

Abstract
Nucleic acid amplification techniques have revolutionised diagnostic and research industries. Current technologies that allow the detection of amplification in real-time are fast becoming industry standards, particularly in a diagnostic context. In this review, we describe and explore the application of numerous real-time detection chemistries and amplification techniques for pathogen detection and identification, including the polymerase chain reaction, nucleic acid sequence-based amplification, strand displacement amplification and the ligase chain reaction. The emergence of newer technologies, such as lab-on-a-chip devices and photo-cleavable linkers, is also discussed.
Keywords: Nucleic acid, Amplification, Detection, Real-time PCR, PCR, NASBA, Biosensor, LCR
1. Introduction
A variety of nucleic acid amplification techniques were developed in the mid to late 1980's. These include the polymerase chain reaction (PCR) (Mullis and Faloona, 1987), ligation-mediated amplification (Wu and Wallace, 1989) and transcription-based amplification (Kwoh et al., 1989). Since then, these techniques have been refined and alternative approaches have been developed for amplification (e.g. transcription-mediated amplification (TMA), nucleic acid sequence-based amplification (NASBA), ligase chain reaction (LCR), strand displacement amplification (SDA), linear linked amplification, see Monis et al., 2002 for an overview). None of these techniques have achieved the same widespread research application as PCR, most likely due to the simplicity and cost-effectiveness of PCR. However, some of these techniques have been incorporated into clinical diagnostic assays (e.g. SDA is a platform technology used by Becton Dickinson for Mycobacteria and Chlamydia detection, NASBA is used by Biomérieux for HIV-1, CMV and Enteroviruses, TMA is used by Gen-Probe for the detection of Mycobacteria, Neisseria and Chlamydia). As an adjunct to the emerging amplification technologies that are rapidly being developed, conventional amplification techniques continue to play an integral role in characterising and genotyping parasites for medical, environmental and epidemiological investigations. Techniques such as amplified restriction fragment length polymorphism (AFLP), PCR-restriction fragment length polymorphism (PCR-RFLP) and random amplified polymorphic DNA (RAPD) are currently employed to answer specific biological and evolutionary questions and these techniques have been the subject of numerous reviews (e.g. Masiga et al., 2000, Monis et al., 2002, Singh, 1997). The greatest recent advancement in amplification technology has been the development of systems that allow monitoring of amplification in real-time. This paper will provide an overview of recent developments in amplification and detection technologies, with a focus on real-time detection chemistries.
1.1. Real-time amplification
The first real-time amplification system used ethidium bromide and a mounted CCD camera to monitor PCR amplification in a closed reaction tube (Higuchi et al., 1992). Since then, significant advancements have been made in technology and software exploiting Higuchi's initial principle of monitoring changes in amplification signal with time. As a result, real-time PCR now provides researchers and diagnostic laboratories with additional tools for disease diagnosis, identification of species, quantifying gene expression, single nucleotide polymorphism (SNP) detection and monitoring infection loads during therapy. There are many fluorescent detection chemistries currently employed in real-time PCR assays and while there may be superficial similarities between some chemistries, specific assay design criteria need to be adhered to for each chemistry application. (Readers are directed to a recently published text book for design criteria (Edwards et al., 2004).)
The advent of real-time PCR has overcome a number of short-comings of conventional PCR. Real-time PCR readily allows quantitation of DNA over a broad dynamic range and it is a closed-tube format that requires no post PCR handling for identification of amplicons, reducing the potential for sample contamination and making the entire process more amenable to high throughput analysis. For quantitation, real-time PCR exploits the proportional relationship between the cycle where exponential amplification is detected (the threshold cycle or C t) and the starting number of copies of the target nucleic acid fragment. In order to do this, standards with defined numbers of copies of the target fragment are used to generate a standard curve (based on C t value versus copy number) and the gene copy number in an unknown sample is estimated by comparison to this standard curve (Saunders, 2004). Closed tube verification of the amplification of the correct fragment can be achieved by DNA melting curve analysis, which is analogous to the detection of a band by conventional gel electrophoresis. In the case where intercalating dyes are used, the dissociation kinetics of the entire amplified fragment is measured, and plotting the first derivative of the melting curve versus temperature allows determination of the melting temperature of the product. Probe:amplicon hybrids can be analysed in a similar fashion, except the melting temperature is determined by measuring the dissociation of the probe from target DNA, rather than measuring the melting temperature of the whole of the amplified fragment. In both cases, the melting temperature is affected by the GC content of the DNA duplex (the higher the GC, the higher the melting temperature), the absolute order of the bases in the sequence and the size of the amplicon or probe:target hybrid.
1.2. Real-time detection chemistries
The initial development of real-time PCR made use of the double-stranded DNA (dsDNA)-specific intercalating dye ethidium bromide (Higuchi et al., 1992). dsDNA-specific intercalating dyes exhibit little or no fluorescence when free in solution (Fig. 1 a) but produce a large quantum yield increase in fluorescence when bound to dsDNA and exposed to the appropriate wavelength of light (Fig. 1b). SYBR Green I (Becker et al., 1996) is currently the industry standard, although the use of other dyes has been described (e.g. BEBO (Bengtsson et al., 2003), LC Green (Wittwer et al., 2003), SYTO9 (Monis et al., 2005)). Intercalating dyes are the most cost effective chemistry and are possibly now the most widely used detection chemistry, particularly for gene expression studies. Caution must be taken when using SYBR Green I, however, as there are disadvantages that limit its ease of use. In general, reaction conditions require further optimisation by including additional reagents to improve reaction efficiency, such as DMSO (Jung et al., 2001), BSA and Triton X-100 (Karsai et al., 2002). Depending on the reaction conditions, SYBR Green I also appears to be inhibitory to PCR in a concentration dependent manner (Monis et al., 2005, Nath et al., 2000, Wittwer et al., 2003) and the degradation products of the dye have also been reported to be inhibitory to PCR (Karsai et al., 2002). In addition, the preferential binding of SYBR Green to specific amplicons during multiplex PCR limits its use for this application (Giglio et al., 2003). Recent work describing the evaluation of SYTO9 for real-time PCR has found that this new dye is much less inhibitory to PCR compared with SYBR Green I and that it does not appear to exhibit preferential binding to specific amplicons, allowing it to be used to analyse multiplex PCRs by DNA melting curve analysis (Monis et al., 2005). In addition, SYTO9 appears to be much more robust and reliable for DNA melting curve analysis compared to SYBR Green I, making melting curve analysis a more reliable tool for genetic discrimination/identification (Monis et al., 2005). Intercalating dyes are cost efficient when compared to probe based detection systems and so are the first choice for many applications in research and diagnostics. One of the main advantages of this chemistry is the confirmation of amplicons by melt curve analysis, with each amplicon having a specific melting temperature value. A disadvantage of using these dyes is that any double stranded DNA will be detected, e.g. non-specific amplicons and primer–dimers, which may cause problems with the accuracy of quantitative PCR if they are not resolved by either re-designing primers or acquiring data above the temperature where non-specific products are observed. The use of SYBR Green I for real-time PCR of parasites (excluding bacteria and viruses) has largely been limited to protozoans, including high through-put antimalarial drug screening (Smilkstein et al., 2004), genotyping of Cryptosporidium parvum (Widmer et al., 2004), and quantitative detection of Leishmania (Nicolas et al., 2002), Neospora (Collantes-Fernandez et al., 2002), Toxoplasma (Contini et al., 2005) and Trypanosoma brucei (Becker et al., 2004). There have been limited reports on the use of SYBR Green I for the detection of nematodes (Madani et al., 2005) and study of nematode gene regulation (Li et al., 2004). There has been a single report of the application of SYTO9 for the identification of Giardia, where amplification and melting curve analysis of a fragment of the gdh locus allowed discrimination of Giardia duodenalis Assemblages A, B and F and Giardia ardeae (Monis et al., 2005).
Fig. 1.
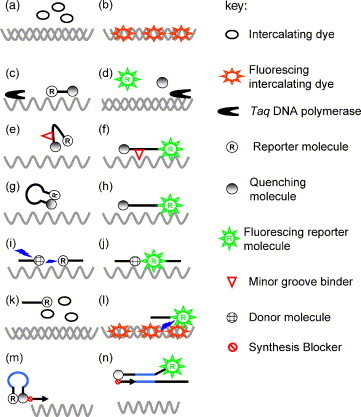
Schematic of commonly used real-time PCR chemistries. Unbound intercalating dye is not fluorescent (a) but will produce a quantum yield increase in fluorescence upon binding to double-stranded DNA (b). Taqman probes cannot fluoresce when intact due to the proximity of the reporter and quencher molecule (c), but will produce fluorescent signal following hydrolysis by Taq polymerase and release of the reporter molecule (d). The secondary structure of MGB Eclipse probes causes the quencher and reporter molecules to be in close proximity so that the reporter will not fluoresce (e), but once bound to target DNA the probe is stabilised by the minor groove binder and separates the quencher and reporter sufficiently to allow fluorescence (f). Molecular beacons hold the quencher and reporter in close proximity via a stem loop structure, preventing fluorescence when not bound (g) but allowing fluorescence once bound to the target DNA (h). FRET probes anneal to target sequences and energy transfer from the donor to reporter molecule (i) results in increased fluorescence of the reporter molecule (j). Intercalating dyes will not fluoresce unless bound to double-stranded DNA and so cannot act as energy donors when in the unbound state for iFRET reactions (k). Upon binding of the intercalator and iFRET probe, energy transfer can occur, resulting in production of fluorescence (l). Scorpion probes are held in a stem-loop structure that prevents probe fluorescence (m), but once incorporated into an amplicon the structure is opened by binding of the loop to complementary sequence within the amplicon (n). Figure adapted from Monis et al. (2005).
Taqman (hydrolysis probes, 5′ nuclease assay) (Heid et al., 1996) and 3′ MGB probe technologies both work by a similar principle. Both systems have a 5′ fluorescent reporter molecule (e.g. carboxyfluorescein (FAM)), and a 3′ quencher molecule (e.g. BHQ1 or TAMRA). Taqman probes are typically 20–30 nucleotides in length and the fluorescence of the reporter molecule is quenched at this proximity between reporter and quencher molecule (Fig. 1c). Hydrolysis of the Taqman probe by the 5′ exonuclease activity of Taq DNA polymerase at 60 °C separates the reporter and quencher molecule and a signal is emitted by the reporter molecule that is detected by the real-time PCR instrument (Fig. 1d). The incubation temperature of DNA synthesis step is critical because at higher temperatures (e.g. 72 °C), which are normally used for DNA synthesis in conventional PCR, the Taq DNA polymerase will displace the probe rather than degrade it. As a result of probe degradation, fluorescent signal increases as a function of the number of amplification cycles and allows specific detection and quantitation of the target DNA. In contrast, 3′ MGB probes have a 3′ minor groove-binding moiety in addition to a reporter and quencher molecule, resulting in improved primer binding (by increasing the effective primer melting temperature) and allowing the use of shorter probes, resulting in better efficiency (Kutyavin et al., 2000). Taqman probes are one of the most widely used chemistries because assay design is relatively simple and assays are generally robust. Taqman probes do not allow confirmation that the correct fragment has been amplified (other than by running a gel), and as a result it is particularly important to validate the specificity of primer/probe combinations. Multiplexing Taqman assays is achieved using probes labelled with different reporter molecules that have distinct fluorescence properties (namely excitation/emission maxima). The combinations of fluorophores that can be used is dependant on the technical specifications of the real-time PCR instrument used (an upper limit of six fluorophores can be detected on some instruments). Taqman probes allow presence/absence detection of a particular target sequence but do not allow genetic discrimination unless used in a multiplex format. Taqman assays have been used for the detection of numerous parasites including Cryptosporidium (Fontaine and Guillot, 2003, Higgins et al., 2001, Keegan et al., 2003), Giardia (Bertrand et al., 2004), Leishmania (Gomez-Saladin et al., 2005, Rolao et al., 2004), Myxobolus cerebralis (Cavender et al., 2004, Kelley et al., 2004), Plasmodium (Blair et al., 2002, Witney et al., 2001), Theileria (Jeong et al., 2003) and Toxoplasma gondii (Jauregui et al., 2001). In these cases the Taqman assays were used for detection but not differentiation of parasites. The only reported use of Taqman probes for parasite differentiation has been for trichostrongyle nematodes, where multiplex assays were developed for the pair-wise differentiation of Haemonchus contortus, Ostertagia leptospicularis, Tricginstohgylus colubriformis and Cooperia curticei (von Samson-Himmelstjerna et al., 2002). Molecular beacons (Piatek et al., 1998) and MGB Eclipse probes (Afonina et al., 2002) both use secondary structure (a stem-loop in the case of molecular beacons) to hold a reporter molecule and quencher in close proximity when the probe is in solution (Fig. 1e for MGB Eclipse probes, Fig. 1g for molecular beacons), preventing the production of any fluorescent signal. When either type of probe anneals to target DNA, they unfold and there is sufficient distance between the reporter and quencher molecules to allow fluorescence (Fig. 1f and h). Unlike Taqman probes, MGB Eclipse probes and molecular beacons are not hydrolysed because the extension temperatures are 72 °C or higher. However, MGB Eclipse probes are claimed to be more stable than molecular beacons (presumably because of the presence of the 5′ MGB moiety) and appear to produce better signal to noise ratio than molecular beacons (http://www.epochbio.com/products/mgbe_how_it_works.htm). Both MGB Eclipse probes and molecular beacons can be used for DNA melting curve analysis (measuring the dissociation kinetics of the release of the bound probe to target DNA), allowing further genetic characterisation of the amplified DNA. The use of MGB Eclipse probes appears to be limited in the literature, although they have been demonstrated to be useful for SNP typing (Belousov et al., 2004). Molecular beacons have been widely used for a variety of pathogens (e.g. Bordetella (Poddar and Le, 2001) and Hepatitis C (Yang et al., 2002)), particularly in combination with NASBA (e.g. Greijer et al., 2002), but have only been applied to Plasmodium (Bustamante et al., 2004, Schneider et al., 2005) and Entamoeba histolytica (Roy et al., 2005) in terms of parasites.
Fluorescence (or forster) resonance energy transfer (FRET)-based assays rely on energy transfer between a 3′ donor fluorophore and 5′ reporter fluorophore on separate probes (Fig. 1i and j), rather than the quenching of a fluorophore as seen with MGB Eclipse, molecular beacon and Taqman probes. Melt curve analysis in FRET assays measure the temperature at which the bound probes are dissociated from the target amplicon and not the melting temperature of the entire amplicon as is the case when using intercalating dyes. Thus a single primer set may be used to amplify a region of interest, with species/strain discrimination possible by designing the FRET probes to bind to variable regions within the region bound by the primer set used for amplification. This technology lends itself to SNP detection, as a single mismatch between the probe and target sequences will yield a sufficiently different melting temperature to allow detection. iFRET (Howell et al., 2002) is a variation of FRET and uses an intercalating dye (SYBR Green I) as the donor fluorophore and only a single probe with a reporter molecule (Fig. 1k and l). iFRET appears to have advantages over FRET in terms of cost and signal strength (Howell et al., 2002) but does not appear to have been widely used. FRET assays have been used for the clinical diagnosis of Toxoplasmagondii infections (Reischl et al., 2003, Simon et al., 2004) and to study the transmission of T. gondii between mother and foetus in an animal model (Flori et al., 2002). In addition, FRET assays have been used to detect and discriminate species/genotypes of Cryptosporidium (Limor et al., 2002), and to detect, quantitate and differentiate the three clinically relevant groups of Leishmania (Schulz et al., 2003). Although a powerful tool, FRET assays are more cumbersome to optimise than Taqman assays and require more care when used in quantitation because excessive levels of target DNA can adversely affect the amplification signal (known as a “hook” effect, which is caused by excess amplicon interfering with probe binding) and melting curve analysis.
Scorpion primers (Solinas et al., 2001) perform the dual function of probe and primer. In structure Scorpion primers are essentially a molecular beacon with a primer linked to the 3′ end of the stem of the beacon (Fig. 1m). The link between the beacon section and the primer has a PCR blocker to prevent replication of the beacon sequence. The loop of the beacon is complimentary to the sequence synthesised immediately 3′ from the primer. Following primer extension, denaturation and annealing, the loop sequence hybridises to the newly synthesised complementary sequence adjacent to the primer, resulting in a separation between the quencher and fluorophore and production of a fluorescent signal (Fig. 1n). Unlike molecular beacons, Scorpion probes cannot be used for DNA melting curve analysis. The proximity of the probe to the target region means that the interaction is more efficient than other probe formats, allowing good sensitivity. Despite this advantage, the application of Scorpion probes has been relatively limited in terms of pathogen diagnosis, with reports on the use of Scorpions for quantitation of HIV-1 (Saha et al., 2001) and differentiation of Giardia assemblages A and B directly from faecal samples using a multiplex assay (Ng et al., 2005).
1.3. Nucleic acid sequence-based amplification
NASBA is an isothermal amplification technique making use of a dual function reverse transcriptase/DNA polymerase, RNA polymerase, RNaseH and a T7 promoter-labelled target-specific primer (Guatelli et al., 1990). TMA works by a similar principle to NASBA, except that the assay relies on the RNaseH activity of the reverse transcriptase, rather than using a separate enzyme with RNaseH activity. This combination of enzymes and primers targets a specific RNA transcript and initially produces an RNA:DNA hybrid fragment with a T7 promoter (Fig. 2 ). The RNA in this hybrid is degraded by the RNaseH and the DNA is extended to form a dsDNA fragment with a T7 promoter, forming a template for the production of more RNA transcripts by T7 RNA polymerase. These transcripts can then be used for the production of additional DNA fragments with T7 promoters. The reaction is self-sustaining, giving rise to an alternative name for this process, self-sustained sequence replication or 3SR. DNA contamination of samples does not affect this process, which means that RNA quantitation can be conducted from crude cell extracts. NASBA is not widely used as the cost of commercial kits is prohibitive and there are difficulties in reliably preparing an in-house NASBA mastermix. To date, NASBA has been most widely used for virus detection, most typically using commercial NASBA kits and probe-based chemiluminescent detection of the amplified RNA (Lanciotti, 2003, van Gemen et al., 1994, Witt et al., 2000, Wu et al., 2001). The application of NASBA to detect bacterial pathogens (Cook, 2003) and Cryptosporidium (Baeumner et al., 2001) has been more limited. More recently, NASBA technology has been coupled with molecular beacons for monitoring product generation in real-time (Leone et al., 1998) and has been used to quantify Plasmodium (Schneider et al., 2004, Schneider et al., 2005) and viruses (Ayele et al., 2004, Capaul and Gorgievski-Hrisoho, 2005, Gulliksen et al., 2004, Moore et al., 2004). The sensitivity of NASBA is often greater than conventional reverse transcription PCR (Loeffler et al., 2001, Wacharapluesadee and Hemachudha, 2001), and NASBA has been reported to be superior for the unambiguous detection of mRNA in the presence of DNA (Simpkins et al., 2000). In addition to real-time detection, NASBA has also been combined with liposome signal amplification technology (http://www.ibi.cc/advantages_of_liposomes.htm) to develop biosensors for Escherichia coli (Baeumner et al., 2004), Bacillus anthracis (Baeumner et al., 2004), Cryptosporidium (Baeumner et al., 2004, Esch et al., 2001a, Esch et al., 2001b) and Dengue virus (Zaytseva et al., 2004).
Fig. 2.
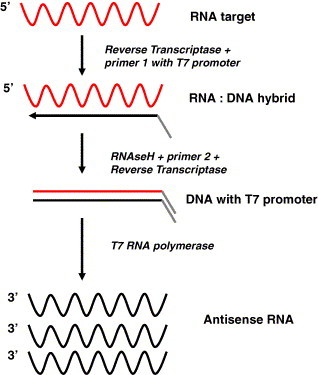
Schematic representation of NASBA. RNA (red wavy line) is converted to double-stranded DNA with a T7 promoter using reverse transcriptase, RNaseH and a primer with a T7 promoter. The DNA is used as a template by T7 RNA polymerase for the production of multiple copies of antisense RNA (black wavy lines). Each transcript can act as a template for the production of additional double-stranded DNA templates.
1.4. Ligase chain reaction
The ligase chain (or detection) reaction (LCR) is another amplification technique developed shortly after PCR (Wu and Wallace, 1989). This technique uses a thermostable DNA ligase and four primers, two adjacent forward primers and their complements. There is typically a gap of 1–3 bases between the adjacent primers, which act as templates for ligation by DNA ligase. DNA ligase is highly specific and intolerant to base mismatches, a property exploited for use in real-time detection of SNPs (Chen et al., 1998) and the detection of intra-individual differentiation of species-specific parasitemia for Plasmodium sp. (McNamara et al., 2004). Uptake of this technology to the routine environment has been slow, possibly due to the market dominance of PCR. Commercial LCR kits (LCx, Abbott laboratories) for Mycobacterium tuberculosis seem to perform adequately (Leon Muinos et al., 2004, Ribeiro et al., 2004), but the Chlamydia trachomatis LCx kit had problems with reproducibility and had to be withdrawn from the market due to “high negative control rates resulting in invalid runs and non-repeating positives” (FDA product recall Z-0859-1/Z-0860-1, http://www.fda.gov/bbs/topics/ENFORCE/2001/ENF00709.html). This technology holds great promise, particularly for SNP detection and use in microchips (Lou et al., 2004) or with universal microarrays (Busti et al., 2002).
1.5. Strand displacement amplification
SDA is another isothermal reaction that was developed at around the same time as NASBA (Walker et al., 1992). This amplification technique makes use of a combination of exonuclease deficient DNA polymerase (Klenow fragment), a restriction endonuclease with a hemiphosphorothioate recognition site, a modified deoxynucleotide to allow the synthesis of hemiphosphorothioated DNA, and two sets of primers (Walker et al., 1992). The first set of primers act in the same way as forward and reverse primers used in PCR, but they have a restriction enzyme recognition site inserted at their 5′-ends. The second set of primers is known as “bumper” primers and these are designed to bind immediately 5′ of the forward and reverse primers. After denaturation of the target DNA, the forward and reverse primers promote the synthesis of hemiphosphorothioated DNA, creating a DNA: hemiphosphorothioated DNA hybrid. These strands are separated by extension of the bumper primers, which displace the newly synthesised hemiphosphorothioated DNA strand. The resulting ssDNA is converted to dsDNA by primer extension using the respective forward or reverse primer. The resulting hemiphosphorothioated dsDNA contains a restriction site and forms the template for SDA. This template is cut by a restriction enzyme to introduce a single stranded nick at the restriction site, which promotes the synthesis of a new strand of DNA by the DNA polymerase as it repairs, the nick in the DNA. The synthesis of the strand of DNA results in the displacement of the old one. Once the hemiphosphorothioated template has been produced, the process is self sustaining.
The original SDA process was not very efficient and it has been improved by incorporating a thermostable polymerase and a different exonuclease to greatly improve the yield and rate of amplification. These new conditions allow a 1010-fold amplification of target after 15 min at 60 °C (Spargo et al., 1996). SDA can also be used to detect RNA by incorporating a reverse transcription step (Nycz et al., 1998). The largest application of SDA has been for clinical diagnosis of pathogenic organisms (HIV-1 (Nycz et al., 1998), M. tuberculosis, (Mazzarelli et al., 2003, Spargo et al., 1996, Visca et al., 2004), Chlamydia and Neisseria (Cosentino et al., 2003, Van Dyck et al., 2001), pathogenic E. coli (Ge et al., 2002)).
1.6. 2005 and beyond
Research continues into the development of new amplification technologies, with several being described in recent years. One of the most recently described amplification techniques makes use of a DNA helicase to allow isothermal DNA amplification (Vincent et al., 2004). A combination of DNA helicase, single-stranded DNA-binding proteins and accessory proteins are used to unwind double-stranded DNA, which can then act as a template for DNA synthesis using primers and a DNA polymerase. This process is the first true isothermal process to be described because there are no other temperature steps required (unlike other isothermal techniques that require an initial denaturation step to initiate the process). Helicase-mediated amplification has been measured in real-time and has been used to detect Treponema denticola and Brugia malayi (Vincent et al., 2004), although further refinements are being made to the assay to improve efficiency and to simplify the reaction by identifying alternative helicases that display activity in the absence of accessory proteins (An et al., 2005).
Another technique, rolling circle amplification, has been used recently for in situ genotyping of individual DNA molecules (Larsson et al., 2004). This process uses restriction digestion and exonuclease digestion to produce single stranded DNA targets, which can then hybridise specific oligonucleotides (padlock probes). The padlock probes circularise following hybridisation and are then ligated to provide a template for rolling circle amplification, resulting in the incorporation of many copies of the padlock probe into the target strand of DNA. The padlock probe has a specific target sequence, which is then detected using a fluorescent complementary probe, allowing the detection of a single gene within a cell. Rolling circle amplification has also been used to detect minicircle DNA by promoting replication branching and restriction enzyme digestion to produce dsDNA fragments that can be detected using a peptide nucleic acid molecular beacon (Smolina et al., 2004). The current technique is rapid but relatively insensitive, although the authors claim that optimisation of the assay volume and detection format should allow detection of 100 copies of target within 50 min. Padlock probes have been used to detect plant pathogens (Szemes et al., 2005).
A more established recently developed technique is multiplex ligation-dependent probe amplification (MLPA), which makes use of both ligation and PCR (Schouten et al., 2002). This process has multiple components and steps (Fig. 3 ). Essentially it uses two DNA fragments, the first with a 5′ fluorescent label, a universal forward primer and a target-specific recognition sequence at the 3′ end, the second with a target-specific recognition sequence at the 5′ end (designed to hybridise next to the specific sequence on the 3′ end of the other fragment), a non-specific stretch of DNA of a defined length termed a “stuffer” sequence and a universal reverse primer at the 3′ end. MLPA has two steps, the first is ligation of the two fragments on binding to the target DNA, and the second is PCR amplification of the ligated probe. Multiplexing is achieved by varying the length of the stuffer sequence for each set of probes used in the assay. Amplification is detected by sequence-grade electrophoresis such as capillary electrophoresis and it is claimed that this approach allows relative quantitation (Schouten et al., 2002). Since the target sequence for this assay is short it is ideal for the detection of fragmented DNA, such as that recovered from formalin preserved material. Although used widely for the detection of human genetic disorders and cancers, MLPA has not been applied in a clinical microbiology context.
Fig. 3.
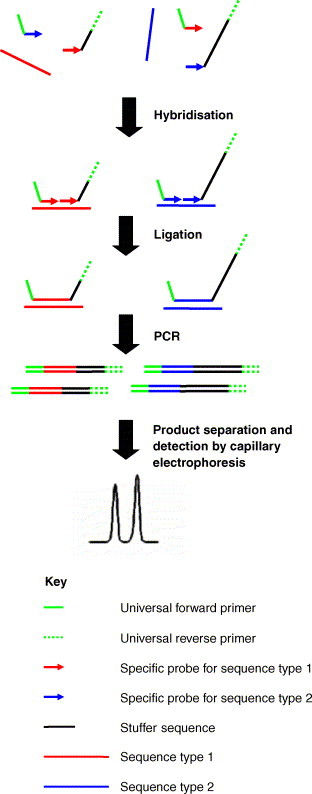
Schematic representation of MLPA. A specific two component probe is used for the detection of each sequence type of interest. Each probe element has a region complementary to the sequence of interest. On binding to the sequence of interest the probe elements are ligated and the intact probe is amplified using universal primers. Probes are differentiated on the basis of size, which is accomplished by varying the length of a “stuffer” sequence that has no similarity to the sequence of interest. Typically the probe is fluorescently labelled to enable detection and quantitation of band intensity by capillary electrophoresis.
In addition to new amplification systems, advances are also being made into new techniques for detecting amplification. Matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry has been used to directly detect amplification products from PCR (Hurst et al., 1996) and LCR (Jurinke et al., 1996), and to analyse restriction digests (Taranenko et al., 2002). Instead of detecting the amplified DNA, an alternative approach has been developed using photo-cleavable linkers (as seen with the Masscode or Masstag systems), providing an interesting alternative to the use of fluorophores for PCR detection. These systems involve linking a small molecular weight molecule to the 5′ end of a nucleotide, which is used as a primer in an allele specific SNP assay, or as a probe for pathogen detection. Detection of these linkers (also called “tags” or “codes”) is achieved by MALDI-TOF mass spectrometry (see http://www1.qiagen.com/literature/qiagennews/0201/1016749_QNews22001p11-12.pdf or http://www.bioserve.com/departments/snp_masscode.cfm for further information). Up to 30 linkers can be used, enabling high throughput screening of SNPs (Haff and Smirnov, 1997, Kokoris et al., 2000, Sauer et al., 2003, Xie et al., 2005). Masscode technology has been applied to the detection of a variety of respiratory pathogens, including Legionella, Influenza and Adenovirus, and levels of detection ranged from 100 to 5000 DNA/RNA copies depending on the pathogen (Briese et al., 2005). The use of MALDI-TOF-based technologies, however, is prohibitive to most laboratories due to the expense associated with purchasing a mass spectrometer, and this is reflected in the relatively limited application of the technology. Considering that there are many more cost effective technologies for high-throughput screening it is debateable whether this technology will become widely adopted, and at the moment the use of such technology is akin to routinely using electron microscopy to examine faecal smears in a clinical laboratory.
Possibly the most rapidly developing area of emerging technology is nanotechnology, which is driving the development of lab-on-a-chip systems (see de Mello, 2001, Gardeniers and van den Berg, 2004 for reviews). Many systems are in development that will miniaturise conventional and real-time amplification systems, allowing extremely rapid analysis of sub-microlitre volume samples (e.g. nanolitre Taqman multiplex PCR, (Matsubara et al., 2004)). There are numerous detection systems ranging from gold nanoparticles tagged with short segments of DNA to multicolour optical coding for biological assays that have been achieved by embedding different-sized quantum dots into polymeric microbeads (reviewed by Jain, 2003). Co-migration electrophoregrams in combination with restriction enzyme digest have been used on chip devices for discrimination and quantitation of PCR products (Xu et al., 2004), and semi-quantitation of SARS-coronavirus has been described (Juang et al., 2004). Certainly many challenges lie ahead in this area, particularly to obtain a cost effective, workable, user friendly interface and end point result analysis system, but if successful its potential applications in the biological setting are far reaching.
2. Summary
Numerous technologies exist for nucleic acid amplification and detection. The choice of which method to use will be dictated by the nature of the sample and the biological question being addressed, as well as the cost and ease of use of the technique, including assay design and ease of data interpretation. For example, NASB A has many advantages over reverse transcription PCR for the detection of RNA, but these need to be balanced against the relative ease of assay design and cost of reagents. There are numerous real-time PCR instruments on the market, and numerous detection chemistries, each with advantages and disadvantages, and choosing the right system is reliant upon its intended use. Lab-on-a-chip devices have the potential to revolutionise medical management and environmental monitoring but more attention is needed to make these systems workable so that they can be taken out of the lab and put into use in field environments. It is almost certain that additional techniques will be developed in the near future that will be faster, cheaper, and easier to use. However, the tools currently available offer a myriad of options to answer specific biological questions.
References
- Afonina I.A., Reed M.W., Lusby E., Shishkina I.G., Belousov Y.S. Minor groove binder-conjugated DNA probes for quantitative DNA detection by hybridisation-triggered fluorescence. Biotechniques. 2002;32 doi: 10.2144/02324pf01. 940–944, 946–949. [DOI] [PubMed] [Google Scholar]
- An L., Tang W., Ranalli T.A., Kim H.J., Wytiaz J., Kong H. Characterisation of a thermostable UVRD helicase and its participation in helicase dependent amplification. J. Biol. Chem. 2005 doi: 10.1074/jbc.M503096200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayele W., Pollakis G., Abebe A., Fisseha B., Tegbaru B., Tesfaye G., Mengistu Y., Wolday D., van Gemen B., Goudsmit J., Dorigo-Zetsma W., de Baar M.P. Development of a nucleic acid sequence-based amplification assay that uses gag-based molecular beacons to distinguish between human immunodeficiency virus type 1 subtype C and C’ infections in Ethiopia. J. Clin. Microbiol. 2004;42:1534–1541. doi: 10.1128/JCM.42.4.1534-1541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeumner A.J., Humiston M.C., Montagna R.A., Durst R.A. Detection of viable oocysts of Cryptosporidium parvum following nucleic acid sequence based amplification. Anal. Chem. 2001;73:1176–1180. doi: 10.1021/ac001293h. [DOI] [PubMed] [Google Scholar]
- Baeumner A.J., Pretz J., Fang S. A universal nucleic acid sequence biosensor with nanomolar detection limits. Anal. Chem. 2004;76:888–894. doi: 10.1021/ac034945l. [DOI] [PubMed] [Google Scholar]
- Becker A., Reith A., Napiwotzki J., Kadenbach B. A quantitative method of determining initial amounts of DNA by polymerase chain reaction cycle titration using digital imaging and a novel DNA stain. Anal. Biochem. 1996;237:204–207. doi: 10.1006/abio.1996.0230. [DOI] [PubMed] [Google Scholar]
- Becker S., Franco J.R., Simarro P.P., Stich A., Abel P.M., Steverding D. Real-time PCR for detection of Trypanosoma brucei in human blood samples. Diag. Microbiol. Infect. Dis. 2004;50:193–199. doi: 10.1016/j.diagmicrobio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Belousov Y.S., Welch R.A., Sanders S., Mills A., Kulchenko A., Dempcy R., Afonina I.A., Walburger D.K., Glaser C.L., Yadavalli S., Vermeulen N.M., Mahoney W. Single nucleotide polymorphism genotyping by two colour melting curve analysis using the MGB Eclipse Probe System in challenging sequence environment. Human Genomics. 2004;1:209–217. doi: 10.1186/1479-7364-1-3-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson M., Karlsson H.J., Westman G., Kubista M. A new minor groove binding asymmetric cyanine reporter dye for real-time PCR. Nucleic Acids Res. 2003;31:e45. doi: 10.1093/nar/gng045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand I., Gantzer C., Chesnot T., Schwartzbrod J. Improved specificity for Giardia lamblia cyst quantification in wastewater by development of a real-time PCR method. J. Microbiol. Meth. 2004;57:41–53. doi: 10.1016/j.mimet.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Blair P.L., Witney A., Haynes J.D., Moch J.K., Carucci D.J., Adams J.H. Transcripts of developmentally regulated Plasmodium falciparum genes quantified by real-time RT-PCR. Nucleic Acids Res. 2002;30:2224–2231. doi: 10.1093/nar/30.10.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese T., Palacios G., Kokoris M., Jabado O., Liu Z., Renwick N., Kapoor V., Casas I., Pozo F., Limberger R., Perez-Brena P., Ju J., Lipkin W.I. Diagnostic system for rapid and sensitive differential detection of pathogens. Emer. Infect. Dis. 2005;11:310–313. doi: 10.3201/eid1102.040492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante L.Y., Crooke A., Martinez J., Diez A., Bautista J.M. Dual-function stem molecular beacons to assess mRNA expression in AT-rich transcripts of Plasmodium falciparum. Biotechniques. 2004;36 doi: 10.2144/04363RN04. 488–492, 494. [DOI] [PubMed] [Google Scholar]
- Busti E., Bordoni R., Castiglioni B., Monciardini P., Sosio M., Donadio S., Consolandi C., Rossi Bernardi L., Battaglia C., De Bellis G. Bacterial discrimination by means of a universal array approach mediated by LDR (ligase detection reaction) BMC Microbiol. 2002;2:27. doi: 10.1186/1471-2180-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaul S.E., Gorgievski-Hrisoho M. Detection of Enterovirus RNA in cerebrospinal fluid (CSF) using NucliSens EasyQ Enterovirus assay. J. Clin. Virol. 2005;32:236–240. doi: 10.1016/j.jcv.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Cavender W.P., Wood J.S., Powell M.S., Overturf K., Cain K.D. Real-time quantitative polymerase chain reaction (QPCR) to identify Myxobolus cerebralis in rainbow trout Oncorhynchus mykiss. Dis. Aquat. Organ. 2004;60:205–213. doi: 10.3354/dao060205. [DOI] [PubMed] [Google Scholar]
- Chen X., Livak K.J., Kwok P.Y. A homogeneous, ligase-mediated DNA diagnostic test. Genome Res. 1998;8:549–556. doi: 10.1101/gr.8.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collantes-Fernandez E., Zaballos A., Alvarez-Garcia G., Ortega-Mora L.M. Quantitative detection of Neospora caninum in bovine aborted fetuses and experimentally infected mice by real-time PCR. J. Clin. Microbiol. 2002;40:1194–1198. doi: 10.1128/JCM.40.4.1194-1198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contini C., Seraceni S., Cultrera R., Incorvaia C., Sebastiani A., Picot S. Evaluation of a real-time PCR-based assay using the lightcycler system for detection of Toxoplasma gondii bradyzoite genes in blood specimens from patients with toxoplasmic retinochoroiditis. Int. J. Parasitol. 2005;35:275–283. doi: 10.1016/j.ijpara.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Cook N. The use of NASBA for the detection of microbial pathogens in food and environmental samples. J. Microbiol. Meth. 2003;53:165–174. doi: 10.1016/s0167-7012(03)00022-8. [DOI] [PubMed] [Google Scholar]
- Cosentino L.A., Landers D.V., Hillier S.L. Detection of Chlamydia trachomatis and Neisseria gonorrhoeae by strand displacement amplification and relevance of the amplification control for use with vaginal swab specimens. J. Clin. Microbiol. 2003;41:3592–3596. doi: 10.1128/JCM.41.8.3592-3596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mello A.J. DNA amplification: does ‘small’ really mean ‘efficient’? Lab. Chip. 2001;1:24N–29N. [Google Scholar]
- Edwards K., Logan J., Saunders N. Horizon Bioscience; Wymondham: 2004. Real-time PCR, An Essential Guide. p. 346. [Google Scholar]
- Esch M.B., Baeumner A.J., Durst R.A. Detection of Cryptosporidium parvum using oligonucleotide-tagged liposomes in a competitive assay format. Anal. Chem. 2001;73:3162–3167. doi: 10.1021/ac010012i. [DOI] [PubMed] [Google Scholar]
- Esch M.B., Locascio L.E., Tarlov M.J., Durst R.A. Detection of viable Cryptosporidium parvum using DNA-modified liposomes in a microfluidic chip. Anal. Chem. 2001;73:2952–2958. doi: 10.1021/ac001508n. [DOI] [PubMed] [Google Scholar]
- Flori P., Hafid J., Bourlet T., Raberin H., Genin C., Sung R.T. Experimental model of congenital toxoplasmosis in guinea-pigs: use of quantitative and qualitative PCR for the study of maternofetal transmission. J. Med. Microbiol. 2002;51:871–878. doi: 10.1099/0022-1317-51-10-871. [DOI] [PubMed] [Google Scholar]
- Fontaine M., Guillot E. An immunomagnetic separation-real-time PCR method for quantification of Cryptosporidium parvum in water samples. J. Microbiol. Meth. 2003;54:29–36. doi: 10.1016/s0167-7012(03)00005-8. [DOI] [PubMed] [Google Scholar]
- Gardeniers J.G., van den Berg A. Lab-on-a-chip systems for biomedical and environmental monitoring. Anal. Bioanal. Chem. 2004;378:1700–1703. doi: 10.1007/s00216-003-2435-7. [DOI] [PubMed] [Google Scholar]
- Ge B., Larkin C., Ahn S., Jolley M., Nasir M., Meng J., Hall R.H. Identification of Escherichia coli O157:H7 and other enterohemorrhagic serotypes by EHEC-hlyA targeting, strand displacement amplification and fluorescence polarisation. Mol. Cell. Probes. 2002;16:85–92. doi: 10.1006/mcpr.2001.0389. [DOI] [PubMed] [Google Scholar]
- Giglio S., Monis P.T., Saint C.P. Demonstration of preferential binding of SYBR Green I to specific DNA fragments in real-time multiplex PCR. Nucleic Acids Res. 2003;31:e136. doi: 10.1093/nar/gng135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Saladin E., Doud C.W., Maroli M. Short report: surveillance of leishmania sp. Among sand flies in sicily (Italy) using a fluorogenic real-time polymerase chain reaction. Am. J. Trop. Med. Hyg. 2005;72:138–141. [PubMed] [Google Scholar]
- Greijer A.E., Adriaanse H.M., Dekkers C.A., Middeldorp J.M. Multiplex real-time NASBA for monitoring expression dynamics of human cytomegalovirus encoded IE1 and pp67 RNA. J. Clin. Virol. 2002;24:57–66. doi: 10.1016/s1386-6532(01)00227-x. [DOI] [PubMed] [Google Scholar]
- Guatelli J.C., Whitfield K.M., Kwoh D.Y., Barringer K.J., Richman D.D., Gingeras T.R. Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1874–1878. doi: 10.1073/pnas.87.5.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulliksen A., Solli L., Karlsen F., Rogne H., Hovig E., Nordstrom T., Sirevag R. Real-time nucleic acid sequence-based amplification in nanoliter volumes. Anal. Chem. 2004;76:9–14. doi: 10.1021/ac034779h. [DOI] [PubMed] [Google Scholar]
- Haff L.A., Smirnov I.P. Multiplex genotyping of PCR products with MassTag-labeled primers. Nucleic Acids Res. 1997;25:3749–3750. doi: 10.1093/nar/25.18.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid C.A., Stevens J., Livak K.J., Williams P.M. Real-time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Higgins J.A., Fayer R., Trout J.M., Xiao L., Lal A.A., Kerby S., Jenkins M.C. Real-time PCR for the detection of Cryptosporidium parvum. J. Microbiol. Meth. 2001;47:323–337. doi: 10.1016/s0167-7012(01)00339-6. [DOI] [PubMed] [Google Scholar]
- Higuchi R., Dollinger G., Walsh P.S., Griffith R. Simultaneous amplification and detection of specific DNA sequences. Biotechnology (NY) 1992;10:413–417. doi: 10.1038/nbt0492-413. [DOI] [PubMed] [Google Scholar]
- Howell W.M., Jobs M., Brookes A.J. iFRET: an improved fluorescence system for DNA-melting analysis. Genome Res. 2002;12:1401–1407. doi: 10.1101/gr.297202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst G.B., Doktycz M.J., Vass A.A., Buchanan M.V. Detection of bacterial DNA polymerase chain reaction products by matrix-assisted laser desorption/ionisation mass spectrometry. Rapid Commun. Mass Spectrom. 1996;10:377–382. doi: 10.1002/(SICI)1097-0231(199602)10:3<377::AID-RCM481>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Jain K.K. Nanodiagnostics: application of nanotechnology in molecular diagnostics. Expert. Rev. Mol. Diagn. 2003;3:153–161. doi: 10.1586/14737159.3.2.153. [DOI] [PubMed] [Google Scholar]
- Jauregui L.H., Higgins J., Zarlenga D., Dubey J.P., Lunney J.K. Development of a real-time PCR assay for detection of Toxoplasma gondii in pig and mouse tissues. J. Clin. Microbiol. 2001;39:2065–2071. doi: 10.1128/JCM.39.6.2065-2071.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong W., Kweon C.H., Kang S.W., Paik S.G. Diagnosis and quantification of Theileriasergenti using TaqMan PCR. Vet. Parasitol. 2003;111:287–295. doi: 10.1016/s0304-4017(02)00388-6. [DOI] [PubMed] [Google Scholar]
- Juang J.L., Chen T.C., Jiang S.S., Hsiung C.A., Chen W.C., Chen G.W., Lin S.M., Lin J.H., Chiu S.C., Lai Y.K. Coupling multiplex RT-PCR to a gene chip assay for sensitive and semiquantitative detection of severe acute respiratory syndrome-coronavirus. Lab. Invest. 2004;84:1085–1091. doi: 10.1038/labinvest.3700136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M., Muche J.M., Lukowsky A., Jung K., Loening S.A. Dimethyl sulfoxide as additive in ready-to-use reaction mixtures for real-time polymerase chain reaction analysis with SYBR Green I dye. Anal. Biochem. 2001;289:292–295. doi: 10.1006/abio.2000.4931. [DOI] [PubMed] [Google Scholar]
- Jurinke C., van den Boom D., Jacob A., Tang K., Worl R., Koster H. Analysis of ligase chain reaction products via matrix-assisted laser desorption/ionisation time-of-flight-mass spectrometry. Anal. Biochem. 1996;237:174–181. doi: 10.1006/abio.1996.0225. [DOI] [PubMed] [Google Scholar]
- Karsai A., Muller S., Platz S., Hauser M.T. Evaluation of a homemade SYBR Green I reaction mixture for real-time PCR quantification of gene expression. Biotechniques. 2002;32 doi: 10.2144/02324st05. 790–792, 794–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan A.R., Fanok S., Monis P.T., Saint C.P. Cell culture-Taqman PCR assay for evaluation of Cryptosporidium parvum disinfection. Appl. Environ. Microbiol. 2003;69:2505–2511. doi: 10.1128/AEM.69.5.2505-2511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley G.O., Zagmutt-Vergara F.J., Leutenegger C.M., Myklebust K.A., Adkison M.A., McDowell T.S., Marty G.D., Kahler A.L., Bush A.L., Gardner I.A., Hedrick R.P. Evaluation of five diagnostic methods for the detection and quantification of Myxobolus cerebralis. J. Vet. Diagn. Invest. 2004;16:202–211. doi: 10.1177/104063870401600305. [DOI] [PubMed] [Google Scholar]
- Kokoris M., Dix K., Moynihan K., Mathis J., Erwin B., Grass P., Hines B., Duesterhoeft A. High-throughput SNP genotyping with the Masscode system. Mol. Diagn. 2000;5:329–340. doi: 10.1007/BF03262094. [DOI] [PubMed] [Google Scholar]
- Kutyavin I.V., Afonina I.A., Mills A., Gorn V.V., Lukhtanov E.A., Belousov E.S., Singer M.J., Walburger D.K., Lokhov S.G., Gall A.A., Dempcy R., Reed M.W., Meyer R.B., Hedgpeth J. 3′-Minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 2000;28:655–661. doi: 10.1093/nar/28.2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwoh D.Y., Davis G.R., Whitfield K.M., Chappelle H.L., DiMichele L.J., Gingeras T.R. Transcription-based amplification system and detection of amplified human immunodeficiency virus type 1 with a bead-based sandwich hybridisation format. Proc. Natl. Acad. Sci. U.S.A. 1989;86:1173–1177. doi: 10.1073/pnas.86.4.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti R.S. Molecular amplification assays for the detection of flaviviruses. Adv. Virus Res. 2003;61:67–99. doi: 10.1016/s0065-3527(03)61002-x. [DOI] [PubMed] [Google Scholar]
- Larsson C., Koch J., Nygren A., Janssen G., Raap A.K., Landegren U., Nilsson M. In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes. Nat. Meth. 2004;1:227–232. doi: 10.1038/nmeth723. [DOI] [PubMed] [Google Scholar]
- Leon Muinos E., Perez Del Molino M.L., Lado Lado F.L., Pazo Nunez M., Pardo F. Use of ligase chain reaction for the rapid diagnosis of lymph node tuberculosis. Scand. J. Infect. Dis. 2004;36:724–726. doi: 10.1080/00365540410020965. [DOI] [PubMed] [Google Scholar]
- Leone G., van Schijndel H., van Gemen B., Kramer F.R., Schoen C.D. Molecular beacon probes combined with amplification by NASBA enable homogeneous, real-time detection of RNA. Nucleic Acids Res. 1998;26:2150–2155. doi: 10.1093/nar/26.9.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.W., Rush A.C., Tan J., Weil G.J. Quantitative analysis of gender-regulated transcripts in the filarial nematode Brugia malayi by real-time RT-PCR. Mol. Biochem. Parasitol. 2004;137:329–337. doi: 10.1016/j.molbiopara.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Limor J.R., Lal A.A., Xiao L. Detection and differentiation of Cryptosporidium parasites that are pathogenic for humans by real-time PCR. J. Clin. Microbiol. 2002;40:2335–2338. doi: 10.1128/JCM.40.7.2335-2338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler J., Hebart H., Cox P., Flues N., Schumacher U., Einsele H. Nucleic acid sequence-based amplification of Aspergillus RNA in blood samples. J. Clin. Microbiol. 2001;39:1626–1629. doi: 10.1128/JCM.39.4.1626-1629.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X.J., Panaro N.J., Wilding P., Fortina P., Kricka L.J. Mutation detection using ligase chain reaction in passivated silicon-glass microchips and microchip capillary electrophoresis. Biotechniques. 2004;37 doi: 10.2144/04373ST03. 392, 394, 396–398. [DOI] [PubMed] [Google Scholar]
- Madani M., Subbotin S.A., Moens M. Quantitative detection of the potato cyst nematode, Globoderapallida and the beet cyst nematode, Heterodera schachtii, using real-time PCR with SYBR Green I dye. Mol. Cell. Probes. 2005;19:81–86. doi: 10.1016/j.mcp.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Masiga D.K., Tait A., Turner C.M. Amplified restriction fragment length polymorphism in parasite genetics. Parasitol. Today. 2000;16:350–353. doi: 10.1016/s0169-4758(00)01706-3. [DOI] [PubMed] [Google Scholar]
- Matsubara Y., Kerman K., Kobayashi M., Yamamura S., Morita Y., Takamura Y., Tamiya E. On-chip nanoliter-volume multiplex TaqMan polymerase chain reaction from a single copy based on counting fluorescence released microchambers. Anal. Chem. 2004;76:6434–6439. doi: 10.1021/ac0497149. [DOI] [PubMed] [Google Scholar]
- Mazzarelli G., Rindi L., Piccoli P., Scarparo C., Garzelli C., Tortoli E. Evaluation of the BDProbeTec ET system for direct detection of Mycobacterium tuberculosis in pulmonary and extrapulmonary samples: a multicenter study. J. Clin. Microbiol. 2003;41:1779–1782. doi: 10.1128/JCM.41.4.1779-1782.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara D.T., Thomson J.M., Kasehagen L.J., Zimmerman P.A. Development of a multiplex PCR-ligase detection reaction assay for diagnosis of infection by the four parasite species causing malaria in humans. J. Clin. Microbiol. 2004;42:2403–2410. doi: 10.1128/JCM.42.6.2403-2410.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monis P.T., Andrews R.H., Saint C.P. Molecular biology techniques in parasite ecology. Int. J. Parasitol. 2002;32:551–562. doi: 10.1016/s0020-7519(01)00352-6. [DOI] [PubMed] [Google Scholar]
- Monis P.T., Giglio S., Saint C.P. Comparison of SYTO9 and SYBR Green I for real-time polymerase chain reaction and investigation of the effect of dye concentration on amplification and DNA melting curve analysis. Anal. Biochem. 2005;370:24–34. doi: 10.1016/j.ab.2005.01.046. [DOI] [PubMed] [Google Scholar]
- Moore C., Hibbitts S., Owen N., Corden S.A., Harrison G., Fox J., Gelder C., Westmoreland D. Development and evaluation of a real-time nucleic acid sequence based amplification assay for rapid detection of influenza A. J. Med. Virol. 2004;74:619–628. doi: 10.1002/jmv.20221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullis K.B., Faloona F.A. Specific synthesis of DNA in vitro via a polymerase-catalysed chain reaction. Meth. Enzymol. 1987;155:335–350. doi: 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- Nath K., Sarosy J.W., Hahn J., Di Como C.J. Effects of ethidium bromide and SYBR Green I on different polymerase chain reaction systems. J. Biochem. Biophys. Meth. 2000;42:15–29. doi: 10.1016/s0165-022x(99)00033-0. [DOI] [PubMed] [Google Scholar]
- Ng C.T., Gilchrist C.A., Lane A., Roy S., Haque R., Houpt E.R. Multiplex real-time PCR assay using Scorpion probes and DNA capture for genotype-specific detection of Giardia lamblia on fecal samples. J. Clin. Microbiol. 2005;43:1256–1260. doi: 10.1128/JCM.43.3.1256-1260.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas L., Prina E., Lang T., Milon G. Real-time PCR for detection and quantitation of leishmania in mouse tissues. J. Clin. Microbiol. 2002;40:1666–1669. doi: 10.1128/JCM.40.5.1666-1669.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nycz C.M., Dean C.H., Haaland P.D., Spargo C.A., Walker G.T. Quantitative reverse transcription strand displacement amplification: quantitation of nucleic acids using an isothermal amplification technique. Anal. Biochem. 1998;259:226–234. doi: 10.1006/abio.1998.2641. [DOI] [PubMed] [Google Scholar]
- Piatek A.S., Tyagi S., Pol A.C., Telenti A., Miller L.P., Kramer F.R., Alland D. Molecular beacon sequence analysis for detecting drug resistance in Mycobacterium tuberculosis. Nat. Biotechnol. 1998;16:359–363. doi: 10.1038/nbt0498-359. [DOI] [PubMed] [Google Scholar]
- Poddar S.K., Le C.T. Bordetellapertussis detection by spectrofluorometry using polymerase chain reaction (PCR) and a molecular beacon probe. Mol. Cell. Probes. 2001;15:161–167. doi: 10.1006/mcpr.2001.0357. [DOI] [PubMed] [Google Scholar]
- Reischl U., Bretagne S., Kruger D., Ernault P., Costa J.M. Comparison of two DNA targets for the diagnosis of toxoplasmosis by real-time PCR using fluorescence resonance energy transfer hybridisation probes. BMC Infect. Dis. 2003;3:7. doi: 10.1186/1471-2334-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro F.K., Dettoni Vdo V., Peres R.L., Vinhas S.A., Co T.R., Dietze R., Palaci M. Evaluation of a commercial test based on ligase chain reaction for direct detection of Mycobacterium tuberculosis in respiratory specimens. Rev. Soc. Bras. Med. Trop. 2004;37:431–435. doi: 10.1590/s0037-86822004000600001. [DOI] [PubMed] [Google Scholar]
- Rolao N., Cortes S., Rodrigues O.R., Campino L. Quantification of Leishmania infantum parasites in tissue biopsies by real-time polymerase chain reaction and polymerase chain reaction-enzyme-linked immunosorbent assay. J. Parasitol. 2004;90:1150–1154. doi: 10.1645/GE-264R1. [DOI] [PubMed] [Google Scholar]
- Roy S., Kabir M., Mondal D., Ali I.K., Petri W.A., Jr., Haque R. Real-time-PCR assay for diagnosis of Entamoeba histolytica infection. J. Clin. Microbiol. 2005;43:2168–2172. doi: 10.1128/JCM.43.5.2168-2172.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha B.K., Tian B., Bucy R.P. Quantitation of HIV-1 by real-time PCR with a unique fluorogenic probe. J. Virol. Meth. 2001;93:33–42. doi: 10.1016/s0166-0934(00)00288-3. [DOI] [PubMed] [Google Scholar]
- Sauer S., Lehrach H., Reinhardt R. MALDI mass spectrometry analysis of single nucleotide polymorphisms by photocleavage and charge-tagging. Nucleic Acids Res. 2003;31:e63. doi: 10.1093/nar/gng062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders N.A. Quantitative real-time PCR. In: Edwards K., Logan J., Saunders N., editors. Real-time PCR, An Essential Guide. Horizon Bioscience; Wymondham: 2004. [Google Scholar]
- Schneider P., Schoone G., Schallig H., Verhage D., Telgt D., Eling W., Sauerwein R. Quantification of Plasmodium falciparum gametocytes in differential stages of development by quantitative nucleic acid sequence-based amplification. Mol. Biochem. Parasitol. 2004;137:35–41. doi: 10.1016/j.molbiopara.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Schneider P., Wolters L., Schoone G., Schallig H., Sillekens P., Hermsen R., Sauerwein R. Real-time nucleic acid sequence-based amplification is more convenient than real-time PCR for quantification of Plasmodium falciparum. J. Clin. Microbiol. 2005;43:402–405. doi: 10.1128/JCM.43.1.402-405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten J.P., McElgunn C.J., Waaijer R., Zwijnenburg D., Diepvens F., Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz A., Mellenthin K., Schonian G., Fleischer B., Drosten C. Detection, differentiation, and quantitation of pathogenic leishmania organisms by a fluorescence resonance energy transfer-based real-time PCR assay. J. Clin. Microbiol. 2003;41:1529–1535. doi: 10.1128/JCM.41.4.1529-1535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A., Labalette P., Ordinaire I., Frealle E., Dei-Cas E., Camus D., Delhaes L. Use of fluorescence resonance energy transfer hybridisation probes to evaluate quantitative real-time PCR for diagnosis of ocular toxoplasmosis. J. Clin. Microbiol. 2004;42:3681–3685. doi: 10.1128/JCM.42.8.3681-3685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins S.A., Chan A.B., Hays J., Popping B., Cook N. An RNA transcription-based amplification technique (NASBA) for the detection of viable Salmonella enterica. Lett. Appl. Microbiol. 2000;30:75–79. doi: 10.1046/j.1472-765x.2000.00670.x. [DOI] [PubMed] [Google Scholar]
- Singh B. Molecular methods for diagnosis and epidemiological studies of parasitic infections. Int. J. Parasitol. 1997;27:1135–1145. doi: 10.1016/s0020-7519(97)00111-2. [DOI] [PubMed] [Google Scholar]
- Smilkstein M., Sriwilaijaroen N., Kelly J.X., Wilairat P., Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolina I.V., Demidov V.V., Cantor C.R., Broude N.E. Real-time monitoring of branched rolling-circle DNA amplification with peptide nucleic acid beacon. Anal. Biochem. 2004;335:326–329. doi: 10.1016/j.ab.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Solinas A., Brown L.J., McKeen C., Mellor J.M., Nicol J., Thelwell N., Brown T. Duplex Scorpion primers in SNP analysis and FRET applications. Nucleic Acids Res. 2001;29:96. doi: 10.1093/nar/29.20.e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spargo C.A., Fraiser M.S., Van Cleve M., Wright D.J., Nycz C.M., Spears P.A., Walker G.T. Detection of M. tuberculosis DNA using thermophilic strand displacement amplification. Mol. Cell. Probes. 1996;10:247–256. doi: 10.1006/mcpr.1996.0034. [DOI] [PubMed] [Google Scholar]
- Szemes M., Bonants P., de Weerdt M., Baner J., Landegren U., Schoen C.D. Diagnostic application of padlock probes-multiplex detection of plant pathogens using universal microarrays. Nucleic Acids Res. 2005;33:e70. doi: 10.1093/nar/gni069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranenko N.I., Hurt R., Zhou J.Z., Isola N.R., Huang H., Lee S.H., Chen C.H. Laser desorption mass spectrometry for microbial DNA analysis. J. Microbiol. Meth. 2002;48:101–106. doi: 10.1016/s0167-7012(01)00314-1. [DOI] [PubMed] [Google Scholar]
- Van Dyck E., Ieven M., Pattyn S., Van Damme L., Laga M. Detection of Chlamydia trachomatis and Neisseria gonorrhoeae by enzyme immunoassay, culture, and three nucleic acid amplification tests. J. Clin. Microbiol. 2001;39:1751–1756. doi: 10.1128/JCM.39.5.1751-1756.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gemen B., van Beuningen R., Nabbe A., van Strijp D., Jurriaans S., Lens P., Kievits T. A one-tube quantitative HIV-1 RNANASBA nucleic acid amplification assay using electrochemiluminescent (ECL) labelled probes. J. Virol. Meth. 1994;49:157–167. doi: 10.1016/0166-0934(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Vincent M., Xu Y., Kong H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004;5:795–800. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visca P., De Mori P., Festa A., Montrone M.L., Amicosante M., Pucillo L.P. Evaluation of the BDProbeTec strand displacement amplification assay in comparison with the AMTD II direct test for rapid diagnosis of tuberculosis. Clin. Microbiol. Infect. 2004;10:332–334. doi: 10.1111/j.1198-743X.2004.00818.x. [DOI] [PubMed] [Google Scholar]
- von Samson-Himmelstjerna G., Harder A., Schnieder T. Quantitative analysis of ITS2 sequences in trichostrongyle parasites. Int. J. Parasitol. 2002;32:1529–1535. doi: 10.1016/s0020-7519(02)00163-7. [DOI] [PubMed] [Google Scholar]
- Wacharapluesadee S., Hemachudha T. Nucleic-acid sequence based amplification in the rapid diagnosis of rabies. Lancet. 2001;358:892–893. doi: 10.1016/S0140-6736(01)06041-X. [DOI] [PubMed] [Google Scholar]
- Walker G.T., Fraiser M.S., Schram J.L., Little M.C., Nadeau J.G., Malinowski D.P. Strand displacement amplification-an isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992;20:1691–1696. doi: 10.1093/nar/20.7.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer G., Feng X., Tanriverdi S. Genotyping of Cryptosporidium parvum with microsatellite markers. Meth. Mol. Biol. 2004;268:177–187. doi: 10.1385/1-59259-766-1:177. [DOI] [PubMed] [Google Scholar]
- Witney A.A., Doolan D.L., Anthony R.M., Weiss W.R., Hoffman S.L., Carucci D.J. Determining liver stage parasite burden by real-time quantitative PCR as a method for evaluating pre-erythrocytic malaria vaccine efficacy. Mol. Biochem. Parasitol. 2001;118:233–245. doi: 10.1016/s0166-6851(01)00372-3. [DOI] [PubMed] [Google Scholar]
- Witt D.J., Kemper M., Stead A., Sillekens P., Ginocchio C.C., Espy M.J., Paya C.V., Smith T.F., Roeles F., Caliendo A.M. Analytical performance and clinical utility of a nucleic acid sequence-based amplification assay for detection of cytomegalovirus infection. J. Clin. Microbiol. 2000;38:3994–3999. doi: 10.1128/jcm.38.11.3994-3999.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittwer C.T., Reed G.H., Gundry C.N., Vandersteen J.G., Pryor R.J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin. Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- Wu D.Y., Wallace R.B. The ligation amplification reaction (LAR)-amplification of specific DNA sequences using sequential rounds of template-dependent ligation. Genomics. 1989;4:560–569. doi: 10.1016/0888-7543(89)90280-2. [DOI] [PubMed] [Google Scholar]
- Wu S.J., Lee E.M., Putvatana R., Shurtliff R.N., Porter K.R., Suharyono W., Watts D.M., King C.C., Murphy G.S., Hayes C.G., Romano J.W. Detection of dengue viral RNA using a nucleic acid sequence-based amplification assay. J. Clin. Microbiol. 2001;39:2794–2798. doi: 10.1128/JCM.39.8.2794-2798.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q., Ratnasinghe L.D., Hong H., Perkins R., Tang Z.Z., Hu N., Taylor P.R., Tong W. Decision forest analysis of 61 single nucleotide polymorphisms in a case-control study of esophageal cancer; a novel method. BMC Bioinformatics. 2005;6(Suppl. 2):4. doi: 10.1186/1471-2105-6-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F., Jabasini M., Zhu B., Ying L., Cui X., Arai A., Baba Y. Single-step quantitation of DNA in microchip electrophoresis with linear imaging UV detection and fluorescence detection through comigration with a digest. J. Chromatogr. A. 2004;1051:147–153. doi: 10.1016/j.chroma.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Yang J.H., Lai J.P., Douglas S.D., Metzger D., Zhu X.H., Ho W.Z. Real-time RT-PCR for quantitation of hepatitis C virus RNA. J. Virol. Meth. 2002;102:119–128. doi: 10.1016/s0166-0934(02)00007-1. [DOI] [PubMed] [Google Scholar]
- Zaytseva N.V., Montagna R.A., Lee E.M., Baeumner A.J. Multi-analyte single-membrane biosensor for the serotype-specific detection of Dengue virus. Anal. Bioanal. Chem. 2004;380:46–53. doi: 10.1007/s00216-004-2724-9. [DOI] [PubMed] [Google Scholar]