

Abstract
Hepatitis C virus (HCV) infection is a major cause of chronic liver disease and hepatocellular carcinoma, yet fully efficacious treatments are missing. In this study, we investigated RNA interference (RNAi), a specific gene silencing process mediated by small interfering RNA (siRNA) duplexes, as an antiviral strategy against HCV. Synthetic siRNAs were designed to target conserved sequences of the HCV 5′ nontranslated region (NTR) located in a functional, stem–loop structured domain of the HCV internal ribosome entry site (IRES), which is crucial for initiation of polyprotein translation. Several siRNAs dramatically reduced or even abrogated the replication of selectable subgenomic HCV replicons upon cotransfection of human hepatoma cells with viral target and siRNAs, or upon transfection of cells supporting autonomous replication of HCV replicon with siRNAs. Importantly, three siRNAs also proved capable of strongly inhibiting virus production in cell culture. One siRNA, targeting a sequence that is highly conserved across all genotypes and forms a critical pseudoknot structure involved in translation, was identified as the most promising therapeutic candidate. These results indicate that the HCV life cycle can be efficiently blocked by using properly-designed siRNAs that target functionally important, highly conserved sequences of the HCV IRES. This finding offers a novel approach towards developing IRES-based antiviral treatment for chronic HCV infections.
Introduction
Hepatitis C virus (HCV) frequently establishes persistent infections in the liver, leading to the development of chronic hepatitis, and, potentially, liver cirrhosis and hepatocellular carcinoma at later stages. Chronic HCV infection affects 2.2% of the world's population and is known to be the leading factor necessitating liver transplantation in patients in developed countries. No vaccine is available for HCV and the current treatment, which consists of administering pegylated interferon α and ribavirin, has limited efficacy against certain HCV genotypes, and also produces significant adverse effects (reviewed in ref. 1). The development of alternative, specific therapies for chronic HCV infection is therefore a major public health objective.
HCV, a member of the Flaviviridae family, contains a single-stranded positive-sense RNA genome that encodes a unique precursor polyprotein; this polyprotein is further processed into structural proteins and nonstructural proteins, thereby ensuring genome replication. The long open reading frame is flanked by 5′ and 3′ nontranslated regions (NTRs) that are highly conserved among the majority of HCV genotypes and contain elements that are essential for genome replication (reviewed in ref. 1). In addition, HCV 5′NTR contains a highly structured element, namely, an internal ribosome entry site (IRES), which is essential for the initiation of HCV polyprotein translation.2 Anti-HCV drug development has been hampered both by the lack of efficient cell culture systems that support virus replication and by the unavailability of accessible animal models. HCV subgenomic RNA replicon systems3 have permitted the assaying of the inhibitory effect of antiviral candidates on HCV genome replication in vitro in human hepatoma cells. The recent development of stable cell culture systems permitting robust production of infectious HCV particles in vitro,based on the JFH-1 strain of HCV genotype 2a,4 will facilitate the investigation and testing of new antiviral strategies. In addition, alternative animal models that have recently been developed seem to show promise in evaluating candidate antiviral therapeutics. These animal models include transgenic mice engrafted with human hepatocytes,5 as well as nonendangered primate species (tamarins, marmosets) infected by GB virus B, a hepatotropic virus that is closely related to HCV,6 , 7 or by GB virus B derivatives containing functional HCV sequences.8
Among possible therapeutic strategies, RNA interference (RNAi) is an attractive path to explore. First described as a natural defense mechanism against plant viruses, RNAi was subsequently shown to be a universal phenomenon of post-transcriptional gene silencing, which is initiated by double-stranded RNA and leads to specific degradation of homologous RNAs. This process involves the generation of 21-nucleotide small interfering RNAs (siRNAs) which, in association with a multiprotein complex named “RNA induced silencing complex”, are used as guides to target specific RNA substrates by Watson–Crick base-pairing. Such siRNAs, when introduced directly into mammalian cells or expressed from viral vectors, can lead to the degradation of targeted sequences (reviewed in ref. 9). During the past few years, RNAi has been extensively investigated as an alternative specific therapy to treat cancers, genetic diseases, and infections by various human pathogens of medical importance, using both in vitro and in vivo model systems (reviewed in refs. 10 , 11). RNA viruses, in particular viruses with liver tropism such as HCV, are ideal candidates for nucleic acid-based therapies that have been shown to efficiently target the liver (reviewed in ref. 12). Recently, various studies have reported variable inhibitory effects of HCV-specific synthetic siRNAs or small hairpin RNAs (shRNAs) expressed from viral vectors, that target sequences encoding various HCV nonstructural proteins as well as sequences within the 5′NTR.13 , 14 , 15 , 16 , 17 , 18 , 19 These studies all relied on the use of HCV subgenomic or genomic replicon models in cell culture.
In this study we designed siRNAs targeting highly conserved sequences within the HCV 5′NTR. The efficiency of these HCV-specific siRNAs was first evaluated using previously described self-replicating HCV RNAs.20 , 21 This allowed us to select four siRNAs that abrogated or substantially reduced genome replication. By using these siRNAs in recently described cell culture systems that support the production of genotype 2a JFH-14 or chimeric 1a/2a (D. Delgrange, A. Pillez, L. Cocquerel, G. Paranhos-Baccala, Y. Rouillé, J. Dubuisson et al., unpublished results) infectious particles, we demonstrated the successful silencing of HCV infection in cell culture.
Results
Selection of siRNAs targeting HCV 5′NTR
In order to investigate the potency of virus-specific siRNAs in inhibiting HCV infection, we selected seven siRNAs that target conserved sequences of the pivotal domain (domain III) of the HCV IRES within the 5′NTR2 (Figure 1a ). This domain is particularly important for initiation of polyprotein translation, as it directly contacts 40S ribosomal subunits22 and binds translation initiation factor 3, eIF3.23 Domain III has also been reported to be involved to some extent in genome replication.24 siRNAs were designed essentially according to previously described criteria; in particular, whenever possible, the asymmetric thermostability of siRNA duplexes was respected.25 , 26 As a messenger, HCV positive strand RNA genome is an ideal target for siRNAs, but it harbors strong secondary and tertiary structures, in particular within the IRES. This makes it difficult to design optimal siRNAs according to the criteria referred to earlier. On the other hand, the high nucleotide conservation of this region permitted the identification of siRNAs that were homologous to several HCV genotypes (Table 1 ). Selected siRNAs were named according to the nucleotide position within the genome of the H77 strain of HCV genotype 1a27 that corresponds to the first 5′ nucleotide targeted by the siRNA antisense strand: si205, si214, si240-1a, si240-1b, si244, si284, si313 (Table 1). siRNA si313 targets the 3′ end of domain III and 5′ end of domain IV involved in a pseudo-knot located upstream from the initiator codon (Figure 1a). The sequences of all siRNAs, with the exception of si240, matched both HCV genotype sequences 1a and 1b that were present in the subgenomic replicons used in the first part of the study (Figure 1b). Two siRNAs si240 (si240-1a and si240-1b) were synthesized with sequences homologous to genotypes 1a and 1b, respectively. When targeted to genotype 1b 5′NTR, si240-1a formed a G:U wobble base-pairing at position 18 of the siRNA antisense strand, whereas si240-1b formed an A:C mismatch with the genotype 1a replicon (Figure 1c). The G:U non-canonical base-pairing is known to be non-disruptive in double-stranded RNA structures and both G:U wobble base-pairing and A:C mismatch, if present at certain positions of the siRNA, were recently reported to be well tolerated for siRNA-mediated gene silencing.28 , 29 , 30 , 31 Therefore, we proceeded to analyze the effects of si240-1a and si240-1b on both homologous and heterologous replicon targets. Finally, as a non-specific, negative control in these experiments, we used an irrelevant siRNA, referred to as siIRR (Table 1), that had previously been used for targeting a sequence within the 5′NTR of a poliovirus strain,30 and that did not share homology with the HCV sequence.
Figure 1.

Hepatitis C virus (HCV)-specific small interfering RNAs (siRNAs) and HCV replicons used. (a) The footprints of the seven HCV-specific siRNAs selected to target the internal ribosome entry site (IRES) are represented on the schematic structure of domains III–IV of the 5′ nontranslated region (NTR) from the H77 strain of HCV genotype 1a.2 siRNA si240 targets a viral sequence that contains a nucleotide change in HCV genotype 1a and 1b sequences, as indicated. (b) The two replicons used in this study, Ntat2Aneo21 and NNeo20 with 5′NTRs derived from HCV genotype 1a and 1b strains, respectively, are schematically represented, with cell culture adaptive mutations (S2005I in NS5A and R2889G in NS5B, respectively) indicated by arrowheads. Both replicons encode neomycine phosphotransferase (Neo) C-terminally fused to either human immunodeficiency virus Tat protein followed by foot-and-mouth disease virus autocatalytically cleaved 2A protein (Ntat2ANeo), or to a few amino acid residues from core protein (ΔC in NNeo). HCV nonstructural proteins NS3-NS5B are translated from the encephalomyocarditis virus (EMCV) IRES. (c) The base-pairing of si240-1a and si240-1b with 5′NTR sequences of replicons Ntat2ANeo (1a genotype) and NNeo (1b genotype), and of si244 with 5′NTR sequence of virus JFH-1 (2a genotype) is shown. Viral RNA/siRNA duplexes are delineated by dotted lines. Nucleotide mismatches between siRNAs and HCV sequences are boxed and their position within the siRNA anti-sense strand is indicated.
Table 1.
Sequences of siRNAs used for targeting domain III of the internal ribosome entry site of HCV genome
| siRNAa | siRNA sense-stranded (5′→3′) | Degree of conservation (%)b |
|---|---|---|
| si205 | CCCGCUCAAUGCCUGGAGATT | 73.6 |
| si214 | UGCCUGGAGAUUUGGGCGUTT | 73.6 |
| si240-1a | CAAGACUGCUAGCCGAGUATT | 28.9 |
| si240-1b | CGAGACUGCUAGCCGAGUATT | 62.3 |
| si244 | ACUGCUAGCCGAGUAGUGUTT | 71.1 |
| si284 | GUACUGCCUGAUAGGGUGCTT | 98.7 |
| si313 | CCCGGGAGGUCUCGUAGACTT | 98.1 |
| siIRR | CGUUUUACUCCUUAACUUATT | NA |
Abbreviations: HCV, hepatitis C virus; NA, not applicable; siIRR, irrelevant siRNA; siRNA, small interfering RNA.
siRNA numbering corresponds to the first nucleotide of the H77 strain of HCV genotype 1a27 targeted by the 21-nucleotide siRNA antisense-strand.
Degree of conservation (%) of each siRNA-targeted sequence among 159 HCV sequences of all genotypes, as found in the Los Alamos National Laboratory database (http://hcv.lanl.gov/content/hcv-db/index).
Effect of siRNAs on the replication of reporter HCV subgenomic replicon
First we evaluated whether selected siRNAs were able to inhibit the replication of an HCV subgenomic replicon (Ntat2ANeo) in cell culture. We did this by using a previously described system that allowed simple monitoring of RNA replication by measurement of an enzymatic activity, namely, secreted alkaline phosphatase (SEAP) activity21 (see Materials and Methods and Figure 1b). All HCV sequences of Ntat2ANeo replicon are derived from the N strain of genotype 1b, with the exception of the 5′NTR, which is derived from the H77 strain of genotype 1a.
Following coelectroporation of En5-3 cells with 5 μg of in vitro-transcribed Ntat2ANeo RNA and 2 μg of each synthetic siRNA, the culture supernatant was replaced daily with fresh medium during the first 4 days after transfection and then on day 7, and SEAP activity was measured in all culture supernatants collected. During the first 2 days, SEAP signals essentially reflected translation of the input RNAs, since SEAP levels were roughly similar in cells transfected with Ntat2ANeo replicon and those transfected with a replication-defective RNA encoding an inactive NS5B RNA polymerase deleted in its active site (ΔGDD, see Materials and Methods). RNA replication was then clearly detected between days 3 and 7 after transfection (Figure 2a , compare SEAP levels in cells transfected with Ntat2ANeo and ΔGDD). Cotransfection of the irrelevant siRNA (siIRR) did not significantly affect Ntat2ANeo replication (Figure 2a). HCV-specific siRNAs si214 and si284 showed only transient and nil effect on HCV replication, respectively, and SEAP expression levels on day 7 after transfection were similar to those induced by transfection of Ntat2ANeo alone (Figure 2a). These two siRNAs were therefore not further utilized in the course of our study. In sharp contrast, siRNAs si205, si240-1a, si240-1b, si244, and si313 proved capable of abolishing HCV replication in this system, yielding SEAP levels as low as those obtained with replication-defective RNA ΔGDD. In the presence of these siRNAs, SEAP levels were even slightly lower at days 1, 2, and 3 after transfection than those obtained with ΔGDD, as a result of siRNA-mediated degradation of input RNAs, rendering them unavailable for translation (Figure 2a). The inhibitory effect of these five siRNAs on HCV replication was shown to be prolonged to 14 days after transfection (data not shown).
Figure 2.
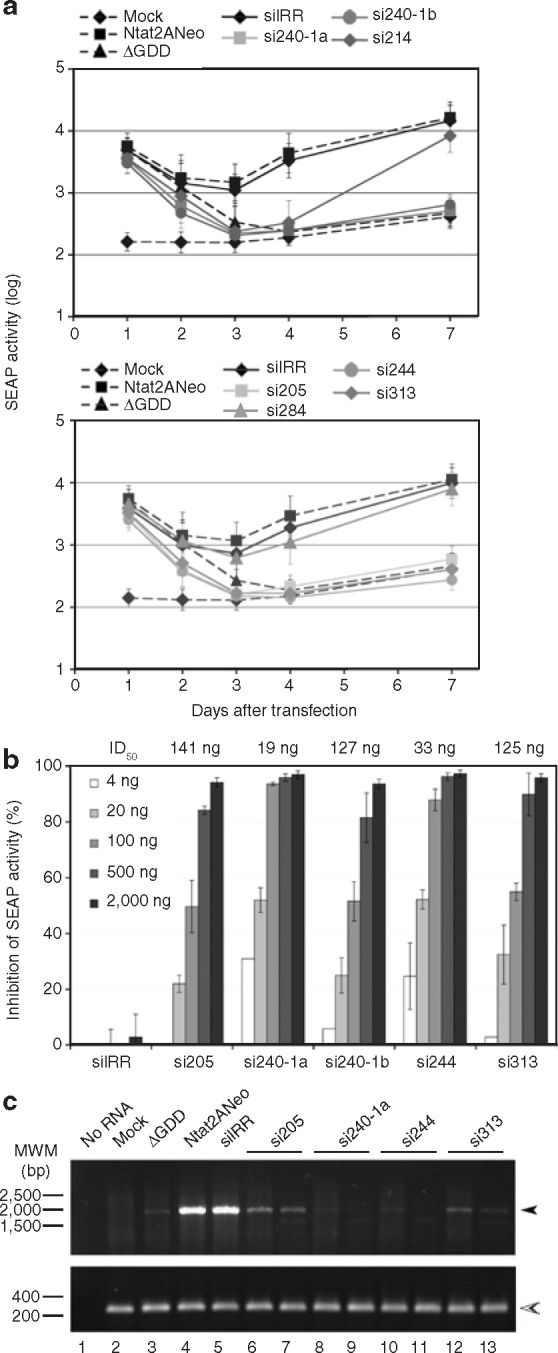
Small interfering RNAs (siRNAs) inhibit efficiently transient replication of reporter hepatitis C virus (HCV) replicon. (a) Graphs represent secreted alkaline phosphatase (SEAP) activities (arbitrary units) in supernatants of En5-3 cell cultures electroporated with no RNA (Mock), 5 μg RNA from HCV Ntat2ANeo replicon (Ntat2ANeo), or a non-replicative HCV RNA (ΔGDD), or coelectroporated with 5 μg of HCV Ntat2ANeo replicon and 2 μg of each indicated siRNA. Each time point corresponds to the amount of SEAP accumulated over 24-hour periods during the first 4 days after transfection, and the last point at day 7 after transfection corresponds to SEAP activity accumulated between days 4 and 7. Each point represents mean ± SD of at least three experiments. Data are represented on a logarithmic scale. (b) Effects of increasing doses of each siRNA (white to dark grey bars for 4–2,000 ng doses) are represented as percentages of SEAP activity inhibition, calculated in proportion to the SEAP activity obtained with replicon Ntat2ANeo RNA alone. Each histogram bar represents mean ± SD of multiple independent experiments. Inhibition doses 50 (ID50s) calculated for each siRNA are indicated on top of the graph. (c) Identical amounts of total RNA extracted from cells transfected with no RNA (Mock), HCV Ntat2ANeo replicon (Ntat2ANeo), or non-replicative HCV RNA (ΔGDD), or coelectroporated with 5 μg of HCV Ntat2ANeo replicon and 2 μg of each indicated siRNA were reverse transcribed and amplified by polymerase chain reaction (PCR) using HCV 5′NTR-specific primers (upper gel) or housekeeping gene GAPDH-specific primers (lower gel). A control reaction performed in the absence of template RNA (No RNA) was analyzed in parallel on 1% agarose gels. PCR products obtained with RNA extracted from two independent cell cultures treated with HCV-specific siRNAs are shown. DNA molecular weight markers are shown on the left of the gels. Filled and open arrowheads point to HCV and GAPDH-specific reverse transcription-PCR products, respectively. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NTR, nontranslated region; siIRR, irrelevant siRNA.
We next monitored dose-response effects of each of the five selected siRNAs using decreasing siRNA doses in the range of 2,000–4 ng (67–0.13 nmol/l per μg replicon) in coelectroporation experiments with 5 μg of Ntat2ANeo replicon (Figure 2b). From these experiments, inhibition doses 50 were calculated for each siRNA (Figure 2b). The siRNAs si240-1a and si244 proved to be the most efficient inhibitors of HCV RNA replication with inhibition doses 50 of 19 ng (0.64 nmol/l) and 33 ng (1.11 nmol/l), respectively, and all siRNAs exhibited inhibition doses 50 within the 19–141 ng range. The inhibition dose 50 of si240-1b (127 ng) was shown to be substantially higher than that of si240-1a (19 ng), in agreement with the fact that si240-1b is not fully homologous to Ntat2ANeo 5′NTR (genotype 1a). This demonstrates the sequence-specificity of these siRNAs. We also found that the co-administration of two siRNAs targeting nonoverlapping HCV sequences, in suboptimal doses, resulted in additive inhibitory effects (data not shown). This might prove useful in future studies in which a strategy based on combined siRNAs is sought to be developed in order to prevent the occurrence of escape mutations. The five siRNAs (si205, si240-1a, si240-1b, si244, and si313) that specifically target domain III of the HCV IRES and strongly inhibit subgenomic RNA replicon replication when used in the nanomolar range were retained in the remaining part of the study.
Viral RNA detection in siRNA-treated cells
Next, in order to determine whether siRNAs are capable of curing replicon-containing cells, we investigated whether HCV replicon RNA was eliminated in siRNA-treated cells in which SEAP activities were at basal level. Total RNA was extracted from cells cotransfected with replicon and siRNA on day 10 after transfection and analyzed for HCV RNA. This was done by semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR) using an HCV primer pair that allows amplification of a 1630 base-pair fragment spanning the NS3 coding region, as well as a primer pair that allows the detection of a housekeeping messenger RNA (glyceraldehyde-3-phosphate dehydrogenase) for the purpose of RNA quantity normalization (Figure 2c). HCV RNA was readily detected in similar abundance in cells transfected with Ntat2ANeo or cotransfected with Ntat2ANeo and siIRR (Figure 2c, lanes 4, 5). Very low levels of residual transfected ΔGDD RNA molecules could be occasionally detected under these experimental conditions (Figure 2c, lane 3). In duplicate experiments, using cells cotransfected with Ntat2ANeo and si240-1a, si244, or si313, viral RNA was either not detected or detected at very low levels, comparable to the results obtained with ΔGDD RNA (Figure 2c, lanes 8–13). HCV RNA was consistently detected in cells cotransfected with Ntat2ANeo and si205 (Figure 2c, lanes 6, 7), but at levels lower than in cells cotransfected with Ntat2ANeo and siIRR. These results correlated well with inhibition levels of reporter SEAP activity, and confirmed that HCV RNA replication is strongly inhibited, if not abolished, in cells treated with 2 μg of the three most efficient siRNAs (si240-1a, si244, and si313).
Effect of siRNAs in a replicon-containing cell line
In order to work with a cell culture system mimicking an established persistent infection, we analyzed the inhibitory capacity of siRNAs in En5-3 cells that stably contain and continuously replicate Ntat2ANeo replicon. The ongoing replication of Ntat2ANeo replicon in these cells was reflected by SEAP levels that were more than tenfold higher than in transient experiments after HCV replicon electroporation. Two micrograms of each siRNA were electroporated into these cells, and their effect on HCV RNA replication was determined at different time-points. Data obtained at day 7 after transfection, corresponding to cumulative SEAP levels secreted between days 4 and 7, are represented in Figure 3 . The SEAP activity generated by the HCV replicon in the presence of either of the homologous siRNAs was reduced by 50% or more relative to that of Ntat2ANeo in mock-transfected cells. When a higher dose (5 μg) of siRNAs was used, SEAP activity was reduced by 70–93% (data not shown). The siRNA-mediated inhibitory effect was not as dramatic as in cotransfection experiments, probably due, in part, to limited transfection efficiency (∼70%, data not shown), thereby permitting ongoing RNA replication in cells that did not receive siRNA in the transfected culture. In addition, viral RNAs undergoing replication within replication complexes may be less accessible than transfected viral RNAs to siRNAs. Nonetheless, we demonstrated strong, dose-dependent inhibitory potency of HCV domain III-specific siRNAs in cells supporting continuous HCV RNA replication.
Figure 3.
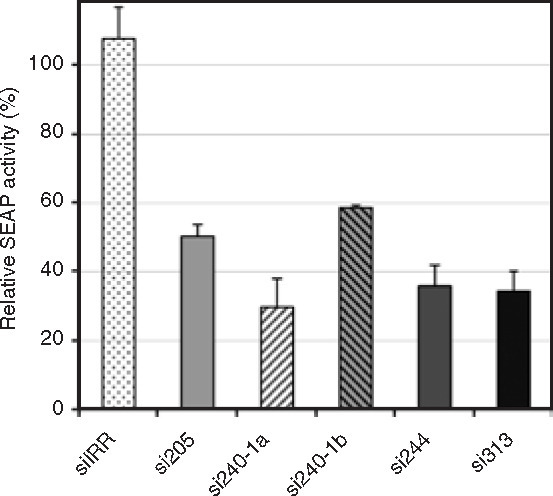
Inhibitory effect of small interfering RNA (siRNA) treatment in cells supporting autonomous replication of hepatitis C virus replicon. Secreted alkaline phosphatase (SEAP) activities in supernatants of En5-3 cells supporting autonomous replication of Ntat2ANeo replicon transfected with 2 μg of each indicated siRNA were determined on day 7 after transfection and represented as a proportion of the SEAP activity present in mock-transfected cells, set at 100%. Each bar represents mean ± SD of four experiments. siIRR, irrelevant siRNA.
Effect of siRNAs on G418-resistant cell clone formation
We next addressed the question of the efficacy of siRNAs in inhibiting HCV genome replication in cells placed under selective pressure, and examined whether there was any HCV escape from specific siRNA treatment. In these experiments, we used the HCV replicon-free subclone 2-3c of Huh7 cells,32 and either Ntat2ANeo or NNeo replicon. In contrast to Ntat2ANeo, NNeo replicon (see Figure 1b and ref. 20) contains a 5′NTR derived from the N strain of genotype 1b and encodes neomycine phosphotransferase gene in the first cistron. 2-3c cells were coelectroporated with either Ntat2ANeo or NNeo replicon and each siRNA, then placed under G418 selective pressure. G418-resistant cell clones were counted on day 21 after transfection. Data from two or three experiments carried out with each replicon were pooled for siRNAs homologous to both replicons (si205, si244, si313), and the mean of these data is shown in Figure 4 . A mean of 34,000 ± 23,800 resistant cell clones was obtained after transfection of HCV replicon alone. The data are represented with respect to 10,000 G418-resistant clones in each experiment. Treatment with each of the HCV-specific siRNAs resulted in a substantial reduction in the number of G418-resistant cell clones (Figure 4). siRNA si205 reduced the formation of G418-resistant cell clones by ∼2 logs, whereas si244 and si313 reduced cell clone formation by 3 logs or more (Figure 4a). This is in good correlation with results obtained in the SEAP reporter system. Both si240-1a and si240-1b were more efficient on their homologous target than on heterologous targets (Figure 4b). The nature and the position of the mismatch between si240 and its target (G:U and A:C for si240-1a on HCV genotype 1b and si240-1b on genotype 1a, respectively, at nucleotide 18 of the siRNA antisense strand) are reasonably well tolerated by the RNAi machinery, resulting in ∼2 log reduction in G418-resistant cell clone formation, an inhibition that was, however, 1–1.5 log less efficient than on homologous targets. These experiments confirmed, as already described,28 , 29 , 31 that perfect base-pairing between siRNAs and targeted messenger RNA is required for most effective silencing.
Figure 4.
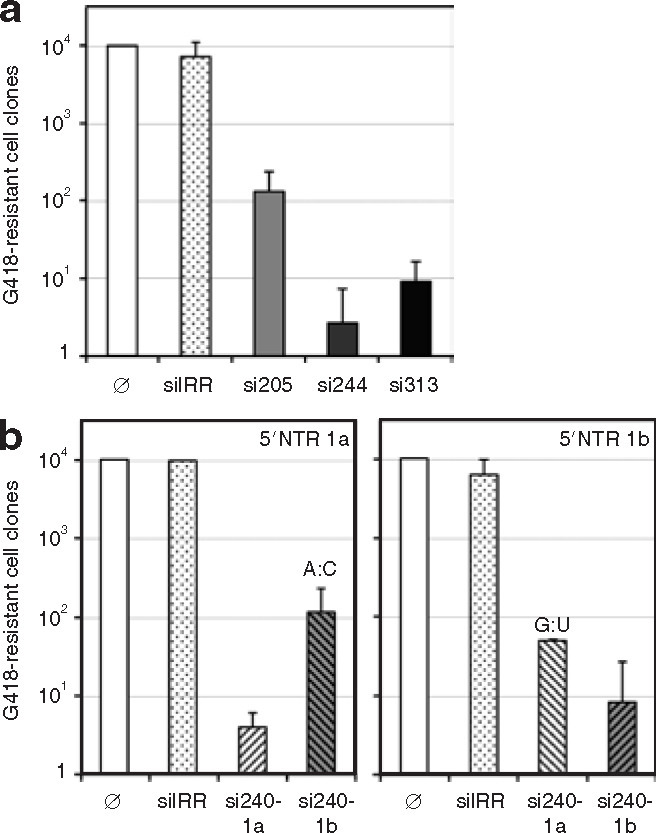
Small interfering RNAs (siRNAs) protect against establishment of hepatitis C virus (HCV) replicon-containing cell clones. (a) Numbers of G418-resistant 2-3c cell clones obtained 3 weeks after cotransfection of 5 μg of Ntat2ANeo or NNeo replicon and 2 μg of each indicated siRNA that is homologous to both HCV replicons (si205, si244, si313) or irrelevant siRNA (siIRR) are expressed relative to 10,000 resistant cell clones obtained after transfection of HCV replicon alone (Ø). Values (means ± SDs of independent transfections) are plotted on a logarithmic scale. (b) The effects of si240-1a or si240-1b treatment on replicon Ntat2ANeo (left) and NNeo (right) are represented separately. Mismatches present at position 18 of the siRNA antisense strand are indicated on top of the bars corresponding to heterologous targets. NTR, nontranslated region.
To look for potential emergence of siRNA escape mutants, several G418-resistant cell clones, obtained after a single treatment with each siRNA, were isolated and independently amplified. The 5′NTRs of the HCV replicon RNAs recovered from 3 to 13 cell clones for each siRNA were reverse-transcribed and PCR-amplified. The resulting PCR products were subjected to sequencing. No nucleotide substitution was found within or in the vicinity of the siRNA-targeted region of the HCV replicon RNAs. These data suggest that either a single siRNA treatment may not be sufficient to select for HCV replicon escape mutants in this system, or that replication competence does not tolerate nucleotide variation in this genomic region.
Inhibitory effect of siRNAs on virus replication in cell culture
We sought to determine whether the siRNAs si240-1a, si244, and si313, shown to be the most efficient in inhibiting the replication of HCV subgenomic replicons both transiently and under selective pressure, are also capable of efficiently blocking virus infection in cell culture. We utilized the JFH-1 strain of HCV genotype 2a that was shown to infect Huh7 and Huh7-derived cell lines and produce infectious particles.4 This in vitro infectious system therefore recapitulates the entire HCV life cycle. Sequences of si240-1a and si313 were perfectly homologous to the 5′NTR sequence of this genotype 2a strain. In contrast, si244 was shown to hybridize to the JFH-1 5′NTR with a C:A mismatch at the third position of the siRNA antisense strand (see Figure 1c). Such a mismatch at the 5′ end of the antisense strand is expected to hamper RNAi.29 , 33 For this reason, we also used an engineered chimeric virus that contains 5′NTR and core-coding sequences derived from genotype 1a within the backbone of the JFH-1 genome (JFH-1/C(+)6-1a; D. Delgrange, A. Pillez, L. Cocquerel, G. Paranhos-Baccala, Y. Rouillé, J. Dubuisson et al., unpublished results). The three siRNA sequences are homologous to the 5′NTR of this chimeric virus. In order to monitor the inhibitory effect of siRNAs on HCV infection, HuAP cells were electroporated with each siRNA and infected with either JFH-1 or the chimeric 1a/2a virus JFH-1/C(+)6-1a. Infected cells were probed by immunofluorescence with anti-core (Figure 5a ) or anti-E2 (data not shown) monoclonal antibodies. Most cells in the control cultures infected with JFH-1/C(+)6-1a (Figure 5a, left panel) or JFH-1 (Figure 5a, right panel) were antigen-positive. Non-specific siIRR had no detectable effect on HCV infection (Figure 5a). In contrast, HCV-specific siRNA si313 considerably reduced the number of cells infected with either JFH-1 or the chimeric virus (Figure 5a). Similarly, si244 efficiently inhibited infection with the 1a/2a chimeric virus (Figure 5a, left), but had only a weak effect, if any, on JFH-1 infection (Figure 5a, right), consistent with the existing mismatch between si244 and genotype 2a 5′NTR (see Figure 1c). Interestingly, si240-1a siRNA also appeared to have a higher inhibitory effect on the infection with the chimeric virus (Figure 5a, left) than with JFH-1 (Figure 5a, right), in spite of perfect homology with the 5′NTR sequences of both viruses. We hypothesized that these differential effects of si240-1a might be linked to delayed replication kinetics of the chimeric 1a/2a virus, as compared to JFH-1 (D. Delgrange, A. Pillez, L. Cocquerel, G. Paranhos-Baccala, Y. Rouillé, J. Dubuisson et al., unpublished results), a phenotype that may impact on target/siRNA ratios at early time-points following infection. The effects of the three siRNAs on both viruses were also monitored by Western blot analysis of E2 expression in infected cells (Figure 5b). The data obtained confirmed that si313 is the most efficient siRNA, causing complete inhibition of E2 expression in cells infected with either JFH-1 or JFH-1/C(+)6-1a.
Figure 5.

Hepatitis C virus (HCV) 5′NTR-specific small interfering RNAs (siRNAs) silence virus replication in cell culture. (a) HuAP cells were transfected with no siRNA (Ø), irrelevant siRNA (siIRR) or the indicated HCV-specific siRNA (si240-1a, si244, si313), and then infected at 16 hours after transfection with either JFH-1 strain of HCV genotype 2a (right panels) or a chimeric JFH-1 virus containing 5′NTR-C sequences from the H77 strain of HCV genotype 1a (JFH-1/C(+)6-1a, left panels). At 42 hours post-infection (p.i.), cells were fixed and processed for core detection by immunofluorescence and counterstained with Hoechst dye to visualize nuclei. Overlaid images (×10) are shown. Control cells that were mock-transfected and mock-infected (Mock) were processed in parallel. (b) Extracts from siRNA-transfected and HCV-infected cells collected at 42 hours p.i. were analyzed by Western blotting for expression of E2 glycoprotein (upper gel) and cellular β-actin (lower gel) as loading controls. (c) Virus yields in supernatants from siRNA-transfected and HCV-infected cells collected at 42 hours p.i. were quantified in genome equivalents/ml by real-time quantitative RT-PCR and expressed relative to virus production in the absence of siRNA treatment, set at 100% (mean ± SD of duplicates in two independent experiments). NTR, nontranslated region; RT-PCR, reverse transcription-polymerase chain reaction.
In order to verify whether virus production from siRNA-treated cells is reduced accordingly, viral particles were titrated in supernatants from cells transfected with each siRNA and infected with either JFH-1 or the chimeric virus, both by real-time quantitative RT-PCR (genome equivalents, Figure 5c) and by determination of infectious focus-forming units (Table 2 ). For each of the siRNAs, quantification of virus production in siRNA-treated cultures (Figure 5c, Table 2) was in good agreement with core and E2 intracellular expression levels (Figure 5a and b). In particular, supernatants from si313-treated, HCV-infected cells exhibited a >92% reduction in genome equivalents/ml titer and a >96% reduction in focus-forming units/ml infectious titer, as compared to mock-treated HCV-infected cells. Taken together, these data demonstrate that HCV 5′NTR-specific siRNAs are capable of strongly inhibiting virus production in cell culture.
Table 2.
Effect of siRNAs on hepatitis C virus production
| Virus productiona | ||
|---|---|---|
| siRNA | JFH-1 | JFH-1/C(+)6-1a-1/C(+)6-1a |
| Ø | 8.4 × 103 (100) | 7.1 × 103 (100) |
| siIRR | 7.8 × 103 (93) | 6.6 × 103 (93) |
| si240-1a | 2.9 × 103 (35) | 1.0 × 103 (14) |
| si244 | 5.1 × 103 (61) | 1.1 × 103 (15) |
| si313 | 1.5 × 102 (2) | 3.0 × 102 (4) |
Abbreviations: siIRR, irrelevant siRNA; siRNA, small interfering RNA.
Viral titers in the supernatants of HuAP cells transfected with each siRNA and infected with the indicated virus are expressed in focus forming units/ml, as well as, in parentheses (%) relative to virus production from mock-transfected cultures (Ø) set at 100%.
Discussion
In exploring RNAi as a potential new therapeutic approach against HCV infection, one of the reasons for our choice to target domain III of the HCV 5′NTR was that domain III contains well-conserved nucleotide sequences across all HCV genotypes (Table 1).34 The high degree of conservation seen in these sequences may be required for preserving virus function, since this 5′NTR domain controls the initiation of polyprotein translation and modulates RNA replication.23 , 24 Recent nuclear magnetic resonance and electron cryomicroscopy studies of domain III22 , 35 , 36 have helped identify the structure–function relationships of the various subdomains of domain III. The results of these studies suggest that subdomains IIIa/c and IIId, as well as subdomain IIIf that forms a pseudoknot structure (Figure 1a), directly contact the 40S subunit body and act synergistically for the proper positioning of the AUG codon. In the present study, si240 and si244 (that hybridize to subdomain IIId) and si313 (that essentially hybridizes to the IIIf pseudoknot), therefore target regions that are essential for IRES structural integrity and functioning. These three siRNAs proved able, at low doses in the nanomolar range, to substantially reduce or even abolish HCV subgenomic RNA replication, as measured by reporter SEAP activities and G418-resistant cell clone formation (Figures 2 and 4), and resulted in the elimination of HCV replicons from treated cells (Figure 2c). Importantly, we also demonstrated that these siRNAs, particularly si313, considerably limit HCV infection in cell culture. We did this using two HCV strains that can be propagated in Huh7-derived cells: (i) the JFH-1 strain of HCV genotype 2a,4 and (ii) a chimeric derivative of JFH-1 carrying 5′NTR and core sequences of HCV genotype 1a (D. Delgrange, A. Pillez, L. Cocquerel, G. Paranhos-Baccala, Y. Rouillé, J. Dubuisson et al., unpublished results). Our data contrast with those obtained by others who concluded that domains II17 and III17 , 19 of HCV 5′NTR are relatively resistant to siRNA, because there is reduced accessibility to these domains, given their association with different proteins and factors involved in translation. Our data indicate that regions involved in complex secondary and tertiary structures should not be disregarded as targets for RNAi. The data also underscore the importance of siRNA design.25 , 26 In contrast, two of the siRNAs tested in the present study (si214 and si284, targeting domains IIIc and IIIe, respectively) exhibited poor or only transient inhibitory effect on HCV RNA replication (Figure 2a). Interestingly, two other studies reported that siRNAs having sequences identical to that of si284 showed relative discrepancies in their effects in cells supporting autonomous replication of HCV subgenomic replicon.13 , 19 These conflicting results may be explained by the different strategies used for siRNA synthesis and/or delivery. In addition, the sensitivity of the replicon systems used may have an impact on the siRNA efficiencies reported.
We observed some differences in siRNA efficiencies between: (i) transient replication systems in which viral target RNA and siRNA were co-introduced into cells (Figures 2 and 4), and (ii) stable replication systems in which siRNA was introduced in cells supporting ongoing replication of the viral target, whether dealing with a subgenomic replicon (Figure 3) or genome-length infectious RNA (Figure 5, Table 2). Two micrograms of siRNA did not inhibit viral RNA replication in the stable subgenomic replicon system as efficiently as in cotransfection experiments (compare Figures 2 and 4). In addition, si240-1a and si244 caused elimination of viral RNA upon cotransfection with subgenomic replicon (Figure 2a), whereas they substantially reduced, but did not abolish the production of virus particles (Figure 5, Table 2). This was in spite of the fact that the molar ratio of siRNA to targeted HCV RNA was higher in cells stably containing HCV replicons or infected with virus than in cells cotransfected with replicon and siRNA. In another study, high doses (4,000 pmol) of siRNAs proved necessary in order to strongly inhibit HCV RNA replication in subgenomic replicon-containing cells.18 We speculate that viral RNA templates engaged in the replicase complex and nascent RNAs are probably not as accessible to the RNAi machinery as transfected viral RNAs are. Our data can also be explained in part by the fact that a proportion of cells in the culture were infected or supported ongoing viral RNA replication but did not receive siRNA, given that the efficiency of electroporation is ∼70% in En5-3 and 2-3c cells (data not shown).
The specificity of HCV siRNAs was demontrated by studying their dose-effect responses (Figure 2b), as well as by using si240 to target the replicon 5′NTR from two HCV genotypes (1a and 1b; Figures 2–4) and si244 to target the 5′NTR of genotype 1a or 2a virus strains (Figure 5, Table 2). When used with heterologous targets, the siRNAs si240-1a and si240-1b exhibited a noncanonical wobble base-pairing and a mismatch at the 3′ end (position 18) of the siRNA antisense strand, respectively (Figure 1c). For both 1a and 1b replicons, perfect base-pairing was preferred for maximum RNAi efficacy, but a G:U wobble and an A:C mismatch were both tolerated (Figure 4). This is consistent with the fact that wobble base-pairing is known to be well tolerated in double-stranded RNA helices, providing stability similar to a Watson–Crick base-pairing. In agreement with our data, wobble base-pairs, especially when located at the 3′ end of the siRNA antisense strand, were recently suggested to have no disruptive effect on RNAi.28 , 29 , 33 More surprisingly, but also consistent with our data, it was reported that an A:C mismatch was not deleterious in double-stranded RNA structures.29 Contrasting with these tolerated mismatches between siRNA and viral target RNA, we found that a C:A mismatch at the 5′ end of the siRNA antisense strand strongly reduces the inhibitory effect of si244 on JFH-1 infection in cell culture (Figure 5, Table 2), and this is consistent with the RNAi mechanism.29 , 37 Taken together, these data provide strong evidence against the involvement of a spurious interferon-mediated mechanism of inhibition in the suppression of viral replication we observed.
The sequence of RNA viruses, such as HCV, is known to evolve continuously, resulting in the production of many quasi-species, because of the high error rate of RNA-dependent RNA polymerases with no proof-reading activity. This property allows RNA viruses to escape rapidly to a selective pressure such as antiviral treatments, when sequence changes are compatible with virus functions. This holds true for anti-protease and anti-polymerase compounds that are currently under development for HCV treatment.38 , 39 However, we did not observe, after a single siRNA treatment, the emergence of escape mutations within siRNA-targeted genomic regions in RNA extracted from several G418-resistant cell clones. In other studies, siRNA-resistant viral mutants harboring single nucleotide substitutions or deletions within or at the vicinity of the siRNA-targeted sequence were reported to have emerged in cells expressing constitutively an shRNA (in the case of human immunodeficiency virus infections),40 , 41 and after repeated treatments with an siRNA targeting the polymerase coding sequence (in the case of HCV subgenomic replicons).16 The lack of escape mutations observed in our study could either be due to the fact that a single siRNA was used in the treatment, or to the fact that siRNAs target highly conserved, functionally important genomic sequences, in which substitutions would not be compatible with translation/replication competence. Additional studies will be needed in order to determine which of these hypotheses holds true.
From the point of view of evaluating the potency of siRNA-based antiviral strategies to eradicate persistent infections, it was recently shown that siRNAs may be used to cure in vitro persistent infections caused by RNA viruses such as poliovirus or lymphocytic choriomeningitis virus.30 , 42 For assaying such a therapeutic strategy in animal models, synthetic siRNAs need to be further chemically stabilized and formulated to be efficiently delivered. Successful utilization of stabilized, lipid-encapsulated siRNAs was reported in a mouse model of hepatitis B virus replication.43 In addition, organ or cell-type specific delivery of siRNAs has been achieved through siRNA binding to cholesterol44 or to antibodies directed against cell surface antigens.45 Synthetic shRNAs also showed a more prolonged inhibitory effect in murine liverthan siRNAs did. This was demonstrated upon co-delivery of shRNA or siRNA targeting domain IV of the HCV IRES and plasmid DNA encoding a reporter protein placed under the translational control of the HCV IRES.46 Alternatively, optimization of siRNA delivery may rely on the use of viral vectors that allow continuous synthesis of shRNAs in cells, thereby leading to sustained RNAi.31 , 47 It is crucial, however, to control intracellular shRNA production levels, since it has recently been reported that constant, high expression of shRNAs in the liver could be lethal in mouse models.48 Nevertherless, controlled doses of siRNA or expression of shRNA have already been shown to be efficient and safe in several animal models of viral infection, including in a mouse model of hepatitis B virus infection,47 a rhesus macaque model of severe acute respiratory syndrome-associated coronavirus infection,49 and mouse models of West Nile virus and Japanese encephalitis virus infections.31
In the present study, we have identified promising anti-HCV siRNAs (si313 and, to a lesser extent, si244 and si240) that target highly conserved sequences in domain III of the 5′NTR and efficiently silence HCV infection in cell culture. It has now become possible to monitor the effect of these siRNAs in vivo using a tamarin/marmoset primate model of infection with a chimeric GB virus B-containing HCV domain III of the HCV IRES, that we have previously described.8
During revision of this manuscript, Kanda et al.50 reported that delivery of an HCV 5′NTR-specific shRNA to hepatoma cell lines infected by HCV resulted in the reduction of viral production.
Materials and Methods
Cells and viruses. Hepatoma cell lines, 2-3c,32 En5-3,21 and HuAP are derived from the hepatocarcinoma cell line Huh7, and were cultured in Dulbecco's modified Eagle medium (Invitrogen, Cergy-Pontoise, France) supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin. En5-3 cells stably express SEAP under the control of the human immunodeficiency virus long terminal repeat promoter,21 and were cultured in the presence of 2 μg/ml blasticidin (InvivoGen, Toulouse, France). En5-3 cells supporting autonomous replication of Ntat2ANeo HCV replicon were cultured in the presence of 2 μg/ml blasticidin and 0.5 mg/ml geneticin (G418, Invitrogen, Cergy-Pontoise, France). Genotype 2a JFH-1 and chimeric JFH-1/C(+)6-1a virus stocks were generated by transfection of HuAP cells with corresponding in vitro transcribed genomic RNAs. Plasmid pJFH-1 containing the genome-length complementary DNA (cDNA) of the JFH-1 isolate of HCV genotype 2a (GenBank accession no. AB047639, ref. 4) was generously provided by T. Wakita. Plasmid pJFH-1/C(+)6-1a was derived from pJFH-1 by substituting nucleotides 154–341 of the 5′NTR, as well as the capsid coding sequence by corresponding sequences of the H77 strain of genotype 1a (D. Delgrange, A. Pillez, L. Cocquerel, G. Paranhos-Baccala, Y. Rouillé, J. Dubuisson et al., will be described elsewhere). Plasmids pJFH-1 and pJFH-1/C(+)6-1a were linearized with XbaI and treated with Mung Bean nuclease prior to in vitro transcription using MEGAscript T7 kit (Ambion, Courtaboeuf, France). HuAP cells were electroporated with in vitro transcribed RNA, as previously described,51 and supernatants collected 8–10 days after transfection were used to re-infect naïve HuAP cells. Supernatants collected at 10 days after infection were used as virus stocks and stored at −80°C.
Design and synthesis of siRNAs. siRNAs targeting HCV IRES were designed using previously described criteria.25 , 26 The sequence of the sense-strand of each siRNA selected is shown in Table 1. siRNAs are referred to by the nucleotide position within the genome of the H77 strain of HCV genotype 1a27 that corresponds to the first nucleotide targeted by the siRNA antisense strand. si240 was synthesized with a sequence homologous to HCV subtype 1a or 1b, i.e., containing a U (si240-1a) or a C (si240-1b) residue at position 18 of the siRNA antisense strand. An irrelevant siRNA, named siIRR, targets the 5′NTR of the Sabin strain of poliovirus type 330 and was used as negative control in this study. The sequences of all siRNAs were compared with known genes by using basic local alignment search tool within the GenBank database, and no significant homology to other genes was found. The two strands of each siRNA were chemically synthesized at the Institut Pasteur (“Plate-forme 7”) and annealed at a final concentration of 500 pmol/μl, as previously described.25 The quality and quantity of hybridized siRNAs were analyzed by electrophoresis on nondenaturing 3% agarose gels.
HCV subgenomic replicons and in vitro transcriptions. Two dicistronic, subgenomic HCV replicons that encode a selectable reporter gene, neomycine phosphotransferase (Neo) were used in this study (see Figure 1). These replicon cDNAs, kindly provided by S.M. Lemon, are inserted downstream of the T7 RNA polymerase promoter and have been previously described as pNNeo/3-5B20 and pNtat2Aneo.21 We will refer to these replicons as NNeo and Ntat2ANeo, respectively, in this study. NNeo and Ntat2ANeo each carries a cell culture adaptive mutation, R2889G and S2005I, respectively. Both replicon cDNAs are derived from the cDNA of the N strain of genotype 1b HCV with the exception of the 5′NTR sequence of the Ntat2ANeo, which is derived from the H77 strain of HCV genotype 1a. Ntat2ANeo RNA encodes human immunodeficiency virus Tat protein fused to the 2A protease of foot-and-mouth disease virus, followed by Neo.21 Upon transfection of En5-3 cells with Ntat2ANeo replicon, the expression of Tat-2A drives the synthesis of secreted SEAP.21 cDNAs bearing a 30-nucleotide deletion, including the three codons (GDD) of the active site of the RNA polymerase NS5B within the backbone of NNeo and Ntat2ANeo,20 , 21 and referred to as ΔGDD, were used as replication-deficient RNAs. Plasmid DNAs were linearized with XbaI prior to in vitro transcription using MEGAscript T7 kit. The DNA template was removed by treatment with TURBO DNase (Ambion, Courtaboeuf, France) and RNAs were purified by phenol-chloroform extractions and precipitated with ethanol. The quality and quantity of replicon RNAs were analyzed by electrophoresis on a non-denaturing 0.8% agarose gel and by optical density measurements.
Cell transfections. En5-3 or 2-3c cells (2 × 106) were electroporated with 5 μg replicon RNA, or coelectroporated with 5 μg replicon RNA and various doses of each siRNA, in a 4 mm-gap width cuvette by applying one pulse at 240 V, 900 μF (EasyjecT Plus, EquiBio, Kent, UK). Cells were resuspended in complete medium immediately after the pulse and seeded at various concentrations. En5-3 cells supporting autonomous replication of Ntat2ANeo replicon were similarly transfected by electroporation with each siRNA.
Measurement of SEAP activity. En5-3 transfected cells (4 × 105) were seeded in 6-well plates and supernatants were collected at 1, 2, 3, 4, and 7 days after electroporation and replaced with fresh medium. SEAP activity was measured in supernatant aliquots with the Phospha-Light System (Applied Biosystems/Tropix, Courtaboeuf, France) using a luminescent substrate according to the manufacturer's recommendations. Luminescent signals were read for 1 second using a Lumat LB 9507 luminometer (Berthold Technologies, Thoiry, France).
G418-resistant cell clone selection. To select for G418-resistant cell clones, variable fractions of 2-3c transfected cells were seeded in 100 mm-diameter dishes and supplemented with NNeo-ΔGDD or Ntat2ANeo-ΔGDD transfected cells to adjust the final number of cells to 5 × 105 cells per dish. Twenty-four to forty-eight hours following transfection, cells were placed under the selective pressure of 0.5 mg/ml geneticin (G418, Invitrogen, Cergy-Pontoise, France) for 3 weeks, with the medium being replaced twice a week. G418-resistant cell clones supporting viral RNA replication were fixed and stained with a 0.1% crystal violet solution, or selected and expanded under selective pressure for viral RNA sequencing.
Viral RNA sequencing. Total RNA was extracted from expanded siRNA-resistant clones with TRIzol reagent (Invitrogen, Cergy-Pontoise, France). Viral RNA was reverse transcribed and amplified with HCV specific primers designed to generate PCR products spanning nucleotides 100–451, using the SuperScript One-Step RT-PCR kit with Platinum Taq (Invitrogen, Cergy-Pontoise, France). Sequencing reactions were carried out on uncloned RT-PCR products using Big Dye Terminator version 1.1 kit (Applied Biosystems, Courtaboeuf, France) and analyzed on a ABI 3700 capillary DNA sequencer (Applied Biosystems, Courtaboeuf, France).
Detection of viral RNA by semi-quantitative RT-PCR. En 5-3 transfected cells (2 × 105) were seeded in 6-well plates (duplicate wells per transfection) and cultured for 4 days, then trypsinized and passaged at a 1:5 dilution in new 6-well plates. On day 10 after transfection, total RNA was extracted independently from duplicate wells using RNeasy mini kit (Qiagen, Courtaboeuf, France). Five micrograms of total cellular RNA were heat-denatured at 65°C for 5 minutes and used as template for reverse transcription with 50 U of Superscript II reverse transcriptase (Invitrogen, Cergy-Pontoise, France) and 250 ng of random hexanucleotide primers (Roche, Meylan, France) for 50 minutes at 42°C. The resulting cDNAs were treated with 2 U of RNaseH (Invitrogen, Cergy-Pontoise, France) for 30 minutes at 37°C, and purified using QIAquick PCR purification kit (Qiagen, Courtaboeuf, France). One fifth of the cDNA was used for programming PCRs with either HCV-specific primers that allowed amplification of a fragment spanning nucleotides 3457–5085 of the N strain of HCV genotype 1b, or primers specific to cellular housekeeping gene glyceraldehyde-3-phosphate dehydrogenase, so as to normalize for total RNA content. PCRs were performed using Platinum Taq DNA polymerase (Invitrogen, Cergy-Pontoise, France) under the following cycling conditions: 1 cycle at 94°C for 3 minutes, followed by 35 cycles of: (i) 30 seconds at 94°C; (ii) 30 seconds at 55°C; and (iii) 1.5 minutes at 72°C. PCR products were analyzed by electrophoresis on 1% agarose gels.
Infection with HCV and indirect immunofluorescence analysis. HuAP cells (4 × 106) were electroporated with 2 μg of siRNA, and 5 × 104 electroporated cells were seeded on coverslips in 24-well plates and subsequently infected at 16 hours after transfection with JFH-1 or JFH-1/C(+)6-1a virus stocks at ∼1 focus forming unit per cell. After 2 hours of incubation at 37°C, virus inoculum was washed off and cells were fed with culture medium. At 42 hours after infection, cells were fixed and processed for core detection by indirect immunofluorescence, as previously described,51 using anti-core monoclonal antibody ACAP27 (kindly provided by J.F. Delagneau, Bio-Rad, Marnes-la-Coquette, France). Cells were counterstained with Hoechst dye to enable nuclei to be detected. For titration of infectious viral particles, 5 × 104 HuAP cells were infected with 1:10 dilutions of supernatants from cells transfected with siRNAs and infected with HCV. The foci of infected cells were detected by immunofluorescence with anti-core monoclonal antibody at 72 hours after infection and counted to determine titers in focus forming units/ml.
Immunoblot analysis of HCV-infected cells. HuAP cells (4 × 106) seeded in 24-well plates were lyzed in 50 mmol/l Tris–HCl (pH 7.5), 150 mmol/l NaCl, 5 mmol/l EDTA, and 0.5% (vol/vol) Igepal buffer containing a mixture of protease inhibitors (Complete, Roche, Meylan, France) and processed for E2 or β-actin detection by immunoblot analysis as previously described,51 using anti-E2 monoclonal antibody 3/1152 or anti-β-actin monoclonal antibody (Sigma-Aldrich, Saint Quentin Fallavier, France), respectively.
Quantification of virus production by quantitative RT-PCR. Viral RNAs were isolated from cell culture supernatants using QIAamp Viral RNA kit (Qiagen, Courtaboeuf, France) and quantified by real-time quantitative RT-PCR using primer pair and probe targeting a sequence spanning nucleotides 130–290 within the HCV 5′NTR : FP 5′-CGGGAGAGCCATAGTGG-3′; RP 5′-AGTACCACAAGGCCTTTCG-3′; Probe 5′-FAM-CTGCGGAACCGGTGAGTACAC-TAMRA-3′. Assays were performed using TaqMan One-Step RT-PCR Master Mix Reagents kit and an ABI PRISM 7700 Sequence Detector instrument (Applied Biosystems, Courtaboeuf, France), according to the manufacturer's instructions.
Acknowledgments
This work was supported in part by grants from the Institut Pasteur (Direction des Applications de la Recherche et des Relations Industrielles), the French National Agency for Research on AIDS and Viral Hepatitis (ANRS), and the National Institutes of Health, USA (R24-RR15081). C.C. was the recipient of a fellowship from the Foundation Pasteur-Weizmann. A.S. was the recipient of a PhD fellowship from the French Ministry of Research (MENESR), then a post-doctoral fellowship from the ANRS. The authors gratefully acknowledge André Pillez (Institut de Biologie de Lille, Lille, France) for technical assistance, Catherine Gouyette (Plate-forme 7, Institut Pasteur, Paris, France) for the synthesis of siRNAs, MinKyung Yi, Masanori Ikeda and Stanley M. Lemon (University of Texas Medical Branch, Galveston, TX) for the gift of hepatitis C virus (HCV) replicons and cells, Jean-François Delagneau (Bio-Rad, Marnes-la-Coquette, France) and Jane McKeating (University of Birmingham Medical School, Birmingham, UK) for the gift of HCV antibodies, and Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for the gift of JFH-1 complementary DNA. We are grateful to Stanley Lemon for critical reading of the manuscript, and to Sylvie van der Werf (Institut Pasteur, Paris, France) for continuous support.
Footnotes
published online 15 May 2007
References
- 1.Bartenschlager R, Frese M, Pietschmann T. Novel insights into hepatitis C virus replication and persistence. Adv Virus Res. 2004;63:71–180. doi: 10.1016/S0065-3527(04)63002-8. [DOI] [PubMed] [Google Scholar]
- 2.Rijnbrand RC, Lemon SM. Internal ribosome entry site-mediated translation in hepatitis C virus replication. Curr Top Microbiol Immunol. 2000;242:85–116. doi: 10.1007/978-3-642-59605-6_5. [DOI] [PubMed] [Google Scholar]
- 3.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 4.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A. Hepatitis C virus replication in mice with chimeric human livers. Nat Med. 2001;7:927–933. doi: 10.1038/90968. [DOI] [PubMed] [Google Scholar]
- 6.Lanford RE, Chavez D, Notvall L, Brasky KM. Comparison of tamarins and marmosets as hosts for GBV-B infections and the effect of immunosuppression on duration of viremia. Virology. 2003;311:72–80. doi: 10.1016/s0042-6822(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 7.Martin A, Bodola F, Sangar DV, Goettge K, Popov V, Rijnbrand R. Chronic hepatitis associated with GB virus B persistence in a tamarin after intrahepatic inoculation of synthetic viral RNA. Proc Natl Acad Sci USA. 2003;100:9962–9967. doi: 10.1073/pnas.1731505100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rijnbrand R, Yang Y, Beales L, Bodola F, Goettge K, Cohen L. A chimeric GB virus B with 5′ nontranslated RNA sequence from hepatitis C virus causes hepatitis in tamarins. Hepatology. 2005;41:986–994. doi: 10.1002/hep.20656. [DOI] [PubMed] [Google Scholar]
- 9.Dykxhoorn DM, Lieberman J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu Rev Med. 2005;56:401–423. doi: 10.1146/annurev.med.56.082103.104606. [DOI] [PubMed] [Google Scholar]
- 10.Colbère-Garapin F, Blondel B, Saulnier A, Pelletier I, Labadie K. Silencing viruses by RNA interference. Microbes Infect. 2005;7:767–775. doi: 10.1016/j.micinf.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonard JN, Schaffer DV. Antiviral RNAi therapy: emerging approaches for hitting a moving target. Gene Ther. 2006;13:532–540. doi: 10.1038/sj.gt.3302645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grimm D, Kay MA. Therapeutic short hairpin RNA expression in the liver: viral targets and vectors. Gene Ther. 2006;13:563–575. doi: 10.1038/sj.gt.3302727. [DOI] [PubMed] [Google Scholar]
- 13.Seo MY, Abrignani S, Houghton M, Han JH. Small interfering RNA-mediated inhibition of hepatitis C virus replication in the human hepatoma cell line Huh-7. J Virol. 2003;77:810–812. doi: 10.1128/JVI.77.1.810-812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapadia SB, Brideau-Andersen A, Chisari FV. Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc Natl Acad Sci USA. 2003;100:2014–2018. doi: 10.1073/pnas.252783999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokota T, Sakamoto N, Enomoto N, Tanabe Y, Miyagishi M, Maekawa S. Inhibition of intracellular hepatitis C virus replication by synthetic and vector-derived small interfering RNAs. EMBO Rep. 2003;4:602–608. doi: 10.1038/sj.embor.embor840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson JA, Richardson CD. Hepatitis C virus replicons escape RNA interference induced by a short interfering RNA directed against the NS5b coding region. J Virol. 2005;79:7050–7058. doi: 10.1128/JVI.79.11.7050-7058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krönke J, Kittler R, Buchholz F, Windisch MP, Pietschmann T, Bartenschlager R. Alternative approaches for efficient inhibition of hepatitis C virus RNA replication by small interfering RNAs. J Virol. 2004;78:3436–3446. doi: 10.1128/JVI.78.7.3436-3446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Randall G, Grakoui A, Rice CM. Clearance of replicating hepatitis C virus replicon RNAs in cell culture by small interfering RNAs. Proc Natl Acad Sci USA. 2003;100:235–240. doi: 10.1073/pnas.0235524100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takigawa Y, Nagano-Fujii M, Deng L, Hidajat R, Tanaka M, Mizuta H. Suppression of hepatitis C virus replicon by RNA interference directed against the NS3 and NS5B regions of the viral genome. Microbiol Immunol. 2004;48:591–598. doi: 10.1111/j.1348-0421.2004.tb03556.x. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda M, Yi M, Li K, Lemon SM. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J Virol. 2002;76:2997–3006. doi: 10.1128/JVI.76.6.2997-3006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi M, Bodola F, Lemon SM. Subgenomic hepatitis C virus replicons inducing expression of a secreted enzymatic reporter protein. Virology. 2002;304:197–210. doi: 10.1006/viro.2002.1652. [DOI] [PubMed] [Google Scholar]
- 22.Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science. 2001;291:1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- 23.Kieft JS, Zhou K, Jubin R, Doudna JA. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA. 2001;7:194–206. doi: 10.1017/s1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friebe P, Lohmann V, Krieger N, Bartenschlager R. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J Virol. 2001;75:12047–12057. doi: 10.1128/JVI.75.24.12047-12057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elbashir SM, Harborth J, Weber K, Tuschl T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods. 2002;26:199–213. doi: 10.1016/S1046-2023(02)00023-3. [DOI] [PubMed] [Google Scholar]
- 26.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 27.Yanagi M, Purcell RH, Emerson SU, Bukh J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci USA. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du Q, Thonberg H, Wang J, Wahlestedt C, Liang Z. A systematic analysis of the silencing effects of an active siRNA at all single-nucleotide mismatched target sites. Nucleic Acids Res. 2005;33:1671–1677. doi: 10.1093/nar/gki312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saulnier A, Pelletier I, Labadie K, Colbère-Garapin F. Complete cure of persistent virus infections by antiviral siRNAs. Mol Ther. 2006;13:142–150. doi: 10.1016/j.ymthe.2005.07.697. [DOI] [PubMed] [Google Scholar]
- 31.Kumar P, Lee SK, Shankar P, Manjunath N. A single siRNA suppresses fatal encephalitis induced by two different flaviviruses. PLoS Med. 2006;3:e96. doi: 10.1371/journal.pmed.0030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scholle F, Li K, Bodola F, Ikeda M, Luxon BA, Lemon SM. Virus-host cell interactions during hepatitis C virus RNA replication: impact of polyprotein expression on the cellular transcriptome and cell cycle association with viral RNA synthesis. J Virol. 2004;78:1513–1524. doi: 10.1128/JVI.78.3.1513-1524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez J, Tuschl T. RISC is a 5′ phosphomonoester-producing RNA endonuclease. Genes Dev. 2004;18:975–980. doi: 10.1101/gad.1187904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simmonds P. Genetic diversity and evolution of hepatitis C virus—15 years on. J Gen Virol. 2004;85:3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 35.Lukavsky PJ, Otto GA, Lancaster AM, Sarnow P, Puglisi JD. Structures of two RNA domains essential for hepatitis C virus internal ribosome entry site function. Nat Struct Biol. 2000;7:1105–1110. doi: 10.1038/81951. [DOI] [PubMed] [Google Scholar]
- 36.Boehringer D, Thermann R, Ostareck-Lederer A, Lewis JD, Stark H. Structure of the hepatitis C Virus IRES bound to the human 80S ribosome: remodeling of the HCV IRES. Structure. 2005;13:1695–1706. doi: 10.1016/j.str.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kneteman NM, Weiner AJ, O'Connell J, Collett M, Gao T, Aukerman L. Anti-HCV therapies in chimeric scid-Alb/uPA mice parallel outcomes in human clinical application. Hepatology. 2006;43:1346–1353. doi: 10.1002/hep.21209. [DOI] [PubMed] [Google Scholar]
- 39.Mo H, Lu L, Pilot-Matias T, Pithawalla R, Mondal R, Masse S. Mutations conferring resistance to a hepatitis C virus (HCV) RNA-dependent RNA polymerase inhibitor alone or in combination with an HCV serine protease inhibitor in vitro. Antimicrob Agents Chemother. 2005;49:4305–4314. doi: 10.1128/AAC.49.10.4305-4314.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das AT, Brummelkamp TR, Westerhout EM, Vink M, Madiredjo M, Bernards R. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005;33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez AB, Perez M, Cornu T, de la Torre JC. RNA interference-mediated virus clearance from cells both acutely and chronically infected with the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol. 2005;79:11071–11081. doi: 10.1128/JVI.79.17.11071-11081.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol. 2005;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- 44.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 45.Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 46.Wang Q, Contag CH, Ilves H, Johnston BH, Kaspar RL. Small hairpin RNAs efficiently inhibit hepatitis C IRES-mediated gene expression in human tissue culture cells and a mouse model. Mol Ther. 2005;12:562–568. doi: 10.1016/j.ymthe.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 47.Uprichard SL, Boyd B, Althage A, Chisari FV. Clearance of hepatitis B virus from the liver of transgenic mice by short hairpin RNAs. Proc Natl Acad Sci USA. 2005;102:773–778. doi: 10.1073/pnas.0409028102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 49.Li BJ, Tang Q, Cheng D, Qin C, Xie FY, Wei Q. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat Med. 2005;11:944–951. doi: 10.1038/nm1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanda T, Steele R, Ray R, Ray RB. Small interfering RNA targeted to hepatitis C virus 5′ nontranslated region exerts potent antiviral effect. J Virol. 2007;81:669–676. doi: 10.1128/JVI.01496-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rouillé Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol. 2006;80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flint M, Thomas JM, Maidens CM, Shotton C, Levy S, Barclay WS. Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J Virol. 1999;73:6782–6790. doi: 10.1128/jvi.73.8.6782-6790.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]