

Abstract
High-throughput screening (HTS) represents the dominant technique for the identification of new lead compounds in current drug discovery. It consists of physical screening (PS) of large libraries of chemicals against one or more specific biological targets. Virtual screening (VS) is a strategy for in silico evaluation of chemical libraries for a given target, and can be integrated to focus the PS process. The present work addresses the integration of both PS and VS, respectively.
Section editors:
Tudor Oprea – University of New Mexico School of Medicine, Albuquerque, USA
Alex Tropsha – University of North Carolina, Chapel Hill, USA
Virtual versus physical (high-throughput) screening
The random physical screening (PS) of compound collections, that is the in-house database of a pharmaceutical company, or the catalogs of various chemicals’ vendors, represented a long-range strategy and was regarded as a substitute for serendipity [1].
Automated compound handling and assaying facilities currently enable tasks such as compound retrieval from storage, dilution and plating of samples; therefore, PS is reaching its potential to identify hits on a timely basis. While random screening has enabled some successes, it has not fulfilled initial expectations. A gross increase in the number of assayed compounds does not guarantee better productivity per se [2]. Also, PS results are not free from errors and different assay formats for the same target can give different results [3, 4].
The in silico screening of (virtual) libraries of compounds is conceptually and economically attractive, as it makes possible the evaluation of an almost unlimited number of chemical structures, only a subset of which will be selected and subsequently assayed in a PS experiment. Typical virtual screening (VS) methods involve, for example, filtering of libraries for compounds containing toxic, reactive, or otherwise undesirable groups, or, by contrast, the search for molecules with preferred lead- or drug-like properties and desired activity [5]. In fact, VS is emerging as a key strategy to help filter out those compounds with poor potency, and biological or pharmacological properties [6, 7, 8]. Recent reviews suggest that while VS is often presented as an alternative to PS, both strategies are highly complementary [5, 9].
Integrating virtual and physical screening
The current trend in the pharmaceutical industry is to integrate computational and experimental technologies early in the drug discovery process [10, 11, 12, 13]. For instance, it is considered that a better and earlier utilization of information, that is (i) genomic, (ii) chemical, (iii) biological, (iv) structural, and (v) molecular property data, would lead to chemical libraries with more desirable chemical and biological properties [14, 15]. This can be done by integrating information from different areas that include: (i) analysis of the gene or protein family for target selection; (ii) absorption, distribution, metabolism, excretion and toxicity evaluation (ADME/Tox); (iii) structural biology; (iv) VS; and (v) medicinal chemistry with parallel synthesis (Fig. 1 ).
Figure 1.
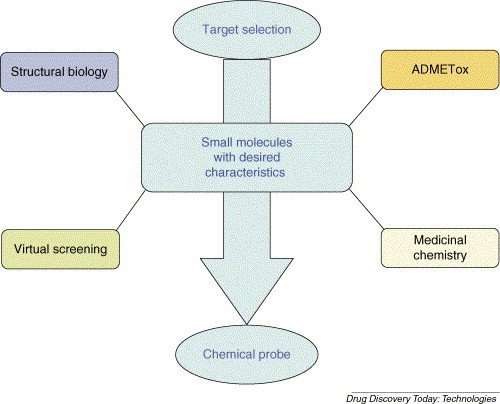
Integration of virtual chemistry and screening. A general scheme that shows how a continuous information exchange between different areas could lead to chemical libraries with more desirable chemical and biological properties. Abbreviation: ADMETox: absorption, distribution, metabolism, excretion and toxicity evaluation.
Literature reports from 2005 to 2006 show an increased dependence on the integration VS and PS for identifying leads in the drug discovery process. Twenty-three representative studies are summarized in Table 1 , which lists (i) the target studied, (ii) the strategy applied for both VS and PS methods, (iii) the size of the library evaluated, and (iv) the results achieved in each project. The last column indicates the original references.
Table 1.
Overview of recent publications that integrate virtual and physical screening (VS and PS)
| Nos. | Target |
Strategy |
Data set (molecules) |
Outcome | Refs | ||
|---|---|---|---|---|---|---|---|
| VS | PS | VS | PS | ||||
|
1 |
FPRa |
Homology modeling + docking + pharmacophore selection |
Hypercyt™ – high-throughput flow cytometry |
∼480,000 |
4324 |
30 potential lead cpds (Kib = 1 ÷ 32 μM); 4 partial agonists; 26 antagonists; VS improved the PS hit rate by fourfold |
[18] |
|
2 |
GPR30c |
2D + shape + field similarity |
Hypercyt™ – high-throughput flow cytometry |
9283 |
100 |
1 low nanomolar GPR30 selective agonist |
[19] |
|
3 |
Caspase-3 |
Self-organizing Kohonen maps |
Fluorescence + microtitration |
18,073 |
1233 |
6 quinoline derivatives as inhibitors; IC50d = 4 ÷ 30 nM |
[31] |
|
4 |
β-Amyloid Peptide |
Docking + scoring |
ThTe fluorescence |
∼278,000 |
125 |
1 potent inhibitor of Aβ1-42 fibrillation (concentration-dependent manner); IC50 = 35.6 μM |
[32] |
|
5 |
fSARS-CoV Mpro |
Docking + scoring + analogue searching |
Fluorescence resonance energy transfer (FRETg) |
58,855 |
50 |
21 inhibitors; IC50 = 0.3 ÷ 50 μM |
[33] |
|
6 |
P-12-LOXh |
Homology modeling + docking + scoring |
Direct measurement of the 12(S)-HETEi formation |
80 |
10 |
4 synthetic curcuminoids as inhibitors; IC50 = 1.7 ÷ 66 μM |
[34] |
|
7 |
FAAHj, MGLk-like in rat cerebellar membranes |
Homology modeling + docking + scoring |
Radiolabeled ligand (FAAH) + HPLC analysis (MGL) |
2376 |
62 |
9 FAAH inhibitors; IC50 = 0.52 ÷ 44 μM; 1 MGL-like inhibitor; IC50 = 31 μM |
[35] |
|
8 |
MDM2l-p53m interaction |
Druglike filtering + pharmacophore searching + docking + scoring |
Fluorescence − polarization-based (FP-based) |
150,000 |
67 |
10 inhibitors (Ki < 10 μM); the most potent has Ki = 120 nM |
[36] |
|
9 |
11β-HSD1n, 11β-HSD2o |
Pharmacophore modeling + data mining + docking |
Radiolabeled ligand in (i) lysates of HEK-293 cellsp, and (ii) intact transfected HEK-293 cells |
1,776,579 |
30 |
7 cpds inhibited more than 70% of the activity of 11β-HSD1 (IC50 < 10 μM); 5 out of those 7 cpds showed significant inhibition of 11β-HSD2 (IC50 < 10 μM) |
[37] |
|
10 |
CypAq |
Focused combinatorial library design + docking + scoring |
Surface plasmon resonance (SPRr) |
85,000 |
40 |
4 potent inhibitors of CypA; IC50 = 2.5 ÷ 6.2 μM |
[38] |
|
11 |
CypA |
Pharmacophore selection + docking + scoring |
Chymotrypsin-coupled colorimetric assay |
296,387 (3129) |
31 |
5 cpds inhibited more than 80% of the isomerase activity of CypA; IC50 = 0.3 ÷ 5 μM |
[39] |
|
12 |
Cysteine protease from P. falciparum (falcipain-2, falcipain-3) and L. donovani |
Filtering + docking + scoring + pharmacophore modeling |
Fluorescence + flow cytometry |
355,000 |
100 |
18 cpds active against falcipain-2 (IC50 = 1.4 ÷ 54.3 μM and falcipain-3 (IC50 = 11.4 ÷ >50 μM); 4 cpds active against Leishmania donovani cysteine protease (IC50 = 23.5 ÷ 43.0 μM) |
[40] |
|
13 |
12-hLOs, 15-hLOt |
Homology modeling + docking + scoring |
Measurement of the rate of formation of the conjugated diene products at 234 nm |
50,000 |
20 |
3 low molar inhibitors of 15-hLO (IC50 = 6.8 ÷ 18.8 μM) and 2 out of 3 are low molar inhibitors of 12-hLO (IC50 = 12.3 ÷ 30.7 μM) |
[41] |
| 14 | ALKu | Pharmacophore selection + docking + scoring + homology modeling | Amplified luminescent proximity homogeneous assay | 60,000 | 2677 | 5-Aryl-pyridone-3-carboxamide derivatives as novel ALK inhibitors; IC50 = 0.4 ÷ 19.5 μM | [42] |
| 724 |
24 |
||||||
|
15 |
ACE2v |
Pharmacophore modeling + docking + scoring |
Fluorescence |
63,307 |
17 |
6 cpds showed an inhibitory effect on ACE2; IC50 = 62 ÷ 179 μM |
[43] |
|
16 |
GSK-3βw |
Docking + scoring + pharmacophore selection |
Kinase-Glo™ luminescent kinase assay + ATPx titration |
16,299 (only 5904 were docked) |
16,299 |
90 validated hits (6 clusters) by PS; VS identified 25–33% of actives in clusters 1–4 and failed to pick up any hit from clusters 5 to 6 |
[44] |
|
17 |
DPP-IVy |
Filtering + pharmacophore selection + docking + scoring |
Fluorescence |
800,000 |
4000 |
51 active compounds (%inhibition ranges from 30 to 81.9 at 30 μM); the hit rate for the actives is 1.28% (VS) and 0.012% (PS) |
[45] |
|
18 |
COX-2z |
Pharmacophore modeling + support vector machine classification + docking |
ELISAaa + monoclonal antibody + microtitration |
∼2,7 millions |
22 (13 cherry picked) |
3 cpds exhibited an inhibitory effect in the activity assay (the most potent had IC50 = 0.2 ± 0.3 μM) |
[46] |
|
19 |
AChEbb |
Pharmacophore modeling + docking + scoring |
Extract screening (LC-MScc) + Modified Ellman's method |
47 |
5 |
2 cpds were confirmed as weak actives: (i) 8-deoxylactucin (IC50 = 308.1 μM), and (ii) lactucopicrin (IC50 = 150.3 μM) |
[47] |
|
20 |
HRVdd coat protein |
Pharmacophore modeling + docking + scoring + PCAee-based clustering |
Multiple-cycle CPEff inhibition + microscopic + spectrophotometric |
∼60,000 |
6 |
6 promising inhibitors; ggEC50 = 7.3 ÷ 247.1 μmol/L (microscopic), EC50 = 4.3 ÷ 245.5 μmol/L (spectrophotometric) |
[48] |
|
21 |
Stat3hh |
Filtering + docking + scoring |
Stat3-dependent luciferase reporter + gel EMSAii |
∼429,000 |
100 |
1 natural product, a deoxytetrangomycin, an angucycline antibiotic, showed remarkable inhibition of Stat3 dimerization, DNA binding, and nucleus translocation as well as the Stat3-regulated genes such as Bcl-XLjj and cyclin D1 |
[49] |
|
22 |
EphB2kk |
Filtering + docking + scoring + pharmacophore modeling |
Frontal affinity chromatography with mass spectrometry detection (FAC-MSll) |
50,452 |
468 |
12 potential inhibitors; IC50 = 5.2 ÷ 250 μM |
[50] |
| 23 | Alpha1Amm | Filtering + pharmacophore modeling + docking + scoring | Radioligand displacement assay | 23,000 | 80 | 37 hits confirmed (Ki < 10 μM) of which 3 cpds showed affinity between 1 and 10 nM | [51] |
Formyl peptide receptor.
Inhibition constant.
G protein-coupled receptor 30.
The half maximal inhibitory concentration, represents the concentration of an inhibitor that is required for 50% inhibition.
Thioflavin T.
Severe acute respiratory syndrome coronavirus main protease.
Fluorescence resonance energy transfer.
Platelet 12-lipoxygenase.
12(S)-hydroxyeicosatetraenoic acid.
Fatty acid amide hydrolase.
Monoglyceride lipase.
Murine double minute 2.
Tumor protein 53.
11β-hydroxysteroid dehydrogenase type 1.
11β-hydroxysteroid dehydrogenase type 2.
Human embryonic kidney epithelial cell line.
Cyclophilin A.
Surface plasmon resonance.
Human platelet-type 12-lipoxygenase.
Human reticulocyte 15-lipoxygenase-l.
Anaplastic lymphoma kinase.
Angiotensin converting enzyme 2.
Glycogen synthase kinase-3β.
Adenosine triphosphate.
Dipeptidyl peptidase IV.
Cyclooxygenase 2.
Enzyme-linked immunosorbent assay.
Acetylcholinesterase.
Liquid chromatography–mass spectrometry.
Human rhinovirus.
Principal components analysis.
Cytopathic effect.
The concentration of a compound where 50% of its maximal effect is observed.
Signal transducers and activators of transcription 3.
Electrophoretic mobility shift assay.
An anti-apoptotic protein of the Bcl-2 (B-cell lymphoma 2) gene family.
Erythropoietin-producing hepatocellular B2.
Frontal affinity chromatography with mass spectrometry detection.
Adrenergic receptor subtype A.
Table 1 reveals the VS workflow used in most of the studies: (i) preliminary filtering of a virtual library (tens to hundreds of thousands of compounds) using various criteria (e.g. Lipinski's rule of five); (ii) homology and pharmacophore modeling; combined with (iii) docking and scoring of the protein–ligand complexes. For PS methods, assays that are particular to each specific target were applied, most of them being based on fluorescence detection. The outcome of these studies indicates that preliminary VS not only drastically reduces the number of chemicals that are physically screened, but also increases the hit rates.
Integration strategy
Target selection
VS is a knowledge-driven approach that depends on the amount and quality of information available about the system under investigation. Knowing the structure of the biological target macromolecule offers many advantages in comparison with the situation where only information about the geometry of a bioactive (reference) ligand is available. When structural information about the target is available from either fact or inference, we consider this target-based, or structure-based, virtual screening (TBVS). All other cases represent ligand-based virtual screening (LBVS), where the reference compound(s) are known substrates, inhibitors, agonists or antagonists, among others. An important issue in target selection is the druggability of the molecule under consideration, and whether the target is amenable to therapeutic intervention via small molecules. One approach is to assess whether members from the same gene family show similar binding affinity towards drug-like ligands with related physicochemical properties [16]; the other approach is sometimes referred as ‘target fishing’ (see J. L. Jenkins, this issue).
With the advent of structural genomics and homology modeling initiatives [17], the number of potential targets is expected to grow. For instance, over the last year (2005–2006), 26 new targets were screened at the NIH Molecular Libraries Initiative (formerly the Molecular Libraries Screening Center Network) (http://mli.nih.gov/); more are expected to be screened within next 2 years.
Our Center (New Mexico Molecular Libraries Screening Center: http://screening.health.unm.edu/) has undertaken projects on several classes of receptors, which include two GPCRs listed in Table 1 and an integrin, LFA-1. For the formyl peptide receptor, we compared several strategies including screening of class-focused (GPCR) and target-focused (FPR) libraries (Table 2 ). In the latter case, we used a homology model with pharmacophore docking to select a compound library [18]. We recorded active molecules at the rate of 1/880 from the Prestwick Chemical Library (http://www.prestwickchemical.com/chem_lib.htm), 17/9993 in a diverse set from the NIH Small Molecule Repository (http://mlsmr.glpg.com/MLSMR_HomePage/), 12/4959 in a GPCR class focused library from ChemDiv (http://www.chemdiv.com/en/products/screening/), and 30/4324 in a target focused library [18].
Table 2.
Screening of various class and target focused libraries: brief comparison
| Library | Library type | Compounds | Confirmed hits <30 μM | Best hit (μM) | % Hits |
|---|---|---|---|---|---|
| PCLa | Specialty set (marketed drugs) | 880 | 1 | 24 | 0.11 |
| MLSCN 10Kb | Diverse (85% DCc, 10% TLd, 5% SSe) | 9993 | 17 | 4 | 0.17 |
| GPCRf | Class focused | 4959 | 12 | 3 | 0.24 |
| FPRg | Target focused (pharmacophore) | 4324 | 30 | 1 | 0.70 |
A library from the Prestwick Chemical Library.
A library from the NIH small molecule repository.
Diverse collection.
Specialty sets.
Targeted libraries.
Library focused for G protein-coupled receptors.
Library focused for formyl peptide receptor.
In the case of GPR30, where no Target-specific ligand was known, we performed LBVS on the latter two libraries, using 17β-estradiol as the reference ligand. We ranked the top 100 structures for PS, finding one selective nM affinity agonist for GPR30 [19] and two ERα/ERβ that do not bind to GPR30 (C.G. Bologa, unpublished).
Workflow
The generic workflow shown in Fig. 2 is currently implemented at the New Mexico Molecular Libraries Screening Center, and follows the general template presented in Fig. 1. The core of the system is represented by the ‘screening database’ where complete information related to the (i) chemical structure data of the compounds, that is existing in-house or purchased from different vendors, and (ii) bioactivity data, that is plate format information and biological screening outcome, is recorded. The workflow steps are well defined. The biological team provides the target and the corresponding assay to the screening team, which is responsible for the PS. Once compounds are registered into the database by the cheminformatics team, plates are created and recorded in the database. The medicinal chemistry team provides chemistry follow-up and property optimization.
Figure 2.
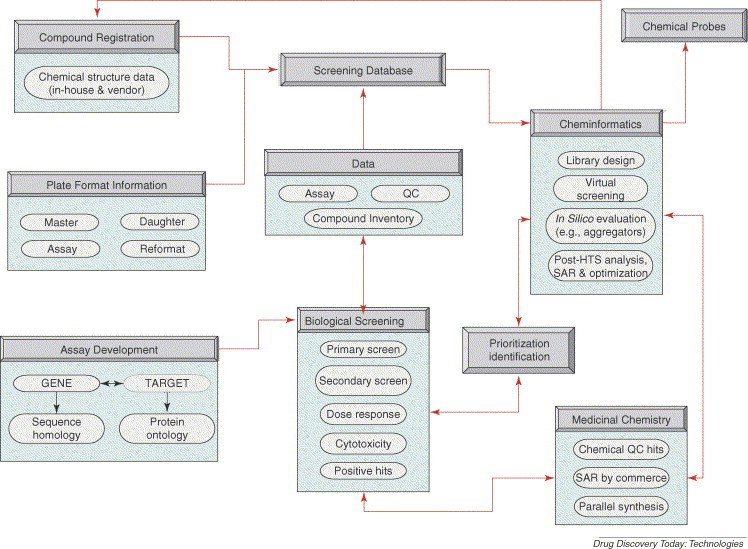
Workflow. The strategy implemented at the New Mexico Molecular Libraries Screening Center for the integration of virtual and physical screening. Abbreviations: QC: qualitative chemistry; SAR, structure–activity relationships.
There is a continuous information exchange between the screening, cheminformatics, and medicinal chemistry teams. Based on the outcome from the screening team, the cheminformatics team applies VS and post-high-throughput screening (HTS) analyses to further identify and prioritize compounds (or chemotypes) for further evaluation. The medicinal chemistry team verifies the chemical qualitative chemistry (QC) of the proposed hits and gives feedback to both the screening and cheminformatics teams. The final result is represented by the so-called ‘chemical probe’, which can be an inhibitor, activator or modulator of the studied target.
VS methods have been comprehensively reviewed [20] (see I. Muegge, this issue). Therefore, only a brief description is given here. Our VS workflow is shown in Fig. 3 . There are two main types of VS: (i) target or structure-based virtual screening (TBVS, or SBVS); and (ii) LBVS.
Figure 3.
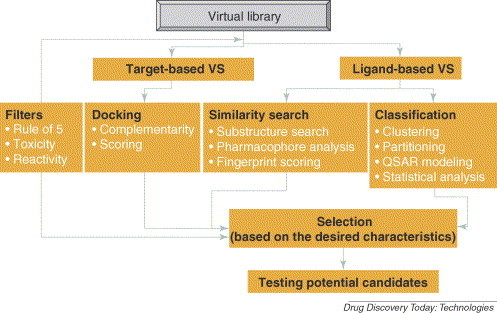
Virtual library. An example of virtual screening (VS) workflow as implemented at the University of New Mexico, Department of Biochemistry and Molecular Biology, Division of Biocomputing (http://biocomp.health.unm.edu/).
TBVS is applied when the target (protein, enzyme) structure is (i) known, based on the crystallographic or other structural methods, or (ii) built, using homology modeling. The procedure consists of docking a large number of molecules into the active site of the target, then scoring the binding affinity. The limiting step in this strategy is accurate scoring. There are four categories of scoring functions:
-
(i)
Knowledge-based methods [21] based on Boltzmann-weighted Potentials of Mean Force (PMF) derived from statistical analyses of ligand–receptor interatomic contacts from Protein Data Bank (PDB: http://www.rcsb.org/) complexes.
-
(ii)
‘Master equation’ approaches [22] estimate the energetic contributions of various interaction types in a semi-quantitative manner.
-
(iii)
Regression-based methods [23, 24] train on bioactives with known binding mode using sets of ligand–receptor complexes from the PDB.
-
(iv)
ZAP is a Poisson-Boltzmann equation solver [25] that scores ligands while incorporating solvent effects.
Because none of the above schemes has been shown to be general, it is preferable to use consensus-scoring methods [26].
LBVS methods start from known bioactives, which can be inhibitors or activators. Similarity search [27] and classification [28] methods can be used to select novel scaffolds (see K. V. Balakin, this issue). Filtering can be applied (i) forward or (ii) backward to the hits obtained in the VS campaign. In forward filtering [29], the selection criteria are used to reduce the size of the initial library, that is from several millions to several hundreds or hundreds of test compounds further to be docked. The backward procedure uses filtering criteria to prioritize candidates for PS during post-HTS analysis [30].
Testing strategies for the selected compounds can include does–response characterization and cytotoxicity of the selected compounds. In our Center, many of the assays are being set up as high throughput multiplexes so that selectivity and specificity information against families of targets is available for individual compounds. The physical properties of the components to be tested contribute to the screening strategy because the presence of molecules which are insoluble, are aggregators, or are fluorescent, can interfere with the identification of their activities in the assays.
When screening 100,000 to more than 1,000,000 compound libraries, it is paramount that the assay be optimized for complexity/simplicity (minimal additions, no wash steps, simple detection schemes), volume (typically no more than 10 μL, permitting use of 384 or 1536 well plates) and reproducibility (because the library is only screened once or twice). However, if VS technologies can reduce the library size to 10's or 100's of compounds, many of these constraints are lifted allowing the use of more physiologically relevant or complex assays, potentially including whole animal-based assays, which are recognized as a viable alternative in today's search for novel pharmaceuticals. Another issue related to assay implementation is the selection of in vitro vs. in vivo assays. Many of the initial hits in enzyme/protein-based in vitro assays may fail at the cellular or animal level due to stability or metabolism issues. However, with the development of VS and post-HTS analysis methods, this can be minimized. Finally, assay selection has a strong influence on the outcome of the physical screen. For example, the use of a simple binding assay screen can yield both agonists and antagonists of target activity. If only agonists are desired, a functional assay based on target activity might be preferentially selected.
Post-HTS analysis
The results of both VS and PS can yield a large number (often in the order of 100–1000) of interesting hits that warrant further attention. The issue of what molecules to select for dose–response confirmation is often left with the medicinal chemist or the biologist, and requires cheminformatics support. In our center, we apply the following post-HTS prioritization scheme [30] for chemotype as well as individual molecule evaluation (See Box 1 ).
Box 1. Evaluation criteria to select molecules for chemistry follow-up.
(i) Chemotype evaluation: This criterion gives higher priority (a) to chemotypes that occur more in active compounds, compared to the overall number of tested chemotypes; (b) to chemotypes that are absent or less present in patents, disclosures and medicinal chemistry (for intellectual property reasons); and (c) to chemotypes that are generally regarded as safe, or are less frequent in toxicity databases.
Chemotype evaluation is used to rank families of HTS hits, and is further applied to (ii) Individual molecule evaluation: Our scheme gives higher priority (a) to molecules that are in the desired physico-chemical property range (using methods to compute, e.g. solubility and permeability); (b) to molecules that have high(er) activity, compared to those that are less active. To the above, (c) we add the chemotype score computed earlier, for a final composite score that ranks all confirmed or presumed actives.
Thus, post-HTS analysis is essentially a practical step designed to assist the decision-makers to evaluate compounds for further experiments. It should be applied only to confirmed hits, both at the structure and purity, as well as at the dose–response level. The final score captures information related not only to actives, but also inactives from the same chemical family, while the intellectual property and toxicity evaluation schemes are aimed at encoding information related to individual chemotypes. At the individual molecule level, the use of estimated physico-chemical properties can assist the final prioritization score.
Summary and outlook
The integration of VS and PS technologies is attractive for both scientific and economic reasons. Scientifically, one can reduce the search space to rapidly find a solution; economically, one is no longer required to screen millions of compounds physically, before identifying hits. Negative aspects of this integration effort are of theoretical and practical nature. Theoretically, the streamlining of PS may result in testing the wrong library subset. This relates to both the limitation of theoretical methods, and to the inappropriate use of VS technologies. Practically speaking, it is possible that the entire effort leads to naught; in this case, doubt is typically cast over in silico approaches, although experimental techniques are not without flaw either. Continuous information exchange and effective team communication, as illustrated in Fig. 2, will avoid such negative situations.
Integration of these technologies is further supported by the increasing amount of valuable information being deposited in target and bioactivity databases (see T.I. Oprea, this issue). Conceivably, one can apply existing information to improve the success rate by using, for example, machine learning techniques to develop target-specific libraries; or to include target-specific or ligand-specific information in the post-HTS analysis process, to give higher priority to high-quality probes; or perhaps to download the entire matrix of target/bioactivity data and use it to profile compounds.
Acknowledgements
This work was supported by National Institutes of Health grant U54 MH074425-01 (National Institutes of Health Molecular Libraries Initiative); and by the New Mexico Tobacco Settlement Fund (D.C.F. and T.I.O.).
References
- 1.Kubinyi H. Chance favors the prepared mind – from serendipity to rational drug design. J. Recept. Signal Tr. R. 1999;19:15–39. doi: 10.3109/10799899909036635. [DOI] [PubMed] [Google Scholar]
- 2.Fox S. High throughput screening 2002: moving toward increased success rates. J. Biomol. Screen. 2002;7:313–316. doi: 10.1177/108705710200700402. [DOI] [PubMed] [Google Scholar]
- 3.Caron P.R. Chemogenomic approaches to drug discovery. Curr. Opin. Chem. Biol. 2001;5:464–470. doi: 10.1016/s1367-5931(00)00229-5. [DOI] [PubMed] [Google Scholar]
- 4.Sills M.A. Comparison of assay technologies for a tyrosine kinase assay generates different results in high throughput screening. J. Biomol. Screen. 2002;7:191–214. doi: 10.1177/108705710200700304. [DOI] [PubMed] [Google Scholar]
- 5.Bajorath J. Integration of virtual and high-throughput screening. Nature Rev. Drug. Discov. 2002;1:882–894. doi: 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- 6.Toledo-Sherman L.M., Chen D. High-throughput virtual screening for drug discovery in parallel. Curr. Opin. Drug Discov. Develop. 2002;5:414–421. [PubMed] [Google Scholar]
- 7.Walters W.P. Virtual screening – an overview. Drug Discov. Today. 1998;3:160–178. [Google Scholar]
- 8.Oprea T.I. Virtual screening in lead discovery: a viewpoint. Molecules. 2002;7:51–62. [Google Scholar]
- 9.Mestres J. Virtual screening: a real screening complement to high-throughput screening. Biochem. Soc. Trans. 2002;30:797–799. doi: 10.1042/bst0300797. [DOI] [PubMed] [Google Scholar]
- 10.Beresford A.P. The emerging importance of predictive ADME simulation in drug discovery. Drug Discov. Today. 2002;7:109–116. doi: 10.1016/s1359-6446(01)02100-6. [DOI] [PubMed] [Google Scholar]
- 11.Johnson D.E., Wolfgang G.H.I. Predicting human safety: screening and computational approaches. Drug Discov. Today. 2000;5:445–454. doi: 10.1016/s1359-6446(00)01559-2. [DOI] [PubMed] [Google Scholar]
- 12.Bajorath J. Rational drug discovery revisited: interfacing experimental programs with bio- and chemo-informatics. Drug Discov. Today. 2001;6:989–995. doi: 10.1016/s1359-6446(01)01961-4. [DOI] [PubMed] [Google Scholar]
- 13.Terstappen G.C., Reggiani A. In silico research in drug discovery. Trends Pharmacol. Sci. 2001;22:23–26. doi: 10.1016/s0165-6147(00)01584-4. [DOI] [PubMed] [Google Scholar]
- 14.Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 15.Leach A.R., Hann M.M. The in silico world of virtual libraries. Drug Discov. Today. 2000;5:326–336. doi: 10.1016/s1359-6446(00)01516-6. [DOI] [PubMed] [Google Scholar]
- 16.Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 17.Stevens R.C. Global efforts in structural genomics. Science. 2001;294:89–92. doi: 10.1126/science.1066011. [DOI] [PubMed] [Google Scholar]
- 18.Edwards B.S. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharmacol. 2005;68:1301–1310. doi: 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- 19.Bologa C.G. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nature Chem. Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 20.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov. Today. 2006;11:580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sippl M.J. Knowledge-based potentials for proteins. Curr. Opin. Struct. Biol. 1995;5:229–235. doi: 10.1016/0959-440x(95)80081-6. [DOI] [PubMed] [Google Scholar]
- 22.Ajay, Murcko M.A. Computational methods to predict binding free energy in ligand-receptor complexes. J. Med. Chem. 1995;38:4953–4967. doi: 10.1021/jm00026a001. [DOI] [PubMed] [Google Scholar]
- 23.Bohm H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J. Comput. Aid. Mol. Des. 1994;8:243–256. doi: 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- 24.Head R.D. VALIDATE: A new method for the receptor-based prediction of binding affinities of novel ligands. J. Am. Chem. Soc. 1996;118:3959–3969. [Google Scholar]
- 25.Grant J.A. A smooth permittivity function for Poisson-Boltzmann solvation methods. J. Comp. Chem. 2001;22:608–640. [Google Scholar]
- 26.Feher M. Consensus scoring for protein–ligand interactions. Drug Discov. Today. 2006;11:421–428. doi: 10.1016/j.drudis.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Hert J. New methods for similarity-based virtual screening. Chemogenomics. 2006:133–156. [Google Scholar]
- 28.Schneider G., Nettekoven M. Ligand-based combinatorial design of selective purinergic receptor (A2A) antagonists using self-organizing maps. J. Comb. Chem. 2003;5:233–237. doi: 10.1021/cc020092j. [DOI] [PubMed] [Google Scholar]
- 29.Oprea T.I. Property distribution of drug-related chemical databases. J. Comput. Aid. Mol. Des. 2000;14:251–264. doi: 10.1023/a:1008130001697. [DOI] [PubMed] [Google Scholar]
- 30.Oprea T.I. Post-HTS compound prioritization analysis: An empirical evaluation scheme. J. Biomol. Screen. 2005;10:419–425. doi: 10.1177/1087057104272660. [DOI] [PubMed] [Google Scholar]
- 31.Kravchencko D.V. Design and synthesis of new nonpeptide caspase-3 inhibitors. Pharma. Chem. J. 2006;40:127–131. [Google Scholar]
- 32.Liu D. Inhibitor discovery targeting the intermediate structure of β-amyloid peptide on the conformational transition pathway: implications in the aggregation mechanism of β-amyloid peptide. Biochemistry. 2006;45:10963–10972. doi: 10.1021/bi060955f. [DOI] [PubMed] [Google Scholar]
- 33.Lu I.-L. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J. Med. Chem. 2006;49:5154–5161. doi: 10.1021/jm060207o. [DOI] [PubMed] [Google Scholar]
- 34.Jankun J. Synthetic curcuminoids modulate the arachidonic acid metabolism of human platelet 12-lipoxygenase and reduce sprout formation of human endothelial cells. Mol. Cancer Ther. 2006;5:1371–1382. doi: 10.1158/1535-7163.MCT-06-0021. [DOI] [PubMed] [Google Scholar]
- 35.Saario S.M. Fatty acid amide hydrolase inhibitors from virtual screening of the endocannabinoid system. J. Med. Chem. 2006;49:4650–4656. doi: 10.1021/jm060394q. [DOI] [PubMed] [Google Scholar]
- 36.Lu Y. Discovery of a nanomolar inhibitor of the human murine double minute 2(MDM2)-p53 interaction through an integrated, virtual database screening strategy. J. Med. Chem. 2006;49:3759–3762. doi: 10.1021/jm060023+. [DOI] [PubMed] [Google Scholar]
- 37.Schuster D. The discovery of new 11β-hydroxysteroid dehydrogenase type 1 inhibitors by common feature pharmacophore modeling and virtual screening. J. Med. Chem. 2006;49:3454–3466. doi: 10.1021/jm0600794. [DOI] [PubMed] [Google Scholar]
- 38.Li J. Discovering novel chemical inhibitors of human cyclophilin A: virtual screening, synthesis, and bioassay. Bioorg. & Med. Chem. 2006;14:2209–2224. doi: 10.1016/j.bmc.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guichou J.-F. Structure-based design, synthesis, and biological evaluation of novel inhibitors of human cyclophilin A. J. Med. Chem. 2006;49:900–910. doi: 10.1021/jm050716a. [DOI] [PubMed] [Google Scholar]
- 40.Desai P.V. Identification of novel parasitic cysteine protease inhibitors by use of virtual screening. 2. The Available Chemical Directory. J. Med. Chem. 2006;49:1576–1584. doi: 10.1021/jm0505765. [DOI] [PubMed] [Google Scholar]
- 41.Kenyon V. Novel human lipoxygenase inhibitors discovered using virtual screening with homology models. J. Med. Chem. 2006;49:1356–1363. doi: 10.1021/jm050639j. [DOI] [PubMed] [Google Scholar]
- 42.Li R. Design and synthesis of 5-aryl-pyridone-carboxamides as inhibitors of anaplastic lymphoma kinase. J. Med. Chem. 2006;49:1006–1015. doi: 10.1021/jm050824x. [DOI] [PubMed] [Google Scholar]
- 43.Rella M. Structure-based pharmacophore design and virtual screening for novel angiotensin converting enzyme 2 inhibitors. J. Chem. Inf. Model. 2006;46:708–716. doi: 10.1021/ci0503614. [DOI] [PubMed] [Google Scholar]
- 44.Polgár T. Comparative virtual and experimental high-throughput screening for glycogen synthase kinase-3β inhibitors. J. Med. Chem. 2005;48:7946–7959. doi: 10.1021/jm050504d. [DOI] [PubMed] [Google Scholar]
- 45.Ward R.A. Structure-based virtual screening for low molecular weight chemical starting points for dipeptidyl peptidase IV inhibitors. J. Med. Chem. 2005;48:6991–6996. doi: 10.1021/jm0505866. [DOI] [PubMed] [Google Scholar]
- 46.Franke L. Extraction and visualization of potential pharmacophore points using support vector machines: application to ligand-based virtual screening for COX-2 inhibitors. J. Med. Chem. 2005;48:6997–7004. doi: 10.1021/jm050619h. [DOI] [PubMed] [Google Scholar]
- 47.Rollinger J.M. Application of the in combo screening approach for the discovery of non-alkaloid acetylcholinesterase inhibitors from Cichorium intybus. Curr. Drug. Discov. Technol. 2005;2:185–193. doi: 10.2174/1570163054866855. [DOI] [PubMed] [Google Scholar]
- 48.Steindl T.M. Pharmacophore modeling, docking, and principal component analysis based clustering: combined computer-assisted approaches to identify new inhibitors of the human rhinovirus coat protein. J. Med. Chem. 2005;48:6250–6260. doi: 10.1021/jm050343d. [DOI] [PubMed] [Google Scholar]
- 49.Song H. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U S A. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toledo-Sherman L. Frontal affinity chromatography with MS detection of EphB2 tyrosine kinase receptor. 2. Identification of small molecule inhibitors via coupling with virtual screening. J. Med. Chem. 2005;48:3221–3230. doi: 10.1021/jm0492204. [DOI] [PubMed] [Google Scholar]
- 51.Evers A., Klabunde T. Structure-based drug discovery using GPCR homology modeling: successful virtual screening for antagonists of the Alpha1A adrenergic receptor. J. Med. Chem. 2005;48:1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]