

Highlights
-
•
We investigated the prevalence of HRSV during 2008–2013.
-
•
Novel HRSV-A ON1 genotype was emerged in August 2011.
-
•
After 1 year of emergence in 2012–2013, 94.6% was replaced with novel ON1 genotype.
-
•
Evolutionary dynamics also drastically increased in 2011.
-
•
The result of epitope prediction shows the possibilities of antigenic variation.
Keywords: Human respiratory syncytial virus (HRSV), Attachment G gene, Duplication, Diversity, Evolution
Abstract
Human respiratory syncytial virus (HRSV) is the main cause of severe respiratory illness in young children and elderly people. We investigated the genetic characteristics of the circulating HRSV subgroup A (HRSV-A) to determine the distribution of genotype ON1, which has a 72-nucleotide duplication in attachment G gene. We obtained 456 HRSV-A positive samples between October 2008 and February 2013, which were subjected to sequence analysis. The first ON1 genotype was discovered in August 2011 and 273 samples were identified as ON1 up to February 2013. The prevalence of the ON1 genotype increased rapidly from 17.4% in 2011–2012 to 94.6% in 2012–2013. The mean evolutionary rate of G protein was calculated as 3.275 × 10−3 nucleotide substitution/site/year and several positively selected sites for amino acid substitutions were located in the predicted epitope region. This basic and important information may facilitate a better understanding of HRSV epidemiology and evolution.
1. Introduction
Human respiratory syncytial virus (HRSV) is recognized by pediatricians as the most common cause of acute respiratory tract infections and is a leading cause of hospital admissions and death among children aged <5 years worldwide (Hall et al., 2013, Cho et al., 2013, Munywoki et al., 2013, Bezerra et al., 2011, Nair et al., 2010). The World Health Organization has estimated that the annual global disease burden is more than 64 million HRSV infections and 160,000 deaths related to HRSV infection (World Health Organization (WHO), 2009). HRSV infection is a major concern in developed and developing countries, but no effective vaccine is available and immunoprophylaxis is the only treatment for preventing HRSV infection, although access is limited (Chang, 2011, Rudraraju et al., 2013, Graham, 2011, Jorquera et al., 2013, Shaw et al., 2013, Wang et al., 2011a, Zeitlin et al., 2013).
Two antigenic groups of HRSV have been differentiated based on antigenic variability in the attachment G gene, i.e., HRSV subgroup A (HRSV-A) and HRSV subgroup B (HRSV-B). Ten HRSV-A and 13 HRSV-B genotypes have been classified in different geographical regions, which are designated as GA1–GA7, SAA1, NA1, and NA2 for HRSV-A, and GB1–GB4, SAB1–SAB3, and BA1–BA6 for HRSV-B (Auksornkitti et al., 2013, Eshaghi et al., 2012, Lee et al., 2012, Khor et al., 2013, Shobugawa et al., 2009). Most previous molecular epidemiological and divergence studies of HRSV have focused on analyses of nucleotide and/or amino acid changes in part of the G protein, which is a type II glycoprotein that mediates attachment of the virus to the cell during virus entry, and is one of the targets of the immune response (Agoti et al., 2010, Cui et al., 2013, Baek et al., 2012, Tan et al., 2013, Murata and Catherman, 2012). These studies have yielded significant volumes of partial genomic information for HRSV and primary analyses based on variation in the G protein.
In particular, the BA genotype of HRSV-B, which was isolated in Buenos Aires, Argentina during 1999, contains a duplication of 60 nucleotides (nt) in the C-terminal third of the G protein gene and is the predominant strain according to global epidemiological studies (Sullender et al., 1991, Trento et al., 2006, Trento et al., 2010, van Niekert and Venter, 2011, Zhang et al., 2010). More recently, a similar duplication was reported in HRSV-A (ON1) isolates from Canada, Germany, Malaysia, Thailand, Kenya, and South Korea, which was characterized as a 72-nt duplication in the C-terminal third of the G gene (Munywoki et al., 2013, Auksornkitti et al., 2013, Eshaghi et al., 2012, Lee et al., 2012, Prifert et al., 2013). The exact mechanism that allows such duplications to play roles during selective pressure and the factors that may increase their fitness to substitute the BA genotype or other HRSV-B viruses remain to be defined. Similarly, the basis of the evolutionary advantage and antigenic dominance of the HRSV-A (ON1) strain due to the introduction of 72 identical bases in the G gene also needs to be clarified.
In this study, we investigated the emergence of the new HRSV-A ON1 genotype and conducted an in-depth analysis of the genetic predisposition of the G protein gene. In addition, we predicted the epitope of the duplicated G protein with an insertion of 23 amino acids, which we compared with ancestral strains. This analysis is of importance for elucidating the antigenic variation of the HRSV G protein and its relationships with clinical manifestations and vaccine development.
2. Materials and methods
2.1. Ethical statement
The clinical samples used in this study were collected as part of the laboratory surveillance system in South Korea, i.e., the Acute Respiratory Infection Network (ARI-NET) conducted until April 2009 and the Korea Influenza and Respiratory Viruses Surveillance System (KINRESS) since May 2009. This study was approved by the Korea National Institute of Health institutional review boards (Approval Nos. 2010-03EXP-1-R, 2011-03CON-04-C, 2011-06EXP01-C, 2012-08EXP-06-3C, and 2012-09CON-03-4C) because it involved anonymization of the remaining respiratory tract samples, which were not related to human gene studies. These samples were collected for respiratory virus diagnosis and written informed consent was obtained from the patients, their parents, or legal guardians.
2.2. Specimen collection and virus detection
This study used nasal aspirate specimens and throat swab samples taken from patients enrolled in ARI-NET and KINRESS with acute respiratory illness who were diagnosed as HRSV-positive including co-infected samples with influenza virus, human rhinovirus, adenovirus, human coronavirus, human bocavirus, human enterovirus, parainfluenza virus or human metapneumovirus. ARI-NET used conventional reverse transcriptase (RT)-PCR (Solgent, Seoul, South Korea) to detect the HRSV-A and -B subgroups simultaneously. By contrast, KINRESS used an improved real-time one-step RT-PCR to distinguish the HRSV-A and -B subgroups (Kogen Bio, Seoul, South Korea) after July 2011, where viral RNA was extracted from 140 μL of each respiratory specimen using QIAamp Viral RNA Mini Kits (Qiagen GmbH, Hilden, Germany).
2.3. PCR amplification of the G gene
HRSV-positive clinical samples were subjected to amplification of the partial G gene using a G gene-specific primer set for sequence analysis, i.e., the forward primer G(151–173)F: CTGGCAATGATAATCTCAACTTC and reverse primer F(3–22)R: CAACTCCATTGTTATTTGCC (da Silva et al., 2008). The cDNA was prepared using the viral RNA extraction method employed by the routine respiratory virus test. The reaction mixture contained 5 μL of RNA, which was mixed with a final concentration of 10 mM dNTPs, 20 μM random primer, 1× RT buffer, 200 U of Superscript III reverse transcriptase (Invitrogen, CA, USA), 40 U of RNase-OUT RNase Inhibitor (Invitrogen), 25 mM MgCl2, and 0.1 mM dithiothreitol (DTT), and RNase-free water was added to make a final volume of 20 μL. The mixture was then incubated at 25 °C for 5 min, 50 °C for 60 min, and 72 °C for 5 min to terminate cDNA synthesis. Next, 5 μL of cDNA was added to a PCR mixture containing 1 μL of SP-Taq DNA polymerase (2.5 U/μL) (Cosmo Genetech, Seoul, South Korea), 36 μL of distilled water, 5 μL of 10× PCR buffer, 1 μL of 10 mM dNTPs, and 1 μL each of the forward and reverse primers (both 20 μM) for the G gene. Primary denaturation was conducted at 95 °C for 10 min, which was followed by 35 cycles of PCR where each cycle comprised denaturation for 40 s at 95 °C, annealing for 40 s at 54 °C, and elongation for 1 min at 72 °C, with a final extension cycle of 5 min at 72 °C. The PCR products were separated by electrophoresis using 1% agarose gel and visualized using 1× SYBR Safe DNA Gel Stain (Invitrogen).
2.4. Nucleotide sequencing and phylogenetic analysis
The amplified PCR products were sequenced bidirectionally with same primers for PCR amplification mentioned in Section 2.3. using an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA). The sequences were edited with SeqMan Pro in the Lasergene 8 software suite (version 8.0; DNAStar, Madison, WI, USA) and aligned using MEGA 5 (ver. 5.05). The 717 nt length fragments of G gene from 181 nt position to stop codon based on A2 strain were used for further analysis. To minimize potential biases in the alignment and to obtain a more tractable representation of the dataset, identical sequences were removed using CD-HIT before performing the alignments (Huang et al., 2010). After clustering sequences with an amino acid sequence similarity of >0.98 into one cluster, one sequence was selected from each cluster and the sequences with no identical sequence were also removed. The final representative dataset comprised 18 sequences, which were submitted to GenBank and assigned accession numbers of AB860223–AB860240. To obtain a comprehensive representation of the diverse HRSV-A subgroup, we downloaded 15 publicly available HRSV sequences: WUE/14576/12 (JX912357), NG-016-04 (AB470478), RSV A2 (M74568), Chiba-C/24031 (AB698559), ON138-0111A (JN257694), MO48 (AF233914), SEL/02/98072 (AF193325), CN2395 (AF233905), CH09 (AF065254), NG-009-02 (AB175815), NY20 (AF233918), MO02 (AF233910), NG-082-05 (AB470479), SA98V603 (AF348807), and WUE/16397/12 (JX912364) (Prifert et al., 2013). In total, 33 sequences were used in the subsequent analysis, which comprised 18 representative sequences and 15 reference sequences. The phylogenetic trees were constructed using MUSCLE and MEGA5 (Tamura et al., 2011). The maximum composite likelihood for nucleotide sequences and the JTT model for amino acid sequences were used with the neighbor-joining (NJ) methods to perform the distance calculations. The trees generated were visualized and edited using EvolView (Zhang et al., 2012).
2.5. Selective pressure analysis
To investigate the selective pressure, we used a dataset that comprised 18 C-terminal regions (secondary hypervariable region) of the G genes and NG-016-04 (GenBank accession No. AB470478) as a reference sequence. In this analysis, we performed a multiple sequence alignment and a phylogenetic tree was generated using CLUSTALW and MEGA5. The nucleotide frequencies in the codon positions were assumed based on unequal codon frequencies. The maximum-likelihood (ML) method was used to analyze the selection pressure with the CODEML program in the phylogenetic analysis ML package (PAML, http://abacus.gene.ucl.ac.uk/software/paml.html) (Yang, 2007). CODEML was used to estimate the numbers of nonsynonymous (dN) to synonymous (dS) codon changes per site. Positive selection was defined as dN > dS (ω ratio >1). Different codon substitution models were tested in this study, i.e., M1 and M7 (neutral), and M2 and M8 (positive). The likelihood rates were calculated as twice the difference between the log-likelihood values (2ΔL) of the models, which were compared using the χ2 distribution (two degrees of freedom).
2.6. Evolution estimation using Bayesian skyline plot analysis
This analysis used the partial G gene sequences of HRSV samples isolated in the present study between 2008 and 2013 (n = 456) and additional sequences (n = 48) reported from South Korea during 1991–2010 (Baek et al., 2012, Choi and Lee, 2000). After removing 100% identical sequences using CD-HIT (Huang et al., 2010), the population dynamics of HRSV were estimated over time using a Bayesian Markov chain Monte Carlo approach (MCMC in BEAST version 1.7), which included the date of virus sampling (Drummond et al., 2012). The dataset was analyzed with an uncorrelated log-normal relaxed uncorrelated clock using the general time-reversible substitution model selected by jModelTest version 2.0 (Posada, 2008). The MCMC chain was run for 200 million steps to achieve convergence, with sampling every 10,000 steps. Convergence was assessed based on the effective sample size (ESS) after a 10% burn-in using Tracer version 1.5 (http://beast.bio.ed.ac.uk/Tracer) and only parameters where ESS > 200 were accepted. The uncertainties of the estimates were indicated by the 95% highest posterior density intervals. The final tree was visualized and edited with FigTree version 1.3.1 (http://tree.bio.ed.ac.uk/software/figtree).
2.7. Epitope prediction
We predicted the epitope for three HRSV-A G gene sequences using seven prediction tools, i.e., BepiPreds (Larsen et al., 2006), LBtope (Singh et al., 2013), BCPRED/FBCPRED (El-Manzalawy et al., 2008), Antigenic (Rice et al., 2000), LEPS (Wang et al., 2011b), and Epitopia (Rubinstein et al., 2009). These tools made the predictions using the initial values of the parameters, and the common epitopes predicted by four or more tools with ⩾10 consecutive amino acids were selected.
3. Results
3.1. General characteristics of HRSV subgroups and the distributions of genotypes
We collected 36,404 samples during 2010–2013 via KINRESS, nationwide surveillance for outpatients with acute respiratory illness covers about 100 hospitals located all over Korea and includes all ages. Of these, 18,112 samples were positive for respiratory viral infections and 1,643 samples (9.1%) had positive results for HRSV. Mean age of entire study group is 3.85 year. Sex ratio and co-infection rate were 46.3% and 21.9 %, respectively and corresponding data per each period is summarized in Table 1 . During the 2010–2011 season, 234 samples (4.58% in respiratory virus-positive patients) were HRSV-positive. During the next two seasons, however, the HRSV-positive ratio increased and 643 (8.41%) and 766 (14.30%) cases were detected during the 2011–2012 and 2012–2013 seasons, respectively, as shown in Fig. 1 A. There were no specific differences in the gender distribution, mean age, or coinfection rate during these three consecutive seasons.
Table 1.
Subject data in this study.
| Period | 2010.5.-2011.4. | 2011.5.-2012.4. | 2012.5.-2013.2. | 2010.5.-2013.2. | |
|---|---|---|---|---|---|
| Patients enrolled in KINRESS | Positive for respiratory infections/ enrolled total no. | 5,108/11,300 | 7,647/14,788 | 5,357/10,316 | 18,112/36,404 |
| HRSV positive patients (n = 1,643) | Total no. (%) |
234 (4.58) |
643 (8.41) |
766 (14.30) |
1,643 (9.1) |
| F/M | 113/121 | 293/350 | 355/409⁎ | 761/880 | |
| Female ratio | 48.3 | 45.6 | 46.5 | 46.3 | |
| Mean age | 4.19 year | 3.71 year | 3.87 year | 3.85 year | |
| Co-infections (%) | 38 (16.2) | 156 (24.3) | 166 (21.7) | 360 (21.9) | |
KINRESS, Korea influenza and respiratory surveillance system; HRSV, human respiratory syncytial virus.
Two samples have no record about gender.
Fig. 1.
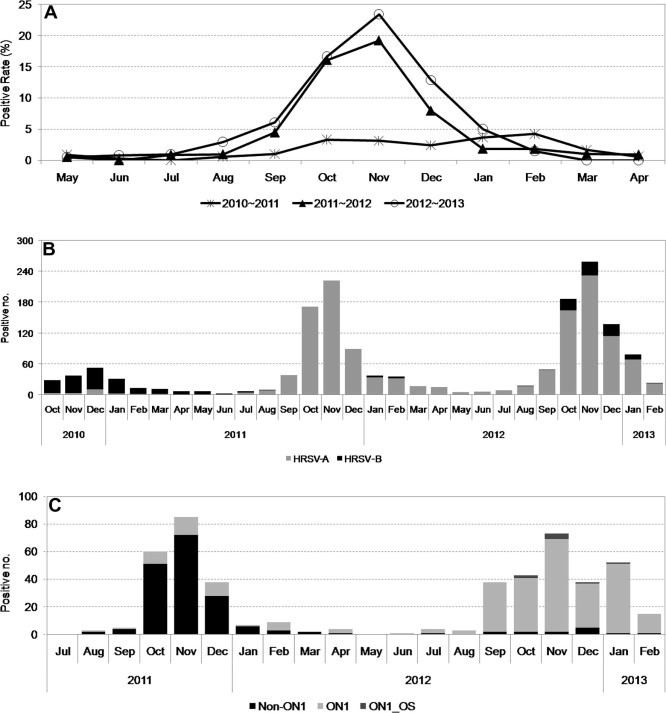
Prevalence and genotype distributions of HRSV-positive samples in South Korea during 2010–2013. (A) Prevalence of HRSV-positive samples, including the HRSV-A and -B subgroups, between May 2010 and February 2013. (B) Distributions of the HRSV-A and -B subgroups between October 2010 and February 2013. (C) Distributions of HRSV-A genotypes between July 2011 and February 2013.
To investigate the distributions of the subgroups, we randomly selected HRSV-positive samples obtained during the 2010–2011 season, which were analyzed by real-time RT-PCR because the RT-PCR methods used in that season could not distinguish subgroups HRSV-A and -B. We tested 172 samples, and only 20 samples (11.6%) belonged to the HRSV-A subgroup. In 2011–2012, the prevalence of the HRSV-A subgroup had increased greatly and over 98% of the samples were found to be HRSV-A, while the prevalence of the HRSV-A subgroup was also high in the following season (89.1%) (Fig. 1B and Table 2 ).
Table 2.
General information and frequencies of HRSV subgroup A and B over 3 consecutive seasons in South Korea.
| Period | 2010.5.–2011.4. | 2011.5.–2012.4. | 2012.5.–2013.2. |
|---|---|---|---|
| Total no. of HRSV positive | 234 | 643 | 766 |
| Sample no. of subgroup determined | 172* | 637 | 764 |
| HRSV-A | 20 | 627 | 681 |
| HRSV-B | 152 | 10 | 83 |
| Ratio of A subgroup (%) | 11.6 | 98.4 | 89.1 |
| Sample no. of genotype† determined in HRSV-A | 1 | 167 | 258 |
| GA5 | 0 | 5 | 0 |
| NA1 | 1 | 133 | 14 |
| ON1 | 0 | 29 | 244 |
| Ratio of ON1 (%) | 0 | 17.4% | 94.6% |
HRSV, human respiratory syncytial virus.
Before July 2011, A/B subgroup was tested to random selected samples.
During October 2008 to April 2009, nine GA5 and 21 NA1 genotype stains were identified from 30 random selected samples.
Eshaghi et al. (2012) reported the discovery of a novel genotype in Canada with a 72-nt duplication in the G gene during the winter season in 2010–2011 and we also found ON1 genotype strains in South Korea, for which we reported the whole-genome sequences (Lee et al., 2012). Thus, extensive genotype and sequence analyses were performed using the HRSV-A subgroup to further study the prevalence of ON1 genotype strains. In total, we investigated 426 HRSV-A samples obtained during 2010–2013, where we analyzed the G gene sequences to determine whether a 72-nt duplication was present or absent. According to the broad sequence analysis, the first ON1 genotype strains were discovered in August 2011. In the season from May 2011 to April 2012, 29 samples (17.4%) had the ON1 genotype in South Korea among 167 HRSV-A samples tested. The major genotype in that season was NA1 (133 samples, 79.6%) and the GA5 genotype was also identified (five samples, 3.0%). These genotype distributions were similar to the results reported by the Canadian group (Eshaghi et al., 2012). In the next season (2012–2013), however, the prevalence of the ON1 genotypes increased significantly to 94.6% (244/258 HRSV-A-positive samples) and the GA5 genotype was not detected (Table 2 and Fig. 1C). NA1 genotype strains were still detected but only at a very low frequency (14 samples, 5.4%). Our analysis of this newly emerged strain showed that the rapid replacement of the non-ON1 genotypes without a 72-nt duplication in the G gene by the ON1 genotype was dramatic compared with the earlier spread of the HRSV BA genotype.
3.2. Phylogenetic analysis and alignment
In a further analysis of the 426 HRSV-A sequences obtained during 2010–2013, we added 30 unpublished HRSV-A sequences collected between October 2008 and April 2009. A clustering approach was applied (as described in the Materials and Methods section) to remove any redundant sequences and to make the data clear and manageable, while ensuring that the data still represented the diversity of HRSV. The numbers of sequences that belonged to the same clusters (amino acid sequence similarity >0.98) were plotted next to the phylogenetic tree. Finally, 18 isolates and 15 reference sequences were analyzed and the clusters in the phylogenetic tree generated four major groups: ON1, NA1, GA5, and others, including the reference sequences. The phylogenies based on the nucleotide and amino acid sequences (Fig. 2 A and B) were in almost complete agreement based on the topology of the phylogeny. The branching patterns differed slightly between groups, but the clade compositions were stable within the groups.
Fig. 2.
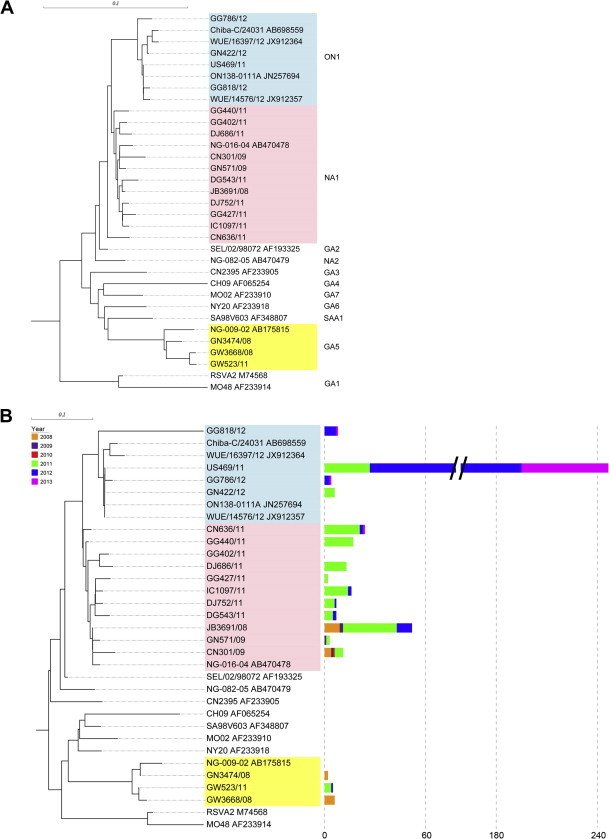
Phylogenetic analysis of the HRSV-A subgroup using partial G sequences based on (A) nucleotide and (B) amino acid sequences. Partial G protein sequences were used to study the phylogenetic relationships between HRSV-A subgroups. The nucleotide and protein sequences were aligned using the MUSCLE algorithm and the phylogenetic trees were generated by the neighbor-joining method with MEGA5 based on 1000 bootstrap replicates. The length of the square bar indicates the number of sequences that belong to the same cluster, while the color indicates the year the sample was isolated. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
However, several sequences (n = 8), which were represented by the GG818/12 sequence, had an apparently different branching structure, although there was low statistical support (bootstrap) for how most of these branches were related. Based on the alignment analysis, these clusters had a 1-nt deletion at nt position 899 in the 72-nt duplicated region. This deletion caused a frame shift from amino acid position 300 in the C-terminal region to the stop codon located at amino acid position 325 (Fig. 3 ). We considered that the GG818/12 clusters represented a subgenotype, which we designated as ON1_OS.
Fig. 3.
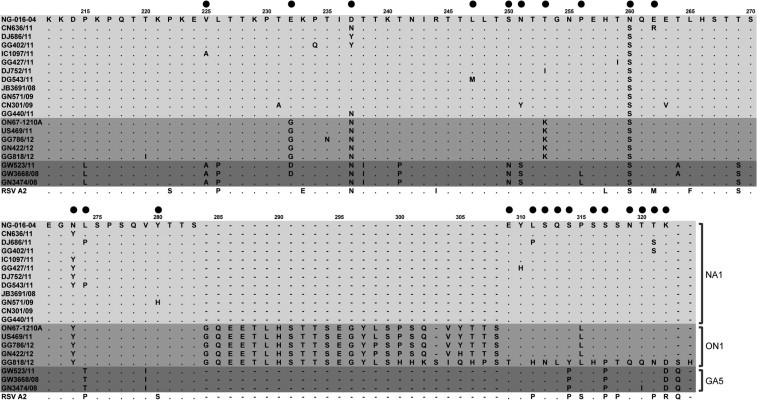
Amino acid alignment of G protein sequences from amino acid position 212 to the C-terminal end. Positively selected sites according to the M8 model are marked as closed black circles.
In addition, the first ON1 sequence, identified i.e., ON67-1210A/2010 from Canada (Eshaghi et al., 2012) and US469/11 in the present study, had the identical duplicated region with the template. However, the L298P and Y305H substitutions in other ON1 clusters were occurred as the same as those reported by Tsukagoshi et al. (2013) from Japan.
3.3. Selective pressure on the amino acid sequence of attachment G protein
The deduced amino acid sequences contained various substitutions in the secondary variable region of the G protein. In total, 18 representative sequences and an NA1 genotype reference sequence (NG-016-04, AB470478) were subjected to selection pressure analysis, as described in the Section 2. The dN/dS (nonsynonymous to synonymous codon changes per site) ratio averaged over all sites ranged from 0.669 to 1.185 with different codon substitution models. Models M2 and M8, which allowed for positive selection, fitted our dataset better than the two neutral models (M1 and M7). Using the NG-016-04 strain as an outgroup, 22 and 24 sites were found to be under positive selection by the M2 and M8 models, respectively, and these positively selected sites are shown in Fig. 3 and Table 3 . The empirical Bayes analysis showed that 3/24 (positions 237, 274, and 322) positively selected sites were under positive selection at the 95% level in the M8 model. Previously, positions 225, 256, 274, and 311 were reported to be flip-flop sites that tend to revert to a previous state over time (Botosso et al., 2009). Thus, we also found that the substitutions of V225A, P256L, and L274P/T were forward replacements, whereas L311H/P was a backward replacement.
Table 3.
Parameter estimates, dN/dS, values of log-Likelihood (l), positive selection sites, and Likelihood Ratio Tests (LRT) in the G gene analysis of HRSV-A isolated in South Korea between 2008 and 2013.
| Model | Parameter estimates | dN/dS | Log-likelyhood (l) | Positively selected sites⁎ | Model comparison |
|---|---|---|---|---|---|
| M1 | w0 = 0.0 | 0.6688 | −818.392077 | 8.71 | |
| w1 = 1.0 | d.f. = 2, p < 0.05 | ||||
| p0 = 0.33115 | |||||
| p1 = 0.66885 | |||||
| M2† | w0 = 0.15815 | 1.1852 | −814.034775 | V225A, E232G/D, D237N/Y, L247M, S250N, N251Y/S, T253K/I, P256L, E262R, L274P/T, Y280H, E309T, L311H/P, S312N, Q313L, S314Y/P, S316H, S317P, N319Q, T320Q/I, T321N/S, K322D | |
| w1 = 1.0 | |||||
| w2 = 2.31504 | |||||
| p0 = 0.52384 | |||||
| p1 = 0.0 | |||||
| p2 = 0.47616 | |||||
| M7 | p = 0.01165 | 0.70 | −817.440963 | 6.81 | |
| q = 0.00500 | d.f. = 2, p < 0.05 | ||||
| M8‡ | p0 = 0.52384 | 1.1849 | −814.035088 | V225A, E232G/D, D237N/Y, L247M, S250N, N251Y/S, T253K/I, P256L, N260S, E262R, N273Y, L274P/T, Y280H, E309T, L311H/P, S312N, Q313L, S314Y/P, S316H, S317P, N319Q, T320Q/I, T321N/S, K322D | |
| p1 = 0.47616 | |||||
| p = 18.69927 | |||||
| q = 99.0 | |||||
| w = 2.31393 | |||||
HRSV, human respiratory syncytial virus.
Sites were numbered as based on ON1 sequence (ON67-1210A, Genbank accession No. JN257693).
Posterior probability of positively selected sites with M2 model: 50% to 74% (232, 317, 225, 253, 280, 319, 256, 250, 313, 251, 247, 321, 312); 75% to 84% (316, 314, 262, 311, 320); 85% to 94% (322, 309, 274, 237); and 95% < (none).
Posterior probability of positively selected sites with M8 model: 50% to 74% (260, 273, 232, 280, 317, 225, 253, 319, 256, 247, 250, 313, 251); 75% to 84% (none); 85% to 94% (321, 312, 316, 314, 262, 311, 320, 309); and 95% < (322, 274, 237).
A frame shift occurred because of the 1-nt deletion at amino acid position 300, but the results of the analysis showed that there was high pressure for positive selection from amino acid position 309 to the stop codon.
3.4. Evolutionary dynamics of the G gene
To estimate the dynamics of the nucleotide substitutions and variation in HRSV-A, all 456 of the HRSV-A sequences obtained from this study during 2008–2013 and 48 previously published sequences from South Korea during 1991–2010 (Baek et al., 2012, Choi and Lee, 2000) were analyzed using the Bayesian skyline plot method after removing homologous sequences (Fig. 4 ). The calculated mean evolutionary rate was 3.275 × 10−3 nucleotide substitutions/site/year, which was faster than the whole-genome evolutionary rate of 6.47 × 10−4 for HRSV-A (Tan et al., 2013) but slower than the rate of 4.7 × 10−3 nucleotide substitutions/site/year in the HRSV-B G gene during the 10 years since the discovery of the 60-nt duplicated BA genotypes (Trento et al., 2010). A previous study reported a similar evolutionary rate for the HRSV-A G gene of 3.57 × 10−3 nucleotide substitutions/site/year (Tsukagoshi et al., 2013).
Fig. 4.
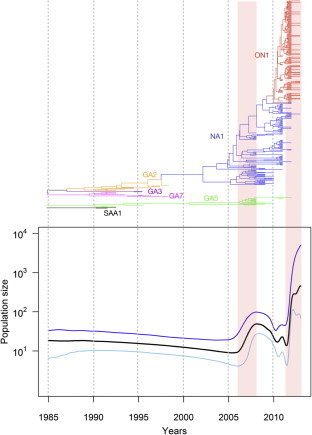
Genealogies and the corresponding Bayesian skyline plot showing the demographic history of HRSV sequences, which are drawn using the same time scale. The y-axes of the skyline plot (lower panel) represent the population size, which is equal to the product of the effective population size (shown as the product of Ne and generation time τ). The black line represents the median estimate and the areas between the blue and sky lines show the 95% highest posterior density limits. The Maximum clade credibility (MCC) tree (upper panel) is represented on the same time scale as the skyline plot. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The time course analysis showed that the genetic diversity was steady during 20 years in the course of NA1 genotype emergence in late 1990’s, consistent with the analysis of Kushibuchi in Japan (Kushibuchi et al., 2013). The relative genetic diversity has increased around 2006 in population size. Following that period, a limited fluctuation was observed in 2010 with a subsequent dramatic increase in the effective virus population size after 2011. The specific selective pressure applied to the virus in 2006 remains to be clarified, but this increasing trend in the relative genetic diversity of HRSV was followed by the initial appearance of the HRSV-A ON1 strain. In this analysis, we could also assume that ON1 genotype was emerged in 2009 year, although first ON1 case was discovered in 2011. The Bayesian skyline plot also demonstrated that the growth phase of the virus population size agreed with the rapid emergence of the HRSV-A ON1 strain.
3.5. Epitope prediction
The G protein is the major antigenic protein expressed on the surface of HRSV, so we assumed that the 72-nt duplication, which caused a 23-amino-acid duplication in secondary hypervariable regions of the ectodomain, may have affected the antigenicity, antigen recognition by antibodies, or virulence. Thus, we performed B-cell epitope prediction using the representative prototype A2 (GenBank accession No. M74568), the ON1 genotype GN435/11 (GenBank accession No. JX627336) (Lee et al., 2012), and the ON1_OS subgenotype GG818/12 sequences (Table 4 ). For each sequence, 5–7 antigenic peptides were identified by computational prediction. The 121–131, 133–146, 188–203, 221–231, and 272–284 epitope positions were determined for all three sequences. The predicted epitopes were similar but the 287–299 epitope located in the duplicated region was predicted only in the GG818/12 sequence.
Table 4.
Epitope prediction results of novel ON1 types (GN435/11, GG818/12) and prototype A2 strains. Bold letter means common epitope regions in three strains.
| Sequences | Position | Predicted epitope regions |
|---|---|---|
| A2 (M74568) | 85–102 | NTTPTYLTQNPQLGISPS |
| 121–131 | GVKSTLQSTTVKT | |
| 133–152 | TKNTTTTQTQPSKPTTKQRQ | |
| 188–231 | RIPNKKPGKKTTTKPTKKPTLKTTKKDPKPQTTKSKEVPTTKPT | |
| 266–291 | HSTSSEGNPSPSQVSTTSEYPSQPSS | |
| GN435/11 (JX627336) | 115–146 | LASTTPSAESTPQSTTVKIKNTTTTQILPSKP |
| 188–203 | RIPNKKPGKKTTTKPT | |
| 221–242 | KPKEVLTTKPTGKPTINTTKTN | |
| 251–265 | NTKGNPEHTSQEETL | |
| 272–284 | GYLSPSQVYTTSG | |
| GG818/12 (AB860239) | 81–94 | NQIKNTTPTYLTQS |
| 115–152 | LASTTPSAESTPQSTTVKIKNTTTTQILPSKPTTKQRQ | |
| 188–212 | RIPNKKPGKKTTTKPTKKPTLKTTK | |
| 221–242 | KPKEVLTTKPTGKPTINTTKTN | |
| 251–265 | NTKGNPEHTSQEETL | |
| 272–284 | GYLSPSQVYTTSG | |
| 287–299 | ETLHSTTSEGYLS |
4. Discussion
In this study, we analyzed the distribution in South Korea of the novel HRSV ON1 genotype, which has a 72-nt duplicated genetic region, and its rapid replacement of the non-duplicated strains. Our results provide up-to-date sequence information on the prevalent HRSV genotypes in South Korea and insights into HRSV evolution.
In South Korea, the first ON1 genotype was discovered in August 2011, which was the next season after Eshaghi et al. identified the novel ON1 genotype in Canada (2010–2011) (Eshaghi et al., 2012). However, the ON1 genotype was estimated as emerged in 2009 with time scale evolution analysis and became the predominant strain in South Korea in 2012–2013. Studies of HRSV-B show that the prevalence of the 60-nt duplicated BA genotype has fluctuated over the last 10 years but all of the HRSV-B subgroups now belong to the BA genotype (Baek et al., 2012, Trento et al., 2010, van Niekert and Venter, 2011). Further long-term molecular epidemiological studies are required during consecutive seasons to determine whether the ON1 genotype will replace the non-ON1 genotypes completely.
In addition to the introduction of a 72-nt duplication, the C-terminal region of the G gene has been the target of various amino acid sequence changes via substitutions and deletion. We found L298P and Y305H substitutions, as well as a 1-nt deletion that caused a frame shift from amino acid position 300 and a finally two amino acids longer than that in previously reported ON1 strains (Munywoki et al., 2013, Auksornkitti et al., 2013, Eshaghi et al., 2012, Lee et al., 2012, Prifert et al., 2013). The nucleotide substitution in the duplicated region was also found in Japan (Tsukagoshi et al., 2013). However, this is the first report of the 1-nt deletion in the duplicated region, and we designated this as the ON1_OS subgenotype in the present study. This ON1_OS strain was isolated from eight patients who were enrolled between October 2012 and January 2013 in five cities in South Korea (Fig. 1C). This finding suggests the replication and spread of these strains. The change of a 25 amino acid region into a novel sequence at the C-terminal is quite long and we still do not know the structure of G protein, so it might be possible that structural transformations occurred because of the 72-nt duplication or other variations. In vitro and in vivo functional studies should be performed using this novel G protein during attachment to host cells, or in interactions with immune responses, to understand the effects of this major change.
Previous reports have shown that a major HRSV type dominates in a given season, although two or more different genotypes cocirculate at the same time with different levels (Trento et al., 2010). A similar prevalence pattern was also observed in the present study. For example, HRSV-A did not lead the infections of patients with acute respiratory illness during the 2010–2011 season, but HRSV-A was prevalent in the following season of 2011–2012, which might be related to the emergence of the novel HRSV-A genotype in the human population. HRSV-A remained prevalent in the following season of 2012–2013, but we found that the prevalent subgroup of the 2013–2014 season is HRSV-B in South Korea (data not shown).
The G protein is the major antigen of HRSV, and it is known to be highly antigenic with high genetic diversity, which may be related to frequent reinfection with HRSV (Johnson et al., 1987, Sullender, 2000, Parveen et al., 2006). Variation and the positive selection for amino acid changes are focused in two hypervariable regions, and the C-terminal third of the G protein contains multiple epitopes (Melero et al., 1997). Immune escape by new variants may contribute to the diversity and prevalence of HRSV. Combinations of novel substitutions and flip-flop changes in amino acids are involved with various epitope transitions, which reflect the immune status of the human population. Our epitope prediction analysis suggested the presence of a new epitope in the GG818/12 strain, where a 1-nt deletion occurred after the 72-nt duplication. We also found that several positively selected sites, i.e., amino acids 237, 274, and 280, were located in common epitopes with a high probability, which also supported our hypothesis. Furthermore, it was known that protective immunity through neutralizing antibody which was important for host defense against HRSV is of short duration (Sande et al., 2013). Immunological evidence is also required to clarify this phenomenon, but periodic substitutions of the prevalent HRSV-A and -B subgroups, as well as variations in the G protein and short-lived immunity, might be accompanied by changes in the herd immune status of human populations with respect to specific genotypes (Collins and Graham, 2008).
We used Bayesian skyline plots to infer the relative genetic diversity based on 318 HRSV-A G genes collected between 1991 and 2013, which may reflect the general trend. Previous reports indicate that the rapid substitution rate of the G gene compared with other genes or the entire HRSV genome have contributed to its high evolutionary rate (Tan et al., 2013). Most recently, Balmaks et al. (2013) also described population dynamics of ON1genotype based on limited information of sequences and showed effective population size of the ON1 genotype was expanded slowly and even decreased before the beginning of the season 2012–2013. Whereas, our data which were encompassed HRSV-A G genes collected in 2013 strongly indicate that the relative genetic diversity patterns of the HRSV-A G genes were significantly correlated with the emergence and prevalence of the HRSV-A ON1 genotype, which was rapidly emerged and maintained as a predominant strain.
In summary, our large-scale analysis of the HRSV-A G gene indicates that the emergence of the HRSV-A ON1 genotype in South Korea was correlated with an increase in genetic diversity. It remains to be clarified whether changes in the antigenicity of the G protein and/or substantial changes in the herd immune status have contributed to the rapid dominance of HRSV-A ON1. Furthermore it will also be crucial to understand determining factor(s) of viral phenotype, fitness of the HRSV-ON1 and its variant such as HRSV-ON1_OS subgenotype or transmission/complete replacement like BA genotype of HRSV-B. Although we conferred emergence of the strain into South Korea might be mediated by overseas transmission through limited genetic analysis (data not shown), cumulative evolutionary information with time scale is still required to verify global spread of the ON1 genotype precisely, Therefore, meticulous and continuous monitoring of the evolutionary trends in the G gene is essential to obtain insights that may facilitate vaccine development and amendment of public health responses against HRSV infection.
5. Conclusion
We investigated the emergence of the new HRSV-A ON1 genotype having 72-nt duplication in G gene and rapid replacement during 3 years. Time scaled evolutionary study support the drastic increase of genetic diversity resulting to the prevalence of the new genotype which was subdivided around 2009. In addition, we predicted the epitope of the duplicated G protein with an insertion of 23 amino acids and the results suggest the antigenic variation of the HRSV G protein.
Acknowledgement
This study was supported by the intramural research fund (2013-NG47001-00) of the Korea National Institute of Health.
References
- Agoti C.N., Mbisa J.L., Bett A., Medley G.F., Nokes D.J., Cane P.A. Intrapatient variation of the respiratory syncytial virus attachment protein gene. J. Virol. 2010;84(19):10425–10428. doi: 10.1128/JVI.01181-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auksornkitti V., Kamprasert N., Thongkomplew S., Suwannakarn K., Theamboonlers A., Samransamruajkij R., Poovorawan Y. Molecular characterization of human respiratory syncytial virus, 2010-2011: identification of genotype ON1 and a new subgroup B genotype in Thailand. Arch. Virol. 2013 doi: 10.1007/s00705-013-1773-9. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Baek Y.H., Choi E.H., Song M.S., Pascua P.N., Kwon H.I., Park S.J., Lee J.H., Woo S.I., Ahn B.H., Han H.S., Hahn Y.S., Shin K.S., Jang H.L., Kim S.Y., Choi Y.K. Prevalence and genetic characterization of respiratory syncytial virus (RSV) in hospitalized children in Korea. Arch. Virol. 2012;157(6):1039–1050. doi: 10.1007/s00705-012-1267-1. [DOI] [PubMed] [Google Scholar]
- Balmaks R., Ribakova I., Gardovska D., Kazaks A. Molecular epidemiology of human respiratory syncytial virus over three consecutive seasons in Latvia. J. Med. Virol. 2013 doi: 10.1002/jmv.23855. [DOI] [PubMed] [Google Scholar]
- Bezerra P.G., Britto M.C., Correia J.B., Duarte M.C., Fonceca A.M., Rose K., Hopkins M.J., Cuevas L.E., McNamara P.S. Viral and atypical bacterial detection in acute respiratory infection in children under five years. PLoS One. 2011;6(4):e18928. doi: 10.1371/journal.pone.0018928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botosso V.F., Zanotto P.M.A., Ueda M., Arruda E., Gilig A.E., Vieira S.E., Stewien K.E., Peret T.C., Jamal L.F., Pardini M.I., Pinho J.R., Massad E., Sant’anna O.A., Holmes E.C., Durigon E.L., VGDN Consortium Positive selection results in frequent reversible amino acid replacements in the G protein gene of human respiratory syncytial virus. PLoS Pathog. 2009;5(1):e1000254. doi: 10.1371/journal.ppat.1000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. Current progress on development of respiratory syncytial virus vaccine. BMB Rep. 2011;44(4):232–237. doi: 10.5483/BMBRep.2011.44.4.232. [DOI] [PubMed] [Google Scholar]
- Cho H.J., Shim S.Y., Son D.W., Sun Y.H., Tchah H., Jeon I.S. Respiratory viruses in neonates hospitalized with acute lower respiratory tract infections. Pediatr. Int. 2013;55:49–53. doi: 10.1111/j.1442-200X.2012.03727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E.H., Lee H.J. Genetic diversity and molecular epidemiology of the G protein of subgroup A and B of respiratory syncytial viruses isolated over 9 consecutive epidemics in Korea. J. Infect. Dis. 2000;181(5):1547–1556. doi: 10.1086/315468. [DOI] [PubMed] [Google Scholar]
- Collins P.L., Graham B.S. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 2008;82(5):2040–2055. doi: 10.1128/JVI.01625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G., Zhu R., Qian Y., Deng J., Zhao L., Sun Y., Wang F. Genetic variation in attachment glycoprotein genes of human respiratory syncytial virus subgroups A and B in children in recent five consecutive years. PLoS One. 2013;8(9):e75020. doi: 10.1371/journal.pone.0075020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva L.H., Spilki F.R., Riccetto A.G., de Almeida R.S., Baracat E.C., Arns C.W. Genetic variability in the G protein gene of human respiratory syncytial virus isolated from the Campinas metropolitan region. Braz. J. Med. Virol. 2008;80(9):1653–1660. doi: 10.1002/jmv.21249. [DOI] [PubMed] [Google Scholar]
- Drummond A.J., Suchard M.A., Xie D., Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Manzalawy Y., Dobbs D., Honavar V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. 2008;21(4):243–255. doi: 10.1002/jmr.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshaghi A., Duvvuri V.R., Lai R., Nadarajah J.T., Li A., Patel S.N., Low D.E., Gubbay J.B. Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One. 2012;7(3):e32807. doi: 10.1371/journal.pone.0032807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham B.S. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 2011;239(1):149–166. doi: 10.1111/j.1600-065X.2010.00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C.B., Weinberg G.A., Blumkin A.K., Edwards K.M., Staat M.A., Schultz A.F., Poehling K.A., Szilagyi P.G., Griffin M.R., Williams J.V., Zhu Y., Grijalva C.G., Prill M.M., Iwane M.K. Respiratory syncytial virus-associated hospitalizations among children less than 24 months of age. Pediatrics. 2013;132(2):341–348. doi: 10.1542/peds.2013-0303. [DOI] [PubMed] [Google Scholar]
- Huang Y., Niu B., Gao Y., Fu L., Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26(5):680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P.R., Spriggs M.K., Olmsted R.A., Collins P.L. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc. Natl. Acad. Sci. U.S.A. 1987;84:5625–5629. doi: 10.1073/pnas.84.16.5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorquera P.A., Oakley K.E., Tripp R.A. Advances in and the potential of vaccines for respiratory syncytial virus. Expert Rev. Respir. Med. 2013;7(4):411–427. doi: 10.1586/17476348.2013.814409. [DOI] [PubMed] [Google Scholar]
- Khor C.S., Sam I.C., Hooi P.S., Chan Y.F. Displacement of predominant respiratory syncytial virus genotypes in Malaysia between 1989 and 2011. Infect. Genet. Evol. 2013;14:357–360. doi: 10.1016/j.meegid.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Kushibuchi I., Kobayashi M., Kusaka T., Tsukagoshi H., Ryo A., Yoshida A., Ishii H., Saraya T., Kurai D., Yamamoto N., Kanou K., Saitoh M., Noda M., Kuroda M., Morita Y., Kozawa K., Oishi K., Tashiro M., Kimura H. Molecular evolution of attachment glycoprotein (G) gene in human respiratory syncytial virus detected in Japan 2008–2011. Infect. Genet. Evol. 2013;18:168–173. doi: 10.1016/j.meegid.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Larsen J.E.P., Lund O., Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.J., Kim Y.J., Kim D.W., Lee H.S., Lee H.Y., Kim K. Complete genome sequence of human respiratory syncytial virus genotype A with a 72-nucleotide duplication in the attachment protein G gene. J. Virol. 2012;86(24):13810–13811. doi: 10.1128/JVI.02571-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero J.A., Garcia-Barreno B., Martinez I., Pringle C.R., Cane P.A. Antigenic structure, evolution and immunobiology of human respiratory syncytial virus attachment (G) protein. J. Gen. Virol. 1997;78:2411–2418. doi: 10.1099/0022-1317-78-10-2411. [DOI] [PubMed] [Google Scholar]
- Munywoki P.K., Ohuma E.O., Ngama M., Bauni E., Scott J.A., Nokes D.J. Severe lower respiratory tract infection in early infancy and pneumonia hospitalizations among children. Kenya Emerg. Infect. Dis. 2013;19(2):223–239. doi: 10.3201/eid1902.120940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y., Catherman S.C. Antibody response to the central unglycosylated region of the respiratory syncytial virus attachment protein in mice. Vaccine. 2012;30(36):5382–5388. doi: 10.1016/j.vaccine.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair H., Nokes D.J., Gessner B.D., Dherani M., Madhi S.A., Singleton R.J., O’Brien K.L., Roca A., Wright P.F., Bruce N., Chandran A., Theodoratou E., Sutanto A., Sedyaningsih E.R., Ngama M., Munywoki P.K., Kartasasmita C., Simões E.A., Rudan I., Weber M.W., Campbell H. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010;375(9725):1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parveen S., Broor S., Kapoor S.K., Fowler K., Sullender W.M. Genetic diversity among respiratory syncytial viruses that have caused repeated infections in children from rural India. J. Med. Virol. 2006;78:659–665. doi: 10.1002/jmv.20590. [DOI] [PubMed] [Google Scholar]
- Posada D. JModelTest: phylogenetic model averaging. Mol. Biol. Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Prifert C., Streng A., Krempl C.D., Liese J., Weissbrich B. Novel respiratory syncytial virus A Genotype, Germany, 2011–2012. Emerg. Infect. Dis. 2013;19(6):1029–1030. doi: 10.3201/eid1906.121582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., Longden I., Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rubinstein N.D., Mayrose I., Martz E., Pupko T. Epitopia: a web-server for predicting B-cell epitopes. BMC Bioinformatics. 2009;10:287. doi: 10.1186/1471-2105-10-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudraraju R., Jones B.G., Sealy R., Surman S.L., Hurwitz J.L. Respiratory syncytial virus: current progress in vaccine development. Viruses. 2013;5(2):577–594. doi: 10.3390/v5020577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sande C.J., Mutunga M.N., Okiro E.A., Medley G.F., Cane P.A., Nokes D.J. Kinetics of the neutralizing antibody response to respiratory syncytial virus infections in a birth cohort. J. Med. Virol. 2013;85:2020–2025. doi: 10.1002/jmv.23696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw C.A., Ciarlet M., Cooper B.W., Dionigi L., Keith P., O’Brien K.B., Rafie-Kolpin M., Dormitzer P.R. The path to an RSV vaccine. Curr. Opin. Virol. 2013;3(3):332–342. doi: 10.1016/j.coviro.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Shobugawa Y., Saito R., Sano Y., Zaraket H., Suzuki Y., Kumaki A., Dapat I., Oguma T., Yamaguchi M., Suzuki H. Emerging genotypes of human respiratory syncytial virus subgroup A among patients in Japan. J. Clin. Microbiol. 2009;47(8):2475–2482. doi: 10.1128/JCM.00115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H., Ansari H.R., Raghava G.P.S. Improved method for linear B-cell epitope prediction using antigen’s primary sequence. PLoS One. 2013;8(5):e62216. doi: 10.1371/journal.pone.0062216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullender W.M. Respiratory syncytial virus genetic and antigenic diversity. Clin. Microbiol. Rev. 2000;13(1):1–15. doi: 10.1128/cmr.13.1.1-15.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullender W.M., Mufson M.A., Anderson L.J., Wertz G.W. Genetic diversity of the attachment protein of subgroup B respiratory syncytial viruses. J. Virol. 1991;65(10):5425–5434. doi: 10.1128/jvi.65.10.5425-5434.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Pterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L., Coenjaerts F.E., Houspie L., Viveen M.C., van Bleek G.M., Wiertz E.J., Martin D.P., Lemey P. The comparative genomics of human respiratory syncytial virus subgroups A and B: genetic variability and molecular evolutionary dynamics. J. Virol. 2013;87(14):8213–8226. doi: 10.1128/JVI.03278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trento A., Casas I., Calderon A., Garcia-Garcia M.L., Calvo C., Perez-Brena P., Melero J.A. Ten years of global evolution of the human respiratory syncytial virus BA genotype with a 60-nucleotide duplication in the G protein gene. J. Virol. 2010;84(15):7500–7512. doi: 10.1128/JVI.00345-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trento A., Viegas M., Galiano M., Videla C., Carballal G., Mistchenko A.S., Melero J.A. Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J. Virol. 2006;80(2):975–984. doi: 10.1128/JVI.80.2.975-984.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukagoshi H., Yokoi H., Kobayashi M., Kushibuchi I., Okamoto-Nakagawa R., Yoshida A., Morita Y., Noda M., Yamamoto N., Sugai K., Oishi K., Kozawa K., Kuroda M., Shirabe K., Kimura H. Genetic analysis of attachment glycoprotein (G) gene in new genotype ON1 of human respiratory syncytial virus detected in Japan. Microbiol. Immunol. 2013;57:655–659. doi: 10.1111/1348-0421.12075. [DOI] [PubMed] [Google Scholar]
- van Niekert S., Venter M. Replacement of previously circulating respiratory syncytial virus subtype B strains with the BA genotype in South Africa. J. Virol. 2011;85(17):8789–8797. doi: 10.1128/JVI.02623-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Bayliss S., Meads C. Palivizumab for immunoprophylaxis of respiratory syncytial virus (RSV) bronchiolitis in high-risk infants and young children: a systematic review and additional economic modelling of subgroup analyses. Health Technol. Assess. 2011;15(5):1–124. doi: 10.3310/hta15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.W., Lin Y.C., Pai T.W., Chang H.T. Prediction of B-cell linear epitopes with a combination of support vector machine classification and amino acid propensity identification. J. Biomed. Biotechnol. 2011:432830. doi: 10.1155/2011/432830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (WHO). 2009. Initiative for Vaccine Research (IVR). Respiratory syncytial virus and parainfluenza virus. Disease burden. Geneva: The Organization.
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zeitlin L., Bohorov O., Bohorova N., Hiatt A., Kim D.H., Pauly M.H., Velasco J., Whaley K.J., Barnard D.L., Bates J.T., Crowe J.E., Jr., Piedra P.A., Gilbert B.E. Prophylactic and therapeutic testing of nicotiana-derived RSV-neutralizing human monoclonal antibodies in the cotton rat model. MAbs. 2013;5(2):263–269. doi: 10.4161/mabs.23281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.Y., Du L.N., Chen X., Zhao Y., Liu E.M., Yang X.Q., Zhao X.D. Genetic variability of respiratory syncytial viruses (RSV) prevalent in southwestern China from 2006 to 2009: Emergence of subgroup B and A RSV as dominant strains. J. Clin. Virol. 2010;48(4):1201–1207. doi: 10.1128/JCM.02258-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Gao S., Lercher M.J., Hu S., Chen W.H. EvolView, an online tool for visualizing, annotating and managing phylogenetic trees. Nucleic Acids Res. 2012;40:W569–572. doi: 10.1093/nar/gks576. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]