

Abstract
Within a few minutes of an intravenous injection of a lipopolysaccharide (LPS) into mice, platelets accumulate, largely in the lung. At higher doses, LPS induces rapid shock (within 10 min), leading to death within 1 h. This type of shock differs from so-called endotoxin shock, in which shock signs and death occur several hours or more later. Here, we found that platelet depletion (by a monoclonal anti-platelet antibody) prevented LPS-induced rapid shock, but increased delayed lethality. In Japan, glycyrrhizin (GL), a compound isolated from licorice, is daily and slowly infused intravenously into chronic hepatitis C patients. A single bolus intravenous injection into mice of GL (200 mg/kg or less) shortly before (or simultaneously with) LPS injection reduced the pulmonary platelet accumulation and the severity of the rapid shock, and prevented death in both the early and later periods. GL itself, at 400 mg/kg, produced no detectable abnormalities in the appearance or activity of mice. Intraperitoneal injection of aspirin or dexamethasone had only marginal effects on LPS-induced platelet responses and lethality. These results suggest that platelets play important roles in the development of both the rapid and delayed types of shock induced by LPS. Although the mechanism by which GL suppresses platelet responses and delayed lethality remains to be clarified, GL might provide a strategy for alleviating the acute respiratory distress syndrome seen in sepsis. Our results may also support the proposal by Cinatl et al. [Cinatl J, Morgenstern B, Bauer G, Chandra P, Ravenau H, Doerr HW. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 2003; 361: 2045–6.] that GL may be an effective drug against severe acute respiratory syndrome.
Keywords: Glycyrrhizin, Lipopolysaccharide, Platelets, Shock, Acute respiratory distress syndrome, Severe acute respiratory syndrome
1. Introduction
Approximately 200,000 patients develop Gram-negative sepsis each year in the USA [1]. Of these, about one quarter develop acute respiratory distress syndrome (ARDS), and mortality is estimated at 60–90%. Despite the current detailed understanding of the pathology of sepsis, there are still few effective strategies against this condition [2], [3]. Interestingly, recent studies suggest that an inhibition of blood coagulation is beneficial [4], [5]. In addition to neutrophils and macrophages, an interaction between platelets and pulmonary endothelial cells may be involved in ARDS [6]. Platelets also play an important role in the development of multiple organ failure in sepsis [7].
Using 5-hydroxytryptamine (5HT) as a marker for platelets, we have recently demonstrated that within a few minutes of an intravenous injection of lipopolysaccharide (LPS) into mice, platelets accumulate in the lung and liver (particularly in the lung) [8]. At lower doses of LPS, these platelets soon return to the circulation, but at higher doses they are broken down, with their large-scale breakdown inducing lethal shock [9], [10]. This response (the magnitude of which depends on the strain of mouse, the structure of the LPS, and its dose) is induced via an activation of the complement system [9], [10]. Thus, the rapid shock induced by LPS differs from the well-known “endotoxin shock” seen in mice in that in the latter type, shock signs appear 2–3 h or more later, with death occurring several hours or more later [11]. It is thought that LPS is involved as a mediator in several of the signs seen in sepsis (including shock) and that various inflammatory cytokines (such as TNF and IL-1) are also involved in mediating these signs [12], [13], [14].
Glycyrrhizin (GL) is a major constituent of aqueous extracts obtained from the root of the licorice plant. Orally administered GL is converted to its aglycon, 18β-glycyrrhetinic acid (GA), and GL and/or GA exhibit a variety of pharmacological activities, including anti-virus activity [15], [16] and anti-hepatitis activity [17]. They also have anti-inflammatory activities via suppressions of prostaglandin production [18], histamine release [19], and histamine synthesis [20], [21]. In addition, GL and/or GA inhibit the complement system [22], [23]. In Japan, GL is given to chronic hepatitis C patients by daily slow intravenous infusions over a long period [24], [25]. Here, we examined the effects of GL and GA on the platelet response and ensuing rapid shock induced in mice by LPS.
2. Materials and methods
2.1. Animals and materials
Male BALB/c mice (6–7 weeks old) were provided by the animal facility of our university. LPS from the Klebsiella O3 strain LEN-1 (S type) was prepared by the phenol-water method [26], dissolved in sterile saline (0.1 or 0.5 mg/ml), then injected (i.v.) via the tail vein (0.1 ml/10 g body weight). A monoclonal anti-mouse platelet antibody, Pm-1, was kindly provided by Dr. T. Nagasawa (Division of Hematology, University of Tsukuba, Japan) [27]. Although the molecular antigen for Pm-1 has not been identified, Pm-1 has been shown to deplete platelets in mice [28]. Control IgG was prepared from normal mouse serum by precipitation with ammonium sulfate, followed by dialysis of the precipitant. Glycyron Injection (Minophagen Pharmaceutical, Tokyo, Japan) (a 26.5 mg/ml solution of ammonium glycyrrhizinate, equivalent to 20 mg/ml of GL) was diluted with saline, then injected intravenously (i.v.) or intraperitoneally (i.p.) (0.1 ml/10 g body weight). GA (Tokyo Kasei, Tokyo, Japan) was suspended in 1% carboxymethyl cellulose and given into the stomach through a syringe (0.1 ml/10 g body weight). Aspirin (Sigma, St. Louis, MO, USA) was dissolved in saline by adding 1 M NaOH, with the pH of the solution being adjusted to 7 with 1 M HCl. Dexamethasone (Sigma) was dissolved in saline. Before the experiment, mice were moved to a room with a temperature of 26–28 °C, and kept there for 1 h after injection of LPS, because the rapid platelet responses to LPS are reduced at room temperatures lower than that (unpublished data). In experiments intended to last a long time, mice were returned to a normal animal room (23±1 °C) at 1 h after the LPS injection. All procedures complied with the Guidelines for the Care and Use of Laboratory Animals in Tohoku University.
2.2. Platelet counting
Two or three drops of blood from each decapitated mouse were directly collected into a pre-weighed test tube containing 1.0 ml of 4 mM EDTA in 0.01 M phosphate-buffered saline (pH 7.0). The tube plus blood was weighed, and the volume of blood was estimated from the weight of the blood. The number of platelets was then ascertained using a cell counter (Sysmex SF-3000; Toa Medical Electronics, Kobe, Japan) and expressed as platelet count/ml of blood.
2.3. Determination of 5HT
The 5HT levels in the blood and lung were determined as previously described [8]. Briefly, after collecting the blood for measuring the platelet count (see above), the next two or three drops of blood from the same mouse were collected into another pre-weighed tube containing 3 ml of 0.4 M HClO4, 0.1% N-acetylcysteine-HCl, and 4 mM EDTA-2Na. After reweighing, the platelets were destroyed by sonication, and each tube was cooled in an ice bath. Lungs were rapidly removed and kept in a jar containing dry ice until needed. The determination of the 5HT level in the blood was carried out soon after the blood was collected, whereas the 5HT levels in the lung were determined within 2 days of collection. After 5HT had been separated by column chromatography, it was measured fluorometrically as previously described [29]. The amount of 5HT in the blood or lung was expressed as nmol/g of blood or lung.
2.4. Scoring of the rapid shock induced by LPS
The severity of the rapid shock seen in each mouse within 60 min of an LPS injection was scored as described elsewhere [10], the scoring system being: 0 (no signs of shock), 1 (staggering), 2 (crawling and prostration), 3 (prostration and weak convulsions), 4 (prostration and strong convulsions), 5 (death). The shock-scoring data are expressed as scores for individual mice, or sometimes as mean±SD, as appropriate. The estimation of the scores was performed by a blinded assistant.
2.5. Statistical analysis
Experimental values for 5HT levels and platelet count are given as mean±S.D. The statistical significance of differences was assessed using a Student's unpaired t-test or a Bonferroni post-hoc test. The difference in shock scores between two experimental groups was analyzed using a Ridit (relative to an identified distribution) test [30], a non-parametric test.
3. Results
3.1. Effects of Pm-1
Pm-1 induced an almost complete depletion of platelets from the blood within 3 h after its intravenous injection (Fig. 1 ). The injection of Pm-1 also decreased 5HT in the blood, indicating that most of the 5HT in the blood is included in platelets. In this experiment, weak signs of shock (score 1–2) appeared at 10–60 min after the injection of Pm-1 itself. Control IgG did not change the platelet count or the 5HT in the blood (Fig. 1).
Fig. 1.
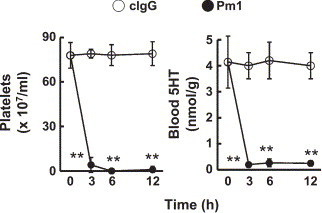
Effects of Pm-1 on platelets and 5HT in the blood. Control IgG or Pm-1 (each, 12 mg/kg) was injected (i.v.) into mice. The blood of each mouse was collected by decapitation at the time indicated, then subjected to assays of platelet count and 5HT. Each value is the mean±S.D. from 4 mice. *P<0.01 vs. control IgG.
In mice given control IgG, an intravenous injection of 1 mg/kg of LPS (given 12 h after the control IgG injection) induced severe shock signs (scores 3–4) within 10 min (Table 1 ). However, most of the mice were alive 24 h later. In contrast, in mice given Pm-1 this dose of LPS induced no apparent shock signs within 1 h after the LPS injection; however, most of these mice died within 24 h (Table 1).
Table 1.
Effects of Pm-1 on shock signs induced by LPS
| Treatment | Shock score in each mouse | Survival after 24 h |
|---|---|---|
| Control IgG | 3,4,4,4,4 (3.8±0.4a) | 4/5 |
| Pm-1 | 0,0,0,0,1 (0.2±0.4*) | 1/5 |
Control IgG or Pm-1 (each, 12 mg/kg) was injected (i.v.) into mice, and 12 h later LPS was injected (i.v.) at a dose of 1 mg/kg. “Shock score” indicates maximum score allocated for the shock signs observed within 60 min after the LPS injection.
Mean±SD.
P<0.01 vs control IgG.
3.2. Effects of GL
Before describing our results on platelets, we should emphasize the following point. As described previously [31], the half-life of the intravenously injected 5HT in the blood is ≪10 s, although we cannot determine this time accurately. When we injected 5HT at a dose of 25 nmol per mouse, we could not detect any increase in 5HT in the blood at 10 s after the 5HT injection. Thus, the value given in the following sections for the amount of 5HT in the blood and lung may represent almost entirely the 5HT present within the platelets themselves.
As shown in Fig. 2 , LPS at 1 mg/kg induced a rapid decline in both platelets and 5HT in the blood within 4 min after its injection, followed by a partial recovery at 10 min. Reciprocal changes were seen in 5HT in the lung. Following these changes, severe shock signs were seen about 6 min after the LPS injection (see inset table in Fig. 2). In Japan, GL is given by an intravenous drip infusion to patients with hepatitis. Thus, we first examined the effects of GL injected intravenously 10 min before an intravenous injection of LPS. In this experiment, GL (200 mg/kg) significantly reduced the changes in the blood and lung as well as the shock signs, that induced by 1 mg/kg of LPS (Fig. 2). GL itself at this dose had no significant effect on either the platelet count or the 5HT in the blood and lung (Fig. 3 ).
Fig. 2.
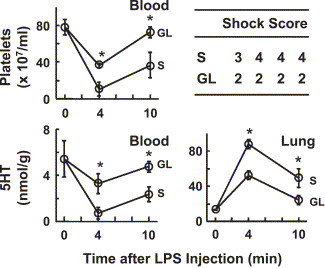
Effects of GL on LPS-induced platelet responses and shock. Mice were given saline (S) or GL (200 mg/kg, i.v.), and 10 min later LPS was injected (1 mg/kg, i.v.). The mice were killed at 4 or 10 min after the injection of LPS. Then, platelet count and 5HT concentrations in the blood and lung were determined. Each value is the mean±S.D. from 4 mice. *P<0.05 vs. saline group. The inset table shows maximum scores allocated for the shock signs observed in each mouse during the experiment. The difference in the scores between the S and GL groups was statistically significant (P<0.05).
Fig. 3.
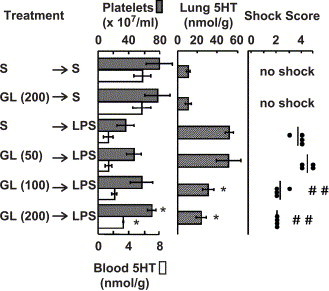
Dose-dependent effects of GL. Mice were injected (i.v.) with saline (S) or the indicated dose of GL, and 10 min later LPS was injected (1 mg/kg, i.v.). The mice were killed at 10 min after the injection of LPS. Then, platelet count, 5HT concentrations in the blood and lung, and shock scores were determined. Each value is the mean±S.D. from 4 mice. *P<0.05 and ##<0.01 vs. S→LPS group.
Intravenous injection of 100 mg/kg of GL at 10 min before an LPS injection (1 mg/kg) also reduced both the 5HT increase in the lung and the severity of the shock (Fig. 3). However, when 200 mg/kg of GL was injected intravenously at an earlier time-point (viz. 30 min before the LPS), there was no significant effect (data not shown). Intraperitoneal injection of GL (100 or 200 mg/kg) at 30 or 60 min before LPS injection was not effective, either (data not shown).
Next, we examined the effects of simultaneous injection of GL and LPS. Lower doses of GL, at 40 and 80 mg/kg, significantly reduced the severity of the rapid shock induced by LPS (1 mg/kg), and they tended to improve the survival rate at 24 h after the injection of LPS (Table 2 ).
Table 2.
Effects of simultaneous injection of GL and LPS on the severity of rapid shock induced by LPS
| GL dose (mg/kg) | Shock score in each mouse | Survival after 24 h |
|---|---|---|
| 0 | 3,3,4,5,5 (4.0±1.0a) | 3/5 |
| 20 | 2,2,3,5,5 (3.4±1.5) | 3/5 |
| 40 | 2,2,3,4,4 (3.0±1.0)* | 4/5 |
| 80 | 2,2,2,3,4 (2.6±0.9)* | 5/5 |
A mixture of GL and LPS was injected (0.25 ml/mouse, i.v.). The dose of GL was 20, 40, or 80 mg/kg, while that of LPS was 1mg/kg. “Shock score” indicates maximum score allocated for the shock signs observed within 60 min after the LPS injection.
Mean±SD.
P<0.05 vs GL 0.
As shown in Table 3 , intravenous injection of 5 mg/kg of LPS caused death in all mice within 1 h after the injection. Pretreatment with GL (200 mg/kg, intravenously) at 10 min before the LPS injection completely prevented the lethal effect of LPS, although LPS still induced shock scores of 2 to 4 in these mice.
Table 3.
Effects of GL on the lethality induced by a lethal dose of LPS
| Treatment | Shock score in each mouse | Survival after 24 h |
|---|---|---|
| Saline+LPS | 5,5,5,5,5 (5.0±0.0a) | 0/5 |
| GL+LPS | 2,3,3,4,4 (3.2±0.8)* | 5/5 |
Saline or GL(200 mg/kg) was injected (i.v.), and 10 min later 5 mg/kg of LPS was injected (i.v.). “Shock score” indicates maximum score allocated for the shock signs observed within 60 min after the LPS injection.
Mean±SD.
P<0.01 vs Saline+LPS.
GL itself was lethal at 600 mg/kg or more, although at 400 mg/kg it produced no detectable abnormalities in the appearance or activity of mice (Fig. 4 ). However, those mice that survived for 1 h after the GL injection (at 600 or 800 mg/kg) were all still alive at 24 h. These mice all looked healthy at 2 h or more after the injection of GL.
Fig. 4.
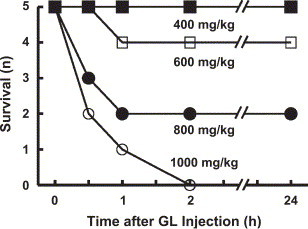
Lethality induced by GL alone. GL was injected i.v. at the doses indicated.
Unlike that of GL, the solubility of GA in water is very low, and so GA is used clinically as an oral drug. We therefore tested the effect of orally administered GA on the LPS-induced rapid shock (Fig. 5 ). Under our conditions, GA slightly reduced the severity of the shock only when it was administered at 500 mg/kg at 1 h before the LPS injection.
Fig. 5.

Effects of GA. Mice were given GA orally at the indicated doses, then injected with LPS (1 mg/kg, i.v.) at the indicated times. “Shock score” indicates maximum score allocated for the shock signs observed within 30 min of the LPS injection. #P<0.05 vs. GA 0.
3.3. Effects of aspirin and dexamethasone, alone or in combination with GL
Non-steroidal anti-inflammatory drugs (NSAIDs) and glucocorticoids are widely used against a variety of inflammatory diseases. Aspirin is a typical NSAID and anti-platelet drug, while dexamethasone is a typical synthetic glucocorticoid. In various murine inflammation models, aspirin and dexamethasone are given intraperitoneally at 50–150 and 0.2–5 mg/kg, respectively. We previously reported that at these doses, aspirin and dexamethasone were effective at reducing the platelet responses induced by a low dose (0.25 mg/kg) of the same LPS as that used in the present study [8] (this dose of the LPS produces shock scores of 0 to 1). Thus, in our next experiment we tested the effects of these drugs on both the platelet response and the severity of the rapid shock induced by 1 mg/kg LPS. At the above doses, these drugs increased the return of platelets to the circulation at 10 min after the injection of LPS, but their effects, although statistically significant, were not large (Fig. 6 ). The effects of these drugs on the shock signs induced by 1 mg/kg of LPS were not significant (Fig. 7 ). When aspirin or dexamethasone was given in combination with GL, however, each combination tended to reduce the severity of shock more effectively than GL alone (Fig. 7).
Fig. 6.
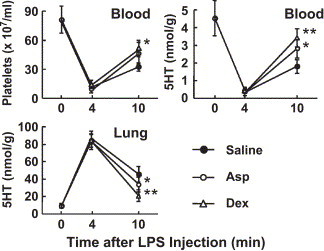
Effects of aspirin and dexamethasone on LPS-induced platelet responses. Saline, aspirin (Asp, 100 mg/kg), or dexamethasone (Dex, 1 mg/kg) was injected (i.p.) at 90 min before an injection of LPS (1 mg/kg, i.v.). The data are the mean±SD from 5 mice. *P<0.05 and **P<0.01 vs. Saline group.
Fig. 7.
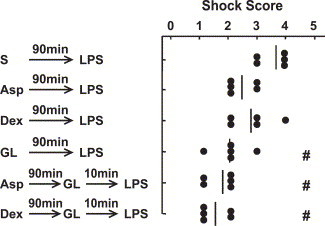
Effects of GL in combination with aspirin or dexamethasone. Saline (S), aspirin (Asp, 100 mg/kg), dexamethasone (Dex, 1 mg/kg), or GL (200 mg/kg) was injected (i.p.) at 90 min before injection of LPS (1 mg/kg, i.v.). In other mice, GL (200 mg/kg) was injected (i.v.) at a time 90 min after the Asp or Dex injection and 10 min before the LPS injection. “Shock score” indicates maximum score allocated for the shock signs observed within 30 min after the LPS injection. The data are individual scores and the mean from 5 mice. #P<0.05 vs. S→LPS.
Finally, we compared the effects of GL, aspirin, and dexamethasone at the above doses on the lethality induced by 5 mg/kg of LPS (Fig. 8 ). Although, as shown in Fig. 7, the effects of aspirin and dexamethasone on the LPS-induced rapid shock were marginal, these drugs prolonged survival time in mice given a lethal dose of LPS. On the other hand, the mice given GL all survived for at least 48 h after the LPS injection.
Fig. 8.
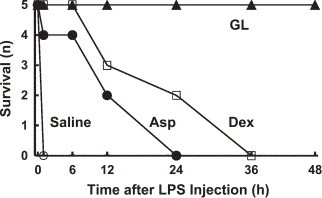
Effects of GL, aspirin, and dexamethasone on the lethality induced by a lethal dose of LPS. Saline or GL (200 mg/kg) was injected (i.v.) 10 min before an injection of LPS (5 mg/kg, i.v.), while aspirin (Asp, 100 mg/kg) or dexamethasone (Dex, 1 mg/kg) was injected (i.p.) 90 min before an injection of LPS (5 mg/kg, i.v.).
4. Discussion
The results obtained from the present experiment using Pm-1, a monoclonal antibody directed toward murine platelets, suggest that platelets are critical for the development of the rapid shock induced by LPS. However, the results suggest that they may also play an important role in helping the animal to resist the delayed lethality induced by LPS, the so-called “endotoxin shock”. Our results demonstrate that GL is effective at suppressing not only the rapid shock induced by LPS, but also the delayed “endotoxin shock”. In experimental models like that employed in the present study, we can distinguish between these rapid and delayed types of shock, because the LPS is given to animals by a single injection. In human patients, however, the level of endotoxin may increase progressively with the increase in bacterial numbers, and the pathology of Gram-negative sepsis may also develop progressively. Therefore, when the two types of shock occur in the same patient, distinguishing between them will be difficult. We speculate that infection with Gram-negative bacteria (or with LPS derived from them) may stimulate mechanisms leading to both types of shock. In the present study, we directed our attention mainly toward the rapid platelet responses to LPS and the associated rapid shock.
We earlier proposed that the lectin pathway that forms C3 convertase from C4 and C2 may be involved in the LPS-induced accumulation of platelets in the lung, while the pathway from C5 to C9 is involved in the subsequent platelet destruction and rapid shock [9], [10]. Interestingly, Kroes et al. [22] reported that both GL and GA inhibit human complement C2 in vitro, while Fujisawa et al. [23] found that GL inhibits human C5 or a later stage of the complement cascade in vitro. These effects are seen at 0.05–0.5 μmol/ml (about 0.05–0.5 mg/ml) of GL (this level of GL might have been attained in the blood in our in vivo experiment, as described below). In the present study, GL inhibited the LPS-induced accumulation of platelets in the lung. In addition, in GL-treated mice the platelet count in the blood had returned almost to its initial level at 10 min after the LPS injection, and the severity of the shock was markedly reduced. These results suggest that GL may inhibit both the early and later stages of the lectin pathway, although other anti-inflammatory effects of GL might also be involved. Interestingly, GL has recently been shown to inhibit thrombin-induced platelet aggregation both in vitro [32] and in vivo models of thrombosis in rats [33]. The dose- and time-dependent effects of GL on in vivo thrombosis that were observed in the latter study were very similar to those seen in the present study. The complement system is composed of several serine proteases [34], [35]. Upon recognition of pathogens, mannose-binding lectin (MBL) and ficolins trigger the activation of the lectin pathway through MBL-associated serine proteases (MASPs) [35]. We have shown that activation of the lectin pathway is involved in the LPS-induced activation of platelets [9], [10]. MASP-1 has thrombin-like activity [36] and thrombin is also a serine protease. Thus, it is likely that GL might exhibit its inhibitory action on the lectin pathway through its inhibitory effect on serine proteases. It should also be noted that many in vivo platelet responses are considered to be mediated, directly or indirectly, by activated complements, and the terminal complement proteins have been suggested to generate thrombin [37]. In any event, we need to clarify the mechanism (possibly involving other anti-inflammatory effects of GL) by which GL suppresses the rapid platelet responses to LPS as well as the delayed lethality induced by LPS.
GL, at 100–200 mg/kg, was effective only when it was injected intravenously shortly before an LPS injection (i.e., at 10 min before, but not at 30 or 60 min before LPS), suggesting that an effective level of GL was not present at the later times. Indeed, lower doses of GL (40 and 80 mg/kg) were effective when injected (i.v.) simultaneously with LPS. These lower doses are equivalent to about 0.5–1 mg/ml in the blood (since the average mouse body weight is about 25 g and its blood volume about 2 ml). In chronic hepatitis C patients, GL is slowly infused intravenously at 2–4 mg/kg/day for a long period [24], [25], which would yield a concentration in the blood of about 0.025–0.05 mg/ml. Compared to this human dose, our murine doses seem to be very high. However, it should be noted that pharmacokinetic profiles differ greatly between mice and humans. This difference may be partly due to the difference in their heart rates: 50–70 and 400–600 beats/min in humans and mice, respectively [38]. Thus, the blood concentration of drugs may decrease more rapidly in mice than in humans, which means that larger doses are needed in the former than in the latter. For example, in various murine inflammation models aspirin and dexamethasone are given intraperitoneally at about 50–150 and 0.2–5 mg/kg, respectively, while in humans the single oral doses of these drugs are roughly 10–20 and 0.01–0.05 mg/kg, respectively. Moreover, more than 80% of GL is cleared from the blood within 10 min of its intravenous bolus injection into mice [39], while the initial half-life of GL in the blood is roughly 1 h in humans [40]. In view of the information about in vitro concentrations given above, the slow intravenous infusion of GL typically used in humans might be expected to provide an effective blood concentration for the suppression or prevention of the platelet responses to LPS in human patients infected with Gram-negative bacteria.
Our results showed that GL itself was neither lethal nor toxic when injected intravenously into mice at 400 mg/kg. Those mice that survived for 1 h after a GL injection (at 600 or 800 mg/kg) were (a) all still alive at 24 h, (b) all apparently healthy at 2 h or more after the injection of GL, and (c) displayed no detectable abnormalities in either appearance or activity. Thus, the hypertonic nature of GL solutions might be involved, at least in part, in the rapid death we observed at 600 mg/kg or more of GL.
In some in vitro experiments, GA has been shown to be more potent than GL [18], [19], [22]. Although we have no relevant data on mice, it has been reported that in rats the plasma levels of GA are <0.1 and <1 μg/ml at 4 and 12 h, respectively, after either oral administration or intravenous injection of GL at 10 mg/kg [41]. However, in the present study the effect of an oral administration of GA (even at a large dose) was only marginal, suggesting that GL acts, not as GA, but as GL itself. It is also likely that if we could inject GA intravenously, GA might be effective at reducing the actions of LPS.
Although NSAIDs and glucocorticoids have been shown to protect experimental animals against endotoxin shock, the clinical usefulness of these drugs is controversial [42], [43], [44]. In the present study, although aspirin and dexamethasone were effective to some degree at promoting the return of platelets to the circulation (suggesting that they reduced the degradation of platelets in the lung and/or liver), their effects were marginal, and their effects, if any, on the rapid shock were also marginal. In addition, it should be noted that glucocorticoids strongly suppress immune responses. In contrast, the side effects of GL are few and can be controlled, and it has been suggested that GL enhances cell-mediated immunity by enhancing the production of Th1 cytokines such as interferon [45] or interleukin-12 [46], [47]. Incidentally, recent studies have demonstrated that histamine suppresses Th1 responses, but enhances Th2 responses [48], [49]. Interestingly, GL suppresses both histamine release [19] and histamine synthesis [20], [21], suggesting that GL may, at least in part, enhance Th1 responses via a suppression of histamine release and/or synthesis.
Pulmonary embolism is a very common condition (about 500,000 events per year in the USA) with a variety of risk factors [50], and platelets are important components of pulmonary emboli [51]. As described in Introduction, many patients develop Gram-negative sepsis, and such patients have a high incidence of ARDS. Intravenous injection of various Gram-positive bacteria themselves also induces rapid shock [52], and we have shown that platelets are involved in this shock, too [53]. Infusion of Streptococcus sanguis into rabbits has been shown to induce a marked accumulation of platelets in the lung [54]. Fungal cells also induce platelet responses similar to those induced by LPS (unpublished data). In view of the above, our findings may help in devising a strategy against not only Gram-negative sepsis, but also the pulmonary thromboembolism induced by a variety of risk factors, including various infectious diseases.
Early studies of severe acute respiratory syndrome (SARS), an infectious disease caused by a new coronavirus, have revealed thrombocytopenia as a common finding [55], [56]. Gram-negative bacterial infection or catheter-related sepsis is often evident in these patients, and a significant percentage (20%) develop ARDS within 3 weeks [56]. Interestingly, Cinatl et al. [57] recently reported that among all the compounds tested (including rivavirin, which is currently used for SARS), GL was the most active at inhibiting the replication of the SARS-associated virus in vitro (although admittedly the effective concentration of GL was markedly higher than those of the other drugs). Rivavirin has many toxic effects, including hemolysis and a drastic reduction in hemoglobin. Since rapid treatment of SARS (and also sepsis) is needed to prevent ARDS developing, and since the side effects of GL are few and can be controlled, Cinatl et al. [57] proposed that GL may be an effective drug against SARS. The present results, including the finding that GL is effective at suppressing so-called “endotoxin shock”, seem to support this proposal.
Acknowledgements
We are grateful to Dr. T. Nagasawa (University of Tsukuba) for providing Pm-1, and to Dr. R. Timms for editing the manuscript.
References
- 1.Martin M.A., Silverman H.J. Gram-negative sepsis and the adult respiratory distress syndrome. Clin. Infect. Dis. 1992;14:1213–1228. doi: 10.1093/clinids/14.6.1213. [DOI] [PubMed] [Google Scholar]
- 2.Opal S.M., Glück T. Endotoxin as a drug target. Crit. Care Med. 2003;31:S57–S64. doi: 10.1097/00003246-200301001-00009. [DOI] [PubMed] [Google Scholar]
- 3.Nasraway S.A. The problems and challenges of immunotherapy in sepsis. Chest. 2003;123:S451–S459. doi: 10.1378/chest.123.5_suppl.451s. [DOI] [PubMed] [Google Scholar]
- 4.Esmon C.T. Protein C pathway in sepsis. Ann. Med. 2002;34:598–605. doi: 10.1080/078538902321117823. [DOI] [PubMed] [Google Scholar]
- 5.Freeman B.D., Buchman T.G. Coagulation inhibitors in the treatment of sepsis. Expert Opin. Investig. Drugs. 2002;11:69–74. doi: 10.1517/13543784.11.1.69. [DOI] [PubMed] [Google Scholar]
- 6.Heffner J.E., Sahn S.A., Repin J.E. The role of platelets in the adult respiratory distress syndrome. Culprits or bystanders? Am. Rev. Respir. Dis. 1987;135:482–492. doi: 10.1164/arrd.1987.135.2.482. [DOI] [PubMed] [Google Scholar]
- 7.Gawaz M., Dickfeld T., Bogner C., Fateh-Moghadam S., Neumann F.J. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med. 1997;23:379–385. doi: 10.1007/s001340050344. [DOI] [PubMed] [Google Scholar]
- 8.Ohtaki Y., Shimauchi H., Yokochi T., Takada H., Endo Y. In vivo platelet response to lipopolysaccharide in mice: proposed method for evaluating new antiplatelet drugs. Thromb. Res. 2002;108:303–309. doi: 10.1016/s0049-3848(03)00092-6. [DOI] [PubMed] [Google Scholar]
- 9.Shibazaki M., Kawabata Y., Yokochi T., Nishida A., Takada H., Endo Y. Complement-dependent accumulation and degradation of platelets in the lung and liver induced following injection of lipopolysaccharides. Infect. Immun. 1999;67:5186–5191. doi: 10.1128/iai.67.10.5186-5191.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao L., Ohtaki Y., Yamaguchi K., Matsushita M., Fujita T., Yokochi T. LPS-induced platelet response and rapid shock in mice: contribution of O-antigen region of LPS and involvement of the lectin pathway of the complement system. Blood. 2002;100:3233–3239. doi: 10.1182/blood-2002-01-0252. [DOI] [PubMed] [Google Scholar]
- 11.Galanos C., Freudenberg M.A., Reutter W. Galactosamine-induced sensitization to lethal effects of endotoxin. Proc. Natl. Acad. Sci. U. S. A. 1979;76:5939–5943. doi: 10.1073/pnas.76.11.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann V., Freudenberg M.A., Galanos C. Lethal toxicity and tumor necrosis factor in normal and D-galactosamine-treated mice. J. Exp. Med. 1987;165:657–663. doi: 10.1084/jem.165.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Endo Y., Kikuchi T., Nakamura M. Ornithine and histidine decarboxylase activities in mice sensitized to endotoxin, interleukin-1 and tumour necrosis factor by D-galactosamine. Br. J. Pharmacol. 1992;107:888–894. doi: 10.1111/j.1476-5381.1992.tb14542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinarello C.A. Anti-cytokine therapeutics and infections. Vaccine. 2003;21:S2/24–S2/34. doi: 10.1016/s0264-410x(03)00196-8. [DOI] [PubMed] [Google Scholar]
- 15.Pompei R., Flore O., Marccialis M.A., Pani A., Loddo B. Glycyrrhizic acid inhibits virus growth and inactivates virus particles. Nature. 1979;281:689–690. doi: 10.1038/281689a0. [DOI] [PubMed] [Google Scholar]
- 16.Utsunomiya T., Kobayashi M., Pollard R.B., Suzuki F. Glycyrrhizin, an active component of licorice roots, reduces morbidity and mortality of mice infected with lethal dose of influenza virus. Antimicrob. Agents Chemother. 1997;41:551–556. doi: 10.1128/aac.41.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okamoto T. The protective effect of glycyrrhizin on anti-Fas antibody-induced hepatitis in mice. Eur. J. Pharmacol. 2000;387:229–232. doi: 10.1016/s0014-2999(99)00807-9. [DOI] [PubMed] [Google Scholar]
- 18.Ohuchi K., Kamada Y., Tsurufuji S. Glycyrrhizin inhibits prostaglandin E2 production by activated peritoneal macrophages from rats. Prostaglandins Med. 1981;7:457–463. doi: 10.1016/0161-4630(81)90033-1. [DOI] [PubMed] [Google Scholar]
- 19.Imanishi N., Kawai H., Hayashi Y., Yatsunami K., Ichikawa A. Effects of glycyrrhizin and glycyrrhetinic acid on dexamethasone-induced changes in histamine synthesis of mouse mastocytoma P-815 cells and in histamine release from rat peritoneal mast cells. Biochem. Pharmacol. 1989;38:2521–2526. doi: 10.1016/0006-2952(89)90097-x. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y.-M., Hirota S., Jippo-Kanemoto T., Kim H.-R., Shin T.-Y., Yeon Y. Inhibition of histamine synthesis by glycyrrhetinic acid in mast cells cocultured with Swii 3T3 fibroblasts. Int. Arch. Allergy Immunol. 1996;110:272–277. doi: 10.1159/000237298. [DOI] [PubMed] [Google Scholar]
- 21.Kai K., Komine K., Asai K., Kuroishi T., Komine Y., Kozutsumi T. Anti-inflammatory effects of intramammary infusion of glycyrrhizin in lactating cows with mastitis caused by coagulase-negative staphylococci. Am. J. Vet. Res. 2003;64:1213–1220. doi: 10.2460/ajvr.2003.64.1213. [DOI] [PubMed] [Google Scholar]
- 22.Kroes B.H., Beukelman C.J., Van Den Berg A.J.J., Wolbink G.J., Van Dijk H., Labadie R.P. Inhibition of human complement by β-glycyrrhetinic acid. Immunology. 1997;90:115–120. doi: 10.1046/j.1365-2567.1997.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujisawa Y., Sakamoto M., Matsushita M., Fujita T., Nishioka K. Glycyrrhizin inhibits the lytic pathway of complement: possible mechanism of its anti-inflammatory effect on liver. Microbiol. Immunol. 2000;44:799–804. doi: 10.1111/j.1348-0421.2000.tb02566.x. [DOI] [PubMed] [Google Scholar]
- 24.Arase Y., Ikeda K., Murashima N., Chayama K., Tsubota A., Koida I. The long term efficacy of glycyrrhizin in chronic hepatitis C patients. Cancer. 1997;79:1494–1500. doi: 10.1002/(sici)1097-0142(19970415)79:8<1494::aid-cncr8>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 25.Van Rossum T.G.J., Vulto A.G., Hop W.C.J., Brouwer J.T., Niesters H.G.M., Schalm S.W. Intravenous glycyrrhizin for the treatment of chronic hepatitis C: a double-blind, randomized, placebo-controlled phase I/II trial. J. Gastroenterol. Hepatol. 1999;14:1093–1099. doi: 10.1046/j.1440-1746.1999.02008.x. [DOI] [PubMed] [Google Scholar]
- 26.Yokochi T., Inoue Y., Yokoo J., Kimura Y., Kato N. Retention of bacterial lipopolysaccharide at the site of subcutaneous injection. Infect. Immun. 1989;57:1786–1791. doi: 10.1128/iai.57.6.1786-1791.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagata Y., Nagahisa H., Aida Y., Okutomi K., Nagasawa T., Todokoro K. Thrombopoietin induces megakaryocyte differentiation in hematopoietic progenitor FDC-P2 cells. J. Biol. Chem. 1995;270:19673–19675. doi: 10.1074/jbc.270.34.19673. [DOI] [PubMed] [Google Scholar]
- 28.Nagata Y., Shozaki Y., Nagahisa H., Nagasawa T., Abe T., Todokoro K. Serumthrombopoietin level is not regulated by transcription but by the total counts of both megakaryocytes and platelets during thrombocytopenia and thrombocytosis. Thromb. Haemost. 1997;77:808–814. [PubMed] [Google Scholar]
- 29.Endo Y., Nakamura M. The effects of lipopolysaccharide, interleukin-1 and tumour necrosis factor on the hepatic accumulation of 5-hydroxytryptamine and platelets in the mouse. Br. J. Pharmacol. 1992;105:613–619. doi: 10.1111/j.1476-5381.1992.tb09028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bross I.D.J. How to use ridit analysis. Biometrics. 1958;4:18–38. [Google Scholar]
- 31.Yoshida A., Ohba M., Wu X., Sasano T., Nakamura M., Endo Y. Accumulation of platelets in the lung and liver and their degranulation following antigen-challenge in sensitized mice. Br. J. Pharmacol. 2002;137:146–152. doi: 10.1038/sj.bjp.0704852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Francischetti I.M.B., Monteiro R.Q., Guimarães J.A. Identification of glycyrrhizin as a thrombin inhibitor. Biochem. Biophys. Res. Commun. 1997;235:259–263. doi: 10.1006/bbrc.1997.6735. [DOI] [PubMed] [Google Scholar]
- 33.Mendes-Silva W., Assafim M., Ruta B., Monteiro R.Q., Guimarães J.A., Zingali R.B. Antithrombotic effect of glycyrrhizin, a plant-derived thrombin inhibitor. Thromb. Res. 2003;112:93–98. doi: 10.1016/j.thromres.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 34.Farries T.C., Atkinson J.P. Evolution of the complement system. Immunol. Today. 1991;12:295–300. doi: 10.1016/0167-5699(91)90002-B. [DOI] [PubMed] [Google Scholar]
- 35.Fujita T., Endo Y., Nonaka M. Primitive complement system: recognition and activation. Mol. Immunol. 2004;41:103–111. doi: 10.1016/j.molimm.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 36.Presanis J.S., Hajela K., Ambrus G., Gál P., Sim R.B. Differential substrate and inhibitor profiles for human MASP-1 and NASP-2. Mol. Immunol. 2003;40:921–929. doi: 10.1016/j.molimm.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 37.Sims P.J., Wiedmer T. The response of human platelets to activated components of the complement system. Immunol. Today. 1991;12:338–342. doi: 10.1016/0167-5699(91)90012-I. [DOI] [PubMed] [Google Scholar]
- 38.Mattson D.L. Comparison of arterial blood pressure in different strains of mice. Am. J. Hypertens. 2001;14:405–408. doi: 10.1016/s0895-7061(00)01285-1. [DOI] [PubMed] [Google Scholar]
- 39.Miyake T., Saito M., Shimura K. Adsorption, excretion and distribution of 3H-glycyrrhizin in mice. Minophagen Med. Rev. 1979;24:263–272. (in Japanese) [Google Scholar]
- 40.Ishiwata S., Nakashita K., Niizeki M., Suzuki N., Kaneko S., Tomioka Y. Determination of serum concentration of glycyrrhizin in humans by semi-micro high-performance liquid chromatography after administration of a therapeutic dose. Biol. Pharm. Bull. 2000;23:904–905. doi: 10.1248/bpb.23.904. [DOI] [PubMed] [Google Scholar]
- 41.Takeda S., Ishihara K., Wakui Y., Amagaya S., Maruno M., Akao T. Bioavailability study of glycyrrhetic acid after oral administration of glycyrrhizin in rats: relevance to the intestinal bacterial hydrolysis. J. Pharm. Pharmacol. 1996;48:902–905. doi: 10.1111/j.2042-7158.1996.tb05998.x. [DOI] [PubMed] [Google Scholar]
- 42.MacLaren R., Jung R. Stress-dose corticosteroid therapy for sepsis and acute lung injury or acute respiratory distress syndrome in critically ill adults. Pharmacotherapy. 2002;22:1140–1156. doi: 10.1592/phco.22.13.1140.33519. [DOI] [PubMed] [Google Scholar]
- 43.Minneci P., Deans K., Natanson C., Eichacker P.Q. Increasing the efficacy of anti-inflammatory agents used in the treatment of sepsis. Eur. J. Clin. Microbiol. Infect. Dis. 2000;22:1–9. doi: 10.1007/s10096-002-0857-3. [DOI] [PubMed] [Google Scholar]
- 44.Freeman B.D., Natanson C. Anti-inflammatory therapies in sepsis and septic shock. Expert Opin. Invest. Drugs. 2000;9:1651–1663. doi: 10.1517/13543784.9.7.1651. [DOI] [PubMed] [Google Scholar]
- 45.Abe N., Ebina T., Ishida N. Interferon induction by glycyrrhizin and glycyrrhetinic acid in mice. Microbiol. Immunol. 1982;26:535–539. doi: 10.1111/j.1348-0421.1982.tb00207.x. [DOI] [PubMed] [Google Scholar]
- 46.Dai J.H., Iwatani Y., Ishida T., Terunuma H., Kasai H., Iwakura Y. Glycyrrhizin enhances interleukin-12 production in peritoneal macrophages. Immunology. 2001;103:235–243. doi: 10.1046/j.1365-2567.2001.01224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Utsunomiya T., Kobayashi M., Ito M., Herndon D.N., Pollard R.B., Suzuki F. Glycyrrhizin restores the impaired IL-12 production in thermally injured mice. Cytokine. 2001;14:49–55. doi: 10.1006/cyto.2001.0847. [DOI] [PubMed] [Google Scholar]
- 48.Elenkov I.J., Chrousos G.P., Wilder R.L. Neuroendocrine regulation of IL-12 and TNF-α/IL-10 balance. Ann. N.Y. Acad. Sci. 2000;917:94–105. doi: 10.1111/j.1749-6632.2000.tb05374.x. [DOI] [PubMed] [Google Scholar]
- 49.Schneider E., Rolli-Derkinderen M., Arock M., Dy M. Trends in histamine research: new functions during immune responses and hematopoiesis. Trends Immunol. 2002;23:255–263. doi: 10.1016/s1471-4906(02)02215-9. [DOI] [PubMed] [Google Scholar]
- 50.Weiss K. Pulmonary thromboembolism: epidemiology and techniques of nuclear medicine. Semin. Thromb. Hemost. 1996;22:27–32. doi: 10.1055/s-2007-998989. [DOI] [PubMed] [Google Scholar]
- 51.Herd C.M., Page C.P. Pulmonary immune cells in health and disease: platelets. Eur. Respir. J. 1994;7:1145–1160. [PubMed] [Google Scholar]
- 52.Isobe Y. “Streptococcus milleri” group induce anaphylactoid reaction and inflammatory cytokines in mice primed with muramyldipeptide. J. Dent. Health. 1998;48:170–182. [abstract in English] [Google Scholar]
- 53.Ohba M., Shibazaki M., Sasano T., Inoue M., Takada H., Endo Y. Platelet responses and anaphylaxis-like shock induced in mice by intravenous injection of whole cells of oral streptococci. Oral Microbiol. Immunol. 2004;19:26–30. doi: 10.1046/j.0902-0055.2002.00107.x. [DOI] [PubMed] [Google Scholar]
- 54.Herzberg M.C., Meyer M.W. Dental plaque, platelets, and cardiovascular diseases. Ann. Periodontol. 1998;3:151–160. doi: 10.1902/annals.1998.3.1.151. [DOI] [PubMed] [Google Scholar]
- 55.Wong R.S.M., Wu A., To K.F., Lee N., Lam C.W.K., Wong C.K. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. Br. Med. J. 2003;326:1358–1362. doi: 10.1136/bmj.326.7403.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peiris J.S.M, Chu C.M., Cheng V.C.C., Chan K.S., Hung I.F.N., Poon L.L.M., members of the HKU/UCH SARS Study Group Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cinatl J., Morgenstern B., Bauer G., Chandra P., Ravenau H., Doerr H.W. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet. 2003;361:2045–2046. doi: 10.1016/S0140-6736(03)13615-X. [DOI] [PMC free article] [PubMed] [Google Scholar]