

Abstract
Human heart disease is a major cause of death and disability. A variety of animal models of cardiac disease have been developed to better understand the etiology, cellular and molecular mechanisms of cardiac dysfunction and novel therapeutic strategies. The animal models have included large animals (e.g. pig and dog) and small rodents (e.g. mouse and rat) and the advantages of genetic manipulation in mice have appropriately encouraged the development of novel mouse models of cardiac disease. However, there are major differences between rodent and human hearts that raise cautions about the extrapolation of results from mouse to human. The rabbit is a medium-sized animal that has many cellular and molecular characteristics very much like human, and is a practical alternative to larger mammals. Numerous rabbit models of cardiac disease are discussed, including pressure or volume overload, ischemia, rapid-pacing, doxorubicin, drug-induced arrhythmias, transgenesis and infection. These models also lead to the assessment of therapeutic strategies which may become beneficial in human cardiac disease.
Section editors:
Ju Chen – University of California, San Diego, Department of Medicine, La Jolla, CA, USA
Robert Ross – University of California, San Diego, Cardiology Section, San Diego, CA, USA
Why rabbit models?
The study of human cardiovascular disease has been greatly facilitated by the use of a wide variety of animal models. Small animal models such as rodents, guinea pigs and hamsters offer many advantages (low cost, short gestation time and short time for disease progression). Furthermore, there is a tremendous amount of historical data over decades in rat models of cardiovascular disease. Moreover, the development of genetically modified mice has transformed medical research, allowing investigators to overexpress, knock-out or knock-in genes of interest to explore the functional consequences of such genetic modulation.
Large animal disease models, such as in dog and pig, offer distinct advantages. Larger hearts provide a considerable amount of tissue (from different chambers and regions of the heart) for biochemical and molecular studies. Hemodynamic assessment and imaging is easier, the heart is large enough for chronic instrumentation or for in vivo cardiac mapping studies, and large size allows assessment of human-scale interventions ranging from left ventricular assist devices (LVADs), implantable cardiac defibrillators (ICDs), cardiac resynchronization therapy (CRT) and cardiac ablative approaches. Moreover, larger animal hearts have physical dimensions more like human hearts, and this larger size may be crucial for some arrhythmogenic mechanisms. However, the cost (both purchase cost and per diem charges) can be prohibitive, especially for long-term chronic studies in disease states.
Because of its intermediate size, the rabbit offers several potential advantages over other species. Although the rabbit heart is smaller than that of dog or pig, it is large enough to easily do surgical and catheter-based interventions at a much lower cost (5–15 times less expensive than those of dogs). At the same time, many ‘adult human scale’ interventions have been or are being scaled down for pediatric use and assessment (e.g. pacemakers and CRT) so that rabbit models can be beneficial (see below). Surgical interventions are still easier than microsurgical approaches in rodents. More importantly, rabbit cardiac physiology has more characteristics that are similar to human cardiac physiology than mouse or rat has.
Indeed, cellular electrophysiology and Ca2+ transport in rabbit are much more like those in human than is the case for either rat or mouse [1]. This is particularly relevant for the studies of heart failure (HF) and arrhythmias because alterations in ion channel and Ca2+ transporter function or expression are thought to contribute directly to depressed contractile performance and arrhythmogenesis [2, 3]. In particular, mouse and rat ventricular action potentials (APs) have very short duration and completely lack the prominent AP plateau phase typical of human, rabbit and most mammals larger than that of rat (Fig. 1D). Furthermore, the main ionic currents underlying repolarization and which greatly influence AP duration (APD) are the same in human and rabbit (delayed rectifier K+ currents I Kr and I Ks). These channels are not present in mouse or rat, where APD is dictated almost entirely by transient outward currents (I to). I to is present in human and rabbit ventricle, but plays only a minor role in APD in human and rabbit. Notably, the electrophysiological characteristics and secondary regulation of I to versus I Kr and I Ks are completely different, making this much more than a quantitative difference.
Figure 1.
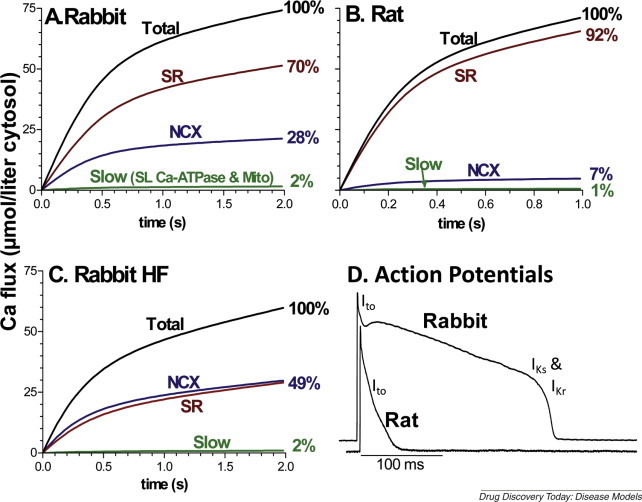
Ca2+ fluxes and action potentials in rabbit and rat ventricular myocytes. (A) and (B) Integrated Ca2+ fluxes during [Ca2+]i decline in normal adult rabbit and rat ventricular myocytes, on the basis of quantitative analysis of Ca2+ removal fluxes by the SR Ca-ATPase (SR) Na/Ca exchange (NCX) and the combined slow action of the plasma membrane Ca-ATPase (SL Ca-ATPase) and mitochondrial uniporter (Mito; on the basis of data in Bassani et al. [105]). (C) In rabbit HF, enhanced NCX function and decreased SR function brings these systems into more equal contribution (based on data in Pogwizd et al. [4]). Human nonfailing and failing hearts exhibit similar balance of fluxes as in rabbit [6]. (D) Action potentials recorded in normal adult rabbit and rat ventricular myocytes (data from Bassani et al. [105]) indicating the K+ currents responsible for repolarization at different phases (Ito, IKs and IKr).
In mouse and rat, almost all of the Ca2+ involved in the activation of contraction is released from the sarcoplasmic reticulum (SR), and during relaxation almost all of that Ca2+ is resequestered by the SR via the SR Ca-ATPase (Fig. 1A, much like in skeletal muscle). However, in human and rabbit ventricular myocytes a considerably larger fraction of activating Ca2+ comes via Ca2+ entry, and the same amount is extruded at each beat by the electrogenic Na/Ca exchange [1]. Under control conditions in rabbit and human this is 24–30% of the Ca2+ involved in excitation–contraction coupling, and in HF the SR and trans-sarcolemmal cycling can be nearly equal (Fig. 1B,C; [4, 5, 6]). Because changes in SR Ca2+ content and release, SR Ca-ATPase function and Na/Ca exchange influence both contractile function (systolic and diastolic) and arrhythmogenesis in HF, this major fundamental species difference may be crucial not only for the end-point phenotype, but also for the adaptive/maladaptive mechanisms involved in disease progression.
Adult mouse and rat ventricles normally express mainly the fast α-myosin isoform (which allows faster crossbridge cycling and muscle shortening than β-myosin), but during hypertrophy and HF there is isoform switching to β-myosin [7]. Adult human and rabbit myocardia express almost entirely β-myosin so there is little room for further isoform switching (although the change may still have functional consequences). These various differences are not arguments against other large animal models (e.g. canine and porcine, which resemble human and rabbit in this regard), but are crucial when extrapolating results from mouse and rat hearts in the context of human disease.
In the rest of this review, we discuss specific rabbit models of heart disease (rabbit models of vascular disease, lipid disorders, diabetes mellitus, thyroid disease, obesity or chronic hypoxia are not addressed). Although constrained by space, we attempt to provide an overview of the range of rabbit heart disease models used, along with some of their methods, advantages, limitations and potential (see Table 1 ).
Table 1.
Rabbit models of heart disease
| Experimental conditions | Refs |
|---|---|
| (a) Volume overload | |
| Arterio-venous shunt formation | [8] |
| Aortic regurgitation |
[9, 10] |
| (b) Models of ventricular hypertrophy | |
| Chronic AV block | [12] |
| Chronic infusion of angiotensin | [18] |
| LV pressure overload | |
| Aortic constriction | [13, 14, 15] |
| Nephrectomy (hypertension) | [16, 17] |
| RV pressure overload | |
| Pulmonary artery constriction – adult | [19] |
| Pulmonary artery constriction – young | [20] |
| Monocrotaline-induced pulmonary hypertension |
[21] |
| (c) Ischemic heart disease models | |
| Myocardial infarction in the rabbit | |
| Coronary artery occlusion | [23, 24] |
| Myocardial ischemia followed by reperfusion | [27] |
| Intracoronary microsphere embolization | [28] |
| Direct current shock | [30] |
| Hibernating myocardium | |
| Coronary artery ligation | [31] |
| Cellular model |
[32] |
| (d) Heart failure | |
| Adriamycin cardiomyopathy | [37, 38, 39] |
| High-dose catecholamines | [42] |
| Pacing-induced heart failure | [44, 45, 46, 47] |
| Combined pressure and volume overload |
[5, 52, 54, 59, 61] |
| (e)In vivoarrhythmia models | |
| Torsades des pointes | [62, 63, 64, 65] |
| Atrial fibrillation | |
| Isolated rabbit atria prep | [67, 68, 69] |
| Ventricular tachypacing-induced HF |
[71] |
| (f) Transgenic rabbit models of heart disease | |
| Long QT syndrome | [72] |
| Hypertrophic cardiomyopathy | [73, 74] |
| Gsα overexpression | [75] |
| Phospholamban overexpression |
[76] |
| (g) Other rabbit cardiac models | |
| Myocarditis | |
| Viral (coronavirus and coxsackie virus) | [77, 78] |
| Fungal (Cryptococcus) | [79] |
| Parasitic (toxoplasmosis) | [80] |
| Bacterial (streptococci and diphtheria) | [81, 82] |
| Chronic Chagas disease (Trypanosoma cruzi) | [83, 84] |
| Bacterial endocarditis (streptococcus, enterococcus and staphylococcus) | [85] |
| Radiation-induced heart disease | [87] |
Volume overload
Arterio-venous (AV) shunt formation can induce volume overload (e.g. by side-by-side anastamosis of the common carotid artery and the external jugular vein). This leads to cardiac hypertrophy, but there are little data on effects on LV contractile function [8].
Aortic regurgitation (AR) also causes volume overload, and can be induced in rabbits by aortic valve cusp perforation with a catheter using a transcarotid approach [9]. Severe AR leads to left ventricular hypertrophy that is followed by LV systolic dysfunction and HF over the course of one to two years. Although not all AR rabbits develop HF, this represents an advantage over AR models in other species such as dog that consistently manifest normal systolic function (for review, see [9]). Chronic AR rabbits exhibit myocardial fibrosis preceding the development of HF, in large part because of cardiac fibroblasts that produce abnormal proportions of noncollagen extracellular matrix, specifically fibronectin, with little change in collagen synthesis [10].
Models of ventricular hypertrophy
Chronic AV block
Chronic AV block in the dog (induced by AV nodal ablation) leads to ventricular remodeling characterized by LV hypertrophy and electrical remodeling [11]. A similar model, developed in the rabbit heart, exhibits electrical remodeling with prolonged QT interval, spontaneous torsades des pointes (TdP) in 75% of bradypaced rabbits, reduced I Ks and I Kr, and downregulated KvLQT1, minK and HERG [12].
Pressure overload
Aortic constriction (ascending or descending) in adult rabbits results in LV hypertrophy (typically on the order of 35–50%) [13], although HF is uncommon. Banding of the descending aorta of infant (ten-day-old) rabbits (a model analogous to coarctation of the aorta seen in children) leads to progressive aortic stenosis, and within six to seven weeks LV mass/volume ratios increases by ∼30–100% (without change in LV systolic function), but with decreased myocyte contractility [14, 15].
Hypertension can induce left ventricular hypertrophy in the rabbit when unilateral nephrectomy is combined with either contralateral renal artery constriction or renal wrapping. As an example, studies in the one-kidney, one-clip (1K,1C) Goldblatt rabbit (removal of the right kidney and partial constriction of the left renal artery) demonstrated severe hypertension, a ∼75% increase in LV mass, and diastolic dysfunction (LV systolic function was preserved) [16]. Mild LV hypertrophy has also been produced in rabbit by a one-kidney, one-wrap method [17]. In other studies, chronic infusion of angiotensin has been used to induce hypertension [18].
RV pressure overload can also induce right heart hypertrophy (and HF). This has been induced in rabbits by several interventions including pulmonary artery constriction in adult [19] or young rabbits [20] and monocrotaline-induced pulmonary hypertension [21].
Ischemic heart disease models
Myocardial infarction (MI) in the rabbit can be induced by coronary artery occlusion, and this is a useful model. There are some differences in coronary anatomy between rabbits and other species. The collateral circulation is not as extensive as it is in the canine heart, and the circumflex supplies most of the LV free wall and is more dominant than the left anterior descending (LAD) coronary artery [22, 23]. As such, the occlusion of the marginal branch of the circumflex coronary artery is often used for MI induction, and leads to mild to moderate degrees of HF, with LV ejection fractions in the range of 40–50%, but in some studies EFs were as low as 27% [23, 24]. MIs are characterized by increased LV end-diastolic dimension and increased left atrial size, similar to that found in LAD infarcts in other species. There are significant alterations in β-adrenergic receptor (β-AR) signaling including global reduction in β-AR density, reduced β-AR coupling and increased protein levels and activity of βARK1 and Gi [25]. There is regional heterogeneity of intracellular Ca2+ handling [23] and impaired synchronization of Ca2+ release events throughout the myocytes [26]. In addition to chronic occlusion, a rabbit model of myocardial ischemia followed by reperfusion has been used [27]. Moreover, other rabbit MI models include intracoronary microsphere embolization [28] (similar to a well-established intracoronary microembolization model in dog) [29], and direct current-shock-induced cardiac muscle injury [30].
Hibernating myocardium models in the rabbit have been developed, for example with coronary artery ligation [31]. Moreover, a cellular model of hibernating myocardium in rabbit cardiac myocytes has been developed [32]. Coculturing of adult rabbit cardiomyocytes with cardiac fibroblasts induced hibernation-like dedifferentiation and serves as a valuable tool to study cellular pathways of hibernating myocardium in vitro.
Rabbit models of heart failure
HF has been induced in the rabbit heart by several approaches. As mentioned above, volume or pressure overload, as well as MI, occasionally leads to HF. However, several HF rabbit models have been developed using approaches that have been successful in other animal species [33, 34, 35].
Doxorubicin (Adriamycin), an anthracycline chemotherapeutic agent, is one of the most prescribed anticancer drugs, primarily because of its effectiveness in a wide variety of hematologic malignancies and solid tumors. However, cardiotoxicity is a major clinical problem, and its cumulative toxicity on myocardium prevents their use at maximum doses that would be needed for optimal treatment [36]. Progressive reduction in ejection fraction is seen during the course of therapy, and life threatening CHF is observed in ∼10% of patients receiving more than 550 mg/m2. This has prompted studies in experimental models. Adriamycin has been used experimentally to induce cardiomyopathy in rabbits [37, 38, 39]. This model has been useful in identifying cardioprotective agents that could limit adriamycin toxicity. It has also been useful as a model of nonischemic cardiomyopathy with severe cardiac dilatation, LV hypertrophy, and decreased LV systolic function that is progressive and irreversible, accompanied by fluid retention and activation of the sympathetic nervous system and the renin–angiotensin systems [37]. However, there is no evidence of β-adrenergic receptor downregulation as occurs in advanced human HF [40]. Cardiotoxicity is due to free radical formation and lipid peroxidation that ultimately alters lysosomes, mitochondria, SR and the sarcolemmal membrane. These changes result in the activation of hydrolytic enzymes, calcium overload and reduced energy production [41]. Pathologic changes including cytosolic vacuolization and myofibrillar loss are typical of adriamycin cardiotoxicity in patients, but are different from those observed in other forms of nonischemic HF in humans (e.g. idiopathic dilated cardiomyopathy (IDCM), long-standing hypertension or valvular heart disease). Other limitations include the variable degree of LV dysfunction produced, undesirable bone marrow and GI toxicity.
High-dose catecholamines (isoproterenol or epinephrine), repetitively infused, induce a cardiomyopathy in rabbits characterized by LV dilation, hypertrophy and depressed systolic function, but a high mortality rate [42].
Rapid pacing induces HF. Tachycardia-induced HF has been described in patients with long-standing tachyarrhythmias such as atrial fibrillation (AF) with rapid ventricular response. As such, rapid pacing (whether ventricular or atrial) has been used to induce HF in several species – most commonly dog, but also pig and rabbit [43]. Rapid ventricular pacing in the rabbit induces HF characterized by cardiac enlargement, systolic dysfunction and ventricular myocytes exhibiting contractile dysfunction. The intermediate size of the rabbit allows implantation of standard human pacemakers. Because the intrinsic heart rates of rabbits are in the range of 220–280 beats/min, pacing to rates in the range of 340–400 beats/min are required [44, 45, 46]. HF develops over the course of two to five weeks and is characterized by dyspnea, appetite loss and body weight loss. Chronic pacing leads to progressive, predictable and time-dependent reduction in LV systolic function and geometry [44], which allow the study of molecular and cellular events during the development of chronic HF. Development of HF is associated with reduced responsiveness to inotropic stimulus, decreased myocyte contractility, and neurohumoral activation [44, 47]. There are changes in Ca2+ homeostatic mechanisms consistent with human HF, including decreases in Ca transient amplitude, L-type Ca2+ channel activity and SERCA expression. However, Yao et al. report decreased Na/Ca exchange current in pacing HF rabbits [45] in contrast to studies in canine pacing HF showing enhanced NCX activity when [Ca2+]i was minimally buffered [48]. As with pacing HF models in other species, LV systolic function returns to normal within approximately one week following cessation of pacing, although adenyl cyclase response to agonists takes two weeks, and β-adrenergic receptor density takes four weeks to return to normal [46]. Moreover, the LV dilatation and dysfunction is not associated with myocardial or cellular hypertrophy [44], so the changes in LV myocardial structure are not similar to those of clinical HF in humans caused by ischemic or hypertensive heart disease. The model has nonetheless provided important information regarding the pathophysiology of the failing heart including electrical remodeling [49], abnormal intracellular Ca2+ cycling [50] and exercise training in HF [51].
Many of the rabbit heart disease models provide investigators with hypertrophied and/or failing myocardium with which to study alterations such as contractile dysfunction, myocardial energetics and gene expression. However, the studies of arrhythmogenesis in the failing heart in these models are limited by the fact that few of these rabbit models are truly arrhythmogenic. In fact, even when one considers other animal models of HF, both large and small, there are few models that exhibit both severe contractile dysfunction and spontaneously occurring and inducible ventricular arrhythmias.
Combining volume overload (AR) with pressure overload (aortic constriction) several weeks later consistently leads to HF (much more so than either volume or pressure overload alone) [52]. We have demonstrated that HF rabbits exhibit severely depressed LV function with LVH (75% increase in HW/BW), spontaneously occurring VT that initiates by a nonreentrant mechanism such as triggered activity, and a 10% incidence of sudden death [5, 53, 54]. We also showed that HF rabbit myocytes exhibit contractile dysfunction from decreased SR Ca load that is associated with altered intracellular Ca handling (↑NCX, ↓RyR and preserved SERCA); preserved β-AR responsiveness with enhanced β2-AR responsiveness; decrease in ion currents (I to, I K1 and I Ks); activation (by catecholamines) of a transient inward current I ti that can initiate delayed after depolarizations (DADs); downregulation and dephosphorylation of the main ventricular gap junctional protein connexin43 (Cx43) increased the expression and activation of CaMKII and increased RyR phosphorylation and SR Ca2+ leak [3, 5, 53, 55, 56]. Many of these findings have been validated by focused studies in failing human hearts including 3D mapping studies showing focal nonreentrant mechanisms underlying VT in patients with nonischemic HF, NCX upregulation with preserved SERCA in a large subset of human HF patients, enhanced β2-adrenergic responsiveness in failing human cardiac myocytes [53, 57, 58], and Cx43 downregulation and dephosphorylation in human HF (both ischemic and nonischemic) [55]. Other studies with this HF model have demonstrated additional insights into arrhythmogenesis [59], sinus node dysfunction in HF [60] and the effects of acute ischemia superimposed on chronic HF [61]. Thus, findings in these models have provided important new insights into human HF. While this arrhythmogenic rabbit model of nonischemic HF is technically challenging and takes months to develop, the similarities of rabbit myocardium to human myocardium and validating studies in humans indicate that this model provides important insights into the contractile dysfunction and arrhythmogenicity of the failing heart.
Other in vivo arrhythmia models
TdP, a polymorphic ventricular tachycardia that occurs in the setting of a prolonged QT interval, can be caused by several pharmaceutical agents (primarily those that inhibit I Kr, the rapid component of the inward rectifying potassium current, I Kr). TdP had been difficult to model in experimental animals, and mouse and rat which lack an AP plateau and I Kr are less useful animal models in this arena. Carlsson et al. [62] first developed an in vivo model of TdP in the anesthetized α-adrenoreceptor (α-AR)-stimulated rabbit (methoxamine plus the Class III antiarrhythmic clofilium). This model has been used extensively by several groups with minor modification [62, 63, 64] to test for drug-induced TdP, to understand the underlying electrophysiological mechanisms [65] or to develop novel antiarrhythmic approaches for TdP [63]. It remains unclear as to how α-AR stimulation facilitates TdP induction, but could involve increases in [Ca2+]i, elevation of blood pressure or reflex vagal nerve activation [64]. Myocardial failure in the rabbit (induced by coronary artery ligation) has also been used as a model to predict the development of TdP [66].
AF can be studied in isolated rabbit atria preparations, and this has provided key insights into the mechanism of AF [67, 68] and the role of atrial dilatation [69]. Rabbit models of heart disease have been used to study atrial conduction and atrial fibrillation. Chronic volume overload from AV shunt formation slowed atrial conduction and enhanced the induction of atrial tachycardia (but not AF) [70]. However, rabbits with ventricular tachypacing-induced HF exhibit atrial fibrosis and enhanced atrial fibrillation induced by burst pacing [71].
Transgenic rabbit models of heart disease
Transgenic mice have provided important insights into cellular physiology, but the small size of murine hearts limits easy assessment of function and precluding assessment of human-size interventions such as pacing, CRT and defibrillation. However, in the past decade, transgenic rabbits have been developed, combining the value of transgenic approaches (overexpression or knock-outs of key genes, previously restricted to the mouse) with the larger animal size, and the scale and physiology of the rabbit.
Transgenic rabbit models created to date include: long QT syndrome [72], hypertrophic cardiomyopathy [73, 74], G sα overexpression [75] and phospholamban overexpression [76]. In some cases, findings in transgenic rabbits appear different from those in transgenic mice. For example, transgenic rabbits overexpressing the G protein G sα do not develop cardiomyopathy like their transgenic mouse counterparts [75]; and phospholamban overexpressing transgenic rabbits have normal cardiac function and response to β-adrenergic stimulation, unlike transgenic mouse models [76]. These findings might arise from species differences in compensatory changes and/or species differences in cellular physiology (rabbit calcium handling and ion channel physiology is closer to that of human than what is observed in mouse). The expense and time for the development of transgenic rabbits has been limiting, but with further advances in the field, transgenic rabbits will offer important insights into cardiac physiology. Moreover, the addition of induced cardiac disease states (such as pressure overload, MI and HF) will provide novel insights into contractile dysfunction, cardiac remodeling and arrhythmogenesis.
Other cardiac models
Infectious diseases affecting the heart have stimulated several experimental models in the rabbit. Myocarditis (with HF and dilated cardiomyopathy) has been induced in rabbits infected with viruses such as coronavirus and coxsackie virus [77, 78], fungi such as Cryptococcus [79], parasites such as toxoplasmosis [80] and bacteria such as streptococci [81] and diphtheria [82]. Chronic Chagas disease has been produced in rabbits by the inoculation of a virulent strain of Trypanosoma cruzi, and is characterized by biventricular (bi-V) dilatation and hypertrophy, apical aneurysm, interstitial fibrosis and focal myocarditis [83, 84]. Bacterial endocarditis has been produced in rabbit with several bacterial pathogens including streptococcus, enterococcus and staphylococcus [85]. Although the focus of these studies is predominantly in response to antimicrobial therapy, these models have provided insight into pathogenesis of both native valve and prosthetic valve endocarditis in humans. Sepsis affects cardiac function, and endotoxin-induced cardiomyopathy has been induced by the infusion of bacterial lipopolysaccharide [86].
Radiation also induces acute myocardial lesions, typically a pancarditis with inflammatory exudates, followed by a latent phase and eventually myocardial and pericardial fibrosis [87]. Studies in rabbits have also shown that radiation enhances adriamycin cardiotoxicity [88].
Assessment of therapeutic approaches in rabbit heart models
Having discussed a large number of rabbit models of heart disease, we finish this review with a brief discussion of how these models have been and can be used in the development of novel therapeutic approaches to heart disease.
Drug therapy with different pharmacologic agents as therapy for HF has been used in a wide range of rabbit heart models – primarily in infarct and combined pressure and volume overload models, but to some degree in pacing-induced and adriamycin-induced HF.
Cardiac ablative therapy in the rabbit heart is limited by the small size for in vivo studies using currently utilized ablation catheters. However, the isolated perfused rabbit heart has been useful for characterizing the effects of radio-frequency ablation (of atrium, ventricle and AV node) on conduction and the underlying anatomy [89, 90].
CRT also known as bi-V pacing is beginning to be applied to the rabbit model of MI. Recent studies have shown that bi-V pacing attenuated LV dilatation, systolic dysfunction and electrical remodeling [91].
ICDs have not been used in rabbit models to date. However, VF inducibility and VF threshold have been assessed in the in vivo rabbit heart [92] as well as in the Langendorff-perfused rabbit heart [93].
Gene therapy approaches have been used in the rabbit models of heart disease. Direct intramuscular injection of viral vectors into rabbit LV myocardium is the simplest approach, but is limited by local transgene expression as well as traumatic effects from needle injections. Myocardial gene delivery approaches to rabbits initially involved thoracotomy-based approaches: delivery of adenoviral transgenes either by ex vivo (retrograde) intracoronary delivery to donor hearts of a heterotopic heart transplant model [94] or LV cavity injection (with ascending aorta cross-clamped) [95]. However, noninvasive techniques such as percutaneous subselective coronary artery catheterization have resulted in the efficient delivery of transgene that is ventricle-specific and targeted to the particular coronary artery that is catheterized [96]. Koch and his colleagues used these approaches to demonstrate that adenoviral gene transfer to overexpress the β2-adrenergic receptor or βARKct (a peptide inhibitor of β-adrenergic receptor kinase 1 (βARK1)) in the rabbit MI model can enhance contractile function and β-adrenergic receptor responsiveness, and delay the onset of HF [95, 96, 97]. Other studies in rabbit include have explored adenoviral gene transfer of sodium–calcium exchanger (NCX) [98]; fibroblast growth factor-2 [28], caspase inhibitor p35 [99], norepinephrine transporter uptake-1 [100], extracellular superoxide dismutase [101] and of the antiapototic factor Bcl-2 [24].
Cell therapy approaches are being tested in rabbit heart. Examples of recent studies include skeletal myoblast transplantation in the infarcted rabbit heart [102], and autologous stem cell transplantation in doxorubicin-induced nonischemic HF [103, 104].
Conclusion
There are no ideal models of heart disease, and every model utilized has both advantages and disadvantages. The rabbit has been used to develop models of a wide variety of human diseases including pressure overload, volume overload, combined pressure and volume overload, pacing-induced HF, toxic cardiomyopathy, toxic cardiomyopathy, MI and transgenic rabbit models. Transgenic rabbit models of heart disease have been developed for several human diseases and more are on the way. Rabbit models of human heart disease can offer distinct advantages over rodent or large animal (dogs and pigs) in terms of intermediate size, intermediate costs and similarities to human physiology, and can provide novel insights into human cardiac disease.
Acknowledgements
This work was supported by NIH grants HL73966 and HL46929 (S.M.P.) and HL64724 and HL80101 (D.M.B.).
Contributor Information
Steven M. Pogwizd, Email: spogwizd@cardmail.dom.uab.edu.
Donald M. Bers, Email: dmbers@ucdavis.edu.
References
- 1.Bers D.M. Kluwer Academic Publishers; Dordrecht, Netherlands: 2001. Excitation–Contraction Coupling and Cardiac Contractile Force. p. 427. [Google Scholar]
- 2.Bers D.M. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. doi: 10.1152/physiol.00019.2006. (PM:17119150) [DOI] [PubMed] [Google Scholar]
- 3.Pogwizd S.M., Bers D.M. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. (PM:15030791) [DOI] [PubMed] [Google Scholar]
- 4.Pogwizd S.M. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. (PM:10571531) [DOI] [PubMed] [Google Scholar]
- 5.Pogwizd S.M. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium–calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ. Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. (PM:11397782) [DOI] [PubMed] [Google Scholar]
- 6.Piacentino V., III Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. (PM:12600875) [DOI] [PubMed] [Google Scholar]
- 7.Gupta M.P. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2007;43:388–403. doi: 10.1016/j.yjmcc.2007.07.045. (PM:17720186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugawara H. Differential alteration of cardiotonic effects of EMD 57033 and beta-adrenoceptor agonists in volume-overload rabbit ventricular myocytes. J. Card. Fail. 2000;6:338–349. doi: 10.1054/jcaf.2000.19237. (PM:11145759) [DOI] [PubMed] [Google Scholar]
- 9.Magid N.M. Heart failure due to chronic experimental aortic regurgitation. Am. J. Physiol. 1994;267:H556–H562. doi: 10.1152/ajpheart.1994.267.2.H556. (PM:8067410) [DOI] [PubMed] [Google Scholar]
- 10.Borer J.S. Myocardial fibrosis in chronic aortic regurgitation: molecular and cellular responses to volume overload. Circulation. 2002;105:1837–1842. doi: 10.1161/01.cir.0000014419.71706.85. (PM:11956128) [DOI] [PubMed] [Google Scholar]
- 11.Oros A. The canine model with chronic, complete atrio-ventricular block. Pharmacol. Ther. 2008;119:168–178. doi: 10.1016/j.pharmthera.2008.03.006. (PM:18514320) [DOI] [PubMed] [Google Scholar]
- 12.Tsuji Y. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. 2006;113:345–355. doi: 10.1161/CIRCULATIONAHA.105.552968. (PM:16432066) [DOI] [PubMed] [Google Scholar]
- 13.Mohammadi K. Protein kinase C activity and expression in rabbit left ventricular hypertrophy. J. Mol. Cell. Cardiol. 1997;29:1687–1694. doi: 10.1006/jmcc.1997.0411. (PM:9220354) [DOI] [PubMed] [Google Scholar]
- 14.Friehs I. Impaired glucose transporter activity in pressure-overload hypertrophy is an early indicator of progression to failure. Circulation. 1999;100:II187–II193. doi: 10.1161/01.cir.100.suppl_2.ii-187. (PM:10567302) [DOI] [PubMed] [Google Scholar]
- 15.Stamm C. Inhibition of tumor necrosis factor-alpha improves postischemic recovery of hypertrophied hearts. Circulation. 2001;104:I350–I355. doi: 10.1161/hc37t1.094851. (PM:11568081) [DOI] [PubMed] [Google Scholar]
- 16.Signolet I. Echocardiography in conscious 1K,1C Goldblatt rabbits reveals typical features of human hypertensive ventricular diastolic dysfunction. Int. J. Cardiol. 2009;132:135–137. doi: 10.1016/j.ijcard.2007.07.143. (PM:18045708) [DOI] [PubMed] [Google Scholar]
- 17.Lorell B.H. Influence of hypertension with minimal hypertrophy on diastolic function during demand ischemia. Hypertension. 1989;13:361–370. doi: 10.1161/01.hyp.13.4.361. (PM:2522416) [DOI] [PubMed] [Google Scholar]
- 18.Burke S.L. Renal sympathetic neuroeffector function in renovascular and angiotensin II-dependent hypertension in rabbits. Hypertension. 2007;49:932–938. doi: 10.1161/01.HYP.0000260251.11364.1f. (PM:17309940) [DOI] [PubMed] [Google Scholar]
- 19.Hamrell B.B., Alpert N.R. The mechanical characteristics of hypertrophied rabbit cardiac muscle in the absence of congestive heart failure: the contractile and series elastic elements. Circ. Res. 1977;40:20–25. doi: 10.1161/01.res.40.1.20. (PM:137085) [DOI] [PubMed] [Google Scholar]
- 20.Gibbs C.L. Mechanical, energetic, and biochemical changes in long-term pressure overload of rabbit heart. Am. J. Physiol. 1990;259:H849–H859. doi: 10.1152/ajpheart.1990.259.3.H849. (PM:2144403) [DOI] [PubMed] [Google Scholar]
- 21.Gunaydin S. The effects of vasoactive intestinal peptide on monocrotaline induced pulmonary hypertensive rabbits following cardiopulmonary bypass: a comparative study with isoproteronol and nitroglycerine. Cardiovasc. Surg. 2002;10:138–145. doi: 10.1016/s0967-2109(01)00126-0. (PM:11888743) [DOI] [PubMed] [Google Scholar]
- 22.Coker S.J. Anesthetized rabbit as a model for ischemia- and reperfusion-induced arrhythmias: effects of quinidine and bretylium. J. Pharmacol. Methods. 1989;21:263–279. doi: 10.1016/0160-5402(89)90064-8. (PM:2755145) [DOI] [PubMed] [Google Scholar]
- 23.Ng G.A. Non-uniform prolongation of intracellular Ca2+ transients recorded from the epicardial surface of isolated hearts from rabbits with heart failure. Cardiovasc. Res. 1998;37:489–502. doi: 10.1016/s0008-6363(97)00255-1. (PM:9614503) [DOI] [PubMed] [Google Scholar]
- 24.Chatterjee S. Viral gene transfer of the antiapoptotic factor Bcl-2 protects against chronic postischemic heart failure. Circulation. 2002;106:I212–I217. (PM:12354736) [PubMed] [Google Scholar]
- 25.Maurice J.P. Molecular beta-adrenergic signaling abnormalities in failing rabbit hearts after infarction. Am. J. Physiol. 1999;276:H1853–H1860. doi: 10.1152/ajpheart.1999.276.6.H1853. (PM:10362663) [DOI] [PubMed] [Google Scholar]
- 26.Litwin S.E. Dyssynchronous Ca(2+) sparks in myocytes from infarcted hearts. Circ. Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. (PM:11090550) [DOI] [PubMed] [Google Scholar]
- 27.Minatoguchi S. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation. 2004;109:2572–2580. doi: 10.1161/01.CIR.0000129770.93985.3E. (PM:15123535) [DOI] [PubMed] [Google Scholar]
- 28.Iwatate M. Effects of in vivo gene transfer of fibroblast growth factor-2 on cardiac function and collateral vessel formation in the microembolized rabbit heart. Jpn. Circ. J. 2001;65:226–231. doi: 10.1253/jcj.65.226. (PM:11266199) [DOI] [PubMed] [Google Scholar]
- 29.Sabbah H.N. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am. J. Physiol. 1991;260:H1379–H1384. doi: 10.1152/ajpheart.1991.260.4.H1379. (PM:1826414) [DOI] [PubMed] [Google Scholar]
- 30.Arnolda L. Systemic and regional effects of vasopressin and angiotensin in acute left ventricular failure. Am. J. Physiol. 1991;260:H499–H506. doi: 10.1152/ajpheart.1991.260.2.H499. (PM:1825456) [DOI] [PubMed] [Google Scholar]
- 31.Driesen R.B. Structural remodelling of cardiomyocytes in the border zone of infarcted rabbit heart. Mol. Cell. Biochem. 2007;302:225–232. doi: 10.1007/s11010-007-9445-2. (PM:17387581) [DOI] [PubMed] [Google Scholar]
- 32.Dispersyn G.D. Adult rabbit cardiomyocytes undergo hibernation-like dedifferentiation when co-cultured with cardiac fibroblasts. Cardiovasc. Res. 2001;51:230–240. doi: 10.1016/s0008-6363(01)00326-1. (PM:11470462) [DOI] [PubMed] [Google Scholar]
- 33.Smith H.J., Nuttall A. Experimental models of heart failure. Cardiovasc. Res. 1985;19:181–186. doi: 10.1093/cvr/19.4.181. (PM:3159477) [DOI] [PubMed] [Google Scholar]
- 34.Arnolda L.F. Animal models of heart failure. Aust. N. Z. J. Med. 1999;29:403–409. doi: 10.1111/j.1445-5994.1999.tb00735.x. (PM:10868512) [DOI] [PubMed] [Google Scholar]
- 35.Monnet E., Chachques J.C. Animal models of heart failure: what is new? Ann. Thorac. Surg. 2005;79:1445–1453. doi: 10.1016/j.athoracsur.2004.04.002. (PM:15797108) [DOI] [PubMed] [Google Scholar]
- 36.Rhoden W. Anthracyclines and the heart. Br. Heart J. 1993;70:499–502. doi: 10.1136/hrt.70.6.499. (PM:8280512) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arnolda L. Adriamycin cardiomyopathy in the rabbit: an animal model of low output cardiac failure with activation of vasoconstrictor mechanisms. Cardiovasc. Res. 1985;19:378–382. doi: 10.1093/cvr/19.6.378. (PM:4016815) [DOI] [PubMed] [Google Scholar]
- 38.Wanless R.B. An experimental model of chronic cardiac failure using adriamycin in the rabbit: central haemodynamics and regional blood flow. Cardiovasc. Res. 1987;21:7–13. doi: 10.1093/cvr/21.1.7. (PM:3664539) [DOI] [PubMed] [Google Scholar]
- 39.Pye M.P., Cobbe S.M. Arrhythmogenesis in experimental models of heart failure: the role of increased load. Cardiovasc. Res. 1996;32:248–257. doi: 10.1016/0008-6363(96)00080-6. (PM:8796111) [DOI] [PubMed] [Google Scholar]
- 40.Woodcock E.A. Ventricular beta-adrenoceptors in adriamycin-induced cardiomyopathy in the rabbit. J. Mol. Cell. Cardiol. 1988;20:771–777. doi: 10.1016/s0022-2828(88)80002-6. (PM:2906708) [DOI] [PubMed] [Google Scholar]
- 41.Gille L., Nohl H. Analyses of the molecular mechanism of adriamycin-induced cardiotoxicity. Free Radic. Biol. Med. 1997;23:775–782. doi: 10.1016/s0891-5849(97)00025-7. (PM:9296455) [DOI] [PubMed] [Google Scholar]
- 42.Muders F. Hemodynamic changes and neurohumoral regulation during development of congestive heart failure in a model of epinephrine-induced cardiomyopathy in conscious rabbits. J. Card. Fail. 1999;5:109–116. doi: 10.1016/s1071-9164(99)90033-7. (PM:10404350) [DOI] [PubMed] [Google Scholar]
- 43.Shinbane J.S. Tachycardia-induced cardiomyopathy: a review of animal models and clinical studies. J. Am. Coll. Cardiol. 1997;29:709–715. doi: 10.1016/s0735-1097(96)00592-x. (PM:9091514) [DOI] [PubMed] [Google Scholar]
- 44.Eble D.M. Myosin heavy chain synthesis is increased in a rabbit model of heart failure. Am. J. Physiol. 1997;272:H969–H978. doi: 10.1152/ajpheart.1997.272.2.H969. (PM:9124461) [DOI] [PubMed] [Google Scholar]
- 45.Yao A. Abnormal myocyte Ca2+ homeostasis in rabbits with pacing-induced heart failure. Am. J. Physiol. 1998;275:H1441–H1448. doi: 10.1152/ajpheart.1998.275.4.H1441. (PM:9746495) [DOI] [PubMed] [Google Scholar]
- 46.Kawai H. Alterations in cardiac adrenergic terminal function and beta-adrenoceptor density in pacing-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2000;278:H1708–H1716. doi: 10.1152/ajpheart.2000.278.5.H1708. (PM:10775152) [DOI] [PubMed] [Google Scholar]
- 47.Spinale F.G. Left ventricular and myocyte structure and function following chronic ventricular tachycardia in rabbits. Basic Res. Cardiol. 1994;89:456–467. doi: 10.1007/BF00788282. (PM:7702537) [DOI] [PubMed] [Google Scholar]
- 48.Hobai I.A., O’Rourke B. Enhanced Ca(2+)-activated Na(+)–Ca(2+) exchange activity in canine pacing-induced heart failure. Circ. Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. (PM:11029405) [DOI] [PubMed] [Google Scholar]
- 49.Rose J. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H2077–H2087. doi: 10.1152/ajpheart.00526.2003. (PM:15637125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Armoundas A.A. Cellular and molecular determinants of altered Ca2+ handling in the failing rabbit heart: primary defects in SR Ca2+ uptake and release mechanisms. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1607–H1618. doi: 10.1152/ajpheart.00525.2006. (PM:17122195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao L. Exercise training normalizes sympathetic outflow by central antioxidant mechanisms in rabbits with pacing-induced chronic heart failure. Circulation. 2007;115:3095–3102. doi: 10.1161/CIRCULATIONAHA.106.677989. (PM:17548725) [DOI] [PubMed] [Google Scholar]
- 52.Leclercq J.F. Experimental cardiac hypertrophy in rabbits after aortic stenosis or incompetence or both. Biomedicine. 1978;28:180–184. (PM:151562) [PubMed] [Google Scholar]
- 53.Desantiago J. Arrhythmogenic effects of beta2-adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ. Res. 2008;102:1389–1397. doi: 10.1161/CIRCRESAHA.107.169011. (PM:18467626) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pogwizd S.M. Nonreentrant mechanisms underlying spontaneous ventricular arrhythmias in a model of nonischemic heart failure in rabbits. Circulation. 1995;92:1034–1048. doi: 10.1161/01.cir.92.4.1034. (PM:7543829) [DOI] [PubMed] [Google Scholar]
- 55.Ai X., Pogwizd S.M. Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ. Res. 2005;96:54–63. doi: 10.1161/01.RES.0000152325.07495.5a. (PM:15576650) [DOI] [PubMed] [Google Scholar]
- 56.Ai X. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. (PM:16269653) [DOI] [PubMed] [Google Scholar]
- 57.Pogwizd S.M. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 1998;98:2404–2414. doi: 10.1161/01.cir.98.22.2404. (PM:9832485) [DOI] [PubMed] [Google Scholar]
- 58.Studer R. Gene expression of the cardiac Na(+)–Ca2+ exchanger in end-stage human heart failure. Circ. Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. (PM:8062418) [DOI] [PubMed] [Google Scholar]
- 59.Janse M.J. Arrhythmogenesis in heart failure. J. Cardiovasc. Electrophysiol. 2001;12:496–499. doi: 10.1046/j.1540-8167.2001.00496.x. (PM:11332576) [DOI] [PubMed] [Google Scholar]
- 60.Opthof T. Changes in sinus node function in a rabbit model of heart failure with ventricular arrhythmias and sudden death. Circulation. 2000;101:2975–2980. doi: 10.1161/01.cir.101.25.2975. (PM:10869272) [DOI] [PubMed] [Google Scholar]
- 61.Vermeulen J.T. Electrophysiologic and extracellular ionic changes during acute ischemia in failing and normal rabbit myocardium. J. Mol. Cell. Cardiol. 1996;28:123–131. doi: 10.1006/jmcc.1996.0012. (PM:8745220) [DOI] [PubMed] [Google Scholar]
- 62.Carlsson L. QTU-prolongation and torsades de pointes induced by putative class III antiarrhythmic agents in the rabbit: etiology and interventions. J. Cardiovasc. Pharmacol. 1990;16:276–285. doi: 10.1097/00005344-199008000-00014. (PM:1697384) [DOI] [PubMed] [Google Scholar]
- 63.Mazur A. Systemic administration of calmodulin antagonist W-7 or protein kinase A inhibitor H-8 prevents torsade de pointes in rabbits. Circulation. 1999;100:2437–2442. doi: 10.1161/01.cir.100.24.2437. (PM:10595957) [DOI] [PubMed] [Google Scholar]
- 64.Farkas A. Importance of vagally mediated bradycardia for the induction of torsade de pointes in an in vivo model. Br. J. Pharmacol. 2008;154:958–970. doi: 10.1038/bjp.2008.154. (PM:18587444) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gbadebo T.D. Calmodulin inhibitor W-7 unmasks a novel electrocardiographic parameter that predicts initiation of torsade de pointes. Circulation. 2002;105:770–774. doi: 10.1161/hc0602.103724. (PM:11839636) [DOI] [PubMed] [Google Scholar]
- 66.Hamlin R.L., Kijtawornrat A. Use of the rabbit with a failing heart to test for torsadogenicity. Pharmacol. Ther. 2008;119:179–185. doi: 10.1016/j.pharmthera.2008.03.011. (PM:18691764) [DOI] [PubMed] [Google Scholar]
- 67.Allessie M.A. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ. Res. 1977;41:9–18. doi: 10.1161/01.res.41.1.9. (PM:862147) [DOI] [PubMed] [Google Scholar]
- 68.Nattel S. Mechanisms of atrial fibrillation: lessons from animal models. Prog. Cardiovasc. Dis. 2005;48:9–28. doi: 10.1016/j.pcad.2005.06.002. (PM:16194689) [DOI] [PubMed] [Google Scholar]
- 69.Ravelli F., Allessie M. Effects of atrial dilatation on refractory period and vulnerability to atrial fibrillation in the isolated Langendorff-perfused rabbit heart. Circulation. 1997;96:1686–1695. doi: 10.1161/01.cir.96.5.1686. (PM:9315565) [DOI] [PubMed] [Google Scholar]
- 70.Hirose M. Mechanism for atrial tachyarrhythmia in chronic volume overload-induced dilated atria. J. Cardiovasc. Electrophysiol. 2005;16:760–769. doi: 10.1046/j.1540-8167.2005.40331.x. (PM:16050835) [DOI] [PubMed] [Google Scholar]
- 71.Shimano M. Pioglitazone, a peroxisome proliferator-activated receptor-gamma activator, attenuates atrial fibrosis and atrial fibrillation promotion in rabbits with congestive heart failure. Heart Rhythm. 2008;5:451–459. doi: 10.1016/j.hrthm.2007.12.010. (PM:18313605) [DOI] [PubMed] [Google Scholar]
- 72.Brunner M. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J. Clin. Invest. 2008;118:2246–2259. doi: 10.1172/JCI33578. (PM:18464931) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marian A.J. A transgenic rabbit model for human hypertrophic cardiomyopathy. J. Clin. Invest. 1999;104:1683–1692. doi: 10.1172/JCI7956. (PM:10606622) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanbe A. Transgenic rabbit model for human troponin I-based hypertrophic cardiomyopathy. Circulation. 2005;111:2330–2338. doi: 10.1161/01.CIR.0000164234.24957.75. (PM:15867176) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nishizawa T. Overexpressed cardiac Gs alpha in rabbits. J. Mol. Cell. Cardiol. 2006;41:44–50. doi: 10.1016/j.yjmcc.2006.03.008. (PM:16678849) [DOI] [PubMed] [Google Scholar]
- 76.Pattison J.S. Phospholamban overexpression in transgenic rabbits. Transgenic Res. 2008;17:157–170. doi: 10.1007/s11248-007-9139-2. (PM:17882530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edwards S. An experimental model for myocarditis and congestive heart failure after rabbit coronavirus infection. J. Infect. Dis. 1992;165:134–140. doi: 10.1093/infdis/165.1.134. (PM:1309370) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kishimoto C. Role of MIP-2 in coxsackie virus B3 myocarditis. J. Mol. Cell. Cardiol. 2000;32:631–638. doi: 10.1006/jmcc.2000.1102. (PM:10756119) [DOI] [PubMed] [Google Scholar]
- 79.Nagai T., Kawai C. Experimental cryptococcal-induced myocarditis. Jpn. Circ. J. 1981;45:539–546. doi: 10.1253/jcj.45.539. (PM:7014958) [DOI] [PubMed] [Google Scholar]
- 80.Henry L. Experimental toxoplasmic myocarditis in rabbits. J. Pathol. 1970;101:xxi. (PM:5504757) [PubMed] [Google Scholar]
- 81.Burova L.A. Induction of myocarditis in rabbits injected with group A streptococci. Indian J. Med. Res. 2004;119(Suppl.):183–185. (PM:15232191) [PubMed] [Google Scholar]
- 82.Zabejinski M.M. New animal model of diphtheritic myocarditis. Exp. Toxicol. Pathol. 2000;52:67–70. doi: 10.1016/S0940-2993(00)80020-2. (PM:10779154) [DOI] [PubMed] [Google Scholar]
- 83.Figueiredo F. The evolution of experimental Trypanosoma cruzi cardiomyopathy in rabbits: further parasitological, morphological and functional studies. Int. J. Cardiol. 1986;10:277–290. doi: 10.1016/0167-5273(86)90009-4. (PM:3514479) [DOI] [PubMed] [Google Scholar]
- 84.Teixeira A.R. The immunology of experimental Chagas’ disease. IV. Production of lesions in rabbits similar to those of chronic Chagas’ disease in man. Am. J. Pathol. 1975;80:163–180. (PM:808136) [PMC free article] [PubMed] [Google Scholar]
- 85.Perdikaris G.S. Successful single-dose teicoplanin prophylaxis against experimental streptococcal, enterococcal, and staphylococcal aortic valve endocarditis. Antimicrob. Agents Chemother. 1997;41:1916–1921. doi: 10.1128/aac.41.9.1916. (PM:9303384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Faivre V. Cardiac and renal effects of levosimendan, arginine vasopressin, and norepinephrine in lipopolysaccharide-treated rabbits. Anesthesiology. 2005;103:514–521. doi: 10.1097/00000542-200509000-00014. (PM:16129976) [DOI] [PubMed] [Google Scholar]
- 87.Fajardo L.F., Stewart J.R. Pathogenesis of radiation-induced myocardial fibrosis. Lab. Invest. 1973;29:244–257. (PM:4724850) [PubMed] [Google Scholar]
- 88.Eltringham J.R. Adriamycin cardiomyopathy: enhanced cardiac damage in rabbits with combined drug and cardiac irradiation. Radiology. 1975;115:471–472. doi: 10.1148/115.2.471. (PM:806934) [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y. Atrioventricular nodal fast pathway modification: mechanism for lack of ventricular rate slowing in atrial fibrillation. Cardiovasc. Res. 2004;61:45–55. doi: 10.1016/j.cardiores.2003.10.023. (PM:14732201) [DOI] [PubMed] [Google Scholar]
- 90.Himel H.D. Translesion stimulus–excitation delay indicates quality of linear lesions produced by radiofrequency ablation in rabbit hearts. Physiol. Meas. 2007;28:611–623. doi: 10.1088/0967-3334/28/6/001. (PM:17664616) [DOI] [PubMed] [Google Scholar]
- 91.Saba S. Prevention of adverse electrical and mechanical remodeling with biventricular pacing in a rabbit model of myocardial infarction. Heart Rhythm. 2008;5:124–130. doi: 10.1016/j.hrthm.2007.08.021. (PM:18180026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.So P.P. I(Ks) block by HMR 1556 lowers ventricular defibrillation threshold and reverses the repolarization shortening by isoproterenol without rate-dependence in rabbits. J. Cardiovasc. Electrophysiol. 2007;18:750–756. doi: 10.1111/j.1540-8167.2007.00812.x. (PM:17578345) [DOI] [PubMed] [Google Scholar]
- 93.Efimov I.R. High-resolution fluorescent imaging does not reveal a distinct atrioventricular nodal anterior input channel (fast pathway) in the rabbit heart during sinus rhythm. J. Cardiovasc. Electrophysiol. 1997;8:295–306. doi: 10.1111/j.1540-8167.1997.tb00792.x. (PM:9083879) [DOI] [PubMed] [Google Scholar]
- 94.Tevaearai H.T. Myocardial gene transfer and overexpression of beta2-adrenergic receptors potentiates the functional recovery of unloaded failing hearts. Circulation. 2002;106:124–129. doi: 10.1161/01.cir.0000020220.79105.fd. (PM:12093781) [DOI] [PubMed] [Google Scholar]
- 95.White D.C. Preservation of myocardial beta-adrenergic receptor signaling delays the development of heart failure after myocardial infarction. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5428–5433. doi: 10.1073/pnas.090091197. (PM:10779554) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shah A.S. In vivo ventricular gene delivery of a beta-adrenergic receptor kinase inhibitor to the failing heart reverses cardiac dysfunction. Circulation. 2001;103:1311–1316. doi: 10.1161/01.cir.103.9.1311. (PM:11238278) [DOI] [PubMed] [Google Scholar]
- 97.Maurice J.P. Enhancement of cardiac function after adenoviral-mediated in vivo intracoronary beta2-adrenergic receptor gene delivery. J. Clin. Invest. 1999;104:21–29. doi: 10.1172/JCI6026. (PM:10393695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Munch G. Functional alterations after cardiac sodium–calcium exchanger overexpression in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H488–H495. doi: 10.1152/ajpheart.01324.2005. (PM:16603685) [DOI] [PubMed] [Google Scholar]
- 99.Laugwitz K.L. Blocking caspase-activated apoptosis improves contractility in failing myocardium. Hum. Gene Ther. 2001;12:2051–2063. doi: 10.1089/10430340152677403. (PM:11747596) [DOI] [PubMed] [Google Scholar]
- 100.Munch G. Cardiac overexpression of the norepinephrine transporter uptake-1 results in marked improvement of heart failure. Circ. Res. 2005;97:928–936. doi: 10.1161/01.RES.0000186685.46829.E5. (PM:16166553) [DOI] [PubMed] [Google Scholar]
- 101.Li Q. Gene therapy with extracellular superoxide dismutase protects conscious rabbits against myocardial infarction. Circulation. 2001;103:1893–1898. doi: 10.1161/01.cir.103.14.1893. (PM:11294809) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McCue J.D. The real estate of myoblast cardiac transplantation: negative remodeling is associated with location. J. Heart Lung Transplant. 2008;27:116–123. doi: 10.1016/j.healun.2007.10.011. (PM:18187097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen M. Effects of autologous stem cell transplantation on ventricular electrophysiology in doxorubicin-induced heart failure. Cell Biol. Int. 2006;30:576–582. doi: 10.1016/j.cellbi.2006.03.002. (PM:16731012) [DOI] [PubMed] [Google Scholar]
- 104.Dhein S. Effects of autologous bone marrow stem cell transplantation on beta-adrenoceptor density and electrical activation pattern in a rabbit model of non-ischemic heart failure. J. Cardiothorac. Surg. 2006;1:17. doi: 10.1186/1749-8090-1-17. (PM:16800896) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bassani R.A. Action potential duration determines sarcoplasmic reticulum Ca2+ reloading in mammalian ventricular myocytes. J. Physiol. 2004;559:591–607. doi: 10.1113/jphysiol.2004.067959. (PM:15243136) [DOI] [PMC free article] [PubMed] [Google Scholar]