

Highlights
-
•
Porcine epidemic diarrhea virus has been reemerged at November 2013 in Korea.
-
•
The reemerging Korean PEDV has been completely sequenced.
-
•
Complete genome sequences were highly diverge between new and old Korean PEDVs.
-
•
The reemerging Korean PEDV is practically identical to the USA PEDV.
Keywords: Animal virus, Complete genome, Coronavirus, Phylogenetic analysis, Porcine epidemic diarrhea virus
Abstract
Porcine epidemic diarrhea virus (PEDV), a member of the Coronaviridae family, is an enveloped, positive-sense, single-stranded RNA virus, which causes severe diarrhea and dehydration in suckling pigs. We detected three PEDV strains from ten small intestine samples from piglets with acute diarrhea and we determined the complete genome sequences of the reemerging Korean PEDV field isolates, except for the noncoding regions from both ends. The complete genome sequences of the strains were identical or almost identical (one synonymous single-nucleotide polymorphism (SNP) in the ORF1a/1b genomic sequence). Interestingly, comparative genome analysis of recent Korean PEDV isolates and other strains revealed that the complete genome sequences of recent Korean strains were almost identical (99.9%) to those of the US PEDV strains isolated in 2013. These results suggest that the three reemerging Korean strains are distinct from previous endemic Korean PEDV strains and has been recently introduced into Korea from oversea with high likelihood.
1. Introduction
Porcine epidemic diarrhea virus (PEDV) is a member of the family Coronaviridae, subfamily Coronavirinae, and genus Alphacoronavirus, which include some human and bat coronaviruses. PEDV containing a positive-sense, single-stranded RNA genome, causes severe diarrhea and dehydration in suckling piglets (Song and Park, 2012). Since the first report of isolation in Europe in 1978 (Pensaert and de Bouck, 1978), PEDV has become an economic concern in the swine industry in Europe and Asia (Song and Park, 2012).
In late 2010, various Chinese strains of PEDV that were clinically more severe than the classical strains, with 80–100% morbidity and 50–90% mortality in suckling piglets, were detected (Li et al., 2012). In April 2013, PEDV outbreaks were confirmed in the US for the first time and the isolates showed very close relationship with the Chinese isolate AH2012. A previous study showed that the emergent US PEDV strains were likely introduced into the US through intercontinental transmission from China (Huang et al., 2013).
In Korea, PEDV was first isolated in 1992, followed by a large two-year-long outbreak. Despite the use of vaccines, frequent occurrence of PEDV was detected across the country, mainly during the winter season (Chae et al., 2000, Kweon et al., 1993). Since late November 2013, PEDV has reemerged in Korea and caused significant economic losses in the swine industry. This study aimed to determine the complete genome sequence of the reemerging Korean PEDV strain and to investigate their genetic relationship with other strains using comparative genome analysis and phylogenetic analysis.
2. Materials and methods
Ten small intestine samples were collected from dead piglets from two commercial pig farms in Korea. The piglets died following acute watery diarrhea. The macroscopic features of the intestines were typical of PEDV infections, including yellowish contents and distended appearance. To detect PEDV genome, M gene-targeted RT-PCR was performed (Kim et al., 2000) using total RNA from mucosal scrapings. Three of ten samples were positive in the PEDV specific RT-PCR. To investigate the origin of the reemerging Korean PEDV strain, complete genome sequences of the three reemerging Korean PEDV strains were determined using Sanger sequencing.
For Sanger sequencing, 18 primer pairs were designed for the highly conserved sites of the PEDV genome using Primer3 (Koressaar and Remm, 2007, Untergasser et al., 2012) or designed manually when Primer3 failed to identify optimal primer sites near the appropriate genomic regions (Table 1 ). Eighteen DNA fragments, which covered the entire genome of PEDV except the noncoding regions from both ends, were amplified using the SuperScript® One-Step RT-PCR System. Sequencing reactions were performed using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). The products were analyzed using ABI 3730xl DNA Analyzer (Applied Biosystems). The sequencing results were assembled using Geneious v5.6.7 software. Complete genome sequences of the three reemerging Korean PEDV strains have been submitted into the GeneBank database under the accession numbers KJ588062, KJ588063, and KJ588064.
Table 1.
Primers used for the amplification of full-length genomes of the reemerging Korean PEDV strains.
| Primer | Sequence (5′-3′) | PCR product size (bp) | Position |
|---|---|---|---|
| 1F | CTTAAAAAGATTTTCTATCTA | 394 | 42–436 |
| 1R | CCTCAGAATAGTATGAGACG | ||
| 2F | GTCGCCTTCTACATACTAGACAAACAGC | 1542 | 237–1778 |
| 2R | CCGACCTTTAAGCAGTCACAGG | ||
| 3F | CTTGGGAGCAGCTTAAGGC | 1600 | 1562–3161 |
| 3R | CCTGTAACCTTGATACGATTACCAACAGC | ||
| 4F | CTATTATGATGGAACACTATACTATCC | 1755 | 2946–4700 |
| 4R | GCATCTACCAAGCCATCC | ||
| 5F | GGTCTTAAGGTCTTTAATGTTGTTGG | 1934 | 4441–6374 |
| 5R | CCCAACGCCTTTGCATTATAGC | ||
| 6F | CAGACGGCTGTTGTGATTAAAGACC | 1849 | 6103–7951 |
| 6R | CAGCAACTATGAACAGACACAAAAACC | ||
| 7F | TTTATGTTGACCTTTAATGATTGTCGTATGC | 1833 | 7813–9645 |
| 7R | TCTAACTGTGCGATAGGTGTACTTAGG | ||
| 8F | CCTTTCTGTTTTACGCCTCC | 1937 | 9495–11431 |
| 8R | GCTTAACCAACTGAGGTGG | ||
| 9F | TATGTTGCAGAGTGTTGC | 1876 | 11313–13188 |
| 9R | TAACGCATTTAAGCATAGC | ||
| 10F | TTACCGAGTATACTATGATGG | 1873 | 12973–14845 |
| 10R | ACGAACTGGTCATCGACG | ||
| 11F | ATGCAACCACCGCATATGC | 1699 | 14671–16369 |
| 11R | AGTGAATCGACCGCTGC | ||
| 12F | AAGCTTATTCTAGCTTAGTGC | 1833 | 16198–18030 |
| 12R | TTATGGCATCACCAGAAGC | ||
| 13F | TACTGTTGTTTCAAACATGC | 1886 | 17874–19759 |
| 13R | AATGTCTGGAGTTTATGATCC | ||
| 14F | ACGAGTTTGTGTCTAGTAATGATAGC | 2324 | 19531–21854 |
| 14R | TGACAGTAGGAGGTAAAACAGCC | ||
| 15F | ATGGAGTTTGTAATGGAGC | 2092 | 21579–23670 |
| 15R | TCAGCAAGCAATTGCTGG | ||
| 16F | GGTGTCATGGTACTACCT | 2156 | 23477–25632 |
| 16R | TAAACTGCGCTATTACACAACC | ||
| 17F | TTCAACTAGACGAGTATGC | 1825 | 25469–27293 |
| 17R | AAGCTGCTACGCTATTTTCG | ||
| 18F | GATGATCTGGTGGCTGC | 980 | 27071–28050 |
| 18R | GGTCTTCAGTTACTAACAGTCC |
Complete genome alignment between the reemerging Korean strains and ten other available strains were performed using Multiple Alignment with Fast Fourier Transformation (MAFFT) v6 (Katoh and Toh, 2008). To study the relationship between the US and Korean PEDV outbreaks, US and Chinese strains (which were epidemic in 2012) were included in genome alignment. In addition, all available complete genome sequences of PEDV isolates from Korea were included in the alignment to compare the recent Korean strains with previous endemic Korean PEDV strains. CV777 strain was included in the alignment as PEDV reference strain. The maximum likelihood phylogenetic trees for the complete genome and the S gene sequence alignments were generated using PhyML version 2.4.4 (Guindon and Gascuel, 2003) with the generalized time reversible (GTR) substitution model (Rodriguez et al., 1990). The best nucleotide substitution model for analysis was confirmed using MEGA 5.2.2 (Tamura et al., 2011).
3. Results and discussions
The complete genome sequences of the reemerging Korean strains showed a typical PEDV gene order of 5′UTR-ORF1a/1b-S-ORF3-E-M-N-3′UTR and were identical or almost identical (one synonymous single-nucleotide polymorphism (SNP) in the ORF1a/1b gene) to each other. Multiple alignment with other PEDV complete genomes indicated that the reemerging Korean strains possess genome sequences, which are distinct from those of previous Korean field strains (Fig. 1 ). A previous study had discussed evidence of frequent recombination events between different genetic lineages or sublineages of PEDV (Huang et al., 2013). However, genomic sequences of the reemerging Korean strains did not show any regions recombined with those of the previous Korean strains during the recombination analysis performed using SimPlot 3.5.1 (Lole et al., 1999) (data not shown). In addition, phylogenetic analysis of the S gene between the reemerging and previous Korean strains of PEDV indicated that the reemerging Korean strains were included into a genetic lineage different from those of previous endemic Korean PEDV strains (Fig. 2 ). These results suggest that the reemerging strains have been recently introduced into Korea from another country.
Fig. 1.

Nucleotide sequence alignment and phylogenetic tree analysis for complete genomes of PEDV strains. (A) Alignment of the complete genome sequences of the reemerging Korean PEDV field strains and other strains was performed using MAFFT. One of the reemerging Korean strain sequences was set as the reference sequence. Vertical lines indicate the SNPs compared to the reference sequence and dashes indicate sequence gaps. Protein-coding regions are indicated with arrows. (B) A maximum likelihood phylogenetic tree was generated using the alignment. One-hundred bootstrap replicates were used to assess the significance of the tree topology. A bar indicates nucleotide substitutions per site.
Fig. 2.
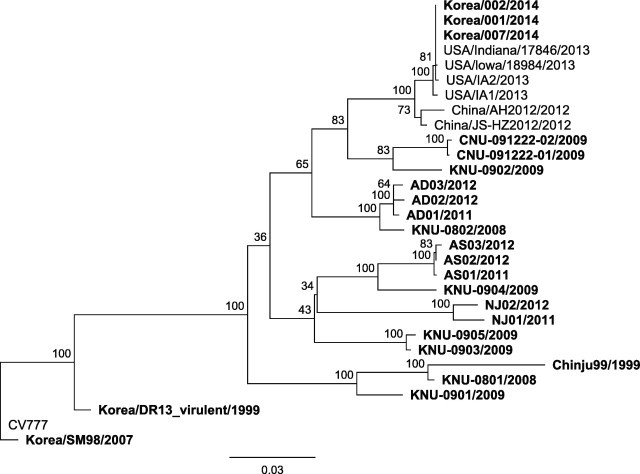
Phylogenetic tree showing the relationship between the reemerging Korean strains from previous endemic Korean strains and strains from overseas, based on the analysis of the S gene. A maximum likelihood phylogenetic tree was generated from the alignment of complete S gene sequences. One-hundred bootstrap replicates were used to assess the significance of the tree topology. A bar indicates nucleotide substitutions per site. Korean PEDV strains are denoted using bold characters.
Interestingly, comparative genome analysis of the reemerging Korean PEDV isolates and other strains revealed that the complete genome sequences of the recent Korean strains were almost identical (99.9%) to those of the US PEDV strains isolated in 2013 (Fig. 1). Compared with the complete genome of the reemerging Korean isolates, genomes of the US strains, USA/Iowa/18984/2013 and USA/Indiana/17846/2013 showed five (three non-synonymous and one synonymous in the ORF1a/1b gene and one non-synonymous in the S gene) and seven (four non-synonymous and two synonymous in the ORF1a/1b gene and one non-synonymous in the ORF3 gene) SNPs, respectively. Both US strains have one insertion sequence causing early termination of the translation of polyprotein encoded in the ORF1a/1b gene. On the other hand, the complete genome of the US strain USA/IA2/2013 did not show any indels, but nine (three non-synonymous and three synonymous in the ORF1a/1b gene, one non-synonymous in the S gene, and one non-synonymous in the N gene) SNPs, when compared with the complete genome of the reemerging Korean isolates. According to the phylogenetic analysis, the reemerging Korean PEDV isolates were closely clustered with the US strains isolated in 2013 and Chinese strains isolated in 2012 (Fig. 1).
Comparative genome analysis and phylogenetic analysis revealed that the reemerging Korean PEDV strains are practically identical to the US strains. A previous study suggested that the three emergent US strains were most closely related to a strain isolated in 2012 from Anhui Province in China. In addition, the genomes of the reemerging Korean PEDV strains did not possess any genetic feature from the genomes of the previously sequenced Korean field and attenuated vaccine strains. These results suggest that the reemerging Korean PEDV strains are not variant strains of old Korean field or attenuated vaccine strains. There are two possible sources of origin of the reemerging Korean PEDV strains. First, the same source of origin of the US strains containing Chinese PEDV-like virus could have been introduced into Korea slightly later than the US outbreak events. This hypothesis can explain why the reemerging Korean PEDV strains are identical to the US strain. Another possibility is that US strain has been directly transmitted into Korea. During the US outbreak of PEDV in 2013, two genetic sublineages of the US strains were isolated. In a previous report, the authors stated that the US strain of PEDV diverged during evolution and that evolution generated two genetic sublineages, namely, IA1-CO/13 and MN-IA2 (Huang et al., 2013). During complete genome alignment as part of this study, one of the US strains, IA1, showed a recombined genomic region in the ORF1a/1b gene, which closely matched that of the Chinese strain JS-HZ2012. All reemerging Korean PEDV strains isolated in this study showed a close relationship with only one of the genetic sublineages of the US strains, namely, MN-IA2. We could not detect a PEDV strain with a close relationship with the IA1-CO/13 sublineage. This possibly suggests that only one sublineage of the US strain has been directly introduced into Korea from the US. To identify the exact source of origin of the reemerging Korean strain, further investigation and surveillance are required. Furthermore, to prevent the introduction of PEDV into Korea from overseas in future, the quarantine policy on feed ingredients should be reinforced.
Acknowledgment
This research was supported by a fund (Project Code No. Z-1541780-2013-13-03) by Research of Animal and Plant Quarantine Agency, South Korea.
References
- Chae C., Kim O., Choi C., Min K., Cho W.S., Kim J., Tai J.H. Prevalence of porcine epidemic diarrhoea virus and transmissible gastroenteritis virus infection in Korean pigs. Vet. Rec. 2000;147:606–608. doi: 10.1136/vr.147.21.606. [DOI] [PubMed] [Google Scholar]
- Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Huang Y.W., Dickerman A.W., Pineyro P., Li L., Fang L., Kiehne R., Opriessnig T., Meng X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio. 2013;4:e00737–00713. doi: 10.1128/mBio.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- Kim O., Choi C., Kim B., Chae C. Detection and differentiation of porcine epidemic diarrhoea virus and transmissible gastroenteritis virus in clinical samples by multiplex RT-PCR. Vet. Rec. 2000;146:637–640. doi: 10.1136/vr.146.22.637. [DOI] [PubMed] [Google Scholar]
- Koressaar T., Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- Kweon C.H., Kwon B.J., Jung T.S., Kee Y.J., Hur D.H., Hwang E.K., Rhee J.C., An S.H. Isolation of porcine epidemic diarrhea virus (PEDV) in Korea. Korean J. Vet. Res. 1993;33:249–254. [Google Scholar]
- Li W., Li H., Liu Y., Pan Y., Deng F., Song Y., Tang X., He Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012;18:1350–1353. doi: 10.3201/eid1808.120002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole K.S., Bollinger R.C., Paranjape R.S., Gadkari D., Kulkarni S.S., Novak N.G., Ingersoll R., Sheppard H.W., Ray S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensaert M.B., de Bouck P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978;58:243–247. doi: 10.1007/BF01317606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez F., Oliver J.L., Marin A., Medina J.R. The general stochastic model of nucleotide substitution. J. Theor. Biol. 1990;142:485–501. doi: 10.1016/s0022-5193(05)80104-3. [DOI] [PubMed] [Google Scholar]
- Song D., Park B. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes. 2012;44:167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G. Primer3 – new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]