

Abstract
Bats carry a great diversity of zoonotic viruses with a high-impact on human health and livestock. Since the emergence of new coronaviruses and paramyxoviruses in humans (e.g. Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and Nipah virus), numerous studies clearly established that bats can maintain some of these viruses. Improving our understanding on the role of bats in the epidemiology of the pathogens they harbour is necessary to prevent cross-species spill over along the wild/domestic/human gradient. In this study, we screened bat faecal samples for the presence of Coronavirus and Paramyxovirus in two caves frequently visited by local people to collect manure and/or to hunt bats in Zimbabwe. We amplified partial RNA-dependent RNA polymerase genes of Alpha and Betacoronavirus together with the partial polymerase gene of Paramyxovirus. Identified coronaviruses were related to pathogenic human strains and the paramyxovirus belonged to the recently described Jeilongvirus genus. Our results highlighted the importance of monitoring virus circulation in wildlife, especially bats, in the context of intense human-wildlife interfaces in order to strengthen prevention measures among local populations and to implement sentinel surveillance in sites with high zoonotic diseases transmission potential.
Keywords: Bat, Coronavirus, Paramyxovirus, Phylogeny, Emerging infectious diseases, Zimbabwe
Highlights
-
•
Coronavirus and Paramyxovirus circulate in Hipposideros bat species in Zimbabwe.
-
•
Importance of widening viral screening in under-investigated countries
-
•
Sentinel surveillance in sites with high zoonotic transmission potential
Bats comprise nearly 1200 species and constitute ≈20% of living mammal species and are distributed on all continents except Antarctic, Artic and a few islands (Simmons, 2005). Due to their unique (only flying mammals) and diverse lifestyles, bats differ from other sylvatic disease mammalian reservoirs and are predisposed for the acquisition and maintenance of viruses (Hayman et al., 2013). During the past two decades, bats (Chiroptera) have been identified as the reservoir host of a number of high-impact zoonotic viruses known to induce highly lethal diseases in humans and domestic animals (Brook and Dobson, 2015). They have been associated with emerging Paramyxovirus (Nipah and hendra viruses), Coronavirus (MERS-CoV and SARS-CoV) and Filovirus (Ebola and Marburg viruses) (Smith and Wang, 2013) which attracted global attention due to their severity and/or large-scale spread. Those emergences have been caused by the ever-increasing interfaces between domestic animals, people and bat communities created by current global and human changes (Brierley et al., 2016). Human activities that increase exposure to bats induce new and more infectious contacts between species and promote the spill over of unknown pathogens from bats to other animals. The identification of the reservoir species is key for the control of these emerging infectious diseases in order to prevent/manage practices at risk of pathogens spill over.
Although numerous studies have been implemented on bat-borne viruses around the world, large gaps still exists concerning the viral diversity among Chiroptera especially in some regions that attracted little disease research until now. The Republic of Zimbabwe is situated southern Africa in the subtropical zone and has an exceptional great diversity of wildlife. To date more than 60 bat species have been recorded in Zimbabwe (Monadjem et al., 2010). Accordingly, Zimbabwe represents a potential hot spot for future emergence of microorganisms from bats that can transmit infections to humans and livestock (Morse et al., 2012). Many cases of rabies, anthrax, African swine fever and foot and mouth diseases have been recorded in the last 20 years. Furthermore, Lyssavirus were demonstrated in bats (Duvenhage virus) and in cats and dogs (Mokola Virus, Lagos bat virus) (Bingham et al., 2001; Foggin, 1982). In the 1970s, a traveller who passed through Zimbabwe was probably infected with the Marburg virus after visiting the Chinhoyi caves about 135 km northwest of Harare, capital of Zimbabwe (Peterson et al., 2006). We report here the first evidence of circulation of Coronavirus and Paramyxovirus in Hipposideros bat species in Zimbabwe.
Between June 2016 and February 2017, 99 and 146 faecal samples were respectively collected in two caves (Fig. 1 ) regularly visited by local people to collect bat guano used as fertiliser. Each cave was visited twice at two different periods. Two square meters plastic sheets were laid down in the caves, underneath the bat colonies for overnight (five plastic sheets per cave). Faeces were collected from each plastic sheet at a rate of ≈ 6 g of pooled faeces in 15 ml tube with 6 ml of homemade RNA stabilisation solution (Pol Scientific, 1999). Samples were stored at −80 °C until laboratory analyses.
Fig. 1.
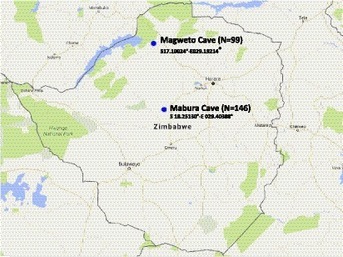
Geographical distribution of bats faecal samples collection sites.
Blues circles represent the caves where bat faecal samples were collected. The name of the caves as well as the GPS location is noted next to the circle. Number of faeces collected per site is shown in brackets.
Bat species were identified by Cytochrome b amplification (Kocher et al., 1989) and sequencing after DNA extraction using Qiamp DNA stool (Qiagen S.A, Courtaboeuf, France). Cytochrome b sequences were then compared to available bat sequences in the GenBank database using Basic Local Alignment Search Tool (BLAST) program and species were confirmed by phylogenetic analysis (supplementary material, Fig. 1S,). Only bats from Hipposideros spp., representing two distinct colonies, were identified. To date, two different Hipposideros bat species have been reported in Zimbabwe; Hipposideros caffer and Hipposideros vittatus (Monadjem et al., 2010). Our samples were closer to Hip. caffer than any other Hipposideros spp. (supplementary material, Fig. 1S,).
Bat species were identified by Cytochrome b amplification (Kocher et al., 1989) and sequencing after DNA extraction using Qiamp DNA stool (Qiagen S.A, Courtaboeuf, France). Cytochrome b sequences were then compared to available bat sequences in the GenBank database using Basic Local Alignment Search Tool (BLAST) program and species were confirmed by phylogenetic analysis (supplementary material, Fig. 1S,). Only bats from Hipposideros spp., representing two distinct colonies, were identified. To date, two different Hipposideros bat species have been reported in Zimbabwe; Hipposideros caffer and Hipposideros vittatus (Monadjem et al., 2010). Our samples were closer to Hip. caffer than any other Hipposideros spp. (supplementary material, Fig. 1S,).
RNA extraction was carried out from all faecal samples collected. Briefly, two sample tubes from the same plastic sheet were pooled and transferred in a 50 ml tube with 20 ml of PBS 1× then vigorously mixed. All together we made 73 (51 in June 2016 and 22 in February 2017) pools from Mabura cave and 50 (35 in June 2016 and 15 in February 2017) pools from Magweto cave respectively. Tubes were centrifuged at 4500 rpm for 10 min. Supernatant was filtered using gauze in order to eliminate faecal matter and transferred in fresh tubes then re-centrifuged at 4500 rpm for 10 min. Supernatant was filtered through a 0.2 μm filter to remove eukaryotic and bacterial sized particles. Seven millilitres of filtered samples were centrifuged at 250,000 g for 2.5 h at 4 °C. The pellets were re-suspended in 600 μl H20 molecular grade and 150 μl were used to extract RNA using NucleoSpin® RNA Kit (Macherey-Nagel, France) according to the manufacturer's protocol. The 123 RNA samples extracted from the pools were then reverse transcribed using random hexamers and screened for Coronavirus (CoV) and Paramyxovirus (ParV) as previously described employing a pan-coronavirus and pan-paramyxovirus nested RT-PCR directed against partial polymerase RNA-dependent RNA polymerase (RdPd) and polymerase gene sequences, respectively (Chu et al., 2011; Tong et al., 2008). PCR products (415 bp for CoV and 531 bp for ParV) were agarose gel purified (Geneclean Turbo Kit, MP Biomedicals, France) and directly sequenced in both 5′ and 3′ directions using cycle sequencing and dye terminator methodologies (Eurofins, Germany). Overlapping sequences were assembled into contiguous sequences using SEQMAN DNASTAR software (lasergene, DNASTAR, Inc., Madison, WI, USA). Partial non-concatenated nucleic acid sequences of the new Coronavirus and Paramyxovirus as well as from Cytochrome B were aligned using MEGA 7 (Kumar et al., 2016), with minor manual adjustments. Sites that could not be unambiguously aligned were excluded and divergent regions were excluded from subsequent analyses. Phylogenies were inferred using both Bayesian methods and Maximum Likelihood (ML) method implemented in MrBayes v3.2.6 and in PhyML respectively (Guindon et al., 2010; Ronquist et al., 2012). Mr. Bayes ran for four million generations for Coronavirus RdRp and Paramyxovirus polymerase genes, respectively, with a 10% burn-in. Bayesian parameters were examined with the Tracer program (Tracer, 2003). Convergence diagnostic for the Estimated sample Size (ESS) values and Potential Scale Reduction Factor (PSRF) were >500 and equal to 1 respectively. In ML method, the reliability of branching orders was tested using the bootstrap approach (1000 replicates). The suited evolution model (GTR + Γ4 + I for Coronavirus and Cytochrome B, and GTR + Γ4 for Paramyxovirus) was selected by Akaie's Information criterion (AIC) using Topali software (Milne et al., 2009). From both phylogenetic analyses, similar tree topologies were obtained (data not shown). Identities analyses were done using ClustalX (Larkin et al., 2007).
We characterised Alphacoronavirus in Mabura cave as well as Betacoronavirus and Paramyxovirus in Magweto cave from roundleaf bats, which was the only bat genus observed in the two visited caves at the time of our samplings. Our new Alphacoronavirus formed a well sustained specific sub-clade close to the human Coronavirus 229E strain (HCoV-229E) (Fig. 2) that circulates in human population worldwide and mostly causes mild respiratory disease (Masters and Perlman, 2013). This close relationship is confirmed by a high percentage (95%) of amino acid identities (Supplementary Material, Table S1). Interestingly, our BtCoV 229E related strains are distinct to those identified in Hip. caffer rufer from Ghana (Pfefferle et al., 2009). Our results are in accord with the recently suggested long evolutionary history of 229E-related CoV in old world hipposiderid bats (Corman et al., 2015). Nonetheless it is unclear whether bats directly transmitted this virus to human or if an intermediate host was involved in the transmission chain such as demonstrated for SARS-CoV and MERS-CoV (Smith and Wang, 2013).
Fig. 2.
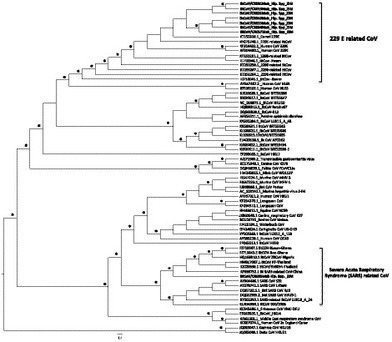
Phylogenetic analysis of partial RNA-dependent RNA polymerase (RdRp) of the newly identified Alphacoronavirus and Betacoronavirus sequences from Zimbabwe. New partial RdRp (415 bp) CoV sequences are represented in bold and were compared to previously identified Alphacoronavirus and Betacoronavirus available in the GenBank. Accession numbers are showed before the strain name. Only Bayesian posterior probabilities are showed. Asterisks at nodes represent posterior probability ≥90%. Scale bars indicate the number of base substitutions per site.
We characterised Alphacoronavirus in Mabura cave as well as Betacoronavirus and Paramyxovirus in Magweto cave from roundleaf bats, which was the only bat genus observed in the two visited caves at the time of our samplings. Our new Alphacoronavirus formed a well sustained specific sub-clade close to the human Coronavirus 229E strain (HCoV-229E) (Fig. 2) that circulates in human population worldwide and mostly causes mild respiratory disease (Masters and Perlman, 2013). This close relationship is confirmed by a high percentage (95%) of amino acid identities (Supplementary Material, Table S1). Interestingly, our BtCoV 229E related strains are distinct to those identified in Hip. caffer rufer from Ghana (Pfefferle et al., 2009). Our results are in accord with the recently suggested long evolutionary history of 229E-related CoV in old world hipposiderid bats (Corman et al., 2015). Nonetheless it is unclear whether bats directly transmitted this virus to human or if an intermediate host was involved in the transmission chain such as demonstrated for SARS-CoV and MERS-CoV (Smith and Wang, 2013).
In Mabura cave, during our first visit during the cold dry season in June 2016 we collected faeces from three plastic sheets and Bat 229-E like virus was amplified from samples issued from each plastic sheet suggesting an important circulation of this virus in the bat colony. Interestingly, no viruses were amplified from the second sampling in this cave during the rainy season in February 2017. Nonetheless, during the second visit we observed a consequent diminution of bats present in the cave and our sampling was lower than expected. This could be due to Hipposideros spp. seasonal movement. Besides, the absence of Alphacoronavirus could also be due to temporal variation in virus shedding in bats (Plowright et al., 2015).
In Magweto cave we amplified Betacoronavirus from only one pooled sample (Fig. 2). It could be due to a low circulation of this virus in the bat colony. Phylogenetic analyses showed that this new virus formed a specific clade with betacoronaviruses isolated in Asia and Africa (Gouilh et al., 2011; Pfefferle et al., 2009; Quan et al., 2010) with 90% to 87% of amino acid identities (Supplementary material, Table S1) and together they formed a sister clade with the described SARS-CoV strains with 77% of amino acid identities (Fig. 2, Supplementary material, Table S1). The SARS-CoV related (SARS-CoVr) sister clade is well sustained and our new Bt SARS-CoVr strain is positioned at the root of this clade. This finding could strengthen the African origin hypothesis of SARS-like group (Pfefferle et al., 2009; Quan et al., 2010). Nonetheless, this hypothesis is controversial and, in order to disentangle the Bt SARS-CoVr origin, future studies should focus on Hipposideridae as well as on Rhinolophidae and Rhinonycteridae since these three bat families diverged from a common ancestor, which potentially hosted the ancestor of SARS-related COVs (Foley et al., 2015; Gouilh et al., 2011). Additionally, SARS-CoVr have been characterised from these three bat families (Pfefferle et al., 2009; Smith et al., 2016; Wu et al., 2016).
In Magweto cave we amplified Betacoronavirus from only one pooled sample (Fig. 2). It could be due to a low circulation of this virus in the bat colony. Phylogenetic analyses showed that this new virus formed a specific clade with betacoronaviruses isolated in Asia and Africa (Gouilh et al., 2011; Pfefferle et al., 2009; Quan et al., 2010) with 90% to 87% of amino acid identities (Supplementary material, Table S1) and together they formed a sister clade with the described SARS-CoV strains with 77% of amino acid identities (Fig. 2, Supplementary material, Table S1). The SARS-CoV related (SARS-CoVr) sister clade is well sustained and our new Bt SARS-CoVr strain is positioned at the root of this clade. This finding could strengthen the African origin hypothesis of SARS-like group (Pfefferle et al., 2009; Quan et al., 2010). Nonetheless, this hypothesis is controversial and, in order to disentangle the Bt SARS-CoVr origin, future studies should focus on Hipposideridae as well as on Rhinolophidae and Rhinonycteridae since these three bat families diverged from a common ancestor, which potentially hosted the ancestor of SARS-related COVs (Foley et al., 2015; Gouilh et al., 2011). Additionally, SARS-CoVr have been characterised from these three bat families (Pfefferle et al., 2009; Smith et al., 2016; Wu et al., 2016).
SARS-CoV emerged at the beginning of 21e century following a human transmission by an intermediary host, a palm civet, in China. More than 8000 human infections were reported around the world with a case fatality rate of up to 10% (Smith and Wang, 2013). To date several studies evidenced different bat species as potential SARS and SARS-like CoV reservoirs worldwide (Li et al., 2005).
In addition, in the same cave we amplified a Paramyxovirus closer to bat Paramyxovirus (77 to 87% of amino acid identities) related to the putative Jeilongvirus genus (Fig. 3, Supplementary material Table S1) than other Paramyxovirus lineages. To date, the pathogenic potential of the viruses from this genus is currently unknown. However, the Beilong virus was discovered on human kidney cell lines and neutralising antibodies against J virus have been detected in rodents, pigs and humans (Audsley et al., 2016). In addition, bat viruses belonging to the related-Jeilongvirus genus were widely detected in China and more recently in Luxembourg in Europe (Pauly et al., 2017). Altogether, these data highlight the need for further studies on the zoonotic potential of these viruses.
Fig. 3.
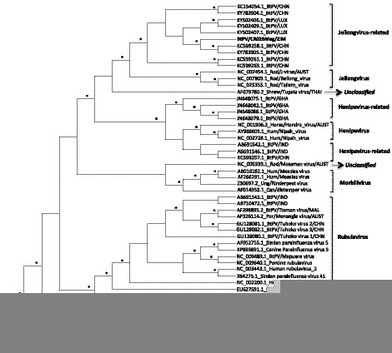
Phylogenetic analysis of partial polymerase gene of the newly identified Paramyxovirus (ParV) sequence from Zimbabwe. New partial pol (531 bp) ParV sequences are represented in bold and were compared to previously identified Paramyxovirus available in the GenBank. Accession numbers are showed before the strain name. Only Bayesian posterior probabilities are showed. Asterisks at nodes represent posterior probability ≥90%. Scale bars indicate the number of base substitutions per site
In addition, in the same cave we amplified a Paramyxovirus closer to bat Paramyxovirus (77 to 87% of amino acid identities) related to the putative Jeilongvirus genus (Fig. 3, Supplementary material Table S1) than other Paramyxovirus lineages. To date, the pathogenic potential of the viruses from this genus is currently unknown. However, the Beilong virus was discovered on human kidney cell lines and neutralising antibodies against J virus have been detected in rodents, pigs and humans (Audsley et al., 2016). In addition, bat viruses belonging to the related-Jeilongvirus genus were widely detected in China and more recently in Luxembourg in Europe (Pauly et al., 2017). Altogether, these data highlight the need for further studies on the zoonotic potential of these viruses.
Although Coronavirus and Paramyxovirus have been widely described in bats around the world (Anthony et al., 2017; Drexler et al., 2012), our results pointed out the need to widen viral screening in under-investigated countries particularly when the country has considerable potential as a hot spot for emerging infectious diseases (Morse et al., 2012). Our study focused on two caves in Zimbabwe with an important bat-human interface throughout guano harvesting and/or bats poaching. Non-invasive sampling provides a rapid approach to target site of interest for in-depth studies on virus prevalence in bats and temporal variation in virus shedding in bats (viral ecology) and provides a first risk assessment of the transmission of bat-borne pathogens to humans. Finally, our study will enable, in agreement with the local health authorities, to carry out a specific communication within the local populations on the risk of contamination and how to prevent it.
The following are the supplementary data related to this article.
Phylogenetic analysis of partial CytB sequences (550 bp). New CytB sequences obtained were compared to representative bat family CytB sequences available in the GenBank. Only Bayesian posterior probabilities are showed. Asterisks at nodes represent posterior probability ≥85%. Scale bars indicate the number of base substitutions per site. New CytB sequences are represented by the red triangle. The following sequences have been used for this analysis: KX467590.1 Miniopterus schreibersii, KX467591.1 Miniopterus magnater, KX467593.1 Tylonycteris pachypus, KX467595.1 Hypsugoal aschanicus, KX467596.1 Nyctalus plancyi, KX467597.1 Pipistrellus abramus, KX467598.1 Murina leucogaster, KX467601.1 Myotis laniger, KX467611.1 Myotis davidii, KX467606.1 Myotis blythii, KX467607.1 Myotis chinensis, KX467608.1 Myotis pequinius, KX467609.1 Myotis bombinus, KX467605.1 Myotis formosus, KX467610.1 Myotis altarium, KX467612.1 Myotis frater, KX467599.1 Myotis ricketti, KX467600.1 Myotis adversus, KX467604.1 Myotis fimbriatus, KX467602.1 Myotis daubentonii, KX467603.1 Myotis macrodactylus, KF218429.1 Taphozous nudiventris, AY591536.1 Otomops martiensseni, AY591537.1 Otomops martiensseni, KX467574.1 Megaderma lyra, KX467575.1 Rhinolophus pusillus, KX467576.1 Rhinolophus lepidus, KX467579.1 Rhinolophus macrotis, KX467580.1 Rhinolophus marshalli, KX467582.1 Rhinolophus rex, KX467578.1 Rhinolophus ferrumequinum, KX467583.1 Rhinolophus sinicus, KX467577.1 Rhinolophus affinis, KX467581.1 Rhinolophus pearsonii, KX467584.1 Hipposideros cineraceus, KX467585.1 Hipposideros pomona, FJ347979.1 Hipposideros caffer, FJ347980.1 Hipposideros caffer, KX467587.1 Hipposideros armiger, KX467588.1 Hipposideros larvatus, KX467589.1 Hipposideros pratti, KX467586.1 Aselliscus stoliczkanus, KT583805.1 Hipposideros vittatus, KT583806.1 Hipposideros vittatus, KT583801.1 Hipposideros gigas, KT583817.1 Hipposideros commersoni, DQ005796.1 Triaenops persicus, DQ005797.1 Triaenops persicus, KX823312.1 Megaerops niphanae, KX823313.1 Cynopterus sp., KX823314.1 Cynopterus sp., KX823317.1 Sphaeria sblanfordi, KX822888.1 Eidolon helvum, KX822889.1 Eidolon helvum, KX823318.1 Macroglossus sobrinus, KX823319.1 Eonycteris spelaea, KX822938.1 Rousettus aegyptiacus, KX822939.1 Rousettus aegyptiacus, KX823315.1 Rousettus leschenaultii, KX823311.1 Plerotes anchietae, KX823011.1 Hypsignathus monstrosus, KX823012.1 Hypsignathus monstrosus, KX823087.1 Epomops franqueti, KX823088.1 Epomops franqueti, KX823233.1 Epomops buettikoferi, KX823234.1 Epomops buettikoferi, KX822985.1 Nanonycteris veldkampii, KX822986.1 Nanonycteris veldkampii, KX823306.1 Epomops dobsonii, KX823307.1 Epomophorus anselli, KX823308.1 Epomophorus crypturus, KX823309.1 Epomophorus labiatus, KX823310.1 Epomophorus minimus, KX822798.1 Epomophorus gambianus, KX822810.1 Micropteropus pusillus, KX822797.1 Epomophorus gambianus, KX822809.1 Micropteropus pusillus.
Percentage Amino Acid identity between A/New alphacoronaviruses identified in Mabura cave and representative Alphacoronavirus strains of interest, B/New betacoronaviruses identified in Magweto cave and representative Betacoronavirus strains of interest, C/New paramyxovirus identified in Magweto cave and representative Paramyxovirus strains of interest. All sequences of interest were obtained from the genbank. Accession numbers are before of the strain name.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.meegid.2018.01.007.
Acknowledgments
Acknowledgements
We thank Billy Butete and Cavin Mandina for their field assistance. We thank Estelle Ména for her technical assistance. This work was supported by grants from the Agence Nationale de la Recherche (grant ANR-14-CE14-0029). We thank the Research Council of Zimbabwe for approving this study (research registration certificate No. 03006) and the Hurunguwe Rural District council and the Zibagwe Rural District Council for their assistance and facilitation. We thank the Animal Research Ethics Committee of Zimbabwe for their approval (ref number 002/2017). This work was conducted within the framework of the Research Platform “Production and Conservation in Partnership” (RP–PCP).
Declaration of interest
We declare that we have no conflicts of interest.
Footnotes
Nucleotide sequence accession number: The new Coronavirus and Paramyxovirus sequences reported in this study are available in GenBank under the following accession numbers: BtCov-Zim001Mab, MG000865; BtCov-Zim015Mab, MG000866; BtCov-Zim019Mab, MG000867; BtCov-Zim021, Mab MG000868; BtCov-Zim037Mab, MG000869; BtCov-Zim040Mab, MG000870; BtCov-Zim034Mab, MG000871; BtCov-Zim035Mag, MG000872; BtPV-Zim026Mag, MG000873.
References
- Anthony S.J., Johnson C.K., Greig D.J., Kramer S., Che X., Wells H., Hicks A.L., Joly D.O., Wolfe N.D., Daszak P., Karesh W., Lipkin W.I., Morse S.S., Consortium P., Mazet J.A.K., Goldstein T. Global patterns in coronavirus diversity. Virus Evol. 2017;3(vex012) doi: 10.1093/ve/vex012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audsley M.D., Marsh G.A., Lieu K.G., Tachedjian M., Joubert D.A., Wang L.F., Jans D.A., Moseley G.W. The immune evasion function of J and Beilong virus V proteins is distinct from that of other paramyxoviruses, consistent with their inclusion in the proposed genus Jeilongvirus. J. Gen. Virol. 2016;97:581–592. doi: 10.1099/jgv.0.000388. [DOI] [PubMed] [Google Scholar]
- Bingham J., Javangwe S., Sabeta C.T., Wandeler A.I., Nel L.H. Report of isolations of unusual lyssaviruses (rabies and Mokola virus) identified retrospectively from Zimbabwe. J. S. Afr. Vet. Assoc. 2001;72:92–94. doi: 10.4102/jsava.v72i2.624. [DOI] [PubMed] [Google Scholar]
- Brierley L., Vonhof M.J., Olival K.J., Daszak P., Jones K.E. Quantifying global drivers of zoonotic bat viruses: a process-based perspective. Am. Nat. 2016;187:E53–64. doi: 10.1086/684391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook C.E., Dobson A.P. Bats as ‘special’ reservoirs for emerging zoonotic pathogens. Trends Microbiol. 2015;23:172–180. doi: 10.1016/j.tim.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D.K., Leung C.Y., Gilbert M., Joyner P.H., Ng E.M., Tse T.M., Guan Y., Peiris J.S., Poon L.L. Avian coronavirus in wild aquatic birds. J. Virol. 2011;85:12815–12820. doi: 10.1128/JVI.05838-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V.M., Baldwin H.J., Tateno A.F., Zerbinati R.M., Annan A., Owusu M., Nkrumah E.E., Maganga G.D., Oppong S., Adu-Sarkodie Y., Vallo P., da Silva Filho L.V., Leroy E.M., Thiel V., van der Hoek L., Poon L.L., Tschapka M., Drosten C., Drexler J.F. Evidence for an ancestral Association of Human Coronavirus 229E with bats. J. Virol. 2015;89:11858–11870. doi: 10.1128/JVI.01755-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler J.F., Corman V.M., Muller M.A., Maganga G.D., Vallo P., Binger T., Gloza-Rausch F., Cottontail V.M., Rasche A., Yordanov S., Seebens A., Knornschild M., Oppong S., Adu Sarkodie Y., Pongombo C., Lukashev A.N., Schmidt-Chanasit J., Stocker A., Carneiro A.J., Erbar S., Maisner A., Fronhoffs F., Buettner R., Kalko E.K., Kruppa T., Franke C.R., Kallies R., Yandoko E.R., Herrler G., Reusken C., Hassanin A., Kruger D.H., Matthee S., Ulrich R.G., Leroy E.M., Drosten C. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012;3:796. doi: 10.1038/ncomms1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foggin C.M. Atypical rabies virus in cats and a dog in Zimbabwe. Vet. Rec. 1982;110:338. doi: 10.1136/vr.110.14.338. [DOI] [PubMed] [Google Scholar]
- Foley N.M., Thong V.D., Soisook P., Goodman S.M., Armstrong K.N., Jacobs D.S., Puechmaille S.J., Teeling E.C. How and why overcome the impediments to resolution: lessons from rhinolophid and hipposiderid bats. Mol. Biol. Evol. 2015;32:313–333. doi: 10.1093/molbev/msu329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouilh M.A., Puechmaille S.J., Gonzalez J.P., Teeling E., Kittayapong P., Manuguerra J.C. SARS-Coronavirus ancestor's foot-prints in South-East Asian bat colonies and the refuge theory. Infect. Genet. Evol. 2011;11:1690–1702. doi: 10.1016/j.meegid.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hayman D.T., Bowen R.A., Cryan P.M., McCracken G.F., O'Shea T.J., Peel A.J., Gilbert A., Webb C.T., Wood J.L. Ecology of zoonotic infectious diseases in bats: current knowledge and future directions. Zoonoses Public Health. 2013;60:2–21. doi: 10.1111/zph.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocher T.D., Thomas W.K., Meyer A., Edwards S.V., Paabo S., Villablanca F.X., Wilson A.C. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. U. S. A. 1989;86:6196–6200. doi: 10.1073/pnas.86.16.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., Thompson J.D., Gibson T.J., Higgins D.G. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H., Zhang J., McEachern J., Field H., Daszak P., Eaton B.T., Zhang S., Wang L.F. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Milne I., Lindner D., Bayer M., Husmeier D., McGuire G., Marshall D.F., Wright F. TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics. 2009;25:126–127. doi: 10.1093/bioinformatics/btn575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monadjem A., Taylor P.J., Cotterill P.D.F., Schoeman M.C. Book, Wits University Press; 2010. Bats of Southern and Central Africa: A Biogeographic and Taxonomic Synthesis. [Google Scholar]
- Morse S.S., Mazet J.A., Woolhouse M., Parrish C.R., Carroll D., Karesh W.B., Zambrana-Torrelio C., Lipkin W.I., Daszak P. Prediction and prevention of the next pandemic zoonosis. Lancet. 2012;380:1956–1965. doi: 10.1016/S0140-6736(12)61684-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly M., Pir J.B., Loesch C., Sausy A., Snoeck C.J., Hubschen J.M., Muller C.P. Novel alphacoronaviruses and paramyxoviruses Cocirculate with type 1 and severe acute respiratory system (SARS)-related Betacoronaviruses in Synanthropic bats of Luxembourg. Appl. Environ. Microbiol. 2017;83 doi: 10.1128/AEM.01326-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters S.P., Perlman S. Fields Virology. Sixth Ed. Lippincott Williams & Wilkins; Philadelphia: 2013. Coronaviridae; pp. 825–858. [Google Scholar]
- Peterson A.T., Lash R.R., Carroll D.S., Johnson K.M. Geographic potential for outbreaks of Marburg hemorrhagic fever. Am. J. Trop. Med. Hyg. 2006;75:9–15. doi: 10.4269/ajtmh.2006.75.1.0750009. [DOI] [PubMed] [Google Scholar]
- Pfefferle S., Oppong S., Drexler J.F., Gloza-Rausch F., Ipsen A., Seebens A., Muller M.A., Annan A., Vallo P., Adu-Sarkodie Y., Kruppa T.F., Drosten C. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 2009;15:1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowright R.K., Eby P., Hudson P.J., Smith I.L., Westcott D., Bryden W.L., Middleton D., Reid P.A., McFarlane R.A., Martin G., Tabor G.M., Skerratt L.F., Anderson D.L., Crameri G., Quammen D., Jordan D., Freeman P., Wang L.F., Epstein J.H., Marsh G.A., Kung N.Y., McCallum H. Ecological dynamics of emerging bat virus spillover. Proc. Biol. Sci. 2015;282 doi: 10.1098/rspb.2014.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol Scientific 1999. http://www.protocol-online.org/prot/Protocols/RNAlater-3999.html
- Quan P.L., Firth C., Street C., Henriquez J.A., Petrosov A., Tashmukhamedova A., Hutchison S.K., Egholm M., Osinubi M.O., Niezgoda M., Ogunkoya A.B., Briese T., Rupprecht C.E., Lipkin W.I. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. MBio. 2010;1 doi: 10.1128/mBio.00208-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A., Hohna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J.P. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons N.B. Evolution. An Eocene big bang for bats. Science. 2005;307:527–528. doi: 10.1126/science.1108871. [DOI] [PubMed] [Google Scholar]
- Smith I., Wang L.F. Bats and their virome: an important source of emerging viruses capable of infecting humans. Curr. Opin. Virol. 2013;3:84–91. doi: 10.1016/j.coviro.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.S., de Jong C.E., Meers J., Henning J., Wang L., Field H.E. Coronavirus infection and diversity in bats in the Australasian region. EcoHealth. 2016;13:72–82. doi: 10.1007/s10393-016-1116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S., Chern S.W., Li Y., Pallansch M.A., Anderson L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008;46:2652–2658. doi: 10.1128/JCM.00192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracer, 2003. v1.6. Available online: http://tree.bio.ed.ac.uk/software/tracer/.
- Wu Z., Yang L., Ren X., Zhang J., Yang F., Zhang S., Jin Q. ORF8-related genetic evidence for Chinese horseshoe bats as the source of human severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2016;213:579–583. doi: 10.1093/infdis/jiv476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic analysis of partial CytB sequences (550 bp). New CytB sequences obtained were compared to representative bat family CytB sequences available in the GenBank. Only Bayesian posterior probabilities are showed. Asterisks at nodes represent posterior probability ≥85%. Scale bars indicate the number of base substitutions per site. New CytB sequences are represented by the red triangle. The following sequences have been used for this analysis: KX467590.1 Miniopterus schreibersii, KX467591.1 Miniopterus magnater, KX467593.1 Tylonycteris pachypus, KX467595.1 Hypsugoal aschanicus, KX467596.1 Nyctalus plancyi, KX467597.1 Pipistrellus abramus, KX467598.1 Murina leucogaster, KX467601.1 Myotis laniger, KX467611.1 Myotis davidii, KX467606.1 Myotis blythii, KX467607.1 Myotis chinensis, KX467608.1 Myotis pequinius, KX467609.1 Myotis bombinus, KX467605.1 Myotis formosus, KX467610.1 Myotis altarium, KX467612.1 Myotis frater, KX467599.1 Myotis ricketti, KX467600.1 Myotis adversus, KX467604.1 Myotis fimbriatus, KX467602.1 Myotis daubentonii, KX467603.1 Myotis macrodactylus, KF218429.1 Taphozous nudiventris, AY591536.1 Otomops martiensseni, AY591537.1 Otomops martiensseni, KX467574.1 Megaderma lyra, KX467575.1 Rhinolophus pusillus, KX467576.1 Rhinolophus lepidus, KX467579.1 Rhinolophus macrotis, KX467580.1 Rhinolophus marshalli, KX467582.1 Rhinolophus rex, KX467578.1 Rhinolophus ferrumequinum, KX467583.1 Rhinolophus sinicus, KX467577.1 Rhinolophus affinis, KX467581.1 Rhinolophus pearsonii, KX467584.1 Hipposideros cineraceus, KX467585.1 Hipposideros pomona, FJ347979.1 Hipposideros caffer, FJ347980.1 Hipposideros caffer, KX467587.1 Hipposideros armiger, KX467588.1 Hipposideros larvatus, KX467589.1 Hipposideros pratti, KX467586.1 Aselliscus stoliczkanus, KT583805.1 Hipposideros vittatus, KT583806.1 Hipposideros vittatus, KT583801.1 Hipposideros gigas, KT583817.1 Hipposideros commersoni, DQ005796.1 Triaenops persicus, DQ005797.1 Triaenops persicus, KX823312.1 Megaerops niphanae, KX823313.1 Cynopterus sp., KX823314.1 Cynopterus sp., KX823317.1 Sphaeria sblanfordi, KX822888.1 Eidolon helvum, KX822889.1 Eidolon helvum, KX823318.1 Macroglossus sobrinus, KX823319.1 Eonycteris spelaea, KX822938.1 Rousettus aegyptiacus, KX822939.1 Rousettus aegyptiacus, KX823315.1 Rousettus leschenaultii, KX823311.1 Plerotes anchietae, KX823011.1 Hypsignathus monstrosus, KX823012.1 Hypsignathus monstrosus, KX823087.1 Epomops franqueti, KX823088.1 Epomops franqueti, KX823233.1 Epomops buettikoferi, KX823234.1 Epomops buettikoferi, KX822985.1 Nanonycteris veldkampii, KX822986.1 Nanonycteris veldkampii, KX823306.1 Epomops dobsonii, KX823307.1 Epomophorus anselli, KX823308.1 Epomophorus crypturus, KX823309.1 Epomophorus labiatus, KX823310.1 Epomophorus minimus, KX822798.1 Epomophorus gambianus, KX822810.1 Micropteropus pusillus, KX822797.1 Epomophorus gambianus, KX822809.1 Micropteropus pusillus.
Percentage Amino Acid identity between A/New alphacoronaviruses identified in Mabura cave and representative Alphacoronavirus strains of interest, B/New betacoronaviruses identified in Magweto cave and representative Betacoronavirus strains of interest, C/New paramyxovirus identified in Magweto cave and representative Paramyxovirus strains of interest. All sequences of interest were obtained from the genbank. Accession numbers are before of the strain name.