

Abstract
Influenza A is a serious respiratory illness that can be debilitating and may cause complications leading to hospitalization and death. The outcome of infection with the influenza A virus is determined by a complex interplay of viral and host factors. With the ongoing threat of seasonal influenza and the potential emergence of new, more virulent strains of influenza viruses, we need to develop a better understanding of genetic variation in the human population and its association with severe outcomes from influenza infection. We propose a list of approximately 100 systems-based candidate genes for future study of the genetic basis of influenza disease and immunity in humans, based on evidence in the published literature for their potential role in the pathogenesis of this infection: binding of the virus to receptors on the host cell surface; cleavability of HA by host proteases; virus replication in host cells; destruction of host cells by apoptosis; state of immunocompetence of the individual host; and viral infections predisposing to bacterial infection.
Keywords: Influenza, Human, Infection, Influenza A virus, Genes
1. Introduction
Influenza is a highly contagious respiratory illness that can be debilitating or cause complications leading to hospitalization and death. The disease's etiological agents, influenza viruses (orthomyxoviruses), are single-stranded, negative-sense RNA viruses (Palese and Shaw, 2007). Of the 3 types (influenza A, B, and C), only A and B cause epidemic human disease. In addition, influenza A virus (IAV) is the precursor of all pandemic viruses and will be the focus of this review. The genome of IAV consists of 8 separate segments, encoding 10–11 proteins covered by the nucleocapsid protein (NP) (McGeoch et al., 1976). Virions are enveloped with two surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA), which are responsible for binding of the virus to host cell receptors and release of progeny virions from host cells, respectively, and are the target of host antibody response. The active RNA-dependent RNA polymerase responsible for replication and transcription consists of three polymerase proteins (PB1, PB2, and PA), which together with each viral genome segment and NP form the eight viral ribonucleoprotein (vRNP) complexes in each virion (Compans et al., 1974). Matrix protein 1 (M1) is a structural protein encasing the viral genome just beneath the envelope, and matrix protein 2 (M2) acts as an ion channel pump that permits ions to enter the virion and undergo uncoating in the endosome (Fujiyoshi et al., 1994, Pinto and Lamb, 2006). The NS gene encodes NS1, which has multiple functions including obstruction of the host antiviral response (Garcia-Sastre, 2006, Krug et al., 2003), and NS2, also known as nuclear export protein, which is important for nuclear export of viral ribonucleic acids (RNAs) (Inglis et al., 1979). By virtue of possessing a segmented genome, influenza virus can easily reassort by rearranging viral segments of two different influenza viruses infecting the same cell and thereby acquire radically different antigenic properties to produce a novel subtype, a phenomenon termed antigenic shift (Cox and Subbarao, 2000, Parrish and Kawaoka, 2005). Antigenic shift may also occur when an animal or avian IAV is transmitted without reassortment directly from an animal reservoir to humans. A pandemic can occur when a new IAV subtype emerges from the reassortment process with the ability to cause disease and spread efficiently in large geographic areas (more than one continent) among humans who have little or no immunity. An example is the current 2009 pandemic swine-origin H1N1 influenza A virus (Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team, 2009, Smith et al., 2009, Zarocostas, 2009), which is slightly more pathogenic than seasonal influenza viruses but much less so than the 1918 pandemic or H5N1 avian IAV in ferret and/or mouse models (Maines et al., 2009, Munster et al., 2009).
During entry into cells, the IAV particle binds to the host cell receptor, sialic acid (SA), through the viral HA (Weis et al., 1988). The virus is taken up into the cell by endocytosis. Acidification of the endocytic vesicle results in a conformational change of HA, leading to fusion of the viral envelope with the endosomal membrane and liberation of vRNPs into the cytoplasm of the host cell (Pinto et al., 1992). The vRNPs are transported to the nucleus for transcription of viral mRNAs and replication of genomic RNA in a complex process, which is delicately regulated by viral and host cellular factors (Boulo et al., 2007, Nagata et al., 2008). Upon translation of viral proteins and assembly of nucleocapsids harboring replicated genomic RNA, progeny virions bud from the cellular membrane, aided by the activity of the NA (Fig. 1 ).
Fig. 1.
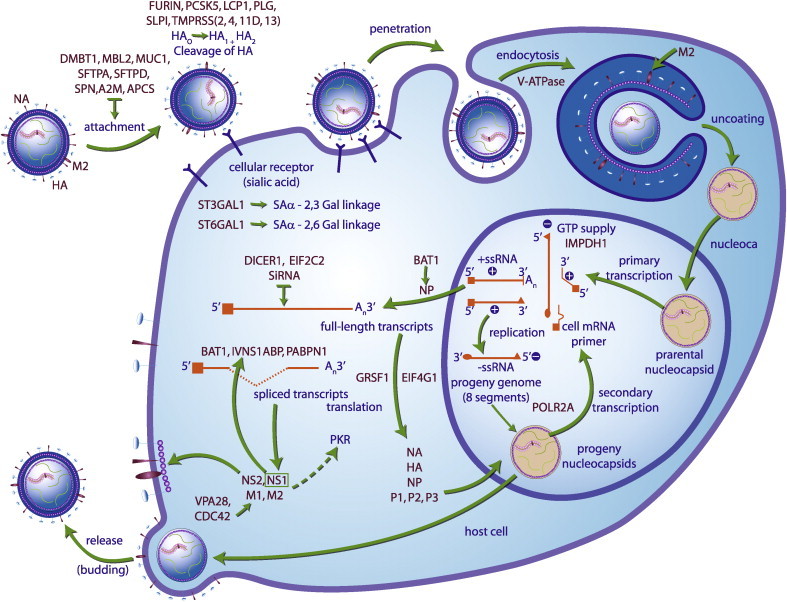
Candidate genes (in purple) involved in IAV replication cycle. Candidate genes controlling virus replication that are discussed in this review are highlighted in purple. During entry into a cell, the IVA particle binds to the host cell receptor SA through its HA. The virus is taken up by the cell by endocytosis. Acidification of the endocytic vesicle results in a conformational change of HA, leading to fusion of the viral envelope with the endosomal membrane and liberation of RNPs into the cytoplasm. The RNPs are transported to the nucleus for transcription of viral mRNAs and replication of genomic RNA in a complex process which is delicately regulated by viral and host cellular factors. Upon translation of viral proteins and assembly of nucleocapsids harboring replicated genomic RNA, progeny virions bud from the cellular membrane by the NA activity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
This figure was adapted from J. Biol. Chem. 2006 March; 281 (13), 8306 with permission.
Based on the antigenicity and amino acid sequences of HA and NA, IAVs are subdivided into 16 HA (H1-H16) and 9 NA (N1-N9) subtypes (Fouchier et al., 2005, World Health Organization, 1980). IAVs can infect people and many other animal species, but wild aquatic birds are the natural hosts for all known subtypes of IAVs. Avian IAV strains are further classified as low pathogenic or highly pathogenic avian influenza (LPAI or HPAI, respectively) on the basis of their intravenous pathogenicity criteria (Swayne and Pantin-Jackwood, 2006, World Animal Health Organization, 2008). HPAI viruses mostly occur among avian H5 and H7 subtypes. In recent years, rare infections among humans with HPAI H5N1 viruses in areas experiencing H5N1 epizootics have resulted in over 60% mortality in humans (EPR, 2009, Uyeki, 2008). H5N1 is considered a significant pandemic threat because human populations lack immunity to it and ongoing genetic variation may allow it to acquire capacity for human-to-human transmission. Avian H7 and H9 subtypes, which have also infected humans, are also considered potential pandemic threats (Werner and Harder, 2006).
The IAV is notorious for its unique ability to cause both recurrent epidemics and global pandemics. Annually, the global burden of influenza epidemics is believed to be 3–5 million cases of severe illness and 300,000–500,000 deaths. In the United States alone, an annual average of approximately 36,000 deaths was estimated for the 1991 through 1999 influenza seasons (Thompson et al., 2003). In the 20th–21st centuries, pandemics occurred in 1918, 1957, 1968, and 2009. The 1918 pandemic was the most severe, with half of the world population infected, of which 20% developed severe disease; estimates of the number of deaths range from 20 to 100 million (Johnson and Mueller, 2002, Oxford, 2000). Fatality after influenza is often caused by secondary bacterial pneumonia and acute respiratory distress syndrome (Grose and Chokephaibulkit, 2004, Muir and Wilson, 1919), as a consequence of highly efficient viral replication, preferred tropism, and a dysfunctional host inflammatory response (Maines et al., 2008). The risk of serious illness and death from influenza is highest in persons aged 65 years or older and in patients with medical conditions that place them at increased risk of developing complications. Recently, a substantial impact of influenza has been recognized in pediatric populations, especially among children who are co-infected with IAV and bacteria; even children who appear to be healthy, infection can progress rapidly to severe illness (Bhat et al., 2005).
Many factors are associated with protection against or increased risk of a fatal outcome caused by a given influenza strain (Behrens and Stoll, 2006). The virulence of IAV is determined by numerous interacting host and viral factors (Baigent and McCauley, 2003). Although characteristics of the host such as age, comorbidity, degree of pre-existing immunity, immunosuppression, pregnancy, and smoking are recognized to influence the acquisition, progression, and resolution of influenza infections, host genetic factors remain a possibility (Nicholson et al., 2003). It has been suggested that individual genetic variation may lead to differential response of the host to IAV infection (Albright et al., 2008). For example, in some recent familial clusters of H5N1 infection, fatal cases curiously clustered among blood relatives (Kandun et al., 2006, Normile, 2007, Olsen et al., 2005).
Host genetic factors determining differential susceptibility or resistance to major infectious diseases of humans (including malaria, HIV/AIDS, tuberculosis, and invasive pneumococcal disease) are well documented (Burgner et al., 2006, Hill, 2006). We carried out a thorough literature review using PubMed and the Human Genome Epidemiology Network Published Literature database (www.hugenavigator.net) (Yu et al., 2008) but could not identify any published epidemiologic or family studies that examined individual genetic variability in relation to susceptibility to IAV disease and its outcomes. In the past decade, a great deal of understanding of the pathogenesis and immune evasion strategies of influenza virus has developed; this could help us propose candidate genes that may be responsible for severe illness. Recently, genome-wide association (GWA) studies have had considerable success in identifying candidate genes for several common, complex diseases; this exploratory and hypothesis-free approach has been applied only twice to the study of infectious disease (Fellay et al., 2007, Jallow et al., 2009). With the annual threat of seasonal influenza and the potential emergence of a novel IAV pandemic strain with an unknown level of virulence, we need to understand genetic variation in the human population and its association with severe outcomes from IAV infection. These efforts will be crucial to the development of tools for targeting interventions in populations (Fauci, 2006).
To identify potential genes for future targeted research, we take a systems-based approach to propose candidate genes that could influence the response of the host to influenza infection based on animal studies, such as global transcriptional profiles and knockout mice, clinical data, or simply biological plausibility. We propose candidate genes whose proteins are implicated in one or more stages in the viral replication cycle and induction of host innate responses, and thus in pathogenesis: binding of the virus to receptors on the host cell surface; cleavability of HA by host proteases; virus replication in host cells; destruction of host cells by apoptosis; state of immunocompetence of the individual host; and viral infections predisposing to bacterial infection. A systems-based approach can provide more complete coverage of candidate genes within a suspected etiologic system, for example, in aberrant host innate immune and cell death responses, which have been suggested by global transcriptional profiling of the host response to IAV infection in mice (Ding et al., 2008, Kash et al., 2006b, Rosseau et al., 2007, Seki et al., 2009), ferrets (Cameron et al., 2008), and macaques (Kobasa et al., 2007). By broadening our proposed candidate genes to include multiple genes in an interconnected system, rather than simply focusing on a single putatively interesting gene, we hope to provide a more complete picture of the impact of the entire system or pathway as we discuss below. We suggest that a systems-based approach to the study of susceptibility to IAV is reasonable, despite the popularity of hypothesis-free GWA studies, which have uncovered “new” candidate genes for a variety of common human diseases; such studies need very large sample sizes and their findings require further replication (Manolio et al., 2008). A systems-based approach takes advantage of understanding the phenotype and biological pathways relating genes to disease; this approach has identified several genes that confer susceptibility to common infectious diseases, e.g. HIV infection (O’Brien and Nelson, 2004, Tabor et al., 2002). A systems-based approach can be successful only if we apply rigorous epidemiological principles to the choice and analysis of candidate genes in the disease studied. A detailed rationale for the selection of each candidate gene we reviewed is listed in Supplementary Table.
2. Binding of the virus to receptors on the host cell surface
The first key step in infection, transmission, and virulence of IVAs is the binding of HA to the host receptor, SA-terminated glycan, on the surface of host cells. The presence, type, and location of the receptor determine both host susceptibility to infection with specific viruses and location of infection. Human IAVs preferentially bind SA in α-2,6 linkage with galactose (Gal), a major glycan of human upper airways; avian IAVs (e.g., H5N1) preferentially recognize a receptor with SAα-2,3Gal linkage, which is common in birds, but is also found in the epithelium of the human lower respiratory tract (Shinya et al., 2006, van Riel et al., 2006) and less commonly in the upper respiratory tract (Nicholls et al., 2007). Recent evidence suggests that the avian H5N1 virus can infect human nasopharyngeal and oropharyngeal epithelia in ex vivo cultures despite of the paucity of SAα-2,3Gal receptors; this finding suggests other binding sites on the epithelium may exist, although the identity of the specific receptors is not yet known (Nicholls et al., 2007, Nicholls et al., 2008). A switch in recognition from avian-like SAα-2,3Gal to human-like SAα-2,6Gal is believed to be one of the changes that must occur before avian IAVs can transmit efficiently among humans and acquire the potential to cause a pandemic (Russell et al., 2006, Stevens et al., 2006, van Riel et al., 2007). This adaption correlates with point mutations changing certain amino acids in viral HA (Vines et al., 1998, Yamada et al., 2006). Genes that control the expression and specificity of oligosaccharide structures on human tissues may contribute to the host's susceptibility and/or to the extent of influenza infection in the upper or lower airways. The host enzyme ST3 beta-galactosamide alpha-2,3-sialyltranferase 1 (ST3GAL1) produces the SAα-2,3Gal structure (Kitagawa and Paulson, 1994), while the SAα-2,6Gal linkage is primarily generated by ST6 beta-galactoside alpha-2,6-sialyltransferase 1(ST6GAL1) (Gagneux et al., 2003, Martin et al., 2002, Weinstein et al., 1987). It has been shown that MDCK cells over-expressing human ST6GAL1 have significantly augmented susceptibility to human influenza viruses and produce more viruses when compared with unmodified cells (Matrosovich et al., 2003, Oh et al., 2008).
The host may prevent attachment of influenza virus by mucociliary clearance or production of mucoproteins that are able to bind to viral HA (Matrosovich and Klenk, 2003). Collectins bind to N-linked glycans on HA and NA and can neutralize the virus by steric hindrance of HA, virus aggregation, opsonisation, and activation of complement-dependent pathways. This group includes surfactant, pulmonary-associated protein A1 (SFTPA1) and D (SFTPD), together with serum mannose-binding lectin 2 (MBL2) (Hartshorn et al., 1993, Lahti et al., 2002, White et al., 2005). The SA-containing glycoprotein or proteoglycans compete with cellular receptors for virus binding, including alpha-2-macroglobulin (A2 M) (Pritchett and Paulson, 1989), amyloid P component, serum (APCS) (Andersen et al., 1997), deleted in malignant brain tumors 1 (DMBT1) (Hartshorn et al., 2006a), and mucins, the major macromolecular component of respiratory mucus encoded by different MUC genes (Matrosovich and Klenk, 2003). Genetic variations at -211 and codon 54 in the promoter region of and within the MBL2 gene have been associated with severe acute respiratory syndrome (SARS) (Ip et al., 2005, Zhang et al., 2005). The common allelic variants of SFTPA1 and SFTPD genes are associated with respiratory distress syndrome (Haataja et al., 2001, Thomas et al., 2007), and respiratory syncytial virus bronchiolitis (Lahti et al., 2002, Lofgren et al., 2002).
3. Cleavability of HA by host proteases
HA-mediated fusion between the host endosomal membrane and the viral envelope requires the cleavage of HA precursor molecule HA0 into HA1 and HA2 subunits by host proteases. Cleavage of HA is indispensable for infectivity and determines viral pathogenicity and tissue tropism (Steinhauer, 1999). The HA0 of human and LPAI has a consensus cleavage site motif and is cleaved by a typsin-like protease; infection is localized to the primary site of infection, such as the respiratory tract in humans or the gastrointestinal tract of aquatic birds, respectively. Some secreted enzymes isolated from rat and swine lungs are shown to support replication of influenza viruses in vitro using mouse-adapted IAV (e.g., WSN strain), including tryptase Clara, mini-plasmin, and ectopic anionic trypsin from rat lungs, and tryptase TC30 from porcine lungs (Kido et al., 2008); however, only a few proteases are present in the human respiratory tract or tissue fluids, including transmembrane protease, serine 2 (TMPRSS2), 11D (TMPRSS11D) (Bottcher et al., 2006), 4 (TMPRSS4) (Chaipan et al., 2009), and plasmin (PLG). These transmembrane serine proteases are identified by gene expression systems in MDCK cells; TMPRSS2 and TMPRSS4 are considered good candidate proteases for the 1918 influenza virus HA (Chaipan et al., 2009). Plasmin is converted from plasminogen into the circulation. WSN NA can sequester plasminogen, leading to increased HA cleavage by plasmin in vitro (Goto et al., 2001). Conversely, the HA0 of HPAI viruses contains genetic insertions at the cleavage site of HA and is cleaved by subtilisin-like proteases Furin (FURIN) (Walker et al., 1994), proprotein convertase subtilisin/kexin type 5 (PCSK5) (Horimoto et al., 1994), and recently identified TMPRSS13 (Kido et al., 2008). Cleavage by these more ubiquitous proteases allows the HPAI viruses to infect various cell types and cause systemic infection. In addition to host cellular proteases, microbial proteases also activate viral HA, which may play a role in synergism of co-infection by IAV and bacteria, as discussed later (Tashiro et al., 1987). Host protease activities can be suppressed by protease inhibitors such as human mucus secretory leukocyte peptidase inhibitor (SLPI) in the upper respiratory tract and SFTPA1 and SFTPD in the lower respiratory tract, which inhibit the interaction between the proteases and viral HA and NA (Kido et al., 2007)
4. Replication of the virus in host cells
Once the viral HA binds to SA receptors on the cell surface, the virus is internalized either by clathrin-mediated or a clarin- and caveolin-independent endocytic pathway (Lakadamyali et al., 2004, Rust et al., 2004). At low pH within endosome, the viral HA undergoes a conformational change that enables fusion of the viral and host endosomal membranes, allowing for the release of vRNPs into the cytoplasm (Pinto et al., 1992). The acidification of endosomes is regulated by host cellular vacuolar H+ ATPases (Guinea and Carrasco, 1995). After transport of the viral genome into the nucleus, viral transcription is performed by the viral RNA-dependent RNA polymerase to produce mRNAs that are exported from the nucleus and translated into viral proteins in the cytoplasm. Trafficking of viral genomic RNA into and out of the nucleus through the nuclear membrane is through interactions with host cell nuclear import and export machinery, including importin alpha and beta, exportin 1, and RAN (Boulo et al., 2007).
For replication and transcription of the influenza virus genome, not only viral factors but also host-derived cellular factors are required. Key among the functional interactions during influenza virus infection is the dependence of the virus on cellular RNA synthesis by DNA-dependent RNA polymerase II (Pol II), as the viral mRNA transcription is initiated by use of short 5′ capped RNA fragments, derived from host cellular Pol II transcripts, as primers (Engelhardt and Fodor, 2006). The virus can alter the distribution of Pol II on cellular genes, leading to a reduction in Pol II elongation, thereby contributing to the well-known phenomenon of host cell shutoff, in which there is a dramatic decrease in the translation of cellular mRNAs while viral transcripts remain efficiently and selectively translated during influenza virus infection (Chan et al., 2006, Engelhardt et al., 2005). Recent functional assays and proteomics have suggested a panel of host cellular proteins may interact with the viral polymerase and vRNP complexes (for review, see Mayer et al., 2007, Nagata et al., 2008). Viral NS1 is also a major player in shutting down host protein synthesis by interacting with host proteins to interfere with the host machinery, such as CPSF4 (cleavage and polyadenylation specific factor 4, 30kDa) (Noah et al., 2003) and PABN1 (poly(A) binding protein, nuclear 1) (Chen et al., 1999) in polyadenylation; IVNS1ABP (influenza virus NS1A binding protein) (Wolff et al., 1998) in splicing; and BAT1(HLA-B associated transcript 1) (Momose et al., 2001) in nucleocytoplasmic transport of cellular mRNAs. NS1 also recruits the cellular initiation factor, EIF4G1 (eukaryotic translation initiation factor 4 gamma, 1), allowing for the preferential translation of the IAV messengers (Aragon et al., 2000). Viral M1 can interact with VPS28 (vacuolar protein-sorting 28 homolog), a component of endosomal complexes required for transport, and CDC42 (cell division cycle 42), a small G protein, playing an important role in budding of the IAV (Hui et al., 2006). A variety of intracellular signaling pathways activated by IAV infection, such as the Raf/MEK/ERK mitogenic kinase cascade, are in part exploited by the virus to ensure efficient replication (Ludwig, 2007). Specific blockade of the Raf/MEK/ERK pathway results in nuclear retention of viral RNPs complexes in late stages of the replication cycle, which significantly impairs growth of IAVs tested in cell culture and Raf-BxB transgenic mice (Olschlager et al., 2004, Pleschka et al., 2001). A number of host proteins, including lipids, microfilaments, G proteins, and some protein kinases, have been shown to be involved in the budding of influenza virus (for a review, see Nayak et al., 2004). For example, casein kinase 2 (CK2) is found in influenza virus particles; increased CK2 activity correlates with the replication cycle of influenza virus and its inhibitors disrupt virus budding in MDCK cells (Hui and Nayak, 2002, Tucker et al., 1991).
IAV has also developed translational control strategies that use both viral and cellular proteins to ensure high levels of viral protein synthesis during infection (Kash et al., 2006a). The short 5′ capped and highly conserved host cellular RNA sequence at the IAV mRNAs can bind with several host cellular proteins such as the RNA-binding protein GRSF1 (G-rich RNA sequence binding factor 1), which acts as a positive regulator of IAV protein synthesis by selectively recruiting viral and host cellular mRNAs containing 5′ UTR binding sites to polyribosomes in vitro (Kash et al., 2002, Park et al., 1999).
Recent studies indicate that exogenously administered synthetic small interfering RNAs (siRNAs) can protect against replication of the influenza virus in infected mice, although the extent to which this occurs naturally upon influenza infection is not clear (Tompkins et al., 2004). RNA interference (RNAi) is a natural host cellular process that inhibits gene expression by causing the degradation of specific RNA molecules or by hindering the transcription of specific genes. The RNAi pathway is initiated by the host cellular endonuclease Dicer1, which cleaves viral long, double-stranded RNA (dsRNA) molecules into short pieces siRNAs. The slicer, eukaryotic translation initiation factor 2C, 2 (EIF2C2), cuts the siRNA strand that is subsequently degraded and prevents viral replication (Rand et al., 2005).
Reducing the nucleotide supply for synthesis of viral RNA can also inhibit replication of the influenza virus. IMP dehydrogenase 1 (IMPDH1) is a key cellular enzyme involved in the biosynthesis of guanine nucleotides and viral RNA synthesis, which is the main target of antiviral action of Ribavirin, an anti-viral drug against both human and avian (H5N1) influenza viruses (De Clercq, 2006).
5. Destruction of host cells by apoptosis
IAV infection can induce apoptosis, or programmed cell death, which has been postulated to be a host defense mechanism to limit the replication and spread of virus. The precise mechanism of virus-induced apoptosis is unclear. Many pathways can be activated, and the initiation of one pathway can induce multiple signal transduction cascades through feedback loops. Such pathways, at the least, include those mediated by Fas (TNF receptor superfamily, member 6), eukaryotic translation initiation factor 2-alpha kinase 2 (EIF2AK2, also known as PKR), or transforming growth factor, beta 1 (TGFB1) (Brydon et al., 2005). PKR is a key regulator in influenza virus-induced apoptosis. Activation of PKR by IAV dsRNA leads to several sequential downstream events, including the activation of EIF2A, BAX, BCL2, and NFKB1. The latter leads to transcriptional induction of many pro-apoptotic genes, including those encoding Fas, TP53, and JUN. PKR can initiate CASP8, a predominant apoptotic pathway in human bronchiolar cells during IAV infection, leading to a caspase cascade (Zhirnov et al., 2002).
Several influenza viral proteins are involved in induction of apoptosis. NA can activate latent TGFB1 on the cell surface to its active form. TGFB1 initiates a signaling cascade, leading to the activation of the JNK or SAPK, which in turn results in the activation of transcription factors and upregulation of pro-apoptotic gene expression. In virus-infected cells, IAV encodes a second nonstructural polypeptide, PB1-F2, which induces apoptosis through mitochondrial solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 6 (SLC25A6) and voltage-dependent anion channel 1 (VDAC1) (Zamarin et al., 2005).
6. Immunocompetence state of the individual host
Influenza virus infection elicits a complex network of host immune responses. Innate immunity represents the first barrier to limit initial viral replication and provides the signals required for the subsequent adaptive cellular and humoral immune responses to develop. The adaptive immune response ultimately helps to reduce the viral burden, to eliminate the virus, and to trigger disease recovery (Behrens and Stoll, 2006). The immunocompetence of the individual host affects the course of the disease outcome.
6.1. Innate antiviral response
The interferon (IFN)-induced cellular antiviral response has a primary protective function in the early stages of influenza virus infection (Garcia-Sastre, 2006). Both single-strand (ss) and dsRNA, viral RNA products generated during infection, act like triggers for the production of type I IFN, IFNA1 and IFNB1, which are recognized by two types of sensors, the transmembrane Toll-like receptors (TLR3, -7, and -8) and cytoplasmic RIG-I-like receptors (RIG-I, MDA5, and LGP2) (Garcia-Sastre and Biron, 2006). The Toll-like receptors signal via their adaptors either TRIF in the case of TLR3 or MyD88 in the case of TLR7 and -8 (Diebold et al., 2004, Guillot et al., 2005). The RIG-I-like receptors signal via mitochondrial antiviral signaling protein (MASP), also known as IPS-1 (Yoneyama and Fujita, 2007). These interactions initiate signaling pathways that differ in their initial steps but converge in the activation of IRF3 and NFKB1, which induce transcription of IFNB1 and IFNB2. Mice lacking both Myd88 and Masp genes fail to up-regulate antiviral responses; Tlr7- and Myd88-deficient mice have no protection against IAV vaccination challenges (Koyama et al., 2007). Tlr3 knockout mice show significantly reduced inflammatory mediators and better survival rates compared with wild-type mice after IAV infection, despite of a higher viral load in the lungs (Le Goffic et al., 2006). A loss-of-function mutant in TLR3 (F303S) was identified in a patient with influenza-associated encephalopathy (Hidaka et al., 2006).
Virus-infected cells synthesize IFN and secrete it into extracellular fluid, where it binds to type 1 IFN receptors, IFNAR1 and IFNAR2, on uninfected neighboring cells. This binding, followed by activation of the Janus kinase (JAK) family and subsequent activation of signal transducers and activators of transcription (STAT) proteins (Murray, 2007), results in the induction of a cellular antiviral response involving the transcriptional upregulation of more than 100 IFN-stimulated genes (such as CXCL10, MX1, PKR, OAS1, RNASEL, and PML), through several signaling pathways, including NFKB1, JUN, and MX1 (Garcia-Sastre and Biron, 2006). IFN also stimulates the production of a form of inducible NOS2A and the MHC class I and II proteins, all of which play important roles in the immune response to infections. The IFNG-induced genes are vigorously expressed in H5N1-infected ferret lungs, in particular, CXCL10, for which blockade of its cognate receptor, CXCR3, reduces the mortality associated with H5N1 infection in ferrets (Cameron et al., 2008). Susceptibility to the SARS coronavirus has been associated with genetic variants in human OAS1 and MX1 genes (Hamano et al., 2005, He et al., 2006). Genetic susceptibility to human respiratory syncytial virus bronchiolitis is predominantly associated with innate immune genes (JUN, IFNA5, and NOS2A) (Janssen et al., 2007).
To establish a productive infection, influenza viruses must first overcome the IFN-induced blocks imposed on viral replication. dsRNA-activated PKR can restrict viral replication through its ability to phosphorylate the protein synthesis initiation factor EIF2A, resulting in a reduced ability of EIF2A to initiate translation. This prevents viral replication and inhibits normal cell ribosome function, killing both the virus and the host cell if the response is active for a sufficient time. To evade the antiviral effects of PKR, IAV has evolved 2 strategies to block PKR activation or action: the virus activates a host cellular inhibitor of PKR, p58IPK, and its NS1 protein blocks PKR activation (Kash et al., 2006a). IAV NS1 protein has been identified as a potent agonist of the innate antiviral signaling, both by interference with the RIG-I induction of IFNB (Guo et al., 2007, Mibayashi et al., 2007), and at a later stage, by modulating processing of pre-mRNA (Krug et al., 2003). A recent in vitro study has shown that IAVs not only suppress IFNB gene production but also inhibit type I IFN signaling via a NFKB1-dependent induction of the suppressor of cytokine signaling 3 (SOCS3) protein, a potent endogenous blocker of JAK/STAT signaling, which results in an impaired antiviral response (Pauli et al., 2008). Evidence from animal studies suggests that evasion of host innate immunity, including the type I interferon response, is a mechanism of virulence of avian H5N1 viruses in mammals including humans (Maines et al., 2008).
While on the one hand the IAVs are capable of suppressing unwanted antiviral signaling events, there is accumulating evidence that the viruses have evolved strategies to benefit from the antiviral signaling activates to ensure efficient replication (Ludwig, 2007). For example, the IAVs appear to be able to switch the antiviral activity of NFKB1 into a virus supportive event (Ludwig and Planz, 2008), as evidenced by two independent studies showing that influenza viruses replicated much better in human cells in vitro where NFKB1 was pre-activated (Nimmerjahn et al., 2004, Wurzer et al., 2004).
The respiratory tract secretes antimicrobial peptides such as the β-defensins and CAMP (Hartshorn et al., 2006b). β-defensins (DEFA1, DEFA2, DEFB3, and DEFT1P) inhibit infectivity of IAV via interactions with SFTPD. Retrocyclin 2, a synthetic theta-defensin based on a human pseudogene, DEFT1P, and DEFB3 can block virus entry of influenza by blocking membrane fusion mediated by the viral HA (Leikina et al., 2005). Numerous plasma acute phase proteins, including CRP, SAA1, and orosomucoid, increase dramatically in concentration in response to IAV infection (Barclay et al., 1969, Falsey et al., 2001).
6.2. Acute inflammatory reaction, cytokine release, and cytokine storm
Rapid replication and distribution of the IAV within the lungs causes local and systemic inflammation and production of chemokines and cytokines, such as CCL2, CCL5, CCR5, CX3CR1, CXCR3, CXCL10, and IL8 (see Supplementary Table for list of cytokine genes). Cytokines, components of the innate immune response, also affect the adaptive immune response and manifestation of the disease (Kaufmann et al., 2001). Cytokines can signal immune cells, including T-cells and macrophages, to travel to the site of infection. In addition, cytokines activate those cells, stimulating them to produce more cytokines. Normally this positive feedback loop is kept in check by negative regulatory elements, including other specific cytokines. In some instances, however, an uncontrolled or excessive immune response to the IAV results in an outpouring of numerous chemokines and cytokines (Osterholm, 2005). Both pro-inflammatory cytokines, such as TNF, IL1B, and IL6, and anti-inflammatory cytokines, such as IL10 and IL1RN, are elevated in the serum. The fierce and often lethal interplay of these cytokines leading to the acute respiratory distress syndrome is referred to as a “cytokine storm”, which has been suggested as an explanation, in addition to highly efficient viral replication, for the devastating nature of the 1918 pandemic flu, particularly in younger adults. Evidence for this hypothesis comes from 1918-virus-infected mouse (Kash et al., 2006b) and 1918-virus-infected macaque (Kobasa et al., 2007) models and may help explain the high rate of severe and fatal disease outcomes in humans infected with highly pathogenic avian H5N1 influenza viruses (de Jong et al., 2006).
6.3. Adaptive immunity
The adaptive immune response requires some days to be effective in a naïve but not a primed or immune individual, then works to contain the viral spread, to eradicate the virus, and finally to establish a memory response resulting in a long-lived resistance to reinfection with homologous infection. Mucosal or systematically produced antibodies to viral HA are the primary mediators of virus neutralization and the main objectives of immunization with vaccine. Like antibodies to NA, anti-HA antibodies can also reduce viral load by non-neutralizing mechanisms, which include lysis of the infected cell via a complement system or antibody-dependent cellular cytotoxicity (ADCC). Antibodies to the M1 and NP proteins are made following influenza infection but they play an unknown role in immunity against influenza. Although antibodies to M2 can protect from severe disease and reduce viral load in experimental animal studies, evidence for a similar role in humans is lacking. Antigen presentation via MHC class I and II molecules by dendritic cells lead to activation, proliferation, and differentiation of the antigen-specific T-cells, CD4+ and CD8+. The influenza-specific CD4+ T helper cells are primarily responsible for promoting a high-quality antibody response. The CD8+ cytotoxic T lymphocytes (CTL) have the ability to recognize specific viral epitopes encoded by multiple viral genes on MHC class I molecules, but depending on an individual's HLA haplotype. CD8+ T cells recognizing only one or a few viral epitopes will dominate the response, producing antiviral cytokines and lysing target cells that present viral determinants for which they bear a specific T-cell receptor. CD8+ and CD4+ effector T cells (Teff cells) can control lung inflammation during acute influenza virus infection by producing Il10 in mice. Blocking the action of the Teff cell-derived Il10 results in increased pulmonary inflammation and lethal injury (Sun et al., 2009). CD8+ CTLs clear IVA-infected epithelial cells mainly by CD69-mediated cytotoxicity or exocytosis granules containing perforin 1 and granzymes, such as granzyme B (Topham et al., 1997). Natural killer (NK) cells have lectin-like ‘nonspecific’ receptors for virus-infected cells (Biassoni et al., 2001). The IAV HA protein can interact with two lysis receptors, NCR1 and NCR2 (Arnon et al., 2001, Mandelboim et al., 2001). Increased binding to influenza virus-infected cells is also observed in 2 inhibitory receptors, KIR2DL1 and LILRBI (Achdout et al., 2003). NK cells can recognize antibody-coated virally infected cells through their Fcγ receptors, FCGR3A and FCGR3B, and kill the targets by ADCC (Hashimoto et al., 1983). Various Fcγ receptors, including Fcgr1a, Fcgr2b, Fcgr1 g, Fcgr3a, and Fcgr3b, are over-expressed in murine lungs after infection with IAV (Rosseau et al., 2007). The list of host genes involved in the adaptive immune response is extensive (Doherty et al., 2006) and is beyond the scope of this review.
7. Viral infections predisposing to bacterial infection
It has been appreciated for more than a century that influenza virus can predispose the host to bacterial infections that lead to disease complications. The influenza pandemic of 1918 claimed 40 to 50 million lives, many because of secondary bacterial pneumonia (Muir and Wilson, 1919). In recent years in the United States, numbers of influenza-associated pediatric deaths, in which both influenza and pneumonia or bacteremia due to Streptococus aureus coinfection were identified, have increased. Most of these young people were in good health before the onset of illness but progressed rapidly to severe illness (CDC, 2007).
Several factors may explain the lethal synergism between influenza virus and bacteria (McCullers, 2006). Destruction of respiratory epithelium by the virus can promote bacterial adhesion. In addition, several bacteria secrete proteases that activate infectivity of IAV by proteolytic cleavage of the viral HA (Tashiro et al., 1987). Inflammatory response to viral infection may up-regulate expression of molecules such as PTAFR (platelet-activating factor receptor) that bacteria can use as receptors, as suggested by studies in mice (McCullers and Rehg, 2002) and Ptafr-knockout mice (van der Sluijs et al., 2006). Recently, a DNA microarray analysis has demonstrated that 15 platelet activating factor-related molecules were specifically induced in the murine lungs co-infected by IAV and Streptococus pneumoniae (Seki et al., 2009). The virus infection can cause bacterial superinfections by impairing the ability of the host to mount a productive immune response against subsequent bacterial infection. Host immunopathology may be increased through the overproduction of inflammatory cytokines, cytokine storm, and apoptosis (Kash et al., 2006b). Rosseau and colleagues have reported inflammatory transcriptional signatures of 1300 genes from murine lungs after infection with either IAV or Streptococus pneumoniae. Of these, approximately 36% were specific for pneumococcus and 30% were specific for the virus; a differential response was observed on the cytokine and chemokine level, although common induction of type I and type II interferon, Tnf, and Il6 underlies both infections (Rosseau et al., 2007). OX40-immunogolubulin, an effective immunosuppressive agent that targets the co-stimulatory molecule TNFRSF4, formerly OX40, allows sufficient activation of the host immune response to clear an IAV infection, but it suppresses the response enough to prevent severe immunopathology in lungs of infected mice (Humphreys et al., 2003).
8. Conclusion
Annual epidemics of influenza and the threat of another influenza pandemic present a unique challenge to public health and biomedical research worldwide. Recent developments in large-scale complete-genome sequencing, antigenic mapping, and epidemiological modeling promise to provide a more comprehensive picture of the evolution of influenza viruses and of their pattern of transmission through human and animal populations (Nelson and Holmes, 2007). Completion of the human genome project, validation of millions of human genetic variants in HapMap (International HapMap Consortium, 2005) and rapid advances in genotyping technologies create a great opportunity to advance human genomic studies of influenza infection in areas such as susceptibility and disease severity. By leveraging biological knowledge to propose an extended selection of candidate genes across different systems or pathways in the pathogenesis of influenza virus infection, we hope to provide a more complete picture of the impact of the entire system, for which the list of candidate genes should not be limited only to those highlighted here. Studies of candidate genes in human host response to IAV infection are an important starting point for understanding the constantly evolving interaction between virus and host, for identifying prognostic indicators, and for developing tools for targeting interventions in populations and novel antiviral therapies (Fauci, 2006). Establishing a DNA bank from patients with influenza infection for study of genetic variation in the human population and its association with the pathogenicity of influenza, its transmissibility, and the effectiveness of vaccines (and their side effects) will be a first step in this process.
Conflict of interest
None of the authors hold any commercial association that might pose or create a conflict of interest with the information presented in the submitted manuscript, including consultancies, stock ownership, or other equity interests, patent-licensing arrangements, and payments for conducting or publicizing a study described in the manuscript.
Acknowledgements
We thank Lori Durand for creating the schematic diagram of candidate genes involved in IAV replication cycle and Sara Bedrosian for editorial assistance.
Footnotes
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.meegid.2009.07.006.
Appendix A. Supplementary data
References
- Achdout H., Arnon T.I., Markel G., Gonen-Gross T., Katz G., Lieberman N., Gazit R., Joseph A., Kedar E., Mandelboim O. Enhanced recognition of human NK receptors after influenza virus infection. J Immunol. 2003;171:915–923. doi: 10.4049/jimmunol.171.2.915. [DOI] [PubMed] [Google Scholar]
- Albright F.S., Orlando P., Pavia A.T., Jackson G.G., Cannon Albright L.A. Evidence for a heritable predisposition to death due to influenza. J Infect Dis. 2008;197:18–24. doi: 10.1086/524064. [DOI] [PubMed] [Google Scholar]
- Andersen O., Vilsgaard Ravn K., Juul Sorensen I., Jonson G., Holm Nielsen E., Svehag S.E. Serum amyloid P component binds to influenza A virus haemagglutinin and inhibits the virus infection in vitro. Scand J Immunol. 1997;46:331–337. doi: 10.1046/j.1365-3083.1997.d01-147.x. [DOI] [PubMed] [Google Scholar]
- Aragon T., de la Luna S., Novoa I., Carrasco L., Ortin J., Nieto A. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol Cell Biol. 2000;20:6259–6268. doi: 10.1128/mcb.20.17.6259-6268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon T.I., Lev M., Katz G., Chernobrov Y., Porgador A., Mandelboim O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol. 2001;31:2680–2689. doi: 10.1002/1521-4141(200109)31:9<2680::aid-immu2680>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Baigent S.J., McCauley J.W. Influenza type A in humans, mammals and birds: determinants of virus virulence, host-range and interspecies transmission. Bioessays. 2003;25:657–671. doi: 10.1002/bies.10303. [DOI] [PubMed] [Google Scholar]
- Barclay G.R., Flewett T.H., Keller E., Halsall H.B., Spragg S.P. Effect of polymerized orosomucoid on some strains of influenza virus. Biochem J. 1969;111:353–357. doi: 10.1042/bj1110353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, G., Stoll, M., 2006. Pathogenesis and Immunology in “Influenza Report 2006”, www.influenzareport.com.
- Bhat N., Wright J.G., Broder K.R., Murray E.L., Greenberg M.E., Glover M.J., Likos A.M., Posey D.L., Klimov A., Lindstrom S.E., Balish A., Medina M.J., Wallis T.R., Guarner J., Paddock C.D., Shieh W.J., Zaki S.R., Sejvar J.J., Shay D.K., Harper S.A., Cox N.J., Fukuda K., Uyeki T.M. Influenza-associated deaths among children in the United States, 2003-2004. N Engl J Med. 2005;353:2559–2567. doi: 10.1056/NEJMoa051721. [DOI] [PubMed] [Google Scholar]
- Biassoni R., Cantoni C., Pende D., Sivori S., Parolini S., Vitale M., Bottino C., Moretta A. Human natural killer cell receptors and co-receptors. Immunol Rev. 2001;181:203–214. doi: 10.1034/j.1600-065x.2001.1810117.x. [DOI] [PubMed] [Google Scholar]
- Bottcher E., Matrosovich T., Beyerle M., Klenk H.D., Garten W., Matrosovich M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol. 2006;80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulo S., Akarsu H., Ruigrok R.W., Baudin F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007;124:12–21. doi: 10.1016/j.virusres.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Brydon E.W., Morris S.J., Sweet C. Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol Rev. 2005;29:837–850. doi: 10.1016/j.femsre.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Burgner D., Jamieson S.E., Blackwell J.M. Genetic susceptibility to infectious diseases: big is beautiful, but will bigger be even better? Lancet Infect Dis. 2006;6:653–663. doi: 10.1016/S1473-3099(06)70601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron C.M., Cameron M.J., Bermejo-Martin J.F., Ran L., Xu L., Turner P.V., Ran R., Danesh A., Fang Y., Chan P.K., Mytle N., Sullivan T.J., Collins T.L., Johnson M.G., Medina J.C., Rowe T., Kelvin D.J. Gene expression analysis of host innate immune responses during Lethal H5N1 infection in ferrets. J Virol. 2008;82:11308–11317. doi: 10.1128/JVI.00691-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC, 2007. Influenza-associated pediatric mortality and the increase of Staphylococcus aureus co-infection. CDC, Atlanta, GA, http://www2a.cdc.gov/HAN/ArchiveSys/ViewMsgV.asp?AlertNum=00259.
- Chaipan C., Kobasa D., Bertram S., Glowacka I., Steffen I., Tsegaye T.S., Takeda M., Bugge T.H., Kim S., Park Y., Marzi A., Pohlmann S. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol. 2009;83:3200–3211. doi: 10.1128/JVI.02205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A.Y., Vreede F.T., Smith M., Engelhardt O.G., Fodor E. Influenza virus inhibits RNA polymerase II elongation. Virology. 2006;351:210–217. doi: 10.1016/j.virol.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Chen Z., Li Y., Krug R.M. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3’-end processing machinery. EMBO J. 1999;18:2273–2283. doi: 10.1093/emboj/18.8.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compans R.W., Meier-Ewert H., Palese P. Assembly of lipid-containing viruses. J Supramol Struct. 1974;2:496–511. doi: 10.1002/jss.400020234. [DOI] [PubMed] [Google Scholar]
- Cox N.J., Subbarao K. Global epidemiology of influenza: past and present. Annu Rev Med. 2000;51:407–421. doi: 10.1146/annurev.med.51.1.407. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong M.D., Simmons C.P., Thanh T.T., Hien V.M., Smith G.J., Chau T.N., Hoang D.M., Chau N.V., Khanh T.H., Dong V.C., Qui P.T., Cam B.V., Ha do Q., Guan Y., Peiris J.S., Chinh N.T., Hien T.T., Farrar J. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold S.S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Ding M., Lu L., Toth L.A. Gene expression in lung and basal forebrain during influenza infection in mice. Genes Brain Behav. 2008;7:173–183. doi: 10.1111/j.1601-183X.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- Doherty P.C., Turner S.J., Webby R.G., Thomas P.G. Influenza and the challenge for immunology. Nat Immunol. 2006;7:449–455. doi: 10.1038/ni1343. [DOI] [PubMed] [Google Scholar]
- Engelhardt O.G., Fodor E. Functional association between viral and cellular transcription during influenza virus infection. Rev Med Virol. 2006;16:329–345. doi: 10.1002/rmv.512. [DOI] [PubMed] [Google Scholar]
- Engelhardt O.G., Smith M., Fodor E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol. 2005;79:5812–5818. doi: 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsey A.R., Walsh E.E., Francis C.W., Looney R.J., Kolassa J.E., Hall W.J., Abraham G.N. Response of C-reactive protein and serum amyloid A to influenza A infection in older adults. J Infect Dis. 2001;183:995–999. doi: 10.1086/319275. [DOI] [PubMed] [Google Scholar]
- Fauci A.S. Emerging and re-emerging infectious diseases: influenza as a prototype of the host-pathogen balancing act. Cell. 2006;124:665–670. doi: 10.1016/j.cell.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellay J., Shianna K.V., Ge D., Colombo S., Ledergerber B., Weale M., Zhang K., Gumbs C., Castagna A., Cossarizza A., Cozzi-Lepri A., De Luca A., Easterbrook P., Francioli P., Mallal S., Martinez-Picado J., Miro J.M., Obel N., Smith J.P., Wyniger J., Descombes P., Antonarakis S.E., Letvin N.L., McMichael A.J., Haynes B.F., Telenti A., Goldstein D.B. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier R.A., Munster V., Wallensten A., Bestebroer T.M., Herfst S., Smith D., Rimmelzwaan G.F., Olsen B., Osterhaus A.D. Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J Virol. 2005;79:2814–2822. doi: 10.1128/JVI.79.5.2814-2822.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyoshi Y., Kume N.P., Sakata K., Sato S.B. Fine structure of influenza A virus observed by electron cryo-microscopy. EMBO J. 1994;13:318–326. doi: 10.1002/j.1460-2075.1994.tb06264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux P., Cheriyan M., Hurtado-Ziola N., van der Linden E.C., Anderson D., McClure H., Varki A., Varki N.M. Human-specific regulation of alpha 2-6-linked sialic acids. J Biol Chem. 2003;278:48245–48250. doi: 10.1074/jbc.M309813200. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A. Antiviral response in pandemic influenza viruses. Emerg Infect Dis. 2006;12:44–47. doi: 10.3201/eid1201.051186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A., Biron C.A. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Goto H., Wells K., Takada A., Kawaoka Y. Plasminogen-binding activity of neuraminidase determines the pathogenicity of influenza A virus. J Virol. 2001;75:9297–9301. doi: 10.1128/JVI.75.19.9297-9301.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose C., Chokephaibulkit K. Avian influenza virus infection of children in Vietnam and Thailand. Pediatr Infect Dis J. 2004;23:793–794. doi: 10.1097/00006454-200408000-00024. [DOI] [PubMed] [Google Scholar]
- Guillot L., Le Goffic R., Bloch S., Escriou N., Akira S., Chignard M., Si-Tahar M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- Guinea R., Carrasco L. Requirement for vacuolar proton-ATPase activity during entry of influenza virus into cells. J Virol. 1995;69:2306–2312. doi: 10.1128/jvi.69.4.2306-2312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z., Chen L.M., Zeng H., Gomez J.A., Plowden J., Fujita T., Katz J.M., Donis R.O., Sambhara S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am J Respir Cell Mol Biol. 2007;36:263–269. doi: 10.1165/rcmb.2006-0283RC. [DOI] [PubMed] [Google Scholar]
- Haataja R., Marttila R., Uimari P., Lofgren J., Ramet M., Hallman M. Respiratory distress syndrome: evaluation of genetic susceptibility and protection by transmission disequilibrium test. Hum Genet. 2001;109:351–355. doi: 10.1007/s004390100574. [DOI] [PubMed] [Google Scholar]
- Hamano E., Hijikata M., Itoyama S., Quy T., Phi N.C., Long H.T., Ha le D., Ban V.V., Matsushita I., Yanai H., Kirikae F., Kirikae T., Kuratsuji T., Sasazuki T., Keicho N. Polymorphisms of interferon-inducible genes OAS-1 and MxA associated with SARS in the Vietnamese population. Biochem Biophys Res Commun. 2005;329:1234–1239. doi: 10.1016/j.bbrc.2005.02.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn K.L., Ligtenberg A., White M.R., Van Eijk M., Hartshorn M., Pemberton L., Holmskov U., Crouch E. Salivary agglutinin and lung scavenger receptor cysteine-rich glycoprotein 340 have broad anti-influenza activities and interactions with surfactant protein D that vary according to donor source and sialylation. Biochem J. 2006;393:545–553. doi: 10.1042/BJ20050695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn K.L., Sastry K., White M.R., Anders E.M., Super M., Ezekowitz R.A., Tauber A.I. Human mannose-binding protein functions as an opsonin for influenza A viruses. J Clin Invest. 1993;91:1414–1420. doi: 10.1172/JCI116345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn K.L., White M.R., Tecle T., Holmskov U., Crouch E.C. Innate defense against influenza A virus: activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J Immunol. 2006;176:6962–6972. doi: 10.4049/jimmunol.176.11.6962. [DOI] [PubMed] [Google Scholar]
- Hashimoto G., Wright P.F., Karzon D.T. Antibody-dependent cell-mediated cytotoxicity against influenza virus-infected cells. J Infect Dis. 1983;148:785–794. doi: 10.1093/infdis/148.5.785. [DOI] [PubMed] [Google Scholar]
- He J., Feng D., de Vlas S.J., Wang H., Fontanet A., Zhang P., Plancoulaine S., Tang F., Zhan L., Yang H., Wang T., Richardus J.H., Habbema J.D., Cao W. Association of SARS susceptibility with single nucleic acid polymorphisms of OAS1 and MxA genes: a case-control study. BMC Infect Dis. 2006;6:106. doi: 10.1186/1471-2334-6-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka F., Matsuo S., Muta T., Takeshige K., Mizukami T., Nunoi H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin Immunol. 2006;119:188–194. doi: 10.1016/j.clim.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Hill A.V. Aspects of genetic susceptibility to human infectious diseases. Annu Rev Genet. 2006;40:469–486. doi: 10.1146/annurev.genet.40.110405.090546. [DOI] [PubMed] [Google Scholar]
- Horimoto T., Nakayama K., Smeekens S.P., Kawaoka Y. Proprotein-processing endoproteases PC6 and furin both activate hemagglutinin of virulent avian influenza viruses. J Virol. 1994;68:6074–6078. doi: 10.1128/jvi.68.9.6074-6078.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E.K., Barman S., Tang D.H., France B., Nayak D.P. YRKL sequence of influenza virus M1 functions as the L domain motif and interacts with VPS28 and Cdc42. J Virol. 2006;80:2291–2308. doi: 10.1128/JVI.80.5.2291-2308.2006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hui E.K., Nayak D.P. Role of G protein and protein kinase signalling in influenza virus budding in MDCK cells. J Gen Virol. 2002;83:3055–3066. doi: 10.1099/0022-1317-83-12-3055. [DOI] [PubMed] [Google Scholar]
- Humphreys I.R., Walzl G., Edwards L., Rae A., Hill S., Hussell T. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J Exp Med. 2003;198:1237–1242. doi: 10.1084/jem.20030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis S.C., Barrett T., Brown C.M., Almond J.W. The smallest genome RNA segment of influenza virus contains two genes that may overlap. Proc Natl Acad Sci U S A. 1979;76:3790–3794. doi: 10.1073/pnas.76.8.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International-HapMap-Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip W.K., Chan K.H., Law H.K., Tso G.H., Kong E.K., Wong W.H., To Y.F., Yung R.W., Chow E.Y., Au K.L., Chan E.Y., Lim W., Jensenius J.C., Turner M.W., Peiris J.S., Lau Y.L. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis. 2005;191:1697–1704. doi: 10.1086/429631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallow M., Teo Y.Y., Small K.S., Rockett K.A., Deloukas P., Clark T.G., Kivinen K., Bojang K.A., Conway D.J., Pinder M., Sirugo G., Sisay-Joof F., Usen S., Auburn S., Bumpstead S.J., Campino S., Coffey A., Dunham A., Fry A.E., Green A., Gwilliam R., Hunt S.E., Inouye M., Jeffreys A.E., Mendy A., Palotie A., Potter S., Ragoussis J., Rogers J., Rowlands K., Somaskantharajah E., Whittaker P., Widden C., Donnelly P., Howie B., Marchini J., Morris A., Sanjoaquin M., Achidi E.A., Agbenyega T., Allen A., Amodu O., Corran P., Djimde A., Dolo A., Doumbo O.K., Drakeley C., Dunstan S., Evans J., Farrar J., Fernando D., Hien T.T., Horstmann R.D., Ibrahim M., Karunaweera N., Kokwaro G., Koram K.A., Lemnge M., Makani J., Marsh K., Michon P., Modiano D., Molyneux M.E., Mueller I., Parker M., Peshu N., Plowe C.V., Puijalon O., Reeder J., Reyburn H., Riley E.M., Sakuntabhai A., Singhasivanon P., Sirima S., Tall A., Taylor T.E., Thera M., Troye-Blomberg M., Williams T.N., Wilson M., Kwiatkowski D.P. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet. 2009;41:657–665. doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen R., Bont L., Siezen C.L., Hodemaekers H.M., Ermers M.J., Doornbos G., van’t Slot R., Wijmenga C., Goeman J.J., Kimpen J.L., van Houwelingen H.C., Kimman T.G., Hoebee B. Genetic susceptibility to respiratory syncytial virus bronchiolitis is predominantly associated with innate immune genes. J Infect Dis. 2007;196:826–834. doi: 10.1086/520886. [DOI] [PubMed] [Google Scholar]
- Johnson N.P., Mueller J. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull Hist Med. 2002;76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- Kandun I.N., Wibisono H., Sedyaningsih E.R., Yusharmen, Hadisoedarsuno W., Purba W., Santoso H., Septiawati C., Tresnaningsih E., Heriyanto B., Yuwono D., Harun S., Soeroso S., Giriputra S., Blair P.J., Jeremijenko A., Kosasih H., Putnam S.D., Samaan G., Silitonga M., Chan K.H., Poon L.L., Lim W., Klimov A., Lindstrom S., Guan Y., Donis R., Katz J., Cox N., Peiris M., Uyeki T.M. Three Indonesian clusters of H5N1 virus infection in 2005. N Engl J Med. 2006;355:2186–2194. doi: 10.1056/NEJMoa060930. [DOI] [PubMed] [Google Scholar]
- Kash J.C., Cunningham D.M., Smit M.W., Park Y., Fritz D., Wilusz J., Katze M.G. Selective translation of eukaryotic mRNAs: functional molecular analysis of GRSF-1, a positive regulator of influenza virus protein synthesis. J Virol. 2002;76:10417–10426. doi: 10.1128/JVI.76.20.10417-10426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash J.C., Goodman A.G., Korth M.J., Katze M.G. Hijacking of the host-cell response and translational control during influenza virus infection. Virus Res. 2006;119:111–120. doi: 10.1016/j.virusres.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Kash J.C., Tumpey T.M., Proll S.C., Carter V., Perwitasari O., Thomas M.J., Basler C.F., Palese P., Taubenberger J.K., Garcia-Sastre A., Swayne D.E., Katze M.G. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann A., Salentin R., Meyer R.G., Bussfeld D., Pauligk C., Fesq H., Hofmann P., Nain M., Gemsa D., Sprenger H. Defense against influenza A virus infection: essential role of the chemokine system. Immunobiology. 2001;204:603–613. doi: 10.1078/0171-2985-00099. [DOI] [PubMed] [Google Scholar]
- Kido H., Okumura Y., Takahashi E., Pan H.Y., Wang S., Chida J., Le T.Q., Yano M. Host envelope glycoprotein processing proteases are indispensable for entry into human cells by seasonal and highly pathogenic avian influenza viruses. J Mol Genet Med. 2008;3:167–175. [PMC free article] [PubMed] [Google Scholar]
- Kido H., Okumura Y., Yamada H., Le T.Q., Yano M. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr Pharm Des. 2007;13:405–414. doi: 10.2174/138161207780162971. [DOI] [PubMed] [Google Scholar]
- Kitagawa H., Paulson J.C. Differential expression of five sialyltransferase genes in human tissues. J Biol Chem. 1994;269:17872–17878. [PubMed] [Google Scholar]
- Kobasa D., Jones S.M., Shinya K., Kash J.C., Copps J., Ebihara H., Hatta Y., Kim J.H., Halfmann P., Hatta M., Feldmann F., Alimonti J.B., Fernando L., Li Y., Katze M.G., Feldmann H., Kawaoka Y. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- Koyama S., Ishii K.J., Kumar H., Tanimoto T., Coban C., Uematsu S., Kawai T., Akira S. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J Immunol. 2007;179:4711–4720. doi: 10.4049/jimmunol.179.7.4711. [DOI] [PubMed] [Google Scholar]
- Krug R.M., Yuan W., Noah D.L., Latham A.G. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology. 2003;309:181–189. doi: 10.1016/s0042-6822(03)00119-3. [DOI] [PubMed] [Google Scholar]
- Lahti M., Lofgren J., Marttila R., Renko M., Klaavuniemi T., Haataja R., Ramet M., Hallman M. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res. 2002;51:696–699. doi: 10.1203/00006450-200206000-00006. [DOI] [PubMed] [Google Scholar]
- Lakadamyali M., Rust M.J., Zhuang X. Endocytosis of influenza viruses. Microbes Infect. 2004;6:929–936. doi: 10.1016/j.micinf.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goffic R., Balloy V., Lagranderie M., Alexopoulou L., Escriou N., Flavell R., Chignard M., Si-Tahar M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikina E., Delanoe-Ayari H., Melikov K., Cho M.S., Chen A., Waring A.J., Wang W., Xie Y., Loo J.A., Lehrer R.I., Chernomordik L.V. Carbohydrate-binding molecules inhibit viral fusion and entry by crosslinking membrane glycoproteins. Nat Immunol. 2005;6:995–1001. doi: 10.1038/ni1248. [DOI] [PubMed] [Google Scholar]
- Lofgren J., Ramet M., Renko M., Marttila R., Hallman M. Association between surfactant protein A gene locus and severe respiratory syncytial virus infection in infants. J Infect Dis. 2002;185:283–289. doi: 10.1086/338473. [DOI] [PubMed] [Google Scholar]
- Ludwig S. Exploited defense: how influenza viruses take advantage of antiviral signaling responses. Future Virol. 2007;2:101–112. [Google Scholar]
- Ludwig S., Planz O. Influenza viruses and the NF-kappaB signaling pathway—towards a novel concept of antiviral therapy. Biol Chem. 2008;389:1307–1312. doi: 10.1515/BC.2008.148. [DOI] [PubMed] [Google Scholar]
- Maines T.R., Jayaraman A., Belser J.A., Wadford D.A., Pappas C., Zeng H., Gustin K.M., Pearce M.B., Viswanathan K., Shriver Z.H., Raman R., Cox N.J., Sasisekharan R., Katz J.M., Tumpey T.M. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science. 2009;325:484–487. doi: 10.1126/science.1177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines T.R., Szretter K.J., Perrone L., Belser J.A., Bright R.A., Zeng H., Tumpey T.M., Katz J.M. Pathogenesis of emerging avian influenza viruses in mammals and the host innate immune response. Immunol Rev. 2008;225:68–84. doi: 10.1111/j.1600-065X.2008.00690.x. [DOI] [PubMed] [Google Scholar]
- Mandelboim O., Lieberman N., Lev M., Paul L., Arnon T.I., Bushkin Y., Davis D.M., Strominger J.L., Yewdell J.W., Porgador A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409:1055–1060. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- Manolio T.A., Brooks L.D., Collins F.S. A hapmap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L.T., Marth J.D., Varki A., Varki N.M. Genetically altered mice with different sialyltransferase deficiencies show tissue-specific alterations in sialylation and sialic acid 9-O-acetylation. J Biol Chem. 2002;277:32930–32938. doi: 10.1074/jbc.M203362200. [DOI] [PubMed] [Google Scholar]
- Matrosovich M., Klenk H.D. Natural and synthetic sialic acid-containing inhibitors of influenza virus receptor binding. Rev Med Virol. 2003;13:85–97. doi: 10.1002/rmv.372. [DOI] [PubMed] [Google Scholar]
- Matrosovich M., Matrosovich T., Carr J., Roberts N.A., Klenk H.D. Overexpression of the alpha-2,6-sialyltransferase in MDCK cells increases influenza virus sensitivity to neuraminidase inhibitors. J Virol. 2003;77:8418–8425. doi: 10.1128/JVI.77.15.8418-8425.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer D., Molawi K., Martinez-Sobrido L., Ghanem A., Thomas S., Baginsky S., Grossmann J., Garcia-Sastre A., Schwemmle M. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J Proteome Res. 2007;6:672–682. doi: 10.1021/pr060432u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers J.A. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19:571–582. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers J.A., Rehg J.E. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186:341–350. doi: 10.1086/341462. [DOI] [PubMed] [Google Scholar]
- McGeoch D., Fellner P., Newton C. Influenza virus genome consists of eight distinct RNA species. Proc Natl Acad Sci U S A. 1976;73:3045–3049. doi: 10.1073/pnas.73.9.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mibayashi M., Martinez-Sobrido L., Loo Y.M., Cardenas W.B., Gale M., Jr., Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose F., Basler C.F., O’Neill R.E., Iwamatsu A., Palese P., Nagata K. Cellular splicing factor RAF-2p48/NPI-5/BAT1/UAP56 interacts with the influenza virus nucleoprotein and enhances viral RNA synthesis. J Virol. 2001;75:1899–1908. doi: 10.1128/JVI.75.4.1899-1908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir R., Wilson G.H. Influenza and its complications. Br Med J. 1919;1:3–5. doi: 10.1136/bmj.1.3027.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster V.J., de Wit E., van den Brand J.M., Herfst S., Schrauwen E.J., Bestebroer T.M., van de Vijver D., Boucher C.A., Koopmans M., Rimmelzwaan G.F., Kuiken T., Osterhaus A.D., Fouchier R.A. Pathogenesis and transmission of swine-origin 2009 A(H1N1) influenza virus in ferrets. Science. 2009;325:481–483. doi: 10.1126/science.1177127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- Nagata K., Kawaguchi A., Naito T. Host factors for replication and transcription of the influenza virus genome. Rev Med Virol. 2008;18:247–260. doi: 10.1002/rmv.575. [DOI] [PubMed] [Google Scholar]
- Nayak D.P., Hui E.K., Barman S. Assembly and budding of influenza virus. Virus Res. 2004;106:147–165. doi: 10.1016/j.virusres.2004.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M.I., Holmes E.C. The evolution of epidemic influenza. Nat Rev Genet. 2007;8:196–205. doi: 10.1038/nrg2053. [DOI] [PubMed] [Google Scholar]
- Nicholls J.M., Chan M.C., Chan W.Y., Wong H.K., Cheung C.Y., Kwong D.L., Wong M.P., Chui W.H., Poon L.L., Tsao S.W., Guan Y., Peiris J.S. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med. 2007;13:147–149. doi: 10.1038/nm1529. [DOI] [PubMed] [Google Scholar]
- Nicholls J.M., Chan R.W., Russell R.J., Air G.M., Peiris J.S. Evolving complexities of influenza virus and its receptors. Trends Microbiol. 2008;16:149–157. doi: 10.1016/j.tim.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Nicholson K.G., Wood J.M., Zambon M. Influenza. Lancet. 2003;362:1733–1745. doi: 10.1016/S0140-6736(03)14854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F., Dudziak D., Dirmeier U., Hobom G., Riedel A., Schlee M., Staudt L.M., Rosenwald A., Behrends U., Bornkamm G.W., Mautner J. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J Gen Virol. 2004;85:2347–2356. doi: 10.1099/vir.0.79958-0. [DOI] [PubMed] [Google Scholar]
- Noah D.L., Twu K.Y., Krug R.M. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3’ end processing of cellular pre-mRNAS. Virology. 2003;307:386–395. doi: 10.1016/s0042-6822(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Normile D. Epidemiology. Indonesia taps village wisdom to fight bird flu. Science. 2007;315:30–33. doi: 10.1126/science.315.5808.30. [DOI] [PubMed] [Google Scholar]
- Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team. Dawood F.S., Jain S., Finelli L., Shaw M.W., Lindstrom S., Garten R.J., Gubareva L.V., Xu X., Bridges C.B., Uyeki T.M. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med. 2009;360:2605–2615. doi: 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- O’Brien S.J., Nelson G.W. Human genes that limit AIDS. Nat Genet. 2004;36:565–574. doi: 10.1038/ng1369. [DOI] [PubMed] [Google Scholar]
- Oh D.Y., Barr I.G., Mosse J.A., Laurie K.L. MDCK-SIAT1 cells show improved isolation rates for recent human influenza viruses compared to conventional MDCK cells. J Clin Microbiol. 2008;46:2189–2194. doi: 10.1128/JCM.00398-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olschlager V., Pleschka S., Fischer T., Rziha H.J., Wurzer W., Stitz L., Rapp U.R., Ludwig S., Planz O. Lung-specific expression of active Raf kinase results in increased mortality of influenza A virus-infected mice. Oncogene. 2004;23:6639–6646. doi: 10.1038/sj.onc.1207883. [DOI] [PubMed] [Google Scholar]
- Olsen S.J., Ungchusak K., Sovann L., Uyeki T.M., Dowell S.F., Cox N.J., Aldis W., Chunsuttiwat S. Family clustering of avian influenza A (H5N1) Emerg Infect Dis. 2005;11:1799–1801. doi: 10.3201/eid1111.050646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterholm M.T. Preparing for the next pandemic. N Engl J Med. 2005;352:1839–1842. doi: 10.1056/NEJMp058068. [DOI] [PubMed] [Google Scholar]
- Oxford J.S. Influenza A pandemics of the 20th century with special reference to 1918: virology, pathology and epidemiology. Rev Med Virol. 2000;10:119–133. doi: 10.1002/(sici)1099-1654(200003/04)10:2<119::aid-rmv272>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Palese P., Shaw M.L. Orthomyxoviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., editors. Fields Virology. Lippincott Williams & Wilkins; 2007. pp. 1648–1689. [Google Scholar]
- Park Y.W., Wilusz J., Katze M.G. Regulation of eukaryotic protein synthesis: selective influenza viral mRNA translation is mediated by the cellular RNA-binding protein GRSF-1. Proc Natl Acad Sci U S A. 1999;96:6694–6699. doi: 10.1073/pnas.96.12.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish C.R., Kawaoka Y. The origins of new pandemic viruses: the acquisition of new host ranges by canine parvovirus and influenza A viruses. Annu Rev Microbiol. 2005;59:553–586. doi: 10.1146/annurev.micro.59.030804.121059. [DOI] [PubMed] [Google Scholar]
- Pauli E.K., Schmolke M., Wolff T., Viemann D., Roth J., Bode J.G., Ludwig S. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008;4:e1000196. doi: 10.1371/journal.ppat.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L.H., Holsinger L.J., Lamb R.A. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- Pinto L.H., Lamb R.A. The M2 proton channels of influenza A and B viruses. J Biol Chem. 2006;281:8997–9000. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- Pleschka S., Wolff T., Ehrhardt C., Hobom G., Planz O., Rapp U.R., Ludwig S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol. 2001;3:301–305. doi: 10.1038/35060098. [DOI] [PubMed] [Google Scholar]
- Pritchett T.J., Paulson J.C. Basis for the potent inhibition of influenza virus infection by equine and guinea pig alpha 2-macroglobulin. J Biol Chem. 1989;264:9850–9858. [PubMed] [Google Scholar]
- Rand T.A., Petersen S., Du F., Wang X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. 2005;123:621–629. doi: 10.1016/j.cell.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Rosseau S., Hocke A., Mollenkopf H., Schmeck B., Suttorp N., Kaufmann S.H., Zerrahn J. Comparative transcriptional profiling of the lung reveals shared and distinct features of Streptococcus pneumoniae and influenza A virus infection. Immunology. 2007;120:380–391. doi: 10.1111/j.1365-2567.2006.02514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R.J., Stevens D.J., Haire L.F., Gamblin S.J., Skehel J.J. Avian and human receptor binding by hemagglutinins of influenza A viruses. Glycoconj J. 2006;23:85–92. doi: 10.1007/s10719-006-5440-1. [DOI] [PubMed] [Google Scholar]
- Rust M.J., Lakadamyali M., Zhang F., Zhuang X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol. 2004;11:567–573. doi: 10.1038/nsmb769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M., Kosai K., Hara A., Imamura Y., Nakamura S., Kurihara S., Izumikawa K., Kakeya H., Yamamoto Y., Yanagihara K., Miyazaki Y., Mukae H., Tashiro T., Kohno S. Expression and DNA microarray analysis of a platelet activating factor-related molecule in severe pneumonia in mice due to influenza virus and bacterial co-infection. Jpn J Infect Dis. 2009;62:6–10. [PubMed] [Google Scholar]
- Shinya K., Ebina M., Yamada S., Ono M., Kasai N., Kawaoka Y. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440:435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- Smith G.J., Vijaykrishna D., Bahl J., Lycett S.J., Worobey M., Pybus O.G., Ma S.K., Cheung C.L., Raghwani J., Bhatt S., Peiris J.S., Guan Y., Rambaut A. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009;459:1122–1125. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- Steinhauer D.A. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology. 1999;258:1–20. doi: 10.1006/viro.1999.9716. [DOI] [PubMed] [Google Scholar]
- Stevens J., Blixt O., Tumpey T.M., Taubenberger J.K., Paulson J.C., Wilson I.A. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- Sun J., Madan R., Karp C.L., Braciale T.J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. 2009;15:277–284. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swayne D.E., Pantin-Jackwood M. Pathogenicity of avian influenza viruses in poultry. Dev Biol (Basel) 2006;124:61–67. [PubMed] [Google Scholar]
- Tabor H.K., Risch N.J., Myers R.M. Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet. 2002;3:391–397. doi: 10.1038/nrg796. [DOI] [PubMed] [Google Scholar]
- Tashiro M., Ciborowski P., Reinacher M., Pulverer G., Klenk H.D., Rott R. Synergistic role of staphylococcal proteases in the induction of influenza virus pathogenicity. Virology. 1987;157:421–430. doi: 10.1016/0042-6822(87)90284-4. [DOI] [PubMed] [Google Scholar]
- Thomas N.J., Fan R., Diangelo S., Hess J.C., Floros J. Haplotypes of the surfactant protein genes A and D as susceptibility factors for the development of respiratory distress syndrome. Acta Paediatr. 2007;96:985–989. doi: 10.1111/j.1651-2227.2007.00319.x. [DOI] [PubMed] [Google Scholar]
- Thompson W.W., Shay D.K., Weintraub E., Brammer L., Cox N., Anderson L.J., Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- Tompkins S.M., Lo C.Y., Tumpey T.M., Epstein S.L. Protection against lethal influenza virus challenge by RNA interference in vivo. Proc Natl Acad Sci U S A. 2004;101:8682–8686. doi: 10.1073/pnas.0402630101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham D.J., Tripp R.A., Doherty P.C. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol. 1997;159:5197–5200. [PubMed] [Google Scholar]
- Tucker S.P., Penn C.R., McCauley J.W. Characterisation of the influenza virus associated protein kinase and its resemblance to casein kinase II. Virus Res. 1991;18:243–261. doi: 10.1016/0168-1702(91)90022-n. [DOI] [PubMed] [Google Scholar]
- Uyeki T.M. Global epidemiology of human infections with highly pathogenic avian influenza A (H5N1) viruses. Respirology. 2008;13(Suppl. 1):S2–S9. doi: 10.1111/j.1440-1843.2008.01246.x. [DOI] [PubMed] [Google Scholar]
- van der Sluijs K.F., van Elden L.J., Nijhuis M., Schuurman R., Florquin S., Shimizu T., Ishii S., Jansen H.M., Lutter R., van der Poll T. Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol Lung Cell Mol Physiol. 2006;290:L194–L199. doi: 10.1152/ajplung.00050.2005. [DOI] [PubMed] [Google Scholar]
- van Riel D., Munster V.J., de Wit E., Rimmelzwaan G.F., Fouchier R.A., Osterhaus A.D., Kuiken T. H5N1 virus attachment to lower respiratory tract. Science. 2006;312:399. doi: 10.1126/science.1125548. [DOI] [PubMed] [Google Scholar]
- van Riel D., Munster V.J., de Wit E., Rimmelzwaan G.F., Fouchier R.A., Osterhaus A.D., Kuiken T. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol. 2007;171:1215–1223. doi: 10.2353/ajpath.2007.070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vines A., Wells K., Matrosovich M., Castrucci M.R., Ito T., Kawaoka Y. The role of influenza A virus hemagglutinin residues 226 and 228 in receptor specificity and host range restriction. J Virol. 1998;72:7626–7631. doi: 10.1128/jvi.72.9.7626-7631.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.A., Molloy S.S., Thomas G., Sakaguchi T., Yoshida T., Chambers T.M., Kawaoka Y. Sequence specificity of furin, a proprotein-processing endoprotease, for the hemagglutinin of a virulent avian influenza virus. J Virol. 1994;68:1213–1218. doi: 10.1128/jvi.68.2.1213-1218.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J., Lee E.U., McEntee K., Lai P.H., Paulson J.C. Primary structure of beta-galactoside alpha 2,6-sialyltransferase. Conversion of membrane-bound enzyme to soluble forms by cleavage of the NH2-terminal signal anchor. J Biol Chem. 1987;262:17735–17743. [PubMed] [Google Scholar]
- Weis W., Brown J.H., Cusack S., Paulson J.C., Skehel J.J., Wiley D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature. 1988;333:426–431. doi: 10.1038/333426a0. [DOI] [PubMed] [Google Scholar]
- Werner, O., Harder, T.C., 2006. Avian Influenza in “Influenza Report 2006”, www.InfluenzaReport.com.
- White M.R., Crouch E., van Eijk M., Hartshorn M., Pemberton L., Tornoe I., Holmskov U., Hartshorn K.L. Cooperative anti-influenza activities of respiratory innate immune proteins and neuraminidase inhibitor. Am J Physiol Lung Cell Mol Physiol. 2005;288:L831–L840. doi: 10.1152/ajplung.00365.2004. [DOI] [PubMed] [Google Scholar]
- Wolff T., O’Neill R.E., Palese P. NS1-Binding protein (NS1-BP): a novel human protein that interacts with the influenza A virus nonstructural NS1 protein is relocalized in the nuclei of infected cells. J Virol. 1998;72:7170–7180. doi: 10.1128/jvi.72.9.7170-7180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Animal Health Organization, 2008. Avian influenza. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, vol. 1, chapter 2.3.4.
- World Health Organization A revision of the system of nomenclature for influenza viruses: a WHO memorandum. Bull World Health Organ. 1980;58:585–591. [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, (2009). Epidemic and Pandemic Alert and Response (EPR). Cumulative Number of Confirmed Human Cases of Avian Influenza A/(H5N1) Reported to WHO, July 1, 2009.
- Wurzer W.J., Ehrhardt C., Pleschka S., Berberich-Siebelt F., Wolff T., Walczak H., Planz O., Ludwig S. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem. 2004;279:30931–30937. doi: 10.1074/jbc.M403258200. [DOI] [PubMed] [Google Scholar]
- Yamada S., Suzuki Y., Suzuki T., Le M.Q., Nidom C.A., Sakai-Tagawa Y., Muramoto Y., Ito M., Kiso M., Horimoto T., Shinya K., Sawada T., Kiso M., Usui T., Murata T., Lin Y., Hay A., Haire L.F., Stevens D.J., Russell R.J., Gamblin S.J., Skehel J.J., Kawaoka Y. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature. 2006;444:378–382. doi: 10.1038/nature05264. [DOI] [PubMed] [Google Scholar]
- Yoneyama M., Fujita T. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 2007;18:545–551. doi: 10.1016/j.cytogfr.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Yu W., Gwinn M., Clyne M., Yesupriya A., Khoury M.J. A navigator for human genome epidemiology. Nat Genet. 2008;40:124–125. doi: 10.1038/ng0208-124. [DOI] [PubMed] [Google Scholar]
- Zamarin D., Garcia-Sastre A., Xiao X., Wang R., Palese P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005;1:e4. doi: 10.1371/journal.ppat.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarocostas J. World Health Organization declares A (H1N1) influenza pandemic. BMJ. 2009;338:b2425. doi: 10.1136/bmj.b2425. [DOI] [PubMed] [Google Scholar]
- Zhang H., Zhou G., Zhi L., Yang H., Zhai Y., Dong X., Zhang X., Gao X., Zhu Y., He F. Association between mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect Dis. 2005;192:1355–1361. doi: 10.1086/491479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhirnov O.P., Ksenofontov A.L., Kuzmina S.G., Klenk H.D. Interaction of influenza A virus M1 matrix protein with caspases. Biochemistry (Mosc) 2002;67:534–539. doi: 10.1023/a:1015542110798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.