

Abstract
The nsp1 protein of the highly pathogenic SARS coronavirus suppresses host protein synthesis, including genes involved in the innate immune system. A bioinformatic analysis revealed that the nsp1 proteins of group I and SARS coronaviruses have similar structures. Nsp1 proteins of group I coronaviruses interacted with host ribosomal 40S subunit and did not inhibit IRF-3 activation. However, synthesis of host immune and non-immune proteins was inhibited by nsp1 proteins at both transcriptional and translational levels, similar to SARS coronavirus nsp1. These results indicate that different coronaviruses might employ the same nsp1 mechanism to antagonize host innate immunity and cell proliferation. However, nsp1 may not be the key determinant of viral pathogenicity, or the factor used by the SARS coronavirus to evade host innate immunity.
Keywords: Group I coronavirus, nsp1, Innate immunity
1. Introduction
Viral infections are sensed by the host innate immune system through Toll-like receptors and retinoic acid-inducible gene-I-like receptors, which trigger innate immune signal transduction, leading to production of type I interferons (IFNs), and hundreds of proinflammatory cytokines that suppress viral spread (Kawai and Akira, 2008). In return, viruses have evolved at least three mechanisms to evade or antagonize host innate immunity. One is blocking IFN induction. Host innate immunity is initiated by recognition of cellular RNA sensors and viral RNA, which in turn interacts with the adaptor IPS-1 to activate the IFN transcription factor IRF-3 and NF-kB through kinases TBK-1/IKKi and IKKα/β (Kawai and Akira, 2008). Many viruses encode proteins to block the IFN induction pathway. Examples include the influenza A virus NS1 protein (Lu et al., 1995, Garcia-Sastre, 2001), the reovirus sigma3 protein (Jacobs and Langland, 1998), the Ebola virus VP35 protein (Cardenas et al., 2006), the poxvirus E3L protein (Xiang et al., 2002), the herpes simplex virus US11 protein (Poppers et al., 2000) and the murine cytomegalovirus m142 and m143 proteins (Valchanova et al., 2006). The second mechanism is interfering with IFN-activated signaling, mainly through interacting with the Janus kinases JAK-1, TYK-2, STAT-1 and STAT-2 to block the JAK-STAT signaling pathway. Examples include the Ebola virus VP24 protein (Reid et al., 2006), the paramyxovirus C and V proteins (Didcock et al., 1999, Gotoh et al., 2003), and the rabies virus P protein (Brzozka et al., 2006). The third method is inhibiting the specific antiviral proteins that mediate the antiviral state. Viral dsRNA-binding proteins are mostly studied for their capability of preventing activation of PKR or the 2–5 OAS/RNaseL system, as demonstrated by the reovirus sigma3 (Imani and Jacobs, 1988), the herpesvirus US11 protein (Poppers et al., 2000), the poxvirus E3L (Langland and Jacobs, 2004), the cytomegaloviruses dsRNA-binding proteins (Hakki and Geballe, 2005), and the influenza virus NS1 protein (Min and Krug, 2006).
Coronaviruses are important human and animal pathogens that are divided into three groups based on serological criteria. The group II coronaviruses severe acute respiratory syndrome coronavirus (SARS-CoV) and mouse hepatitis coronavirus (MHV) encode a number of proteins that antagonize host innate immunity. The ORF 3b and ORF 6 proteins inhibit both IFN synthesis and signaling (Kopecky-Bromberg et al., 2007). The nucleocapsid protein inhibits NFkB promoter and IFN synthesis (Kopecky-Bromberg et al., 2007, Ye et al., 2007). The papain-like protease interacts with IRF-3 and inhibits its phosphorylation and nuclear translocation (Devaraj et al., 2007). The 3a protein causes endoplasmic reticulum stress, and antagonizes IFN responses and innate immunity (Minakshi et al., 2009). Finally, the M protein associates with RIG-I, TBK1, IKKepsilon, and TRAF3 to inhibit the activation of IRF-3/IRF-7 transcription factors (Siu et al., 2009). Nsp1 is another intensively studied viral protein. The nsp1 proteins of SARS-CoV and MHV promote host mRNA degradation and suppress host gene expression (Kamitani et al., 2006, Zust et al., 2007, Narayanan et al., 2008). Nuclear magnetic resonance (NMR) analysis revealed that SARS-CoV nsp1 has a novel β-barrel structure mixed with α-helixes (Almeida et al., 2007). More recently, SARS-CoV nsp1 was found to block host translational machinery function by binding to the ribosome small subunit (Kamitani et al., 2009). These studies suggest that coronavirus nsp1 is a major virulence and pathogenicity factor (Kamitani et al., 2006, Wathelet et al., 2007, Zust et al., 2007).
Little is known about the biological function of group I coronavirus nsp1 proteins. In this study, we conducted a comparative study of nsp1 proteins from groups I and II coronaviruses. We compared models for nsp1 proteins of HCoV-229E and HCoV-NL63 with models of SARS-CoV. We analyzed the interaction between nsp1 proteins and cellular proteins by co-immunoprecipitation (Co-IP). Finally, we systematically investigated the mechanism by which group I coronavirus nsp1 proteins suppress host protein synthesis, including in the innate immune system.
2. Materials and methods
2.1. Cells, viruses and plasmids
The 293 cell line was maintained at 37 °C in a 5% CO2 incubator in Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (ExCell Bio, New Zealand). Newcastle disease virus (NDV) was harvested from chicken embryos. Plasmids pCDNA-SARSnsp1, pCDNA-229Ensp1 and pCDNA-NL63nsp1 were constructed by reverse-transcription PCR amplification of nsp1 genes from SARS-CoV strain Tor2, HCoV-229E strain ATCC VR-740 and HCoV-NL63 strain Amsterdam I genomic RNA and inserted into pCDNA3.1 vector (Invitrogen, Carlsbad, CA) under the control of the human cytomegalovirus (CMV) promoter. An HA tag was added at the C-terminus for all constructs. Plasmids pGL4.73, pGL4.74 and pGL4.75, which contain Renilla luciferase reporter genes driven by the SV40, herpes simplex virus thymidine kinase (HSV-TK) and CMV promoters, respectively, were from Promega (Madison, WI). Plasmids pGL4-hIFNb-P and pGL4-hISG15-P were constructed by PCR amplification of the human IFN-β gene promotor region (−110 to +20) or the human interferon stimulated gene 15 (ISG15) promotor region (−120 to +21) from 293 genomic DNA, and cloned into pGL4.17 (Promega).
2.2. Bioinformatics
Structures for the nsp1 proteins of HCoV-229E and HCoV-NL63 were computed with Modeller software (Marti-Renom et al., 2000) using the solution structure of NSP113–128 from the SARS-CoV (PDB code 2HSX) as a template structure (Almeida et al., 2007). Sequence alignment of the nsp1 proteins from HCoV-229E, HCoV-NL63 and SARS-CoV was performed with the program ClustalW and refined manually.
2.3. Co-immunoprecipitation
293 cells in six-well plate were transfected with 3 ug of nsp1-expressing plasmids or pCDNA3.1 control plasmid (with HA tag) using Lipofectamin 2000 (Invitrogen). Cells were harvested 36 h after transfection and washed three times with phosphate-buffered saline (PBS). Half the cells were treated with native lysis buffer (Promega) for 30 min on ice, and centrifuged, and the supernatant was used for immunoprecipitation using anti-HA antibody-coupled agarose beads (Pierce) according to the manufacturer's instructions. Eluted proteins were separated by 10% SDS-PAGE followed by immunoblotting analysis using anti-S6 polyclonal antibodies (GenScript). Proteins from the other aliquot of cells were separated by 10% SDS-PAGE followed by immunoblotting using anti-HA or anti-β-actin antibodies (Abcam, HongKong).
2.4. ELISA and immunoblotting
Transfection of 293 cells was as above for 24 h, followed by NDV infection for 24 h. IFN-β levels in cell supernatants were measured using a human IFN-β enzyme-linked immunosorbent (ELISA) kit according to the manufacturer's instructions (PBL Interferonsource, NJ). Cells were washed three times with PBS. Proteins were separated by 10% SDS-PAGE gel and immunoblotted with anti-IRF-3 or anti-phospho-IRF-3 (Ser396) antibodies (Cell Signaling, Beverly, MA).
2.5. Quantitative real-time PCR
Transfection of 293 cells was as above for 24 h, followed by NDV infection for 24 h. Alternatively, 293 cells were co-transfected with nsp1-expressing plasmids or pCDNA3.1 control plasmid together with pGL4-hIFNb-P or pGL4.75 for 24 h. Cell RNA was extracted using a cell total RNA isolation kit (Axygen), followed by DNase I (Fermentas) treatment for 30 min at 37 °C to remove genomic DNA. RNA was reverse transcribed using a PrimeScript first-strand cDNA synthesis kit (Takara, Dalian, China), and SYBR green-based quantitative real-time PCR was performed in a CFX96 machine (Bio-Rad) using a Perfect real-time PCR kit (Takara). Primers were listed in Table 1 . Cycle threshold (Ct) values were normalized to 18S rRNA levels, resulting in a ΔCt value (Roth-Cross et al., 2007).
Table 1.
Primers used in quantitative real-time PCR assay.
| Primers | Sequence |
|---|---|
| Human IFN-β | GATTCATCTAGCACTGGCTGG (forward) |
| CTTCAGGTAATGCAGAATCC (reverse) | |
| Firefly luciferase | CAACTGCATAAGGCTATGAAGAGA (forward) |
| ATTTGTATTCAGCCCATATCGTTT (reverse) | |
| Renilla luciferase | GAGCATCAAGATAAGATCAAAGCA (forward) |
| CTTCACCTTTCTCTTTGAATGGTT (reverse) | |
| Human 18S rRNA | CAGCCACCCGAGATTGAGCA (forward) |
| TAGTAGCGACGGGCGGTGTG (reverse) | |
2.6. Reporter assays
Lipofectamine 2000 (Invitrogen) was used to transfect 293 cells with plasmids pGL4-hIFNb-P or pGL4-hISG15-P, and stable cell lines were selected with 1.3 mg/ml G418 (Invitrogen) and subcloned. The 293 stable cell lines were transfected with nsp1-expressing plasmids for 12 h and then challenged with NDV for 24 h or were co-transfected with nsp1-expressing plasmids and poly (I:C) (Sigma) for 24 h. Cells in microplates were disrupted with 20 μl of passive lysis buffer (Promega) and firefly luciferase activity measured using a dual-luciferase reporter assay system (Promega) and a Synergy 4 Hybrid Multi-Mode Microplate Reader (BioTek, Winooski). Alternatively, normal 293 cells were co-transfected with nsp1-expressing plasmids with pGL4.73, pGL4.74 or pGL4.75 for 24 h, and Renilla luciferase activity was measured. Data were analyzed with the paired Student's t-test assuming that the values followed a Gaussian distribution. A p-value of <0.05 was considered significant.
3. Results
3.1. Group I coronavirus nsp1 proteins have a structure similar to SARS-CoV nsp113–128
The three-dimensional structure of group I coronavirus nsp1 has not been previously solved. To evaluate the general fold of these proteins, we built models for nsp1 proteins of HCoV-229E and HCoV-NL63 based on the solution structure of nsp113–128 from the SARS-CoV (Almeida et al., 2007).
Sequence alignment showed only 21% identity and 23% similarity between the HCoV-229E and SARS-CoV nsp1 proteins. Similarly, the nsp1 proteins of HCoV-NL63 and SARS-CoV share only 20% identity and 23% similarity (Fig. 1A). Nevertheless, a careful inspection of several nsp1 regions revealed conserved hydrophobic clusters counterbalancing the regions of low identity and low similarity. Thus, 36% of the hydrophobic residues of HCoV-229E nsp1, and 38% of the hydrophobic residues of HCoV-NL63 nsp1 are conserved, or replaced by other hydrophobic residues, in the SARS-CoV nsp1 (Fig. 1A).
Fig. 1.
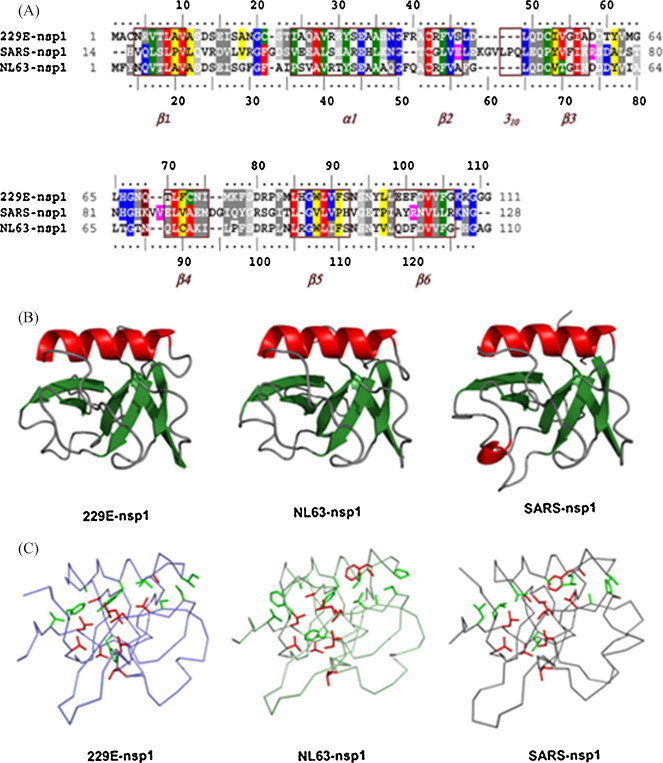
Structural modeling of nsp1 proteins from HCoV-229E and HCoV-NL63. (A) Sequence alignment of nsp1 proteins. Residues identical and important for the β-barrel structure in SARS-CoV protein are in red, other identical residues are in blue, similar residues are in grey, residues conserving hydrophobicity and important for the nsp1 β-barrel structure in SARS (Almeida et al., 2007) are in green, other positions of conserved hydrophobicity are in yellow, and residues important for the nsp1 β-barrel structure in SARS-CoV but not conserved in the compared sequences are in magenta. Boxed and labeled in marron are the regular secondary structure elements of nsp113–128 from SARS-CoV. (B) Conserved overall fold of coronavirus nsp1. 229E-nsp1, ribbon diagram of the modeled structure from HCoV-229E nsp1; NL63-nsp1, structure from HCoV-NL63; SARS-nsp1, template solution structure from SARS-CoV. Molecules are colored according to secondary structure assignment. (C) Comparison of essential residues in the nsp1 structures from SARS-CoV and HCoV-229E. Hydrophobic residues crucial for the β-barrel SARS-CoV nsp1 are relatively well conserved in both structures (red, identical; green, similar or important hydrophobic). Panels B and C were made with Pymol (DeLano, 2002). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
These conserved or similar hydrophobic residues are not randomly positioned. Most are found within, or very close to the regular secondary structure elements of nsp113–128 of SARS-CoV (Fig. 1A). Moreover, the hydrophobic residues crucial for the original β-barrel of SARS-CoV nsp113–128 (Almeida et al., 2007) are conserved or replaced by other hydrophobic residues in the HCoV-229E and HCoV-NL63 nsp1 proteins (Fig. 1A and C).
The hydrophobic cluster analysis identified stronger signatures of the nsp1 fold, centered on the secondary structure elements. Modeling of the HCoV-229E and HCoV-NL63 nsp1 proteins led to three-dimensional structures that conserved the regular secondary structure elements of the unique β-barrel of SARS-CoV nsp113–128, while the loops linking the secondary structures were less similar (Fig. 1B). Intriguingly, HCoV-229E nsp1 contains five cysteine residues, four of which are potentially exposed to solvent. Taking into account the distances separating the cysteines, formation of disulfide bridges is unlikely. Taken together, these findings reinforce the hypothesis that HCoV-229E and HCoV-NL63 nsp1 proteins fold in a manner similar to SARS-CoV nsp113–128, and suggest that these proteins have similar structural and functional relationships.
3.2. Interaction of nsp1 proteins with ribosomal 40S subunit
SARS-CoV nsp1 suppresses host cell protein synthesis by binding to the host cell ribosomal 40S subunit (Kamitani et al., 2009). Based on the bioinformatics results, we examined whether nsp1 proteins of group I coronaviruses also interacted with the ribosomal proteins. HA-tagged nsp1 proteins of HCoV-229E, HCoV-NL63 and SARS-CoV were expressed in 293 cells, and immunoprecipitated with anti-HA antibody. Immunoblotting with anti-S6 antibody detected a band of ∼32 kDa from all three nsp1 protein-expressing cells, which corresponds to the predicted size for the ribosomal protein S6. The nsp1 proteins were detected using anti-HA antibody. The S6 and nsp1 proteins were not detected in mock-transfected cells. As controls, β-actin was detected in all cell lysates (Fig. 2A).
Fig. 2.
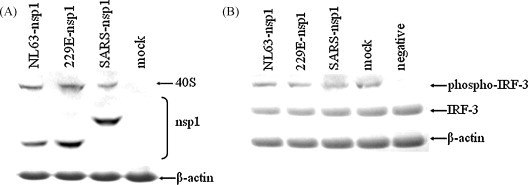
Immunoblot of coronavirus nsp1 proteins interacting with ribosomal protein, and lack of IRF-3 phosphorylation inhibition. (A) 293 cells transfected with nsp1-expressing plasmids or control plasmid and immunoprecipitated with anti-HA antibody. Immunoblotting was with anti-S6, anti-HA or anti-β-actin antibodies. Bands for ribosomal 40S subunit, nsp1 and β-actin proteins are shown. (B) 293 cells transfected as in (A) and cell innate immunity stimulated with NDV. Immunoblotting was with anti-phospho-IRF-3 (Ser396), anti-IRF-3 or anti-β-actin antibodies.
3.3. IRF-3 phosphorylation was not inhibited by nsp1 proteins
We next examined whether the activation of the IFN transcriptional factor IRF-3 was inhibited by nsp1 proteins. Innate immune signal transduction was stimulated by NDV infection in cells transfected with plasmids-expressing nsp1 from HCoV-229E, HCoV-NL63 or SARS-CoV, or with a control plasmid. Immunoblotting with anti-phospho-IRF-3 (Ser396) antibody showed a consistent, homogenous band for the phosphorylated IRF-3 protein in cells expressing the three nsp1 proteins, and in mock-transfected cells, but not in negative control cells that were not stimulated with NDV. Moreover, no obvious inhibition effect was observed in nsp1-transfected cells compared to the mock-transfected cells. Expression of IRF-3 and β-actin proteins was consistently detected in all cell lysates (Fig. 2B).
3.4. Inhibition of IFN-β and luciferase mRNA transcription by nsp1 proteins
We further investigated whether group I coronavirus nsp1 proteins inhibit IFN-β and luciferase transcription. Nsp1-plasmids were transfected into 293 cells, and IFN-β or luciferase transcription was stimulated by NDV or IFN-β promoter- or CMV promoter-driven plasmids, followed by quantification of IFN-β mRNA by real-time PCR. As shown in Fig. 3 , the mRNA levels for cell IFN-β, IFN-β promoter-driven firefly luciferase or CMV promoter-driven Renilla luciferase were inhibited by 0.5–2 ΔCt values by nsp1 proteins from HCoV-229E, HCoV-NL63 and SARS-CoV.
Fig. 3.
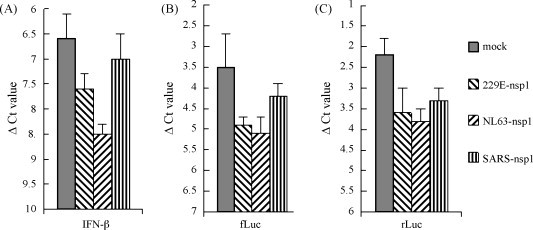
Real-time PCR showing the inhibition of IFN-β and luciferase gene transcription by coronavirus nsp1 proteins. Samples were analyzed using primers specific for human IFN-β gene, firefly or Renilla luciferase genes. Numbers represent ΔCt values normalized to endogenous 18S rRNA. The y-axis is inverted to reflect the inverse relationship between ΔCt and mRNA levels. Error bars indicate standard errors of triplicate treatments in three individual experiments. (A) 293 cells were transfected with nsp1-expressing plasmids or control plasmids and cells were stimulated with NDV for 24 h. B, 293 cells were co-transfected with IFN-β promoter-driven firefly luciferase-expressing plasmid and nsp1-expressing plasmids or control plasmids for 24 h. (C) 293 cells were co-transfected with CMV promoter-driven Renilla luciferase-expressing plasmid and nsp1-expressing plasmids or control plasmids for 24 h.
3.5. Suppression of IFN-β and luciferase proteins synthesis by nsp1 proteins
Suppression of host protein synthesis by SARS-CoV nsp1 protein, including in the innate immune system, has been seen in several studies (Kamitani et al., 2006, Zust et al., 2007, Narayanan et al., 2008). Since the group I coronavirus nsp1 proteins also interact with the cellular translational machinery, we examined the influence of HCoV-229E and HCoV-NL63 nsp1 proteins on host immune and non-immune protein synthesis. Luciferase reporter assays showed that synthesis of the innate immune promoter IFN-β- and ISG15-driven genes was suppressed by 5–20-folds in HCoV-229E and HCoV-NL63 nsp1-expressing 293 cells (Fig. 4A). Synthesis of non-immune promoter-driven genes, including for SV40, HSV-TK and CMV promoters, was inhibited to a similar extent by the two group I coronavirus nsp1 proteins (Fig. 4B). In contrast, SARS-CoV nsp1 suppressed promoter activity by only 3–5-folds by all assays (Fig. 4). Titration of the released IFN-β proteins in cell supernatant by ELISA revealed similar results. The IFN-β levels in coronavirus nsp1-expressing cells were 4–15-folds lower than in control cells. (Fig. 4C).
Fig. 4.
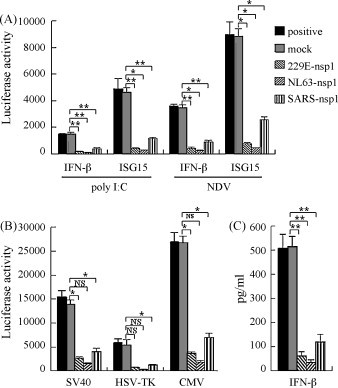
Suppression of host immune and non-immune promoter activities and IFN-β protein synthesis by coronavirus nsp1 proteins. (A) 293 cells stably transfected with IFN-β or ISG15 promoter-driven reporters were transfected with nsp1-expressing plasmids or control plasmids. Cells were stimulated with Poly I:C or NDV for 24 h and firefly luciferase activities were measured. (B) Normal 293 cells were co-transfected with nsp1-expressing plasmids and SV40-, HSV-TK- or CMV-promoter-driven reporter plasmids for 24 h, and Renilla luciferase activity measured. (C) Normal 293 cells transfected with nsp1-expressing plasmids or control plasmids, and then stimulated by NDV for 24 h. Titers of IFN-β in cell supernatants were measured by ELISA. Error bars indicate standard errors of triplicate treatments in three individual experiments. Statistical analysis was performed using paired Student's t-test (**, p < 0.01; *, p < 0.05; NS, not significant, p > 0.05).
4. Discussion
Studies on nsp1 proteins of group II coronavirus SARS-CoV and MHV revealed a novel mechanism for interaction between host and viral proteins. Group I coronaviruses also encode nsp1 proteins of only about 110 amino acids, with relatively high conservation within the group, but with a high degree of polymorphism with SARS-CoV nsp1, which has 179 amino acids. However, the NMR analysis revealed that the segment from residue 13 to 128 of SARS-CoV nsp1 determined its core structure (Almeida et al., 2007), and this corresponded to the overall length of the group I coronavirus nsp1 proteins. Therefore, we built models for the nsp1 proteins of HCoV-229E and HCoV-NL63, two low-pathogenic group I human coronaviruses, for comparison with nsp1 from the highly pathogenic SARS-CoV. Careful inspection of the sequence alignment showed that most of the structurally important hydrophobic residues are conserved or similar for nsp1 proteins from HCoV-229E, HCoV-NL63 and SARS-CoV. Computational modeling demonstrated overall similarity in the three-dimensional nsp1 structures from the two group I coronavirus and SARS-CoV.
Bioinformatics suggest that group I coronavirus and SARS-CoV nsp1 proteins might have similar biological functions. Recent studies revealed that SARS-CoV nsp1 suppresses host protein synthesis by interacting with the ribosomal 40S subunit (Kamitani et al., 2009). In this study, we demonstrated that the group I coronavirus nsp1 proteins also bound to the ribosomal 40S subunit, as shown by co-IP. Further analysis showed that activation of IRF-3 was not affected, but synthesis of host immune and non-immune proteins was potently suppressed by group I coronavirus nsp1 proteins. It seemed that synthesis of these proteins was more strongly inhibited at translational level than transcriptional level. These results indicate that group I coronaviruses have evolved a mechanism strikingly similar to SARS-CoV for antagonizing host cell proliferation and innate immunity using nsp1. Our results are in general consistent to that of Kamitani et al. (2006), who firstly reported that SARS-CoV nsp1 inhibited IFN-β mRNA transcription and IFN-β promoter-driven luciferase protein synthesis. However, in our study, the inhibition of IFN-β mRNA transcription and luciferase protein synthesis was less strong than that reported by Kamitani et al. (2006), most probably due to different expression levels of nsp1. In their study, the SARS-CoV nsp1 was driven by a chicken β-actin/rabbit β-globin hybrid promoter (AG promoter) in PCAGGS vector, which is known for its robust expression in eukaryote cells. In this study, nsp1 was driven by a CMV promoter, which was probably less efficient than the hybrid AG promoter.
Most human coronaviruses are low-pathogenic viruses that often cause mild lower respiratory tract infections like common cold (Thiel and Weber, 2008). However, SARS-CoV infection appears highly pathogenic and causes severe pneumonia and acute respiratory distress syndrome, characterized by the presence of diffuse alveolar damage (Weiss and Navas-Martin, 2005). Surprisingly, few viral particles are isolated from lung tissues of SARS-CoV infected patients, but levels of inflammatory cytokines and chemokines are greatly elevated in the lung. A hyperinflammatory response is presumed to be the key determinant of the high pathogenicity of SARS-CoV, rather than rapid viral spread (De Lang et al., 2009). The specific mechanism for SARS-CoV pathogenicity is not known, but a number of viral coding proteins may be involved (Weiss and Navas-Martin, 2005). The nsp1 protein of SARS-CoV is defined as a major pathogenicity factor (Kamitani et al., 2006, Wathelet et al., 2007). However, our results showed that the nsp1 proteins from low-pathogenic coronaviruses suppressed host protein synthesis more strongly than nsp1 from SARS-CoV, indicating that the nsp1 protein is actually a virulence factor for facilitating viral spread, but is not a major determinant in coronavirus pathogenicity. Although an engineered SARS-CoV with a mutated nsp1 was greatly attenuated in an animal model (Wathelet et al., 2007), caution should be taken in developing this mutant virus as human vaccine because of the pathogenicity factors still present in the virion.
SARS-CoV and MHV fail to induce or induced only weak and delayed innate immune responses (Spiegel et al., 2005, Spiegel and Weber, 2006, Roth-Cross et al., 2007, Zhou and Perlman, 2007). The underlying mechanism for this phenomenom is not fully understood, but nsp1 is proposed to be a major factor in the ability of the viruses to antagonize host innate immunity (Kamitani et al., 2006, Wathelet et al., 2007, Zust et al., 2007, Narayanan et al., 2008). However, surprisingly, activation of host innate immunity and induction of type I IFN were observed for the group I coronavirus TGEV (La Bonnardiere and Laude, 1981, Charley and Laude, 1988, Charley and Lavenant, 1990). We also found that group I coronaviruses HCoV-229E steadily activated IFN transcription factors, and infected cells produced robust IFN-β (unpublished data). This study clearly showed that the HCoV-229E and HCoV-NL63 nsp1 proteins also potently suppressed host innate immune protein synthesis like nsp1 from SARS-CoV, implying that group II coronaviruses do not employ nsp1 to evade host innate immunity. The mechanism underlying the different innate immune responses for the two groups of coronaviruses remains to be further investigated.
Acknowledgments
This work was supported by grants from the Fundamental Research Funds for the Central Universities (No. 0904010), Shanghai Municipal Science and Technology Commission (No. 07DZ22940) and Shanghai Municipal Wildlife Administration (No. SBHZ2006_01).
References
- Almeida M.S., Johnson M.A., Herrmann T., Geralt M., Wuthrich K. Novel beta-barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:3151–3161. doi: 10.1128/JVI.01939-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzozka K., Finke S., Conzelmann K.K. Inhibition of interferon signaling by rabies virus phosphoprotein P: activation-dependent binding of STAT1 and STAT2. J. Virol. 2006;80:2675–2683. doi: 10.1128/JVI.80.6.2675-2683.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas W.B., Loo Y.M., Gale M., Jr, Hartman A.L., Kimberlin C.R., Martinez-Sobrido L., Saphire E.O., Basler C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charley B., Laude H. Induction of alpha interferon by transmissible gastroenteritis coronavirus: role of transmembrane glycoprotein E1. J. Virol. 1988;62:8–11. doi: 10.1128/jvi.62.1.8-11.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charley B., Lavenant L. Characterization of blood mononuclear cells producing IFNa following induction by coronavirus-infected cells (porcine transmissible gastroenteritis virus) Res. Immunol. 1990;141:141–151. doi: 10.1016/0923-2494(90)90133-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lang A., Baas T., Smits S.L., Katze M.G., Psterhaus A.D.M.E., Haagmans B.L. Unraveling the complexities of the interferon response during SARS-CoV infection. Future Virol. 2009;4:71–78. doi: 10.2217/17460794.4.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W.L. DeLano Scientific; Palo Alto, CA, USA: 2002. The PyMOL User's Manual. [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.K., Baker S.C., Li K. Regulation of IRF-3 dependent innate immunity by the papain-like protease domain of the SARS coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didcock L., Young D.F., Goodbourn S., Randall R.E. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 1999;73:9928–9933. doi: 10.1128/jvi.73.12.9928-9933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A. Inhibition of interferon-mediated antiviral responses by Influenza A viruses and other negative-strand RNA viruses. Virology. 2001;279:375–384. doi: 10.1006/viro.2000.0756. [DOI] [PubMed] [Google Scholar]
- Gotoh B., Takeuchi K., Komatsu T., Yokoo J. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J. Virol. 2003;77:3360–3370. doi: 10.1128/JVI.77.6.3360-3370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakki M., Geballe A.P. Double-stranded RNA binding by human cytomegalovirus pTRS1. J. Virol. 2005;79:7311–7318. doi: 10.1128/JVI.79.12.7311-7318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imani F., Jacobs B.L. Inhibitory activity for the interferon-induced protein kinase is associated with the reovirus serotype 1 sigma 3 protein. Proc. Natl. Acad. Sci. USA. 1988;85:7887–7891. doi: 10.1073/pnas.85.21.7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs B.L., Langland J.O. Reovirus sigma 3 protein: dsRNA binding and inhibition of RNA-activated protein kinase. Curr. Top. Microbiol. Immunol. 1998;233:185–196. doi: 10.1007/978-3-642-72092-5_9. [DOI] [PubMed] [Google Scholar]
- Kamitani W., Huang C., Narayanan K., Lokugamage K.G., Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009;16:1134–1141. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N., Kubo H., Makino S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann. N. Y. Acad. Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Sars coronavirus proteins Orf 3b,Orf 6, and nucleocapsid function as interferon antagonists. J. Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Bonnardiere C., Laude H. High interferon titer in newborn pig intestine during experimentally induced viral enteritis. Infect. Immun. 1981;32:28–31. doi: 10.1128/iai.32.1.28-31.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland J.O., Jacobs B.L. Inhibition of PKR by vaccinia virus: role of the N- and C-terminal domains of E3L. Virology. 2004;324:419–429. doi: 10.1016/j.virol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Lu Y., Wambach M., Katze M.G., Krug R.M. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology. 1995;214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- Marti-Renom M.A., Stuart A., Fiser A., Sánchez R., Melo F., Sali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Min J.Y., Krug R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 20–50 oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA. 2006;103:7100–7105. doi: 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakshi R., Padhan K., Rani M., Khan N., Ahmad F., Jameel S. The SARS coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-dependent downregulation of the type I interferon receptor. PLoS One. 2009;4:e8342. doi: 10.1371/journal.pone.0008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Huang C., Lokugamage K., Kamitani W., Ikegami T., Tseng C.K., Makino S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 2008;82:4471–4479. doi: 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppers J., Mulvey M., Khoo D., Mohr I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 2000;74:11215–11221. doi: 10.1128/jvi.74.23.11215-11221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid S.P., Leung L.W., Hartman A.L., Martinez O., Shaw M.L., Carbonnelle C., Volchkov V.E., Nichol S.T., Basler C.F. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 2006;80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth-Cross J.K., Martinez-Sobrido L., Scott E.P., Garcia-Sastre A., Weiss S.R. Inhibition of the Alpha/Beta interferon response by mouse hepatitis virus at multiple levels. J. Virol. 2007;81:7189–7199. doi: 10.1128/JVI.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K.L., Kok K.H., Ng M.H., Poon V.K., Yuen K.Y., Zheng B.J., Jin D.Y. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 2009;284:16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel M., Pichlmair A., Martinez-Sobrido L., Cros J., Garcia-Sastre A., Haller O., Weber F. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 2005;79:2079–2086. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel M., Weber F. Inhibition of cytokine gene expression and induction of chemokine genes in non-lymphatic cells infected with SARS coronavirus. Virol. J. 2006;3:17. doi: 10.1186/1743-422X-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Weber F. Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev. 2008;19:121–132. doi: 10.1016/j.cytogfr.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valchanova R.S., Picard-Maureau M., Budt M., Brune W. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J. Virol. 2006;80:10181–10190. doi: 10.1128/JVI.00908-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005;69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Condit R.C., Vijaysri S., Jacobs B., Williams B.R., Silverman R.H. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002;76:5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Hauns K., Langland J.O., Jacobs B.L., Hogue B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 2007;81:2554–2563. doi: 10.1128/JVI.01634-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Perlman S. Mouse hepatitis virus does not induce Beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J. Virol. 2007;81:568–574. doi: 10.1128/JVI.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zust R., Cervantes-Barragan L., Kuri T., Blakqori G., Weber F., Ludewig B., Thiel V. Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007;3:1062–1072. doi: 10.1371/journal.ppat.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]