

Abstract
The seventh novel human infecting Betacoronavirus that causes pneumonia (2019 novel coronavirus, 2019-nCoV) originated in Wuhan, China. The evolutionary relationship between 2019-nCoV and the other human respiratory illness-causing coronavirus is not closely related. We sought to characterize the relationship of the translated proteins of 2019-nCoV with other species of Orthocoronavirinae. A phylogenetic tree was constructed from the genome sequences. A cluster tree was developed from the profiles retrieved from the presence and absence of homologs of ten 2019-nCoV proteins. The combined data were used to characterize the relationship of the translated proteins of 2019-nCoV to other species of Orthocoronavirinae. Our analysis reliably suggests that 2019-nCoV is most closely related to BatCoV RaTG13 and belongs to subgenus Sarbecovirus of Betacoronavirus, together with SARS coronavirus and Bat-SARS-like coronavirus. The phylogenetic profiling cluster of homolog proteins of one annotated 2019-nCoV protein against other genome sequences revealed two clades of ten 2019-nCoV proteins. Clade 1 consisted of a group of conserved proteins in Orthocoronavirinae comprising Orf1ab polyprotein, Nucleocapsid protein, Spike glycoprotein, and Membrane protein. Clade 2 comprised six proteins exclusive to Sarbecovirus and Hibecovirus. Two of six Clade 2 nonstructural proteins, NS7b and NS8, were exclusively conserved among 2019-nCoV, BetaCoV_RaTG, and BatSARS-like Cov. NS7b and NS8 have previously been shown to affect immune response signaling in the SARS-CoV experimental model. Thus, we speculated that knowledge of the functional changes in the NS7b and NS8 proteins during evolution may provide important information to explore the human infective property of 2019-nCoV.
Keywords: 2019-nCoV, Novel proteins, Phylogenetic tree, Phylogenetic profile
Abbreviations: 2019-nCoV, 2019 novel coronavirus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; HCoV-NL63, human coronavirus NL63; HCoV-229E, human coronavirus 229E; HCoV-OC43, human coronavirus OC43; ECoV-HKU1, human coronavirus HKU1; MERS-CoV, Middle East respiratory syndrome coronavirus; NS, Nonstructural protein; ORF, open reading frame; NF-κB, nuclear factor kappa B
Highlights
-
•
2019-nCoV is most closely related to BatCoV RaTG13.
-
•
2019-nCoV belongs to subgenus Sarbecovirus of Betacoronavirus.
-
•
Phylogenetic profiling analysis and structural characterization of 2019-nCoV genes.
-
•
Functional changes in NS7b and NS8 proteins help understand 2019-nCoV.
1. Introduction
In December 2019, the seventh human coronavirus, termed 2019 novel coronavirus (2019-nCoV) or severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was found in Wuhan, China. On February 8, 2020, the total number of infections and deaths due to 2019-nCoV globally was 34,439 and 720, respectively, according to the Johns Hopkins University Center for Systems Science and Engineering.
Coronaviruses are enveloped RNA viruses that infect many species, including humans, other mammals, and birds. After infection, the host may develop respiratory, bowel, liver, and neurological diseases (Weiss and Leibowitz, 2011; Cui et al., 2019). Coronaviruses are members of the order Nidovirales and subfamily Orthocoronavirinae. This subfamily is divided into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus. Generally, Alphacoronavirus and Betacoronavirus tend to infect mammals, while Gammacoronavirus and Deltacoronavirus typically infect birds. However, some Gammacoronavirus and Deltacoronavirus can infect mammals under specific conditions (Woo et al., 2012).
In immunocompromised individuals, infection with one of the four human coronaviruses—human coronavirus NL63 (HCoV-NL63), human coronavirus 229E (HCoV-229E), human coronavirus OC43 (HCoV-OC43), and human coronavirus HKU1 (ECoV-HKU1)—usually results in cold-like symptoms. These viruses can cause severe infections in some infants and the elderly. Due to the frequent interaction between wild animals and humans, wild animals are a common source of human zoonotic infections. SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) are zoonotic coronaviruses that can cause severe respiratory diseases in humans; both belong to Betacoronavirus (Su et al., 2016; Forni et al., 2017; Cui et al., 2019; Luk et al., 2019; Ramadan and Shaib, 2019). 2019-nCoV is the seventh coronavirus discovered that infects humans. It causes acute respiratory disease in respiratory infections. Immediately after its discovery, the complete genome sequence of 2019-nCoV was determined. The sequence (MN908947) was released by GenBank on 05 January 2020 (Lu et al., 2020). The sequence of 2019-nCoV is 96% identical, at the whole-genome level, to a bat coronavirus (Zhou et al., 2020).
The genomic characteristics and epidemiology of 2019-nCoV have been analyzed (Lu et al., 2020). Nine inpatient culture isolates were subjected to next-generation sequencing, and individual complete and partial 2019-nCoV genomic sequences were obtained. Phylogenetic analysis of these 2019-nCoV genomes and other coronaviruses was performed to determine the evolutionary history of the virus and to explore the origin of 2019-nCoV. At the first onset, homology modeling investigated the potential receptor-binding properties of the virus. However, SARS-CoV and MERS-CoV showed approximate similarities of 79% and 50% with 2019-nCoV, respectively. These findings indicated that there is not a close evolutionary relationship of 2019-nCoV with SARS-CoV and MERS-CoV. Thus, 2019-nCoV is considered the seventh novel human Betacoronavirus (Lu et al., 2020).
In this study, we comprehensively characterized the relationship of the translated proteins of 2019-nCoV to other species of Orthocoronavirinae. This was done using a combination of the phylogenetic tree constructed from the genome sequences and the cluster tree developed from the profiles retrieved from the presence and absence of homologs of ten 2019-nCoV proteins.
2. Methods
The genomes and the combination of genome and protein sequences were used to develop a phylogenetic tree and phylogenetic profiling, respectively. The dataset of the genomes of the Orthocoronavirinae subfamily was collected from the Refseq database using the Orthocoronavirinae NCBI taxonomy ID (txid2501931). This dataset contains representative complete genomes from each species of that subfamily. (Pruitt et al., 2007; Federhen, 2012) (Supplementary Table 1). Additionally, we collected genome sequences from Bat SARS-like coronavirus (MG772934 and MG772933) from NCBI and BetaCoV/bat/Yunnan/RaTG13/2013 (EPI_ISL_402131) from GISAID (http://www.GISAID.org). One species of the Okanivirinae subfamily, the yellow head virus, was also collected as an outgroup (Supplementary Table 1). The genome sequences were aligned using MAFFT multiple sequence alignment program provided at the XSEDE portal in the CIPRES Science Gateway with an automatic selection strategy (Miller et al., 2012; Katoh and Standley, 2013). A phylogenetic tree was constructed using the maximum likelihood method with RAxML-HPC BlackBox in the CIPRES Science Gateway (Stamatakis, 2006). The analysis used an automatic bootstrapping option using a general time-reversible substitution model with a gamma-shape parameter (GTR+ G). The model was selected as the best-fit model under the Akaike information criterion using ModelTest-NG (Darriba et al., 2020). Phylogenetic trees were viewed using FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
The annotated protein sequences of 2019-nCoV were collected from the data of one representative genome from NCBI (MN996527). We built a BLAST database with the retrieved genome sequences data using BLAST+ version 2.2.30 (Camacho et al., 2009). We then determined the presence and absence of homolog proteins of one representative set of annotated 2019-nCoV proteins against other genome sequences in a database using tblastn with a threshold of >50 and > 25 bits score for protein sequences >50 amino acids (aa) and < 50 aa in length, respectively. The results of the presence and absence of homolog proteins were converted into a binary matrix and used to build a clustering tree using ward hierarchical clustering method (Ward Jr, 1963) (Supplementary Table 2). Nonstructural protein (NS) 7b and NS8 local alignments were only positive in the Sarbecovirus subgenus sample, excluding the SARS coronavirus. Additionally, we predicted the structural properties of the 2019-nCoV NS7b protein, including the secondary structure and order-disorder propensity, using Jpred4 and DICHOT, respectively (Fukuchi et al., 2014; Drozdetskiy et al., 2015). We also predicted the structure using the contact assisted protein structure prediction (C-I TASSER) composite approach (Zhang et al., 2018). Additionally, we specifically collected the sequences that produced significant alignments of NS7b using the MEGA X software (Kumar et al., 2018).
3. Results and discussion
3.1. Phylogenetic tree
The phylogenetic analysis using complete genome sequences showed that 2019-nCoV was the most closely related to BatCoV RaTG13 and belonged to the Sarbecovirus subgenus of Betacoronavirus, together with SARS coronavirus and Bat-SARS-like coronavirus (BAT-SL-CoVZXC21 and BAT-SL-CoVZC45) with the full support of reliability (Fig. 1 ). Additionally, Hibecovirus with Bat Hp-betacoronavirus/Zhejiang2013, as the representative species, was the most closely related subgenus of Betacoronavirus to Sarbecovirus as compared to other subgenera, including Merbecovirus (under which MERS-CoV has been classified), Nobecovirus, and Embecovirus. These findings agree with previous phylogenetic tree and similarity plot data (Paraskevis et al., 2020). 2019-nCoV was found to be more closely related to the bat-infecting Sarbecovirus species, Bat SARS-like coronavirus, and BetaCoV RaTG13 than to the SARS coronavirus that infects humans. This indicated that 2019-nCoV more likely originated from bats. However, the Wuhan outbreak was first detected in December, which is a time of year when most bat species hibernate. Moreover, the Huanan seafood market, which is considered as ground zero of the outbreak, does not sell bats. Instead, it has been suggested that there is an animal mediator for virus transmission from bats to humans, similar to the previous cases of SARS-CoV and MERS-CoV, wherein the masked palm civet (Paguma larvata) and dromedary camel (Camelus dromedarius) act as intermediate hosts, respectively (Lu et al., 2020). Although coronaviruses can exchange genetic material during coinfection, a recent report described the lack of a mosaic relationship of 2019-nCoV to the closely related Sarbecovirus, indicating the lack of a recombination event in the emergence of 2019-nCoV (Paraskevis et al., 2020). Hence, 2019-nCoV likely emerged from the accumulation of mutations responding to altered selective pressures or from the infidelity of RNA polymerase perpetuated as replication-neutral mutations. These speculations need to be studied further.
Fig. 1.

Phylogenetic tree of representative species from the Orthocoronavirinae subfamily with Okavirus as an outgroup, constructed using maximum likelihood method. The asterisk indicates the fully supported reliability of maximum likelihood.
3.2. Phylogenetic profiling
A previously reported comprehensive similarity plot revealed notable mutational hotspots and conserved regions of the genome nucleotide positions of 2019-nCoV against closely related coronaviruses (Lu et al., 2020; Paraskevis et al., 2020). The present findings provide a different perspective of the similarity among Orthocoronavirinae species, using a cluster tree developed from the profiles retrieved from the presence and absence of homologs of ten 2019-nCoV proteins. This cluster was combined with the cladogram of a previously constructed phylogenetic tree (Fig. 2 ). Both the trees were consistent in their heatmap distributions. The tree of 2019-nCoV proteins comprised two clades. The first, indicated with a blue bar in Fig. 2, contained a group of conserved proteins in most Orthocoronavirinae species. These comprised Orf1ab polyprotein, Nucleocapsid protein, Spike glycoprotein, and Membrane protein. Spike and Orf1a regions of 2019-nCoV were previously shown to have the lowest sequence identity as compared to the closely related coronavirus species (Lu et al., 2020; Paraskevis et al., 2020). However, since the translated Spike glycoprotein and Orf1ab polyprotein from these regions are very long, the sequence similarity is still sufficient to classify them as homologs. In contrast, another clade, indicated by the green bar in Fig. 2, comprised proteins specific to Sarbecovirus for all proteins in this clade and Hibecovirus for envelope protein only. This clade included proteins that were not completely conserved by all Orthocoronavirus. Two (NS7b and NS8) of five nonstructural proteins were specific for 2019-nCoV and its closely related species, BatCoV RaTG13 and Bat-SARS-like coronavirus (BAT-SL-CoVZXC21 and BAT-SL-CoVZC45). The other three nonstructural proteins (NS3, NS6, and NS7a) were also detected in the SARS coronavirus. Based on these results, we propose that the comprehensive analysis of nonstructural proteins, especially NS7b and NS8, may provide new insight into the properties of 2019-nCoV.
Fig. 2.

Phylogenetic profiling of the annotated 2019-nCoV proteins combined with the maximum likelihood cladogram of the Orthocoronavirinae subfamily with Okavirus as the outgroup. The heatmap indicates the binary matrix of the homolog proteins of 2019-nCoV against other species in the dataset, with black and white colors as presence and absence, respectively. The bit pattern was arranged following the vertical and horizontal trees. The vertical tree is a phylogenetic profiling tree constructed from a binary matrix of the presence and absence of homolog proteins. It has two clades, indicated by blue and green bars. The horizontal tree is the cladogram of the maximum likelihood tree, as shown in Fig. 1, with a collapsed clade of 2019-nCoV. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
As shown in Fig. 2, NS7b and NS8 of 2019-nCoV, BatCoV RaTG13, and Bat-SARS-like coronavirus were distinct from other species of Orthocoronavirus. In SARS-CoV, NS7b is an integral protein localized in the Golgi compartment. The protein is packaged into SARS-CoV particles (Schaecher et al., 2007). Interestingly, open reading frame (ORF) 7b, but not ORF 7b deletion, induces interferon (IFN)-dependent reporter gene expression as well as apoptosis and the type I IFN response (Pfefferle et al., 2009). Moreover, the deletion of ORF 7b enhance virus growth (Pfefferle et al., 2009). Thus, we speculate that the property of the non-conserved NS7b in 2019-nCoV may affect the human infective property of the virus. Similarly, the existence of 29 nucleotide deletions in ORF 8b has been described in SARS-CoV (Oostra et al., 2007). A study involving MERS-CoV described that ORF 8b strongly antagonizes the INF-beta (β) promoter and ORF4b and 8b significantly suppress IFN induction (Lee et al., 2019b). Accessory proteins 8b and 8ab of SARS-CoV can suppress the INF-β signaling pathway (and thus interferon production) by their participation in the ubiquitin-mediated rapid degradation of INF regulatory factor 3 (IRF3) (Wong et al., 2018). In contrast, when we focused on MERS-CoV from bats and camels, ORF 8b antagonized melanoma differentiation-associated protein 5-mediated nuclear factor kappa B (NF-κB) activation. ORF 8b strongly inhibited TANK-binding kinase 1-mediated induction of NF-κB signaling, but not IκB kinase epsilon and IRF3-mediated activations (Lee et al., 2019a). Thus, we speculate that the properties of the accessory proteins, NS7b and NS8, in 2019-nCoV may affect its ability to infect humans. Further studies are required to confirm this speculation.
NS7b is a short peptide of 43 residues. A three-dimensional structure is often difficult to obtain from such a short peptide. We predicted the three-dimensional structure of the queried NS7b amino acid sequence using DICHOT and C-I-TASSER (Supplementary Fig. 1, Fig. 3 ) (Fukuchi et al., 2014; Zhang et al., 2018); a protein family (PF11395) was found, but no known three-dimensional structure was found. The secondary structure of this query was also predicted using Jpred4 (Supplementary Fig. 2) (Drozdetskiy et al., 2015). The secondary structure was predicted to be an α-helix, but that this very likely does not occur depending on the environment (Fig. 3). The alignment of this protein revealed three polymorphism sites between 2019-nCoV, BatCoV RaTG13, and Bat-SARS-like coronavirus sequences (BAT-SL-CoVZXC21 and BAT-SL-CoVZC45) (Supplementary Fig. 3).
Fig. 3.
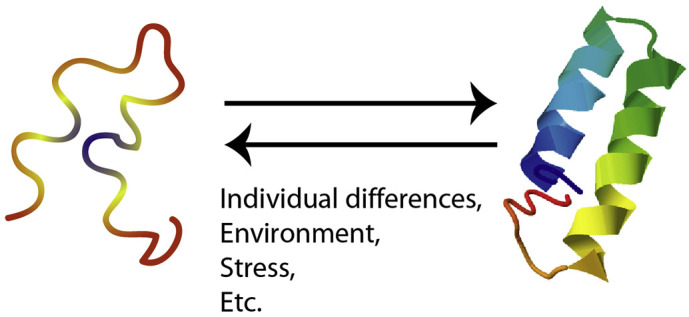
The model of nonstructural-structural transition of 2019-nCoV nonstructural protein 7b. The predicted protein structure of 2019-nCoV nonstructural protein 7b by C-I TASSER is shown as the helix structure protein.
In summary, some nonstructural proteins were conserved and others were not conserved between 2019-nCoV and SARS-CoV. By focusing on the 2019-nCoV-specific proteins, NS7b and NS8, we proposed a combination of phylogenetic profiling analysis and structural characterization of the genes that were specifically expressed in 2019-nCoV and the closely related bat coronavirus. The data provide insight for further characterization of the infective properties of this virus.
The following are the supplementary data related to this article.
Dataset used in this study including accession or ID, taxonomy, and name of data.
Binary matrix of profiling data.
Supplementary material
Funding
This work was supported by the MEXT-Supported Program for the Strategic Research Foundation at Private Universities (grant number S1511028 to T.I) and the Takeda Science Foundation.
Declaration of Competing Interest
The authors declare no competing interest.
Acknowledgments
We want to thank Dr. Motonori Ota, Dr. Satoshi Fukuchi, Dr. Kota Kasahara, and Dr. Takeshi Kikuchi for their support and helpful comments.
References
- Camacho C., Coulouris G., Avagyan V. BLAST+: architecture and applications. BMC Bioinform. 2009;10(1):421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D., Posada D., Kozlov A.M. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020;37(1):291–294. doi: 10.1093/molbev/msz189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdetskiy A., Cole C., Procter J. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federhen S. The NCBI taxonomy database. Nucleic Acids Res. 2012;40(D1):D136–D143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forni D., Cagliani R., Clerici M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017;25:35–48. doi: 10.1016/j.tim.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi S., Amemiya T., Sakamoto S. IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014;42:D320–D325. doi: 10.1093/nar/gkt1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Li M. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.Y., Bae S., Myoung J. Middle East respiratory syndrome coronavirus-encoded accessory proteins impair MDA5-and TBK1-mediated activation of NF-κB. J. Microbiol. Biotechnol. 2019;29(8):1316–1323. doi: 10.4014/jmb.1908.08004. [DOI] [PubMed] [Google Scholar]
- Lee J.Y., Bae S., Myoung J. Middle East respiratory syndrome coronavirus-encoded ORF8b strongly antagonizes IFN-β promoter activation: its implication for vaccine design. J. Microbiol. 2019;57(9):803–811. doi: 10.1007/s12275-019-9272-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;S0140-6736(20):30251–30258. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk H.K.H., Li X., Fung J. Molecular epidemiology, evolution, and phylogeny of SARS coronavirus. Infect. Genet. Evol. 2019;71:21–30. doi: 10.1016/j.meegid.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.A., Pfeiffer W., Schwartz T. 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the Extreme to the Campus and beyond. 2012. The CIPRES science gateway: enabling high-impact science for phylogenetics researchers with limited resources. In proceedings of the; pp. 1–8. [Google Scholar]
- Oostra M., de Haan C.A., Rottier P.J. The 29-nucleotide deletion present in human but not in animal severe acute respiratory syndrome coronaviruses disrupts the functional expression of open reading frame 8. J. Virol. 2007;81(24):13876–13888. doi: 10.1128/JVI.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraskevis D., Kostaki E.G., Magiorkinis G. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect. Genet. Evol. 2020;104212 doi: 10.1016/j.meegid.2020.104212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferle S., Krähling V., Ditt V. Reverse genetic characterization of the natural genomic deletion in SARS-coronavirus strain Frankfurt-1 open reading frame 7b reveals an attenuating function of the 7b protein in-vitro and in-vivo. Virol. J. 2009;6(1):131. doi: 10.1186/1743-422X-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35(suppl_1):D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan N., Shaib H. Middle East respiratory syndrome coronavirus (MERS-CoV): a review. Germs. 2019;9:35–42. doi: 10.18683/germs.2019.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaecher S.R., Mackenzie J.M., Pekosz A. The ORF7b protein of severe acute respiratory syndrome coronavirus (SARS-CoV) is expressed in virus-infected cells and incorporated into SARS-CoV particles. J. Virol. 2007;81(2):718–731. doi: 10.1128/JVI.01691-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Su S. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J.H., Jr. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963;58(301):236–244. [Google Scholar]
- Weiss S.R., Leibowitz J.L. Coronavirus pathogenesis. Adv. Virus Res. 2011;81:85–164. doi: 10.1016/B978-0-12-385885-6.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong H.H., Fung T.S., Fang S. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology. 2018;515:165–175. doi: 10.1016/j.virol.2017.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012;86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Mortuza S.M., He B., Wang Y., Zhang Y. Template-based and free modeling of I-TASSER and QUARK pipelines using predicted contact maps in CASP12. Proteins. 2018;86:136–151. doi: 10.1002/prot.25414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G. A pneumonia outbreak associated with a new coronavirus of probable vat origin. Nature. 2020 doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset used in this study including accession or ID, taxonomy, and name of data.
Binary matrix of profiling data.
Supplementary material