

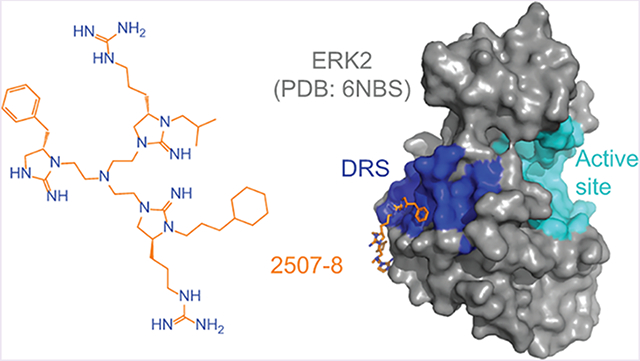
Abstract
Extracellular signal-regulated kinases (ERK1/2) are mitogen-activated protein kinases (MAPKs) that play a pro-tumorigenic role in numerous cancers. ERK1/2 possess two protein-docking sites that are distinct from the active site: the D-recruitment site (DRS) and the F-recruitment site. These docking sites facilitate substrate recognition, intracellular localization, signaling specificity, and protein complex assembly. Targeting these sites on ERK in a therapeutic context may overcome many problems associated with traditional ATP-competitive inhibitors. Here, we identified a new class of inhibitors that target the ERK DRS by screening a synthetic combinatorial library of more than 30 million compounds. The screen detects the competitive displacement of a fluorescent peptide from the DRS of ERK2. The top molecular scaffold from the screen was optimized for structure–activity relationship by positional scanning of different functional groups. This resulted in 10 compounds with similar binding affinities and a shared core structure consisting of a tertiary amine hub with three functionalized cyclic guanidino branches. Compound 2507–1 inhibited ERK2 from phosphorylating a DRS-targeting substrate and prevented the phosphorylation of ERK2 by a constitutively active MEK1 (MAPK/ERK kinase 1) mutant. Interaction between an analogue, 2507–8, and the ERK2 DRS was confirmed by nuclear magnetic resonance and X-ray crystallography. 2507–8 forms critical interactions at the common docking domain residue Asp319 via an arginine-like moiety that is shared by all 10 hits, suggesting a common binding mode. The structural and biochemical insights reported here provide the basis for developing new ERK inhibitors that are not ATP-competitive but instead function by disrupting critical protein–protein interactions.
Graphical Abstract
The human protein kinome consists of 535 identified members to date, of which at least 356 are known to phosphorylate serine or threonine residues of their substrates.1 Extracellular signal-regulated kinases (ERK1/2) are Ser/Thr-directed kinases that alone have more than 250 recognized cellular substrates.2 Thus, ERK1/2 must execute specific interactions, despite having highly conserved mechanisms among other kinases in cells. As a result, extensive regulation of the ERK pathway and other mitogen-activated protein kinase (MAPK) signaling networks is required. One level of MAPK signaling specificity is achieved through protein-docking sites. Docking sites are interfaces of protein–protein interaction that are distinct from the catalytic site. In the case of MAPKs, docking sites can bind substrates to localize specific Ser/Thr-Pro sequences near the active site to promote their phosphorylation.3,4 For example, the transcription factor Ets-1 engages both the F-recruitment site (FRS) and D-recruitment site (DRS) of ERK2 to position Thr38 near the ERK2 active site for efficient phosphorylation.5–7 In comparison, the substrate Elk-1 binds the FRS to direct ERK2 to phosphorylate Ser383, but a different set of residues are phosphorylated when Elk-1 engages the DRS.4,8 The number of protein-docking sites, the order in which they are engaged, and their positions and arrangement on an enzyme can all, in principle, influence binding interactions and substrate phosphorylation. Therefore, inhibitors that block these sites can potentially disrupt particular MAPK binding interactions and signaling events while leaving others unaffected. An additional advantage to targeting these docking sites is that inhibitors do not have to compete with ATP, which is present in cells at millimolar concentrations and binds to MAPKs with a relatively high affinity.9,10 Instead, kinase substrates that utilize these docking sites are typically present in cells at or below their Km values, so high-affinity inhibitors may not be required to block such interactions.11,12 Consequently, protein-docking sites on ERK1/2 and other MAPKs offer an alternative approach to classical kinase inhibition that could improve selectivity and block specific signaling events in a tunable manner.
The ERK signaling cascade controls a variety of cellular processes, such as proliferation and survival. Thus, ERK plays a critical role in the development of diseases associated with dysregulated cell proliferation, like cancer.13,14 ERK is also implicated in cardiac hypertrophy, neurodegenerative diseases, and diabetes.15–18 Consequently, ERK inhibitors potentially provide significant therapeutic value. Here, we focus on identification of ERK inhibitors that target the DRS, a protein–protein interaction surface that is distal from the active site of ERK. Classical linear motifs that bind to the DRS consist of the D-site sequence Ψ1–3X3–7ΦXΦ, where Ψ is a positively charged residue, Φ is a hydrophobic residue, and X can be any amino acid. The DRS is composed of a hydrophobic groove located between an ED domain (Thr157/Thr158 on ERK2, named for the corresponding Glu/Asp residues of p38 MAPKs) and an acidic common docking (CD) domain (includes Asp316 and Asp319 on ERK2) (Figure S1). Though MAPK sequences are highly conserved among the ERK1/2, p38, and c-Jun N-terminal kinase (JNK) subfamilies19 and all of these subfamily members possess a DRS, it is evident that DRS inhibitors that are specific to each subfamily can be designed.20 Specifically, differences among the D-recruitment sites, including variations within the ED domain, can help confer substrate specificity.21 Though protein–protein interaction sites like the DRS have larger and more shallow features than typical druggable pockets, small-molecule inhibitors have been shown to successfully disrupt expansive protein-binding interfaces much larger than the inhibitors themselves.22–25
Several small-molecule ERK DRS inhibitors have been discovered using computer-aided drug design (CADD)22,26 or in silico screening methods27 (Figure S2). This pool of inhibitors was expanded by further in silico screening and computational searches for similar compounds and by synthesis of analogues for structure–activity relationship (SAR) studies.23,28,29 CADD and in silico screens have also been used to identify inhibitors of the other known docking site of ERK2, the FRS30 (Figure S2). Rather than using in silico methods here, we took a biochemical approach and developed a competition-based screen to identify inhibitors that displace a fluorescent peptide from the DRS of ERK2. The compounds identified from this screening method represent a new structural class of ERK inhibitors. Nuclear magnetic resonance (NMR) spectroscopy and macromolecular X-ray crystallography data confirmed that these compounds indeed bind to the DRS of ERK2, opening avenues for the future chemical optimization of ERK inhibitors that target the DRS.
RESULTS AND DISCUSSION
Screening Identified Tripodal Cyclic Guanidino Scaffolds as DRS Inhibitors.
To identify reversible inhibitors that bind the DRS of ERK2, we first developed a screening method suitable for combinatorial ligand libraries. This library format allows high-throughput screening with a low level of resource consumption; i.e., only several hundred samples are needed to screen millions of compounds.31–33 In our screen, a fluorescein isothiocyanate (FITC)-tagged D-site-containing peptide (FITC-X-Lig-D) is competitively displaced from the DRS by library compounds, registering as a change in fluorescence anisotropy.
The screening proceeded in three phases that allowed for inherent optimization of the candidate inhibitors (Figure 1). In phase I, a mixture-based library of 78 scaffold ranking samples, each representing a distinct chemical structure with varied functional groups, was screened to identify the active scaffolds that displaced the peptide FITC-X-Lig-D from the DRS (Figures S3 and S4). Together, these 78 mixtures contained a total of more than 30 million possible individual compounds. The top five most active scaffolds that caused the greatest displacement of the fluorescent peptide all contained cyclic guanidino moieties (Table 1). Scaffold 2408 was the most potent hit, showing an at least 2-fold greater percent signal change in the displacement assay than the rest of the scaffold samples, so it was selected for the second screening phase. Scaffold 2408 consists of a tripodal tris(2-aminoethyl)amine backbone that connects three cyclic guanidino moieties. It qualitatively has an additional plane of symmetry compared to the other active scaffolds, which could increase its productive binding modes and account for its higher potency. Scaffolds 2407, 2157, 2353, and 2354 contain two cyclic guanidino moieties, and all had lower potencies of comparable magnitude against ERK2 (Table 1), further suggesting the importance of the third cyclic guanidino group in 2408.
Figure 1.
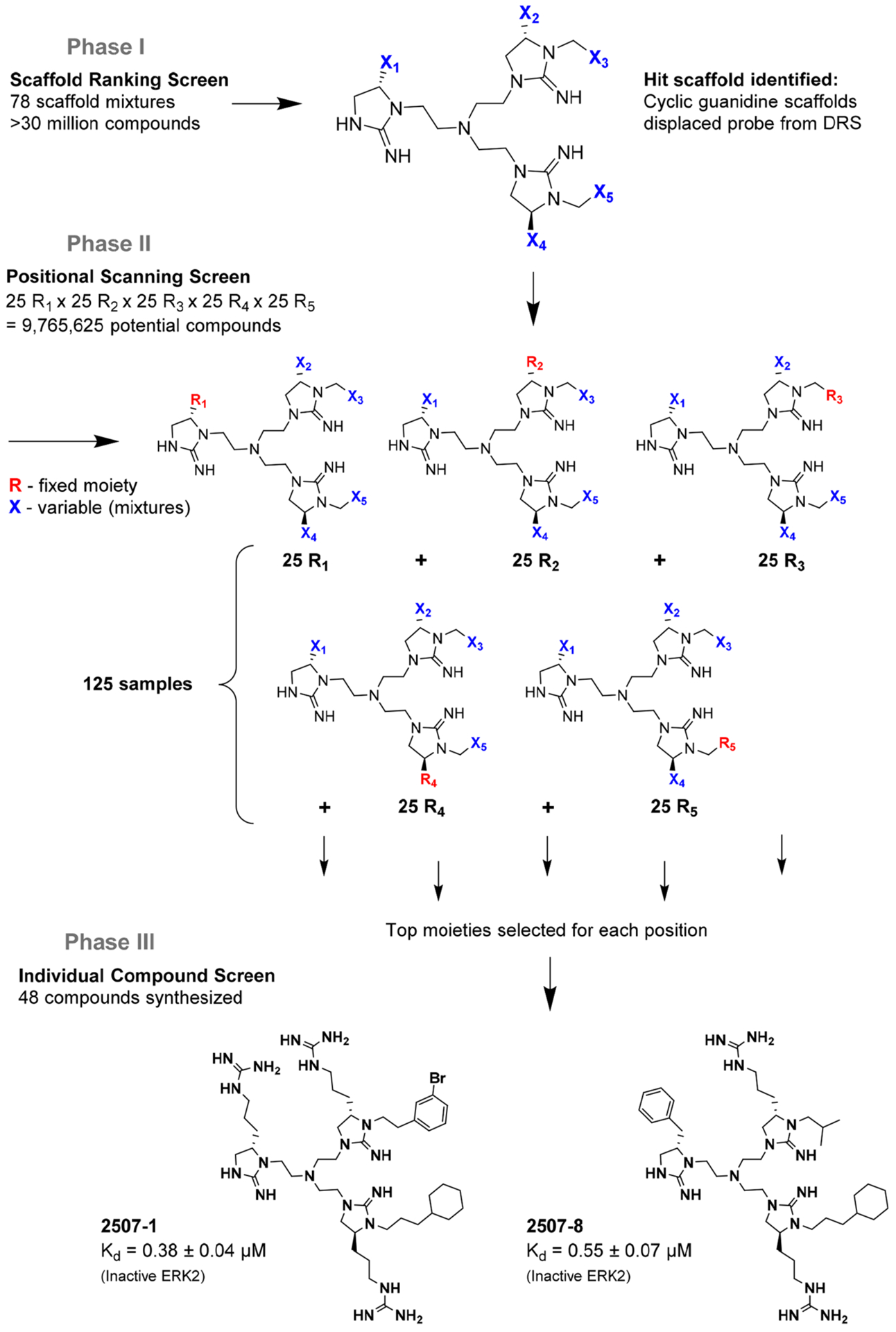
Deconvolution of the tripodal cyclic guanidino libraries. The fluorescence anisotropy-based screening for ERK DRS inhibitors proceeded in three phases: (I) scaffold mixture screen, (II) mixture-based positional scanning screen of each functional group of the top scaffold, and (III) testing of individual compounds with optimized R groups from phase II.
Table 1.
Top Scaffold Samples That Exhibited the Greatest Displacement of the Fluorescent Probe during Scaffold Screeninga
| Scaffold Sample # | Structure | Percent (%) signal change |
|---|---|---|
| 2408 | 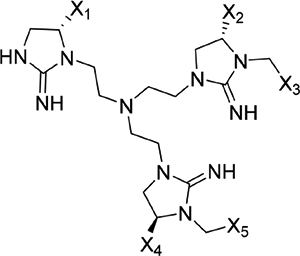 |
55.7 |
| 2407 | 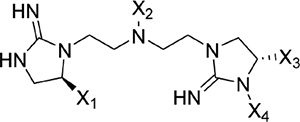 |
21.2 |
| 2353 | 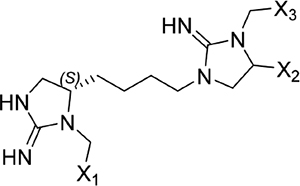 |
19.5 |
| 2157 | 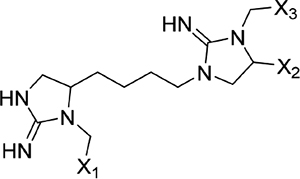 |
19.4 |
| 2354 | 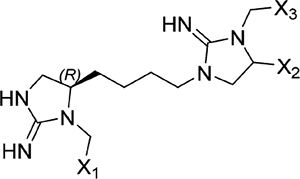 |
16.3 |
Screening results for the mixture-based scaffold ranking library at concentrations of 25 μg mL−1. Scaffold samples were ranked by their average percent change in anisotropy signal relative to controls (free probe in solution = 100%; probe maximally bound to ERK2 = 0%), indicating displacement of the FITC-X-Lig-D probe from the DRS of ERK2. Data reported as the average of two replicate samples at a mixture dose of 25 μg mL−1. Variable functional groups (X1–5) on each scaffold structure indicate that mixtures of different moieties are present.
In phase II of the screening, a mixture-based compound library of scaffold 2408 was screened using the FITC-X-Lig-D displacement assay. All of the compounds in the 2408 mixture-based library had five functional groups, and the 125 mixture samples were arranged into five sublibraries based on the positions of the functional groups. In each mixture sample, one position was held constant with a single known R group (R) while the remaining positions were varied (X) (see Figure 1). This led to a library size of 9765625 compounds (25 possible variants for each of five functional groups, or 255 compounds). On the basis of the results of screening this library shown in Figure S5 and Table S1, the hit functional groups for each position on the scaffold were selected to generate 48 individual compounds. The individual compounds (designated 2507-#) were then rescreened in phase III (Figures S6 and S7), and the resulting top 10 most active compounds were selected as hits and purified for further characterization (Table 2).
Table 2.
Binding Affinities of Top 10 Compoundsa
| Compound | 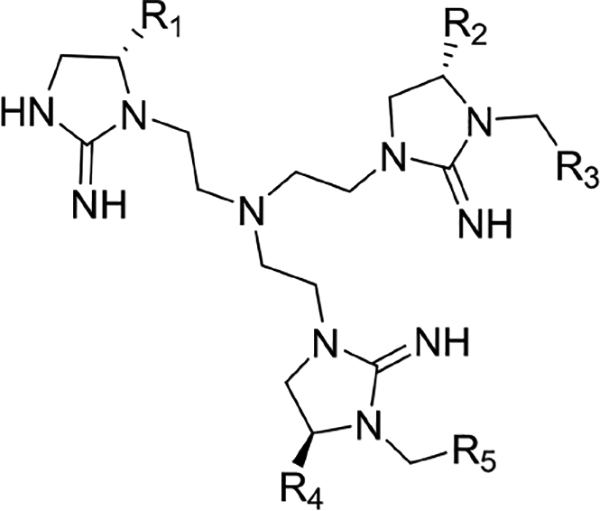 |
Dose- response data | ||||||
|---|---|---|---|---|---|---|---|---|
| Inactive ERK2 Ki, μM | Active ERK2 Ki, μM | Approx. Ki ratio (Active/Inactive) | ||||||
| R1 | R2 | R3 | R4 | R5 | ||||
| 8 | benzyl | guanidinopropyl | isopropyl | guanidinopropyl | cyclohexylethyl | 0.549 ± 0.066 | 0.384 ± 0.048 | 0.7 |
| 2 | 3’-bromobenzyl | 0.247 ± 0.026 | 0.584 ± 0.104 | 2.36 | ||||
| 26 | 4’-methylbenzyl | 0.501 ± 0.075 | 0.404 ± 0.061 | 0.81 | ||||
| 14 | 4’-fluorobenzyl | cyclohexylethyl | 0.933 ± 0.110 | 0.660 ± 0.112 | 0.71 | |||
| 1 | guanidinopropyl | guanidinopropyl | 0.383 ± 0.042 | 0.567 ± 0.122 | 1.48 | |||
| 25 | 4’-methylbenzyl | 0.611 ± 0.064 | 0.861 ± 0.138 | 1.41 | ||||
| 13 | 4’-fluorobenzyl | cyclohexylethyl | 0.351 ± 0.045 | 0.598 ± 0.085 | 1.7 | |||
| 37 | 4’-methylbenzyl | 0.567 ± 0.066 | 0.564 ± 0.078 | 0.99 | ||||
| 16 | methylthioethyl | cyclohexylethyl | 1.48 ± 0.11 | 1.273 ± 0.203 | 0.86 | |||
| 40 | 4’-methylbenzyl | 0.938 ± 0.109 | 0.783 ± 0.092 | 0.83 | ||||
Compounds are organized according to structural similarities. Ki values were determined by an anisotropy assay in the same manner as the original screening. Active or inactive ERK2 at 1 μM was incubated with 0–200 μM compounds for 45 min at room temperature prior to anisotropy measurements as conducted in the main screening. Ki values were calculated as described in the Supporting Information. Values shown indicate the Ki ± the standard error of the fit.
Hit Compounds Inhibit DRS Interactions in a Dose-Dependent Manner.
Next, we measured the dose-dependent change in anisotropy signal for each of the top 10 compounds upon competitive displacement of FITC-X-Lig-D from the DRS of ERK2. We tested both inactive (unphosphorylated) and active (bis-phosphorylated) forms of ERK2 in the anisotropy assay to evaluate if the conformational changes upon its activation have any effect on DRS binding affinity (Kd) (Figure S8). We found that the Kd values for binding of FITC-X-Lig-D to active and inactive ERK2 were very similar at 1.4 ± 0.2 and 1.0 ± 0.1 μM, respectively. We used these data to evaluate the apparent affinities of the compounds (Ki values) for both active and inactive ERK2 and found these values ranged from approximately 0.24 to 1.5 μM for each enzyme activation state (Table 2 and Figures S9–S12). These binding affinities are approximately 3–20-fold tighter than the most potent values of other previously identified DRS inhibitors, as determined by biochemical assays26,28 (Figure S2). The activation state of ERK2 did not appear to have a substantial effect on the inhibitor binding affinities; the largest observed difference between inhibitor affinity values for active and inactive forms of ERK2 was 2.4-fold [in the case of compound 2507–2 (see Table 2)]. These results indicated that all compounds effectively bind to both forms of the enzyme with similar affinities under the assay conditions. Previous findings showed that the proteins Ets-1 and PEA-15 (phosphoprotein enriched in astrocytes-15) both interact at the DRS and have similar binding affinities when comparing active and inactive ERK2, suggesting that the inhibitors here act independently of ERK activity and may inhibit noncatalytic functions.34,35
Characterization of 2507–1 Selectivity.
Given that the top 10 compounds selected in the phase III screen produced similar Ki values [~1 μM (Table 2)], compound 2507–1 (Figure 2A; Ki ~ 0.4 and 0.6 μM toward inactive and active ERK2, respectively) was selected as a representative of the group for mechanistic evaluation. In [γ−32P]ATP-based dose–response assays, we found that 2507–1 potently inhibited ERK2 phosphorylation of Ets-1, a bona fide cellular substrate, with an IC50 of 5.6 ± 1.0 μM (Figure 2B). Consistent with the fact that Ets-1 utilizes both the ERK2 DRS and the ERK2 FRS to dock onto the kinase,6 2507–1 was found to be a partial inhibitor of Ets-1 phosphorylation (~77% maximum inhibition). Knowing that other kinases like p38α and JNK1/2 share similarities in their DRS regions with ERK2, we tested the ability of 2507–1 to inhibit JNK2 phosphorylation of GST-tagged c-Jun in vitro (Figure 2B). This IC50 was measured to be 43 ± 6 μM, which indicated that 2507–1 has less potency for JNK2 when compared to ERK2 phosphorylation of Ets-1.
Figure 2.
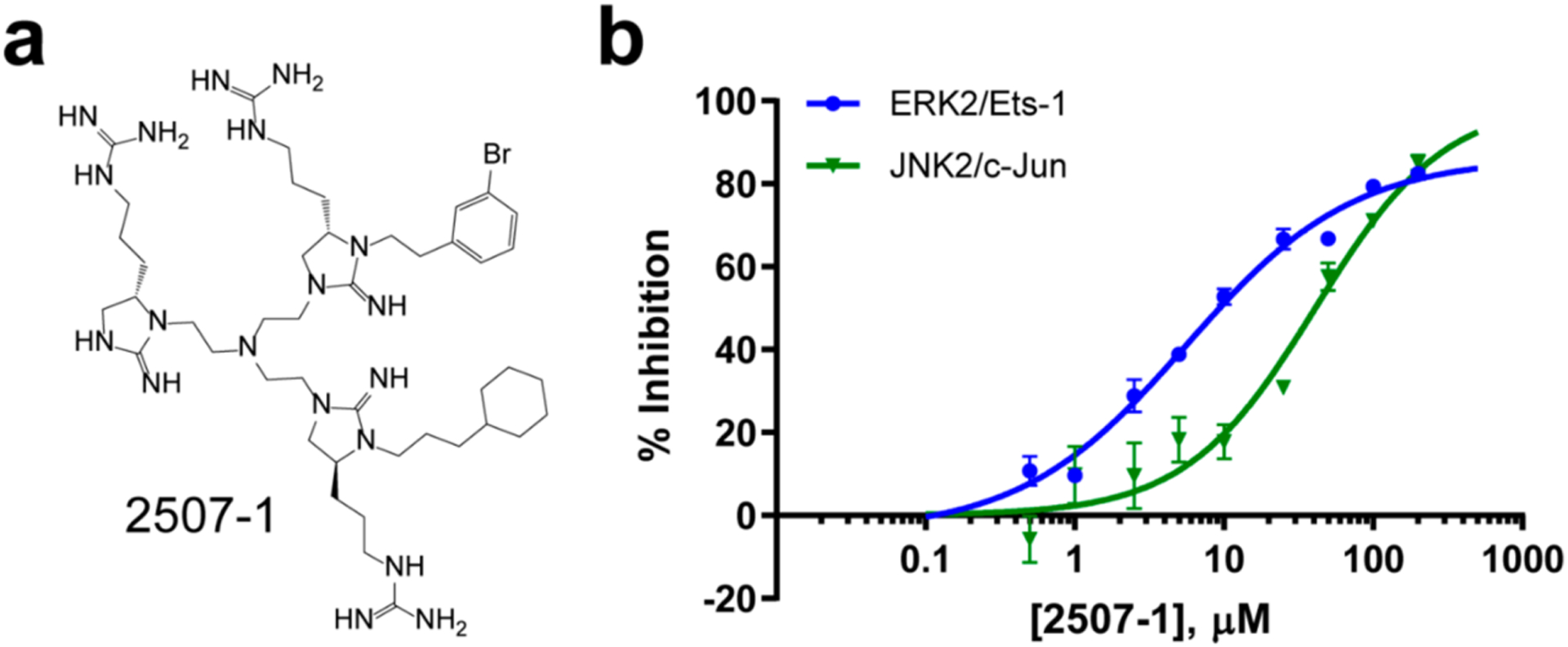
Potency and selectivity of 2507–1. (a) Structure of lead compound 2507–1. (b) Ability of 2507–1 to inhibit 2 nM ERK2 phosphorylation of 10 μM Ets-1 as well as 25 nM JNK2 phosphorylation of 2 μM c-Jun, as assessed by a 32P assay. Enzymes were incubated with 0–200 μM 2507–1 for 30 min prior to the initiation of reaction by addition of MgATP and the substrate. Initial rates ± standard errors are shown fit to standard IC50 models.
2507–1 Inhibited ERK2 Activation by Constitutively Active MEK1.
Because MEK1 is known to interact at the DRS of ERK2,36 we tested whether 2507–1 would prevent activation of ERK2 by the constitutively active MEK1 mutant, MKK1G7B.37 We found that 2507–1 blocks ERK2 activation in vitro in a dose-dependent manner, with an IC50 of 9.9 ± 1.9 μM (Figure 3). Thus, this small molecule can block protein–protein interactions in vitro by both upstream (MEK1) and downstream (Ets-1) effectors of ERK2. D-Site sequences frequently contain basic residues, such as arginine, that are important for engaging the acidic CD region of the DRS. CD domain mutations, namely the sevenmaker mutation D319N, are known to cause a significant decrease in the level of binding of MEK1 to ERK2 and, in cells, can act as gain-of-function mutations.38,39 This gain in ERK function has been attributed to signaling feedback and other effects, including ineffective targeting of ERK by deactivating phosphatases.38,39 Thus, it is evident that even minor variations in this region can disrupt protein docking. 2507–1 significantly blocked MKK1G7B activation of ERK2, suggesting that its arginine-like guanidinopropyl moieties likely function by a “neutralizing” effect on the CD residue Asp319 that is functionally similar to the D319N mutation. The 10 hit compounds listed in Table 2 contain the same guanidinopropyl moiety and likely function in a comparable fashion.
Figure 3.
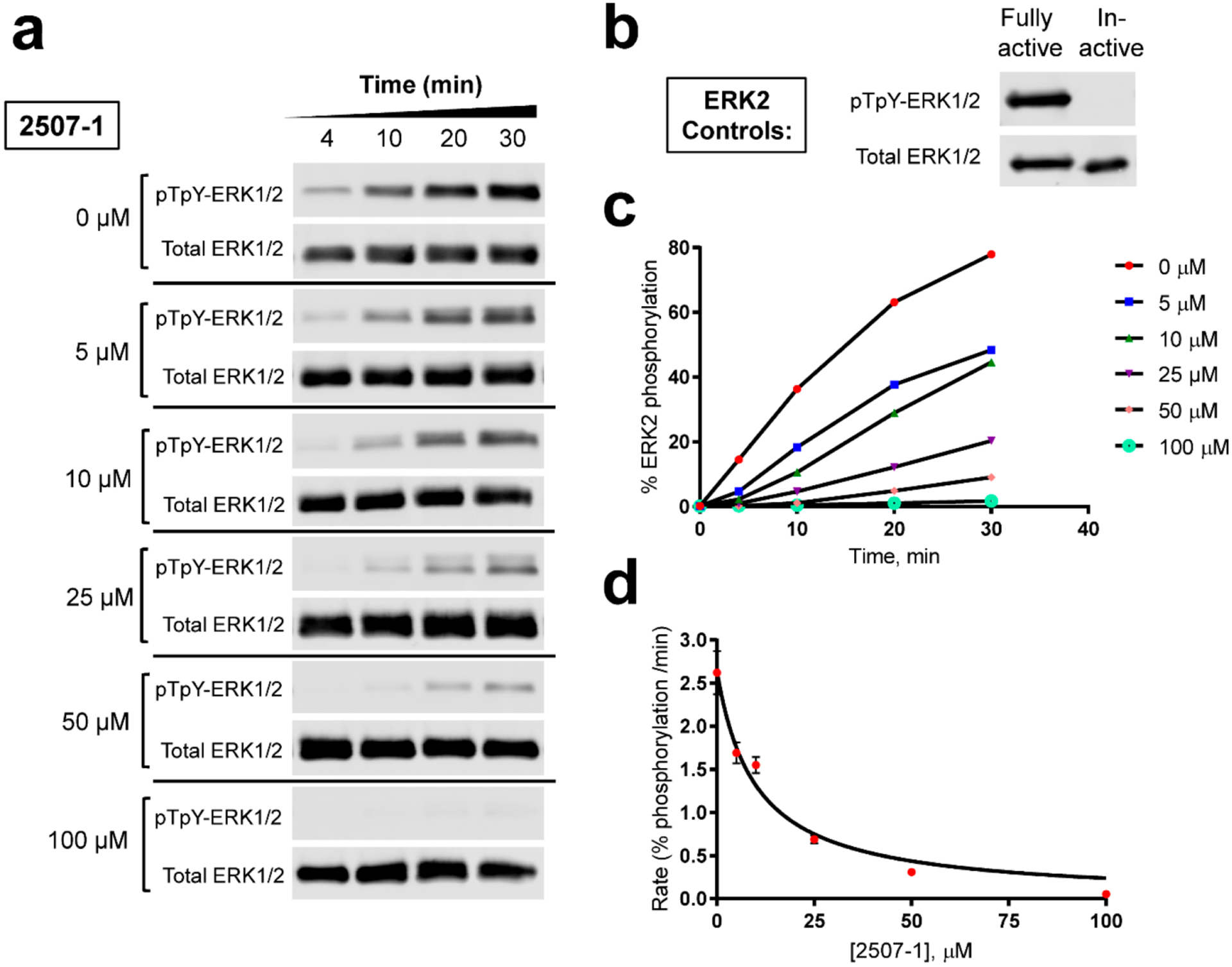
2507–1 inhibits MKK1G7B phosphorylation of ERK2 in vitro. Recombinant ERK2 was incubated with varied concentrations of 2507–1 (0–100 μM) for 10 min prior to reaction with MKK1G7B and MgATP. (a) Resulting ERK2 phosphorylation at Thr183/Tyr185 (pTpY) measured by a Western blot. Controls included detection of total ERK protein per lane and (b) doubly phosphorylated active ERK2 and unphosphorylated inactive ERK2. (c) Densitometry of the Western blot in panel a, where the phospho-ERK signal is normalized to the total ERK signal. (d) IC50 curve generated from initial rate measurements of the percent ERK2 phosphorylation from the densitometry in panel c.
Confirmation of DRS Binding by Structural Data.
To confirm the binding mode of the hit compounds, we used a combination of NMR and macromolecular X-ray crystallography. For the NMR experiments, inhibitor interactions at particular residues of ERK2 were detected by measuring chemical shift perturbations (CSPs) in response to addition of an inhibitor.40 Given that the Ki values were similar for inactive and active ERK2 (Table 2), inactive ERK2 was used in the NMR experiments. We tested three of the top 10 hit compounds, 2507–1, 2507–8, and 2507–26, by this method to observe binding interactions for different R groups on the scaffold. As shown in Figure 4, the CSPs for all three compounds were concentrated in and around the DRS region of ERK2. The 15N–1H TROSY spectra for three example residues found within the DRS further illustrate the common effects of the three compounds (Figure S13). Importantly, all three compounds induced CSPs above two standard deviations for the following key DRS residues: two common docking domain residues (Asp316 and Asp319), Thr158 of the ED domain, the hydrophobic residues Leu119, Leu155, His123, and Tyr126, and a solvent-exposed cysteine residue Cys159 (Tables S2–S4 and Figure S14). These results indicated that the compounds likely target the DRS in a similar fashion, despite their differing R groups.
Figure 4.
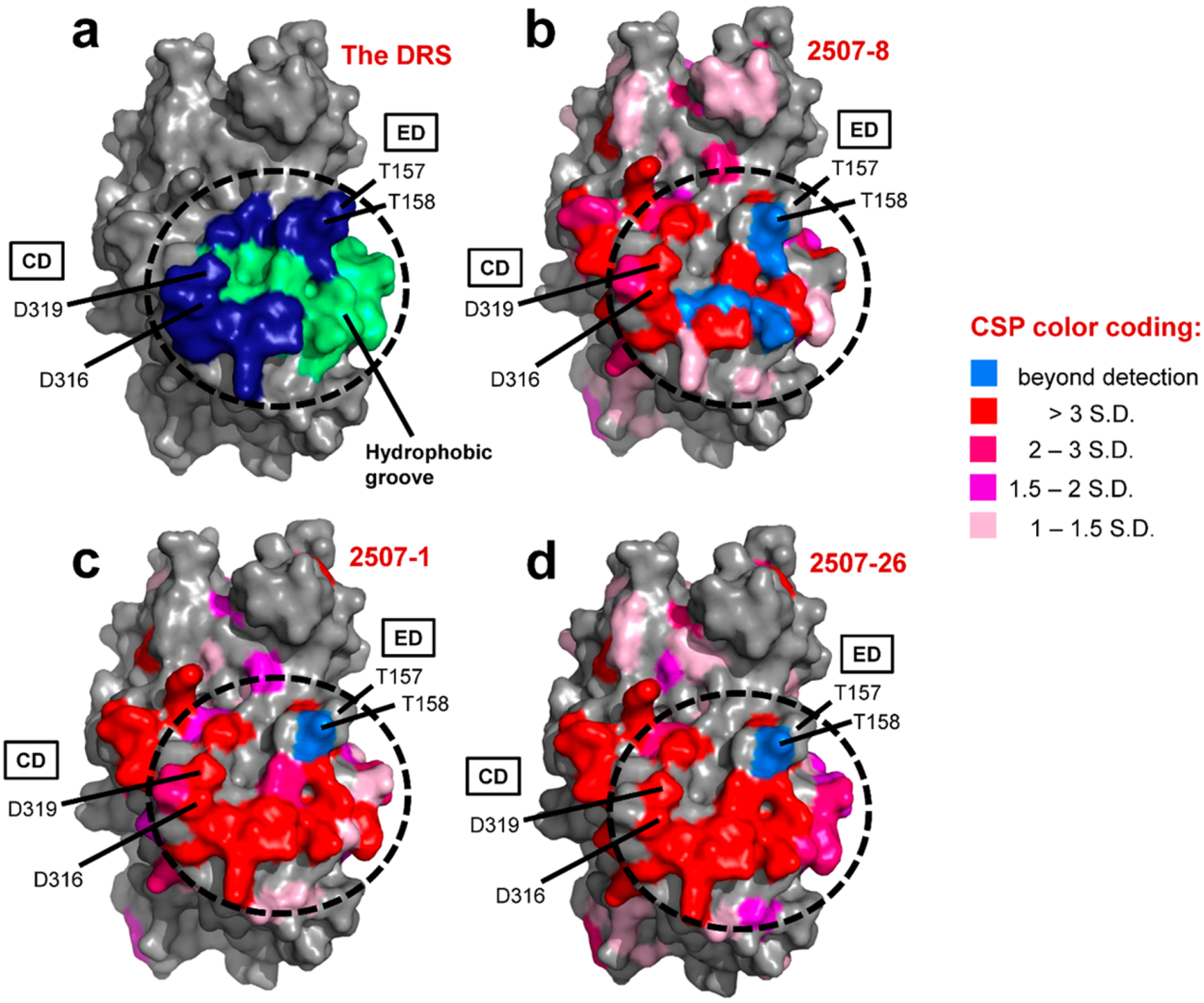
2507–8, 2507–1, and 2507–26 interact with residues of the DRS. The binding of 2507–8, 2507–1, and 2507–26 to ERK2 was analyzed by NMR. In each panel, the location of the DRS is highlighted by a dotted borderline. (a) Structure of ERK2 [Protein Data Bank (PDB) entry 3ERK] showing the different regions of the DRS residues: the ED domain (Thr157/Thr158) and CD domain (Asp316/Asp319) in dark blue and the hydrophobic docking groove in green. The other three panels show NMR results displaying spectral perturbations mapped onto the same surface representation of ERK2 shown in panel a (PDB entry 3ERK). Residues affected by the binding of (b) 2507–8, (c) 2507–1, and (d) 2507–26 are shown according to the legend. Colors indicate chemical shift perturbations (CSPs) with standard deviation (SD) beyond the mean [1–1.5 SD (light pink), 1.5–2 SD (magenta), 2–3 SD (hot pink), >3 SD (red)] and resonances broadened to beyond detectable limits or otherwise unidentifiable upon ligand binding (blue).
To ensure that the chemical shift perturbations detected by NMR were not caused by allosteric effects, conformational changes in the protein, or nonspecific binding of the inhibitors, we crystallized compound 2507–8 (Figure 5A) in complex with inactive ERK2 and determined the structure with 1.9 Å resolution (Figure 5B and Table S5). We observed the appearance of new electron density near ERK2 residues 311–318 [a region around which significant CSPs were measured (see Table S3 and Figure S14)]. This electron density was not fully contiguous and was difficult to model with a single molecule of 2507–8 (Figure 5C,D). Instead, our best interpretation of this electron density used two fragments of 2507–8, termed fragment A and fragment B (see Figure 5A,B). When considering this model, one possibility is that one or both of the fragments are portions of two intact inhibitor molecules but only part of each molecule is resolvable. A second possibility is that one or both of the fragments are impurities or degradation products of 2507–8. This may be likely for fragment B, given that liquid chromatography–mass spectrometry analysis of the 2507–8 inhibitor identified fragment B [(S)-4-benzylimidazoli-din-2-imine] as an impurity (Figure S15). Finally, although it is unlikely given the distance between the two regions, it is possible that this density corresponds to a single inhibitor molecule, where the structural elements of the inhibitor that link these regions are too flexible for visualization. Nevertheless, both modeled 2507–8 fragments indicate that binding is localized at the DRS and does not occur at the active site of ERK2 (Figure 5B) or with the active site of the closest crystallography symmetry mate (Figure S16).
Figure 5.
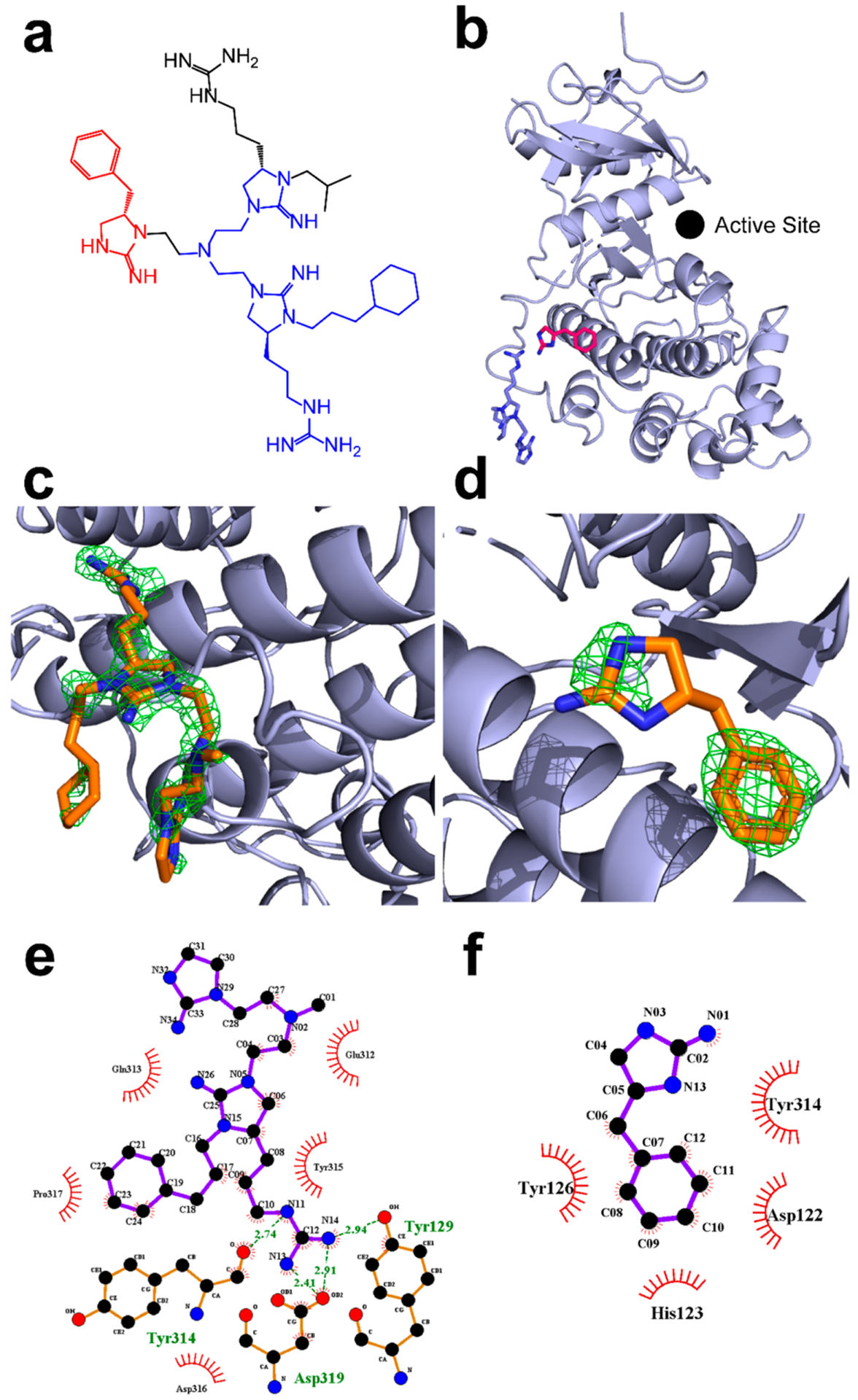
1.9 Å resolution structure of ERK2 in complex with inhibitor 2507–8. (a) Chemical structure of inhibitor 2507–8. (b) Structure of ERK2 (light blue) in complex with inhibitor 2507–8, in which the latter was resolved as two fragments termed A and B, which are colored blue and red, respectively, corresponding to the colored atoms in panel a. The Fo – Fc composite omit map contoured at 3.0σ and calculated in Phenix after the removal of ligands is shown for (c) fragment A and (d) fragment B. LigPlot projections of hydrophobic and ionic interactions are shown for (e) fragment A and (f) fragment B. Ionic interactions are shown using green dashed lines. Hydrophobic interactions are shown using circular red bursts.
As predicted, the arginine-like group on fragment A is located sufficiently close to form ionic interactions with the common docking domain residue Asp319, in addition to Tyr129 and Tyr314 (Figures 5E and 6). These interactions may stabilize the binding position of this functional group, which is consistent with the observation of clear electron density for fragment A. Additional hydrophobic interactions engage fragment A with Asp316 of the common docking domain as well as Glu312, Gln313, Tyr315, and Pro317. In contrast, fragment B is bound in the hydrophobic groove between the common docking and ED domains. It forms hydrophobic interactions at the key DRS residues His123 and Tyr126 and with Asp122 and Tyr314 (Figure 5F). Both fragment A and fragment B appear to participate in binding interactions similar to those of a peptide derived from hematopoietic protein tyrosine phosphatase (HePTP), which has previously been crystallized in complex with ERK2 and is known to bind to the DRS.41 This peptide binds in the hydrophobic region between the CD and ED domains (Figure S1), a region that has been used as a basis for identifying inhibitors of the DRS via computer-aided drug design.26,28 In particular, an arginine residue of the HePTP peptide aligns in the same manner as a guanidinopropyl group of fragment A to form ionic interactions with Asp319 (Figure S17). Interestingly, the phenyl carbon at the meta position of fragment B is approximately 4.5 Å from the Cys159 side chain. It is therefore possible that further functionalization of the phenyl ring with a cysteine-reactive substituent could generate future covalent ERK inhibitors with potency greater than that of the reversible inhibitors identified here.
Figure 6.
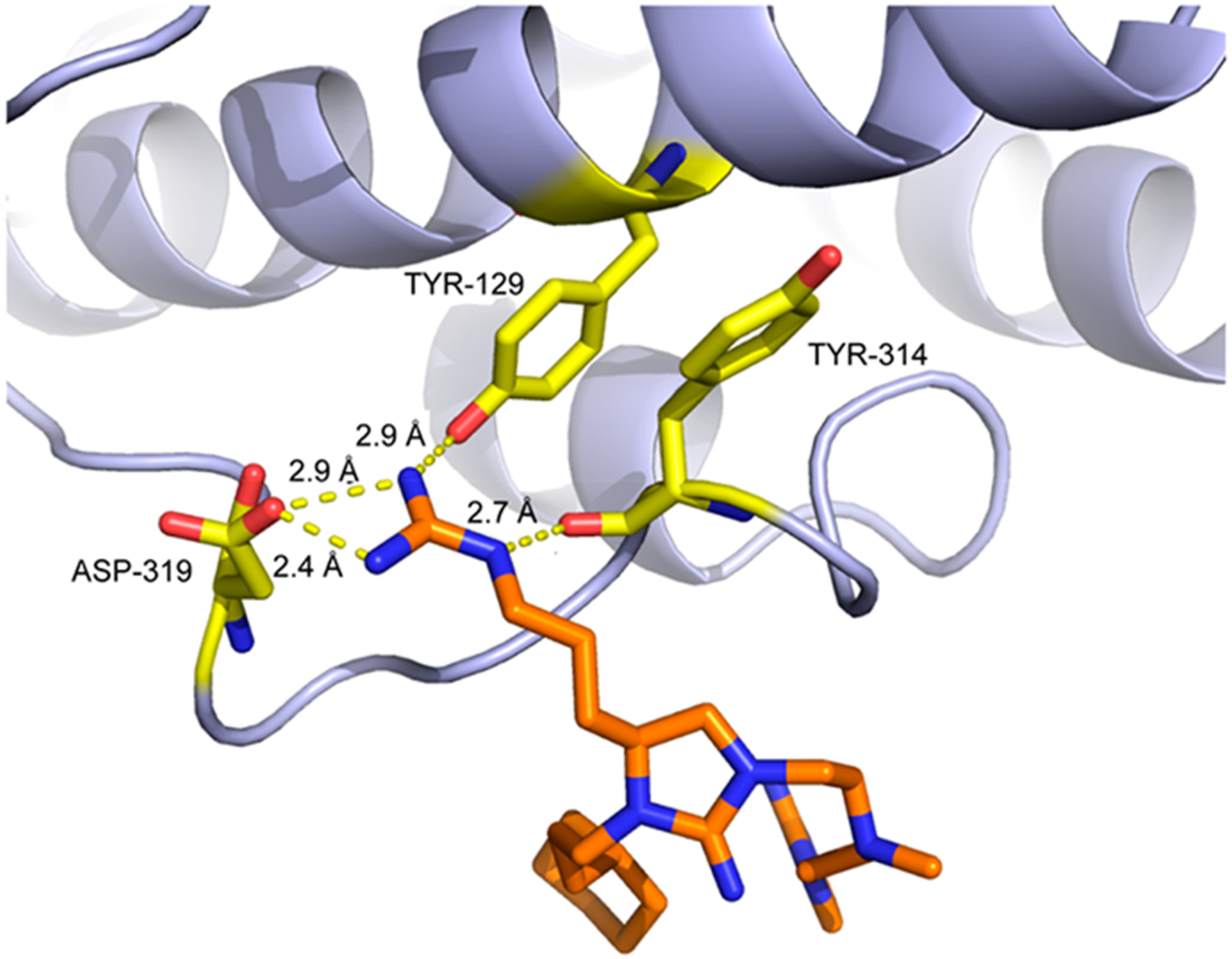
Ionic and hydrogen-bonding interactions between fragment A and the DRS of ERK2. Three nitrogen atoms of inhibitor 2507–8 are within hydrogen-bonding distance of the side chains of residues Tyr129, Tyr314, and Asp319 in ERK2.
Conclusions.
We have developed a class of inhibitors that targets the DRS and can block protein–protein interactions that involve this region. The biophysical and structural studies described here showed that small-molecule interactions in the acidic CD region and the hydrophobic residues adjacent to the ED domain of the DRS are likely the basis of this inhibition. We expect that further modification of these compounds could improve selectivity for ERK1/2 either through optimization of reversible binding interactions, through the introduction of reactive groups that could promote covalent attachment to reactive residues, e.g., a conserved cysteine that is found in this region, or through the development of a PROTAC strategy.42 The continued development of these inhibitors can potentially lead to ERK-targeted drugs that do not compete with ATP but instead function by disrupting critical protein–protein interactions.
METHODS
Proteins, Peptides, and Buffers.
Tag-less ERK2 was expressed, purified, and activated essentially as described previously.43 Erk2 DNA (NM_053842) was ligated into a pet28a (+) vector that was modified to include a TEV cleavage site for His6-tag removal. Vector modification and cleavage of the His6 tag with TEV protease were performed as described previously.44 ERK2 was activated using the constitutively active MKK1G7B mutant.37 The ERK substrate Ets-1 (residues 1–138) (EtsΔ138) was expressed and purified as previously described.45 GST-c-Jun and active JNK2 were prepared as previously reported.44,46 ERK2 for crystallization was expressed from plasmid Npt7–5H6ERK2 and purified in a manner similar to that used for ERK2 described above (see the Supporting Information for details).
FITC-labeled Lig-D (FITC-X-Lig-D) was prepared by labeling the C-terminus of Lig-D47 with FITC via a 6-aminohexanoic acid linker (X) on a cysteine residue (FITC-X-FQRKTLQRRNLKGLNLNL).48
The following buffers were used for all in vitro ERK2 assays: buffer A [25 mM HEPES (pH 7.5), 50 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1.3% (v/v) glycerol, 2 mM DTT, 10 μg mL−1 BSA, and 0.01% (w/v) Triton X-100] and buffer B [25 mM HEPES (pH 7.5), 50 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 10 mM MgCl2, 2 mM DTT, 10 μg mL−1 BSA, 0.01% (w/v) Triton X-100, and 2% (v/v) DMSO].
Combinatorial Library Screening.
All samples for the anisotropy-based biochemical assay were prepared in 384-well low-volume black polystyrene plates (Corning) with final volumes of 30 μL per well. Samples were tested in buffer A with 1 μM ERK2 and 10 nM FITC-X-Lig-D with 0.5% (v/v) DMF. All anisotropy readings were conducted at room temperature after incubation for 1 h. Controls included the maximal anisotropy signal (full binding of 10 nM FITC-X-Lig-D to 1 μM ERK2 with no inhibitor present) and the minimum signal (free 10 nM FITC-X-Lig-D with no ERK2 present). Phase I of the screening tested 78 scaffold ranking samples at 50 and 25 μg mL−1, in duplicate. Each phase I sample comprised a mixture derived from a distinct scaffold with varied functional groups in equimolar amounts, leading to a total library size of more than 30 million compounds with 78 different core structures. The mixture samples were also tested for intrinsic fluorescence in buffer A to rule out potential effects on the anisotropy signal (Figures S3 and S4).
The 2408 library in phase II contained 9765625 possible unique compounds formatted into 125 systematically arranged mixture samples. All samples were methodical variations of functional groups on the top scaffold from phase I, formulated in the same manner as described previously31–33 (see Figure 1). The synthetic methods for creating these scaffold 2408-based samples (including the individual compounds denoted by “2507-#” in phase III below) are described in the Supporting Information (Figure S18). In phase II of the screening, the mixture samples were screened at 50 and 25 μg mL−1 in duplicate, with the original hit mixture from phase I as an additional control.
In phase III, 48 individual compounds, along with controls (the core scaffold with all R groups removed and the original mixture sample 2408-X), were screened at 12.5 and 25 μM. The top 31 compounds from those results were rescreened at 3.125, 6.25, and 12.5 μM. The top 10 compounds were selected for further characterization.
Anisotropy Dose–Response Assays.
Active or inactive ERK2 (1 μM) was preincubated with 10 nM FITC-X-Lig-D and each compound at 0–200 μM at room temperature for 45 min prior to anisotropy measurements as described for the primary screening. The anisotropy dose–response curves were fit to an equilibrium binding model for conditions under which the ERK2 concentration is in excess of the FITC-X-Lig-D concentration (Supporting Information).34 The affinities of active and inactive ERK2 for FITC-X-Lig-D were evaluated for use in the anisotropy binding model (Figure S8).
Quantifying Inhibition of ERK2 Activation in Vitro.
The ability of 20 nM MKK1G7B to phosphorylate and activate 1 μM ERK2 was tested in the presence of 0, 50, 100, and 200 μM compound 2507–1 with 10 mM MgATP in buffer B (with 0.5 mM MgCl2). ERK2 was incubated with 2507–1 for 10 min at 28 °C prior to initiation with MKK1G7B and MgATP. Reactions were quenched in sodium dodecyl sulfate (SDS) loading buffer at time points ranging from 0 to 30 min. All quenched samples were loaded into 10% (w/v) SDS–polyacrylamide gels at a density of 52 ng of ERK2 per lane. Fully active and inactive ERK2 at a density of 52 ng per lane were used as controls. The gels were transferred to Immobilon-FL membranes overnight at 4 °C. Membranes were then blocked in 4× diluted Odyssey Blocking Buffer (PBS) (LI-COR) for 1 h at room temperature. Membranes were incubated in TBST [5% (w/v) milk] with 1:2000 antiphospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (E10) mouse mAb (Cell Signaling Technology) or TBST [5% (w/v) BSA] with 1:1000 anti-p44/42 MAPK (Erk1/2) (137F5) rabbit mAb (Cell Signaling Technology) for 1 h at room temperature and washed with TBST. The membranes were incubated with appropriate secondary antibodies [1:15000 IRDye 800CW goat (polyclonal) anti-rabbit IgG or IRDye 680RD goat (polyclonal) anti-mouse IgG (LI-COR)] for 30 min at room temperature. After samples had again been washed with TBST and Tris-buffered saline, fluorescence was detected on an Odyssey Sa imaging system (LI-COR).
32P Assay for Evaluating 2507–1 Selectivity.
2507–1 was tested for its ability to inhibit ERK2 (2 nM) phosphorylation of Ets-1 (10 μM) and JNK2 (25 nM) phosphorylation of c-Jun (2 μM). Enzymes were preincubated with 0–200 μM 2507–1 at room temperature for 30 min prior to the initiation of reaction by addition of the substrate and [γ−32P]MgATP (1 mM for ERK2 reactions and 500 μM for JNK2 reactions). All reactions were carried out at 28 °C in buffer B with MgCl2 (0.5 mM). The phosphorylated substrate was quantified at 0.5, 1, 1.5, 2, and 4 min time points as previously described (32P protocol)49 on a Tri-Carb 2910TR liquid scintillation analyzer (PerkinElmer).
NMR Spectroscopy.
The protocol for producing uniformly labeled [2H,13C,15N]ERK2 has been described elsewhere.40 The NMR data were collected at 25 °C on a 300 μL sample containing 100 μM inactive ERK2, 20 mM potassium phosphate (pH 6.8), 150 mM KCl, 200 μM EDTA, and 5.0 mM DTT (10% 2H2O was used for locking) using a 700 MHz Bruker Avance III spectrometer equipped with a triple-resonance cryogenic probe and capable of applying pulsed field gradients along the z-axis. 1H–15N TROSY50 experiments with 128 transients, a recycling delay of 1.5 s, and digital resolutions of 8.9 and 18.3 Hz/point in the direct and indirect dimensions, respectively, were performed in the absence and presence of a 2-fold excess (200 μM) of the ligand (compounds 2507–1, 2507–8, or 2507–26). Data were processed using NMRPipe51 and analyzed using Sparky.52 Chemical shift perturbations based on the previously published partial assignment of ERK240 were calculated as previously described.6
Crystallization, Structure Determination, and Analysis.
Crystals were grown using the hanging-drop vapor diffusion method with droplets containing 1 μL of protein [8–10 mg mL−1 ERK2 in 20 mM Tris-HCl (pH 8.0) with 0.1 mM EDTA, 0.1 mM EGTA, 200 mM NaCl, and 10% glycerol] and 1 μL of the reservoir solution [33% PEG 5000 MME, 0.25 M ammonium sulfate, and 0.1 M MES (pH 6.5)] in the presence of an arrestin-3 peptide (CH3CO-AVPETDAPVDTNL-IEFE-NH2) at a molar ratio of 1:2. Droplets were allowed to equilibrate over the reservoir solution at room temperature (approximately 22 °C). The 2507–8 inhibitor was soaked into the crystals at a molar ratio of 1:3 (ERK2:2507–8) for 24 h prior to flash cooling. Crystals were cryoprotected with a solution comprised of the reservoir solution and 15% glycerol.
X-ray diffraction data were collected at Advance Photon Source (APS) beamline 21-ID-F at –173 °C using a wavelength of 0.9798 Å and a MarMosaic225 CCD detector. Data were processed and scaled using HKL2000.53 The structure was determined using molecular replacement in Phaser-MR54 through the Phenix interface,55 with wild-type ERK2 (PDB entry 1ERK56) as the search model. As 1ERK was reported prior to the widespread use of Rfree for cross-validation, Rfree reflections were randomly selected for ERK2. However, because the original model (1ERK) was refined against these reflections, Rfree is of limited utility in this case. Model building was performed in Coot,57 and refinement was conducted using Phenix.55 Residues 328–332 are not included in the final model as the electron density corresponding to these residues was not readily interpretable. Protein–inhibitor interactions were analyzed using the program LigPlot58 (version 1.4.5), which builds schematic diagrams of protein–ligand interactions using PDB files. All structural figures were made using PyMOL Molecular Graphics System59 unless otherwise indicated. Data collection and refinement statistics are summarized in Table S5.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge financial support from the National Institutes of Health (5T32EB007507, GM123252, GM122491, GM120569, DA043680, and EY011500), the Cancer Research and Prevention Institute of Texas (RP180880, RP160657, and RP140108), the Welch Foundation (F-1390), the Florida Drug Discovery Acceleration Program by the State of Florida, Department of Health, the American Heart Association (18PRE34030017), and the Cornelius Vanderbilt Chair, Vanderbilt University.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.9b00093.
Figures S1–S18, Tables S1–S5, methods for expression and purification of ERK2 for crystallization, mathematical anisotropy model for dose–response curves, and chemical synthesis of library compounds (PDF)
Accession Codes
The atomic coordinates of the 2507–8–ERK2 complex have been deposited in the Protein Data Bank as entry 6NBS.
The authors declare no competing financial interest.
REFERENCES
- (1).Wilson LJ, Linley A, Hammond DE, Hood FE, Coulson JM, MacEwan DJ, Ross SJ, Slupsky JR, Smith PD, Eyers PA, and Prior IA (2018) New Perspectives, Opportunities, and Challenges in Exploring the Human Protein Kinome. Cancer Res. 78, 15–29. [DOI] [PubMed] [Google Scholar]
- (2).Eblen ST (2018) Extracellular-Regulated Kinases: Signaling From Ras to ERK Substrates to Control Biological Outcomes. Adv. Cancer Res 138, 99–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sheridan DL, Kong Y, Parker SA, Dalby KN, and Turk BE (2008) Substrate Discrimination Among Mitogen-Activated Protein Kinases Through Distinct Docking Sequence Motifs. J. Biol. Chem 283, 19511–19520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fantz DA, Jacobs D, Glossip D, and Kornfeld K (2001) Docking Sites on Substrate Proteins Direct Extracellular Signal-Regulated Kinase to Phosphorylate Specific Residues. J. Biol. Chem 276, 27256–27265. [DOI] [PubMed] [Google Scholar]
- (5).Callaway K, Waas WF, Rainey MA, Ren P, and Dalby KN (2010) Phosphorylation of the Transcription Factor Ets-1 by ERK2: Rapid Dissociation of ADP and Phospho-Ets-1. Biochemistry 49, 3619–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Piserchio A, Warthaka M, Kaoud TS, Callaway K, Dalby KN, and Ghose R (2017) Local Destabilization, Rigid Body, and Fuzzy Docking Facilitate the Phosphorylation of the Transcription Factor Ets-1 by the Mitogen-Activated Protein Kinase ERK2. Proc. Natl. Acad. Sci. U. S. A 114, E6287–E6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rainey MA, Callaway K, Barnes R, Wilson B, and Dalby KN (2005) Proximity-Induced Catalysis by the Protein Kinase ERK2. J. Am. Chem. Soc 127, 10494–10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Piserchio A, Ramakrishan V, Wang H, Kaoud TS, Arshava B, Dutta K, Dalby KN, and Ghose R (2015) Structural and Dynamic Features of F-Recruitment Site Driven Substrate Phosphorylation by ERK2. Sci. Rep 5, 11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Scapin G (2006) Protein Kinase Inhibition: Different Approaches to Selective Inhibitor Design. Curr. Drug Targets 7, 1443–1454. [DOI] [PubMed] [Google Scholar]
- (10).Knight ZA, and Shokat KM (2005) Features of Selective Kinase Inhibitors. Chem. Biol 12, 621–637. [DOI] [PubMed] [Google Scholar]
- (11).Lawrence DS, and Niu J (1998) Protein Kinase Inhibitors: The Tyrosine-Specific Protein Kinases. Pharmacol. Ther 77, 81–114. [DOI] [PubMed] [Google Scholar]
- (12).Breen ME, and Soellner MB (2015) Small Molecule Substrate Phosphorylation Site Inhibitors of Protein Kinases: Approaches and Challenges. ACS Chem. Biol 10, 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Roberts PJ, and Der CJ (2007) Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment of Cancer. Oncogene 26, 3291–3310. [DOI] [PubMed] [Google Scholar]
- (14).Liu QH, Shi ML, Sun C, Bai J, and Zheng JN (2015) Role of the ERK1/2 Pathway in Tumor Chemoresistance and Tumor Therapy. Bioorg. Med. Chem. Lett 25, 192–197. [DOI] [PubMed] [Google Scholar]
- (15).Chico LK, Van Eldik LJ, and Watterson DM (2009) Targeting Protein Kinases in Central Nervous System Disorders. Nat. Rev. Drug Discovery 8, 892–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tanti JF, and Jager J (2009) Cellular Mechanisms of Insulin Resistance: Role of Stress-Regulated Serine Kinases and Insulin Receptor Substrates (IRS) Serine Phosphorylation. Curr. Opin. Pharmacol 9, 753–762. [DOI] [PubMed] [Google Scholar]
- (17).Kim EK, and Choi EJ (2010) Pathological Roles of MAPK Signaling Pathways in Human Diseases. Biochim. Biophys. Acta, Mol. Basis Dis 1802, 396–405. [DOI] [PubMed] [Google Scholar]
- (18).Muslin AJ (2008) MAPK Signalling in Cardiovascular Health and Disease: Molecular Mechanisms and Therapeutic Targets. Clin. Sci 115, 203–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Schnieders MJ, Kaoud TS, Yan C, Dalby KN, and Ren P (2012) Computational Insights for the Discovery of Non-ATP Competitive Inhibitors of MAP Kinases. Curr. Pharm. Des 18, 1173–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Garai A, Zeke A, Gogl G, Toro I, Fordos F, Blankenburg H, Barkai T, Varga J, Alexa A, Emig D, Albrecht M, and Remenyi A (2012) Specificity of Linear Motifs that Bind to a Common Mitogen-Activated Protein Kinase Docking Groove. Sci. Signaling 5, No. ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tanoue T, Maeda R, Adachi M, and Nishida E (2001) Identification of a Docking Groove on ERK and p38 MAP Kinases that Regulates the Specificity of Docking Interactions. EMBO J. 20, 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Burkhard K, Smith S, Deshmukh R, MacKerell AD Jr., and Shapiro P (2009) Development of Extracellular Signal-Regulated Kinase Inhibitors. Curr. Top. Med. Chem 9, 678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Boston SR, Deshmukh R, Strome S, Priyakumar UD, MacKerell AD Jr., and Shapiro P (2011) Characterization of ERK Docking Domain Inhibitors That Induce Apoptosis by Targeting Rsk-1 and Caspase-9. BMC Cancer 11, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cerchietti LC, Ghetu AF, Zhu X, Da Silva GF, Zhong S, Matthews M, Bunting KL, Polo JM, Fares C, Arrowsmith CH, Yang SN, Garcia M, Coop A, Mackerell AD Jr., Prive GG, and Melnick A (2010) A Small-Molecule Inhibitor of BCL6 Kills DLBCL Cells in Vitro and in Vivo. Cancer Cell 17, 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Arkin MR, Tang Y, and Wells JA (2014) Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing Toward the Reality. Chem. Biol 21, 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hancock CN, Macias A, Lee EK, Yu SY, MacKerell AD Jr., and Shapiro P (2005) Identification of Novel Extracellular Signal-Regulated Kinase Docking Domain Inhibitors. J. Med. Chem 48, 4586–4595. [DOI] [PubMed] [Google Scholar]
- (27).Kinoshita T, Sugiyama H, Mori Y, Takahashi N, and Tomonaga A (2016) Identification of Allosteric ERK2 Inhibitors Through in Silico Biased Screening and Competitive Binding Assay. Bioorg. Med. Chem. Lett 26, 955–958. [DOI] [PubMed] [Google Scholar]
- (28).Chen F, Hancock CN, Macias AT, Joh J, Still K, Zhong S, MacKerell AD Jr., and Shapiro P (2006) Characterization of ATP-Independent ERK Inhibitors Identified Through in Silico Analysis of the Active ERK2 Structure. Bioorg. Med. Chem. Lett 16, 6281–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Li Q, Al-Ayoubi A, Guo T, Zheng H, Sarkar A, Nguyen T, Eblen ST, Grant S, Kellogg GE, and Zhang S (2009) Structure-Activity Relationship (SAR) Studies of 3-(2-Amino-ethyl)-5-(4-ethoxy-benzylidene)-thiazolidine-2,4-dione: Development of Potential Substrate-Specific ERK1/2 Inhibitors. Bioorg. Med. Chem. Lett 19, 6042–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Samadani R, Zhang J, Brophy A, Oashi T, Priyakumar UD, Raman EP, St John FJ, Jung KY, Fletcher S, Pozharski E, MacKerell AD Jr., and Shapiro P (2015) Small-Molecule Inhibitors of ERK-Mediated Immediate Early Gene Expression and Proliferation of Melanoma Cells Expressing Mutated BRaf. Biochem. J 467, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Houghten RA (1994) Combinatorial Libraries. Finding the Needle in the Haystack. Curr. Biol 4, 564–567. [DOI] [PubMed] [Google Scholar]
- (32).Ostresh JM, Husar GM, Blondelle SE, Dorner B, Weber PA, and Houghten RA (1994) Libraries from Libraries“: Chemical Transformation of Combinatorial Libraries to Extend the Range and Repertoire of Chemical Diversity. Proc. Natl. Acad. Sci. U. S. A 91, 11138–11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Houghten RA, Pinilla C, Giulianotti MA, Appel JR, Dooley CT, Nefzi A, Ostresh JM, Yu YP, Maggiora GM, Medina-Franco JL, Brunner D, and Schneider J (2008) Strategies for the Use of Mixture-Based Synthetic Combinatorial Libraries: Scaffold Ranking, Direct Testing, in Vivo, and Enhanced Deconvolution by Computational Methods. J. Comb. Chem 10, 3–19. [DOI] [PubMed] [Google Scholar]
- (34).Callaway K, Rainey MA, and Dalby KN (2005) Quantifying ERK2-Protein Interactions by Fluorescence Anisotropy: PEA-15 Inhibits ERK2 by Blocking the Binding of DEJL Domains. Biochim. Biophys. Acta, Proteins Proteomics 1754, 316–323. [DOI] [PubMed] [Google Scholar]
- (35).Callaway KA, Rainey MA, Riggs AF, Abramczyk O, and Dalby KN (2006) Properties and Regulation of a Transiently Assembled ERK2·Ets-1 Signaling Complex. Biochemistry 45, 13719–13733. [DOI] [PubMed] [Google Scholar]
- (36).Tanoue T, Adachi M, Moriguchi T, and Nishida E (2000) A Conserved Docking Motif in MAP Kinases Common to Substrates, Activators and Regulators. Nat. Cell Biol 2, 110–116. [DOI] [PubMed] [Google Scholar]
- (37).Waas WF, and Dalby KN (2002) Transient Protein-Protein Interactions and a Random-Ordered Kinetic Mechanism for the Phosphorylation of a Transcription Factor by Extracellular-Regulated Protein Kinase 2. J. Biol. Chem 277, 12532–12540. [DOI] [PubMed] [Google Scholar]
- (38).Bott CM, Thorneycroft SG, and Marshall CJ (1994) The Sevenmaker Gain-of-Function Mutation in p42 MAP Kinase Leads to Enhanced Signalling and Reduced Sensitivity to Dual Specificity Phosphatase Action. FEBS Lett. 352, 201–205. [DOI] [PubMed] [Google Scholar]
- (39).Brenan L, Andreev A, Cohen O, Pantel S, Kamburov A, Cacchiarelli D, Persky NS, Zhu C, Bagul M, Goetz EM, Burgin AB, Garraway LA, Getz G, Mikkelsen TS, Piccioni F, Root DE, and Johannessen CM (2016) Phenotypic Characterization of a Comprehensive Set of MAPK1/ERK2Missense Mutants. Cell Rep. 17, 1171–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Piserchio A, Warthaka M, Devkota AK, Kaoud TS, Lee S, Abramczyk O, Ren P, Dalby KN, and Ghose R (2011) Solution NMR Insights into Docking Interactions Involving Inactive ERK2. Biochemistry 50, 3660–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhou T, Sun L, Humphreys J, and Goldsmith EJ (2006) Docking Interactions Induce Exposure of Activation Loop in the MAP Kinase ERK2. Structure 14, 1011–1019. [DOI] [PubMed] [Google Scholar]
- (42).Gu S, Cui D, Chen X, Xiong X, and Zhao Y (2018) PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. BioEssays 40, No. e1700247. [DOI] [PubMed] [Google Scholar]
- (43).Kaoud TS, Devkota AK, Harris R, Rana MS, Abramczyk O, Warthaka M, Lee S, Girvin ME, Riggs AF, and Dalby KN (2011) Activated ERK2 is a Monomer in Vitro With or Without Divalent Cations and When Complexed to the Cytoplasmic Scaffold PEA-15. Biochemistry 50, 4568–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Yan C, Kaoud T, Lee S, Dalby KN, and Ren P (2011) Understanding the Specificity of a Docking Interaction Between JNK1 and the Scaffolding Protein JIP1. J. Phys. Chem. B 115, 1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Waas WF, and Dalby KN (2001) Purification of a Model Substrate for Transcription Factor Phosphorylation by ERK2. Protein Expression Purif. 23, 191–197. [DOI] [PubMed] [Google Scholar]
- (46).Madsen JA, Kaoud TS, Dalby KN, and Brodbelt JS (2011) 193-nm Photodissociation of Singly and Multiply Charged Peptide Anions for Acidic Proteome Characterization. Proteomics 11, 1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lee S, Warthaka M, Yan C, Kaoud TS, Piserchio A, Ghose R, Ren P, and Dalby KN (2011) A Model of a MAPK*Substrate Complex in an Active Conformation: A Computational and Experimental Approach. PLoS One 6, No. e18594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kaoud TS, Mitra S, Lee S, Taliaferro J, Cantrell M, Linse KD, Van Den Berg CL, and Dalby KN (2011) Development of JNK2-Selective Peptide Inhibitors that Inhibit Breast Cancer Cell Migration. ACS Chem. Biol 6, 658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hastie CJ, McLauchlan HJ, and Cohen P (2006) Assay of Protein Kinases Using Radiolabeled ATP: A Protocol. Nat. Protoc 1, 968–971. [DOI] [PubMed] [Google Scholar]
- (50).Pervushin K, Riek R, Wider G, and Wuthrich K (1997) Attenuated T2 Relaxation by Mutual Cancellation of Dipole-Dipole Coupling and Chemical Shift Anisotropy Indicates an Avenue to NMR Structures of Very Large Biological Macromolecules in Solution. Proc. Natl. Acad. Sci. U. S. A 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NMRPipe: a Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (52).Goddard TGK, and Kneller DG (2008) SPARKY3, University of California, San Francisco. [Google Scholar]
- (53).Otwinowski Z, and Minor W (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (54).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser Crystallographic Software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zhang F, Strand A, Robbins D, Cobb MH, and Goldsmith EJ (1994) Atomic Structure of the MAP Kinase ERK2 at 2.3 Å Resolution. Nature 367, 704–711. [DOI] [PubMed] [Google Scholar]
- (57).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wallace AC, Laskowski RA, and Thornton JM (1995) LIGPLOT: A Program to Generate Schematic Diagrams of Protein-Ligand Interactions. Protein Eng., Des. Sel 8, 127–134. [DOI] [PubMed] [Google Scholar]
- (59).PyMOL: The PyMOL Molecular Graphics System, version 1.5.04, Schrö dinger, LLC, 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.