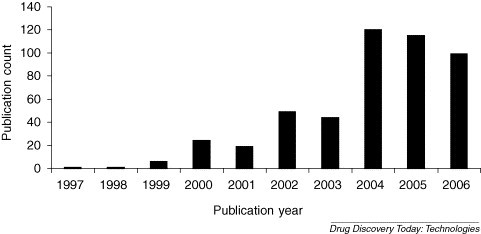
Analysis of VS publications obtained from a PubMed search performed on July 9, 2006 using ‘virtual screening’ as a keyword.
Synergies between structure-based and ligand-based virtual screening
SBVS and LBVS have been considered almost mutually exclusive suggesting LBVS to be used primarily in the absence of protein target structure(s) and SBVS to be used if target structure(s) are available. Especially when a target protein structure at high atomic resolution is available, SBVS is often considered as the first choice strategy ignoring possible LBVS alternatives. Until recently, only sporadic studies have pointed to the fact that LBVS offers a strong alternative to SBVS even in the presence of protein structural information. For instance, a comparison of LBVS and SBVS methods using the recall of known HIV-1 protease inhibitors as test examples has shown how ligand similarity methods can outperform molecular docking as a VS tool [16]. Recently a more systematic comparative study between SBVS and LBVS involving seven different drug targets for which structural information is available has been published [17]. Figure 2 illustrates that in most cases LBVS techniques measuring compound similarity to known potent molecules outperforms molecular docking, a more computationally intensive SBVS technique that generates and scores putative protein–ligand complexes according to their calculated binding affinities [18]. Similar results have recently been reported in the field of GPCRs using homology models for SBVS and 2D-qsar, decision trees, and PLS as LBVS methods [19]. Although studies about scaffold hopping through SBVS are scarce [20] it is recognized that SBVS techniques have a better potential to identify compounds with a novel core scaffold. Therefore, SBVS and LBVS should not be applied independently but rather in concert to increase the chances of finding novel hits [21, 22]. Some software approaches such as SDOCKER have begun integrating both strategies by including ligand similarity as a part of the scoring functions used in the docking algorithm [23]. Bologa et al. has also reported the integration of both strategies to aid in the identification of the first selective GPR30 agonist [24].
Figure 2.
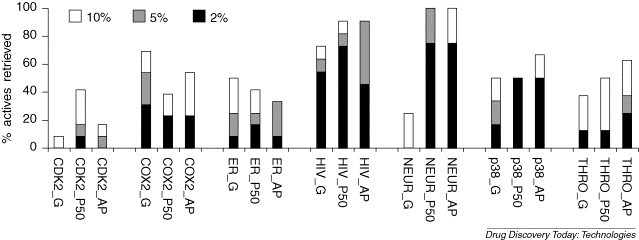
Comparison of percent of actives retrieved for seven protein targets (CDK2, COX2, estrogen receptor, HIV-1 protease, neuramindase, p38 MAP kinase and thrombin). The suffix _G refers to Glide2.5 docking experiments. _P50 and _AP refer to ligand-similarity VS methods using 3D pharmacophore fingerprints with 50 conformations per molecule and to atom pair descriptors, respectively [17].
Machine learning algorithms in virtual screening
An LBVS approach that is quickly gaining popularity in VS, called machine learning, builds predictive compound activity models that are based on available assay data. Machine learning approaches have been reviewed less before in the context of VS. Therefore, we include here a short synopsis of the most prominent techniques in addition to highlighting their applications in VS. Several recent success stories have been reported in the literature and a few examples of the more popular machine learning techniques are listed in Table 1 . Each method has its own advantages and disadvantages that should be understood to select the best approach for a particular LBVS. The first approach listed in the table, a self-organizing map (SOM), is quite simple and easy to visualize. A dataset of compounds are mapped on to a 2D grid such that most similar compounds are grouped together. Compounds found in the same vicinity of those with a desired biological property are considered potential hits in a virtual screen; however the SOM approach in general has a very high false positive rate. It is a simple and easy way to visualize if compounds that have the same biological properties group together. SOMs have been used recently to identify several purinergic receptor agonists [25]. Another approach called Binary QSAR uses all compounds in the training set to predict the biological property of test compound(s) in a virtual screen rather than just the most similar training compounds. Although not providing an image of the training data, this approach is fast and like SOMs it works well if the training data is highly similar to the test compounds being screened. Compounds that are significantly different from the training set are not expected to be predicted accurately and are commonly missed in a virtual screen [26].
Table 1.
Overview of popular regression and learning algorithms used for virtual screening
| Classification | Regression | Variable selection | Explanatory | Virtual screening | |
|---|---|---|---|---|---|
| SOM | Yes, useful for visualizing global data trends | No | Various techniques show success extracting pertinent dependent variables by grouping compounds in the same target class together | Yes, when the pertinent dependent variables are optimized | Identified purinergic receptor antagonists from a virtual combinatorial library [25] |
| Binary QSAR | Yes | No | No | No | Showed superior enrichment rates when compared to Bayesian Classifiers and PLS [26] |
| Bayesian Classifier | Yes | Yes | Descriptors are weighted based on how well each divides the training data | Yes if the significance of each descriptor can be extracted | Performed poorly compared to SVM, kNN, ANN and Decision trees [27] |
| Decision trees | Yes | No | Descriptors that best divide one class from another are used to separate the data | Variables used in the tree(s) suggest activity dependency | Slightly outperformed a Bayes Classifier in a comparison study [27] |
| PLS variants | Yes | Yes | Variable selection techniques are commonly added above PLS model building | Yes, when a variable selection technique is incorporated | Ligands for various GPCR targets were successfully enriched from a test database [19] |
| ANN | Yes | Yes | Performed internally | No | Comparable enrichment rates in a direct comparison to SVM and kNN [27] |
| SVM | Yes | Yes | Performed internally | Yes, if the weights of each descriptor are explicitly solved. | Identified previously characterized Dopamine D1 Inhibitors and suggested new hits [29] |
| kNN | Yes | Yes | Commonly a genetic algorithm or simulated annealing is used | Descriptors selected by multiple models imply relevance to the target property | Identified several anticonvulsant compounds that were experimentally confirmed [28] |
Other approaches require a pre-built model to perform a virtual screen. These techniques form correlations between training data and descriptors that describe training compounds to predict a biological property for a virtual set of compounds. The Bayesian Classifier requires a pre-built model and is somewhat similar to Binary QSAR except it identifies specific descriptors that best distinguish compounds with a desired biological property from others. This search for pertinent descriptors is called variable selection and helps the model eliminate descriptors that are not relevant to the current problem and cloud the separation of one biological property class from another. The Bayesian Classifier algorithm has been shown to not perform as well as even more sophisticated approaches [27]; however, it does handle large training sets much easier.
Decision trees (or Forests) incorporate the simplest form of variable selection and can be considered as a set of Boolean functions. Descriptors that capture molecular features of the training compounds are systematically added to a Decision tree model one at a time until compounds with different biological properties are adequately separated. This approach allows the researcher to easily determine the chemical features most relevant to the target biological property. This information can be used in the design of future molecules. Virtual screening with Decision trees is quite easy as well. Comparison studies have shown it slightly outperforms methods such as the Bayesian Classifier however other more advanced approaches show higher enrichment rates in a virtual screen [27].
In yet another class of machine learning approaches, mathematical function(s) are used to correlate descriptor values with a biological property. The simplest of these builds a linear correlation and is called multiple linear regression. A very popular extension called partial least squares (PLS) helps simplify the model optimization so larger training sets can be easily used. Variable selection techniques are also commonly added above the PLS algorithm to optimize the descriptors used in the linear model. This approach has shown success enriching a virtual database for various GPCR ligands [19]; however, it has an obvious drawback. There is not always a linear correlation between the property modeled and the descriptors describing your dataset. The artificial neural network (ANN) and support vector machine (SVM) approaches allow one to build non-linear correlations. ANN and SVM have become popular tools for model building and virtual screening. In a side-by-side comparison both ANN and SVM show similar enrichment rates for several virtual screens [27].
The k-nearest neighbors approach (kNN) does not require the use of a mathematical function to split one property class from another, which can be very useful when the problem is complex. Compounds in a virtual screen are predicted based on known activities of the most similar training compounds. Similarity between compounds is calculated using only a small set of most pertinent descriptors that are optimized during model building (this optimization can be quite slow for large datasets). When used in a virtual screen kNN shows database enrichment rates similar to both ANN and SVM [27]. A domain of applicability can be used in virtual screening to improve the enrichment rates in a virtual screen by only allowing the model to predict compounds that have the highest chance of being predicted correctly. Applicability domain techniques have been applied very successfully for both kNN [28] and SVM [29]. However, limitations lie in the fact that a very narrow applicability domain only identifies potential compound hits that are highly similar to the training set compounds. Such hits could have possibly been found using a simpler, less time consuming approach also.
A new forum has been created, the comparative evaluation of prediction algorithms (CoEPrA), which compares how predictive different machine learning approaches are for blind test cases. This forum illustrates which techniques consistently work best and should be an interesting media for testing new machine learning approaches. A link to CoEPrA can be found together with other links of interest in the Links section.
Recent successes of virtual screening
A few examples of recent VS applications are highlighted below. In addition, Table 2 shows a collection of reports of recent successful virtual screens.
Table 2.
Recent success stories
| Method used | Protein target | Identified hits |
|---|---|---|
| Multiple linear regression | CCR5 | Several new derivatives of active molecules were proposed [43] |
| Pharmacophore modeling | Fetal hemoglobin | Novel inhibitors were identified from a large chemical database [44] |
| Consensus scoring using multiple docking approaches | CK2 | Identified a highly potent inhibitor from a chemical database[45] |
| rDock | Chk1 | 10 Novel inhibitors were identified with 9 different scaffolds [46] |
| Catalyst | PPARγ | 2 Partial agonists were found among a large chemical database and validated in vivo [47] |
| Pharmacophore modeling and FlexX docking | GSK-3 | 9 New inhibitors were predicted from three large chemical databases [48] |
Structure-based virtual screening
Gold [30] docking and subsequent scoring with the PMF scoring function [31] has identified novel inhibitors of the potential cancer target erythropoietin-producing hepatocellular B2 receptor tyrosine kinase domain with measured K d of 3.3 μM. Docking and scoring results have been combined with pharmacophore modeling aspects and ‘high content’ wet screening techniques using affinity chromatography [32].
Structure-based virtual screening against the target dipeptidyl peptidase IV (DPPIV) has identified chemical starting points for medicinal chemistry follow-up. Docking of compound collections pre-filtered by physical property and medicinal chemistry considerations as well as matching pharmacophores to known DPPIV inhibitors has resulted in 51 compounds with activities between 30% and 82% at 30 μM concentration in an enzyme inhibition assay [33].
Through the combination of homology modeling and docking methods, several successful VS applications have been published recently. They include the discovery of novel lipoxygenase inhibitors [34] as well as a novel cannabinoid CB2 receptor agonist [35].
Combining SBVS and LBVS techniques has resulted in the discovery of a novel family of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) protease inhibitors [36]. Gold docking and mapping CoMFA/CoMSIA models onto the protein active site have been employed to screen through the Maybridge database of 59,363 compounds in search for novel hits. Twenty-one compounds tested have exhibited inhibition below 30 μM IC50. By following up with analog searching through other databases, an additional 25 inhibitors could be identified. This example illustrates the iterative nature of virtual screening. Although a first VS iteration identifies novel classes of actives but does not necessarily contain the most potent compound within a given class, the second iteration focusing on analogs of the newly found class often leads to more potent hits.
Ligand-based virtual screening
Recall experiments using SVMs trained on known cyclooxygenase 2 and thrombin inhibitors have been reported recently. Topological pharmacophore-point triangles have been used as molecular descriptors. In a validation study, 50–90% of the known active compounds could be recalled within the first 0.1% of the ranked databases containing the known actives and a list of arbitrary screening compounds. Following on this positive validation, a subsequent VS study has identified several potential COX-2 inhibitors that have been tested in a cellular activity assay. A newly found benzimidazole derivative has exhibited significant inhibitory activity better than that of Celecoxib [37].
QSAR models have been developed to discover new anti-malaria agents. Specifically, virtual screens for finding Ras farnesyltransferase inhibitors with antimalarial activity have been reported. Following successful recall experiments of known inhibitors QSAR models have been used to identify previously unknown antimalarials. A new arylaminomethylenemalonate has been found through VS with antimalarial activity [38].
Ligand-derived pharmacophore models in concert with cell-based activity assays have been used to discover selective 11beta-hydroxysteroid dehydrogenase (11beta-HSD) inhibitors shown to block subsequent cortisol-dependent activation of glucocorticoid receptors [39].
An extension to the feature tree approach [40] called MTree has been reported [41]. Here topological molecular graphs of several ligands are combined to a common feature tree that allows for matching corresponding functional groups. These functional groups are derived akin to pharmacophore queries from a set of diverse but active ligands against a given target protein. Applying this new multiple feature tree approach to recall experiments of known angiotensin converting enzyme inhibitors and α1a receptor antagonists has led to significant enrichments of known active compounds validating the concept of MTrees.
Conclusions
The prevailing opinion has been for a long time that in the presence of a high-resolution target protein structure one should use SBVS whereas in cases where only ligand information is known LBVS should be used. Recent publications have somewhat challenged this view focusing on using LBVS even in the presence of target structure information. Advancements in VS have therefore been made in understanding the strengths and weaknesses of existing methods and how to use them rather than coming up with new approaches. The majority of reported VS successes make the best use of several informational sources. For instance, pharmacophore models using known ligands are combined with homology models; QSAR models are combined with docking approaches. Using all available information in concert is essential for obtaining optimal VS results making each VS experiment unique. Consequently, an increasing number of successful VS applications use more than one VS technique.
Machine learning techniques have been increasingly applied to virtual screening. This is not surprising as ligand-based virtual screening approaches have experienced growth in general through expansions in available chemical libraries, published compound assay data, and the surge of new molecular descriptors and techniques used in similarity comparisons. As it becomes available, experimental data is incorporated into models that are used to aid the design of new compounds. This helps to reduce redundant compounds from being synthesized and to identify molecular features that are important for the biological property of interest.
Although VS has been in a race with experimental high throughput screening techniques in the past decade to increase the speed of processing more and more compounds in less time, this race has slowed in recent years. Some companies such as Aventis have decided to limit the number of compounds to be tested in high throughput screens to increase the quality of hits [42]. Likewise, the focus of VS is now on increasing reliability, hit rates, and the number and quality of novel scaffolds to be discovered rather than speed. Several future challenges are highlighted in the Outstanding issues box.
Several of the most recent virtual screens have resulted from a combination of different approaches utilizing multiple sources of information rather than just structural or ligand assay data (examples in Table 3 ). Such a combined approach uses ligand-based methods to identify compounds with features important for the target property. Structure-based techniques are used to ensure that the shape, size and energetic interaction potential of the putative ligands complement that of the target protein. Recent successes illustrate the advantage of utilizing all available information in concert making each VS experiment a unique endeavor.
Table 3.
Comparison of VS approaches working in concert
| Method | Structure-based & pharmacophore query | Structure-based & machine learning | Pharmacophores & machine learning |
|---|---|---|---|
| Specific examples | 51 DPP IV inhibitors were identified in a VS using a pharmacophore and docking filter | New SHBG ligands were found using a combined 2D-QSAR and docking filter | New COX-2 inhibitors were found using SVM's with pharmacophoric descriptors |
| Pros | Not dependent on scaffolds within the training set | Works well for compounds similar to the training set | Very fast for screening large databases |
| Cons | Reduced accuracy for training set-like compounds compared to ligand-based methods | VS hits are biased towards training set-like compounds | Limited scaffold hopping ability |
| References | [33] | [49] | [37] |
Links
Outstanding issues
-
•
The reliability of virtual screening needs to be improved: For SBVS, more reliable scoring functions are needed. LDVS descriptors that reliably facilitate scaffold hopping are essential. For regression and classification approaches applicability domains need to be extended.
-
•
Synergies between all VS methods need to be realized.
-
•
Rather than finding ‘ligand-like’ compounds VS needs to focus on improving methods to find ‘lead-like’ compounds. A compound with good molecular potency alone rarely results in a good lead or drug candidate.
Related articles
Shoichet, B.K. (2004) Virtual screening of chemical libraries. Nature 423, 862–865
Lengauer, T. et al. (2004) Novel technologies for virtual screening. Drug Discov. Today 9, 27–34
Alvarez, J. and Shoichet, B.K. (2005) Virtual Screening in Drug Discovery, CRC
Glossary
- Building blocks
chemical reagents to be attached as substituents in combinatorial library design and synthesis.
- Decision trees
a common classification technique that systematically identifies the descriptors that best separate one compound class from another.
- Descriptors
numerical values that describe specific compound features so that compounds can be represented in a mathematical function.
- Enrichment rate
ratio of percentage of true actives and percentage of total compounds retrieved as hits from a database of compounds screened in a VS.
- Machine learning
computer algorithms used to build models of training data and predict future outcomes based on past experiences.
- Pharmacophore
a group of chemical features that are responsible for a compound's biological activity.
- QSAR
(quantitative structure activity relationships) computer-based models that correlate descriptor variations to quantitative changes in biological activity.
- Recall
percentage of known actives retrieved in a VS experiment.
- Scaffold hopping
identification of compounds with different core structure based on other compounds with similar activity.
- Scoring function
mathematical function to rank protein-ligand complexes according to their predicted binding affinity.
- Variable selection
the selection of descriptors most pertinent to the biological property of interest from a larger pool of descriptors.
References
- 1.Stahl M. Integrating molecular design resources within modern drug discovery research: the Roche experience. Drug Discov. Today. 2006;11:326–333. doi: 10.1016/j.drudis.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Martin Y.C. Searching databases of three-dimensional structures. In: Lipkowitz K.B., Boyd D.B., editors. Vol. 1. VCH Publishers; 1990. pp. 213–263. (Reviews in Computational Chemistry). [Google Scholar]
- 3.Good A.C., Mason J.S. Three dimensional structure database searches. In: Lipkowitz K.B., Boyd D.B., editors. Reviews in Computational Chemistry. VCH; 1995. pp. 67–117. [Google Scholar]
- 4.Burns R.F. Virtual screening as a tool for evaluating chemical libraries. Exploiting Molecular Diversity, Symposium Proceedings 2; Cambridge Healthtech Institute; 1995. [Google Scholar]
- 5.Walters W.P. Virtual screening – an overview. Drug Discov. Today. 1998;3:160–178. [Google Scholar]
- 6.Martin Y.C. Challenges and prospects for computational aids to molecular diversity. Perspect. Drug Discov. Design. 1997;7/8:159–172. [Google Scholar]
- 7.Edwards B.S. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharmacol. 2005;68:1301–1310. doi: 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- 8.Fara, D.C. et al. (2006) Integration of Virtual and Physical Screening. This issue. [DOI] [PMC free article] [PubMed]
- 9.Muegge I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003;23:302–321. doi: 10.1002/med.10041. [DOI] [PubMed] [Google Scholar]
- 10.Muegge I., Enyedy I. Virtual screening. In: Abraham D., editor. Burger's Medicinal Chemistry. 5th edn. Wiley; 2003. pp. 243–280. [Google Scholar]
- 11.Rollinger J.M. Strategies for efficient lead structure discovery from natural products. Curr. Med. Chem. 2006;13:1491–1507. doi: 10.2174/092986706777442075. [DOI] [PubMed] [Google Scholar]
- 12.Good A. Structure-based virtual screening protocols. Curr. Opin. Drug Discov. Dev. 2001;4:301–307. [PubMed] [Google Scholar]
- 13.Lyne P.D. Structure-based virtual screening: an overview. Drug Discov. Today. 2002;7:1047–1055. doi: 10.1016/s1359-6446(02)02483-2. [DOI] [PubMed] [Google Scholar]
- 14.Oprea T.I., Matter H. Integrating virtual screening in lead discovery. Curr. Opin. Chem. Biol. 2004;8:349–358. doi: 10.1016/j.cbpa.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Willet P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today. 2006;11:1046–1053. doi: 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Sheridan R.P., Kearsley S.K. Why do we need so many chemical similarity search methods? Drug Discov. Today. 2002;7:903–911. doi: 10.1016/s1359-6446(02)02411-x. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q., Muegge I. Scaffold hopping through virtual screening using 2D and 3D similarity descriptors: ranking, voting, and consensus scoring. J. Med. Chem. 2006;49:1536–1548. doi: 10.1021/jm050468i. [DOI] [PubMed] [Google Scholar]
- 18.Muegge I., Rarey M. Small molecule docking and scoring. In: Boyd D.B., Lipkowitz K.B., editors. Reviews in Computational Chemistry. Wiley-VCH; 2001. pp. 1–60. [Google Scholar]
- 19.Evers A. Virtual screening of biogenic amine-binding G-protein coupled receptors: comparative evaluation of protein- and ligand-based virtual screening protocols. J. Med. Chem. 2005;48:5448–5465. doi: 10.1021/jm050090o. [DOI] [PubMed] [Google Scholar]
- 20.Good A.C. Analysis and optimization of structure-based virtual screening protocols 2. Examination of docked ligand orientation sampling methodology: mapping a pharmacophore for success. J. Mol. Graph. Model. 2003;22:31–40. doi: 10.1016/S1093-3263(03)00124-4. [DOI] [PubMed] [Google Scholar]
- 21.Bissantz C. Focused library design in GPCR projects on the example of 5-HT(2c) agonists: comparison of structure-based virtual screening with ligand-based search methods. Proteins. 2005;61:938–952. doi: 10.1002/prot.20651. [DOI] [PubMed] [Google Scholar]
- 22.Nettles J.H. Bridging chemical and biological space: ‘Target Fishing’ using 2D and 3D molecular descriptors. J. Med. Chem. 2006;49:6802–6810. doi: 10.1021/jm060902w. [DOI] [PubMed] [Google Scholar]
- 23.Wu G., Vieth M. SDOCKER: a method utilizing existing X-ray structures to improve docking accuracy. J. Med. Chem. 2004;47:3142–3148. doi: 10.1021/jm040015y. [DOI] [PubMed] [Google Scholar]
- 24.Bologa C.G. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 25.Schneider G., Nettekoven M. Ligand-based combinatorial design of selective purinergic receptor (A2A) antagonists using self-organizing maps. J. Comb. Chem. 2003;5:233–237. doi: 10.1021/cc020092j. [DOI] [PubMed] [Google Scholar]
- 26.Prathipati P., Saxena A.K. Evaluation of binary QSAR models derived from LUDI and MOE scoring functions for structure based virtual screening. J. Chem. Inf. Model. 2006;46:39–51. doi: 10.1021/ci050120w. [DOI] [PubMed] [Google Scholar]
- 27.Plewczynski D. Assessing different classification methods for virtual screening. J. Chem. Inf. Model. 2006;46:1098–1106. doi: 10.1021/ci050519k. [DOI] [PubMed] [Google Scholar]
- 28.Shen M. Application of predictive QSAR models to database mining: identification and experimental validation of novel anticonvulsant compounds. J. Med. Chem. 2004;47:2356–2364. doi: 10.1021/jm030584q. [DOI] [PubMed] [Google Scholar]
- 29.Oloff S. Application of validated QSAR models of D1 dopaminergic antagonists for database mining. J. Med. Chem. 2005;48:7322–7332. doi: 10.1021/jm049116m. [DOI] [PubMed] [Google Scholar]
- 30.Jones G. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;207:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 31.Muegge I., Martin Y.C. A general and fast scoring function for protein-ligand interactions: a simplified potential approach. J. Med. Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 32.Toledo-Sherman L. Frontal affinity chromatography with MS detection of EphB2 tyrosine kinase receptor. 2. Identification of small-molecule inhibitors via coupling with virtual screening. J. Med. Chem. 2005;48:3221–3230. doi: 10.1021/jm0492204. [DOI] [PubMed] [Google Scholar]
- 33.Ward R.A. Structure-based virtual screening for low molecular weight chemical starting points for dipeptidyl peptidase IV inhibitors. J. Med. Chem. 2005;48:6991–6996. doi: 10.1021/jm0505866. [DOI] [PubMed] [Google Scholar]
- 34.Kenyon V. Novel human lipoxygenase inhibitors discovered using virtual screening with homology models. J. Med. Chem. 2006;49:1356–1363. doi: 10.1021/jm050639j. [DOI] [PubMed] [Google Scholar]
- 35.Salo O.M. Virtual screening of novel CB2 ligands using a comparative model of the human cannabinoid CB2 receptor. J. Med. Chem. 2005;48:7166–7171. doi: 10.1021/jm050565b. [DOI] [PubMed] [Google Scholar]
- 36.Tsai K.C. Discovery of a novel family of SARS-CoV protease inhibitors by virtual screening and 3D-QSAR studies. J. Med. Chem. 2006;49:3485–3495. doi: 10.1021/jm050852f. [DOI] [PubMed] [Google Scholar]
- 37.Franke L. Extraction and visualization of potential pharmacophore points using support vector machines: application to ligand-based virtual screening for COX-2 inhibitors. J. Med. Chem. 2005;48:6997–7004. doi: 10.1021/jm050619h. [DOI] [PubMed] [Google Scholar]
- 38.Marrero-Ponce Y. Ligand-based virtual screening and in silico design of new antimalarial compounds using nonstochastic and stochastic total and atom-type quadratic maps. J. Chem. Inf. Model. 2005;45:1082–1100. doi: 10.1021/ci050085t. [DOI] [PubMed] [Google Scholar]
- 39.Schuster D. The discovery of new 11beta-hydroxysteroid dehydrogenase type 1 inhibitors by common feature pharmacophore modeling and virtual screening. J. Med. Chem. 2006;49:3454–3466. doi: 10.1021/jm0600794. [DOI] [PubMed] [Google Scholar]
- 40.Rarey M., Dixon J.S. Feature Trees: a new molecular similarity measure based on tree matching. J. Comp. Aided Mol. Design. 1998;12:471–490. doi: 10.1023/a:1008068904628. [DOI] [PubMed] [Google Scholar]
- 41.Hessler G. Multiple-ligand-based virtual screening: methods and applications of the MTree approach. J. Med. Chem. 2005;48:6575–6584. doi: 10.1021/jm050078w. [DOI] [PubMed] [Google Scholar]
- 42.Mullin R. DRUG DISCOVERY as high-throughput screening draws fire, researchers leverage science to put automation into perspective. Chem. Eng. News. 2004;82:23–32. [Google Scholar]
- 43.Afantitis A. Investigation of substituent effect of 1-(3,3-diphenylpropyl)-piperidinyl phenylacetamides on CCR5 binding affinity using QSAR and virtual screening techniques. J. Comp. Aided Mol. Design. 2006;20:83–95. doi: 10.1007/s10822-006-9038-2. [DOI] [PubMed] [Google Scholar]
- 44.Bohacek R. Identification of novel small-molecule inducers of fetal hemoglobin using pharmacophore and ‘PSEUDO’ receptor models. Chem. Biol. Drug Design. 2006;67:318–328. doi: 10.1111/j.1747-0285.2006.00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cozza G. Identification of ellagic acid as potent inhibitor of protein kinase CK2: a successful example of a virtual screening application. J. Med. Chem. 2006;49:2363–2366. doi: 10.1021/jm060112m. [DOI] [PubMed] [Google Scholar]
- 46.Foloppe N. Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening. Bioorg. Med. Chem. 2006;14:4792–4802. doi: 10.1016/j.bmc.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 47.Lu I.L. Structure-based drug design of a novel family of PPARgamma partial agonists: virtual screening, X-ray crystallography, and in vitro/in vivo biological activities. J. Med. Chem. 2006;49:2703–2712. doi: 10.1021/jm051129s. [DOI] [PubMed] [Google Scholar]
- 48.Patel D.S., Bharatam P.V. New leads for selective GSK-3 inhibition: pharmacophore mapping and virtual screening studies. J. Comp. Aided Mol. Design. 2006;20:55–66. doi: 10.1007/s10822-006-9036-4. [DOI] [PubMed] [Google Scholar]
- 49.Cherkasov A. Successful in silico discovery of novel nonsteroidal ligands for human sex hormone binding globulin. J. Med. Chem. 2005;48:3203–3213. doi: 10.1021/jm049087f. [DOI] [PubMed] [Google Scholar]